
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contorting the hetero phosphaquinoid: synthesis and electronic insights into a non-planar, ferrocenyl phosphaquinoid†
Rajesh
Deka
 *a,
Samir
Chattopadhyay
b and
Andreas
Orthaber
*a
*a,
Samir
Chattopadhyay
b and
Andreas
Orthaber
*a
aSynthetic Molecular Chemistry, Department of Chemistry Ångström Laboratory, Uppsala University, BOX 523, 75120 Uppsala, Sweden. E-mail: andreas.orthaber@kemi.uu.se; rajeshdeka03@gmail.com
bPhysical Chemistry, Department of Chemistry Ångström Laboratory. Uppsala University, BOX 523, 75120 Uppsala, Sweden
First published on 22nd January 2025
Abstract
We report a highly contorted phosphaquinoid by substituting one of the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of an anthraquinodimethane unit with a phosphaalkene unit (–CP-Mes*, Mes* = 2,4,6-tri-tbutylbenzene) and end-capping the opposite terminus with ‘C(Fc)Ph’. Both isomers (E,Z) exhibit butterfly-like distortion of the anthracene core and demonstrate remarkable stability towards air and moisture.
C bonds of an anthraquinodimethane unit with a phosphaalkene unit (–CP-Mes*, Mes* = 2,4,6-tri-tbutylbenzene) and end-capping the opposite terminus with ‘C(Fc)Ph’. Both isomers (E,Z) exhibit butterfly-like distortion of the anthracene core and demonstrate remarkable stability towards air and moisture.
Quinoidal molecules, composed of planar and non-planar frameworks, have long fascinated researchers with their interesting electronic properties and reactivities, proving invaluable across diverse fields such as organic/inorganic chemistry, materials science, and biology.1–10 A continuous endeavour in this domain involves the functionalization of one or both termini of the quinoidal system with appropriate functional groups to acquire distinct properties and reactivities. Concurrently, substituting one or both the exocyclic C
C double bonds with isolobal main group fragment(s) afforded a wide range of main group-based quinoidal molecules.11–17 These compounds have unveiled a plethora of unique bonding scenarios and chemical reactivities that are unattainable with their hydrocarbon counterparts. Despite these advancements, the inherent high reactivity of main group-based quinoidal molecules poses significant challenges, hindering the pace of research in this field. While low-coordinate phosphorus compounds have been extensively studied, research on phosphaquinoids remains sparse, with only a handful of compounds that have been reported to date. This scarcity is largely attributable to the absence of straightforward synthetic protocols and suitable structural frameworks capable of enhancing stability.
Märkl and coworkers reported the first example of planar 1,4-diphosphabenzoquinone, which– despite the presence of sterically demanding Mes* (2,4,6-tri-tbutylbenzene) substituents – exhibited high reactivity, necessitating additional stabilization in the form of a bis(pentacarbonylchromium) complex, A (Fig. 1).18 Subsequently, Yoshifuji and coworkers reported an isolable hybrid phosphabenzoquinone B. The presence of the tert-butyl substituent alongside the electronegative oxygen atom in B was found to be critical for its stabilization, a stabilization strategy often observed in organic quinoidal motifs.19 Further contributions from Yoshifuji's group included planar diphosphaquinones, namely diphosphathienoquinone (C–D) and the mixed phosphaquinone phosphathieno-quinomethane (E), achieved by replacing the phenylene core with a heteroaromatic thiophene bridge.20,21
 | ||
| Fig. 1 Early examples of (a) planar, and (b) non-planar phosphaquinoids, illustrating the use of diverse structural cores, each highlighted with different colours. (c) This work: ferrocene-embedded anthraquinodimethane based phosphaquinoid. | ||
In our recent work, by utilizing an anthracene-based secondary phosphine synthon, we report a facile synthetic procedure for anthraquinodimethane (AQD)-based phosphaquinoids within the overcrowded ethylene (OCE) family.22 This approach has allowed us to explore the previously uncharted territory of non-planar phosphaquinoids. The anthracene core provides not only kinetic stability through steric protection—surpassing the stabilization typically offered by a phenylene core—but also significant thermodynamic stability via two benzenoid sextets. Importantly, by incorporating a (thio)xanthyl unit at one terminus of the phosphaquinoidal moiety (F–G), we demonstrated that upon oxidation, the molecule undergoes conformational change from a folded to a twisted structure, thus exhibiting a dynamic redox behaviour. It is worth noting that despite the widespread applications of ferrocene,23 and its prominent role in low coordinate and low valent phosphorous compounds,24–27 ferrocene units that are embedded into quinoidal molecules are rare. For instance, Diederich and coworkers functionalized p-quinodimethane with electron-withdrawing cyano group at one terminus and a donor ferrocene group at the other, creating a push–pull chromophore with prominent charge transfer characteristics.28 Building on our previous findings, and expanding the scope of the synthetic protocol, we now report the synthesis of a highly contorted ferrocene-embedded AQD-based phosphaquinoidal molecule 2, and provide a detailed analysis of its electronic structure. This newly synthesized compound represents the first example of a non-planar, main group-based quinoidal molecule embedding a ferrocenyl unit.
Synthesis of anthracene based secondary phosphine precursor 1 was previously reported through the reaction of 10-bromo-9-anthryllithium with Mes*PCl2 followed by LiAlH4 reduction.22 For the present study, the lithium-halogen exchange of 1, followed by nucleophilic addition to benzoylferrocene, resulted in the formation of a ferrocenyl-containing alcoholate intermediate (Scheme 1). Without isolation, addition of 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) and chlorotrimethylsilane (TMSCl) triggered the elimination of TMS-O-TMS and desired phosphaalkene 2 was obtained. Purification of the crude product by column chromatography using pentane as the eluent produced a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis and trans isomers of 2, with an overall yield of 87%. Notably, compound 2 demonstrated remarkable stability under ambient conditions, showing no signs of decomposition in either solid or solution state even after several months. The 31P NMR spectrum of compound 2 displayed two sharp resonances at 233.5 and 233.3 ppm in a 1:1 ratio, attributable to the cis and trans isomers. These chemical shift values are indicative of a normally polarized phosphaalkene (Pδ+Cδ−) and are consistent with those previously reported for phosphaquinoidal molecules.22,29,30 The 1H NMR spectra of the isomeric mixture are complex due to magnetically inequivalent signals of the Mes* and ferrocenyl units, arising from their partially hindered rotation. However, some diagnostic peaks clearly support the formation of a cis/trans isomeric mixture, i.e. two signals integrating to five protons (3.62 and 3.68 ppm, 5H each) and two deshielded aromatic signals (doublets at 5.82 and 5.93 ppm, 1H each) corresponding to the unsubstituted cyclopentadienyl (cp) ring and the 1-anthryl-hydrogen, respectively. A detailed analysis of the data is given in the ESI (Fig. S4–S6†).
:1 mixture of cis and trans isomers of 2, with an overall yield of 87%. Notably, compound 2 demonstrated remarkable stability under ambient conditions, showing no signs of decomposition in either solid or solution state even after several months. The 31P NMR spectrum of compound 2 displayed two sharp resonances at 233.5 and 233.3 ppm in a 1:1 ratio, attributable to the cis and trans isomers. These chemical shift values are indicative of a normally polarized phosphaalkene (Pδ+Cδ−) and are consistent with those previously reported for phosphaquinoidal molecules.22,29,30 The 1H NMR spectra of the isomeric mixture are complex due to magnetically inequivalent signals of the Mes* and ferrocenyl units, arising from their partially hindered rotation. However, some diagnostic peaks clearly support the formation of a cis/trans isomeric mixture, i.e. two signals integrating to five protons (3.62 and 3.68 ppm, 5H each) and two deshielded aromatic signals (doublets at 5.82 and 5.93 ppm, 1H each) corresponding to the unsubstituted cyclopentadienyl (cp) ring and the 1-anthryl-hydrogen, respectively. A detailed analysis of the data is given in the ESI (Fig. S4–S6†).
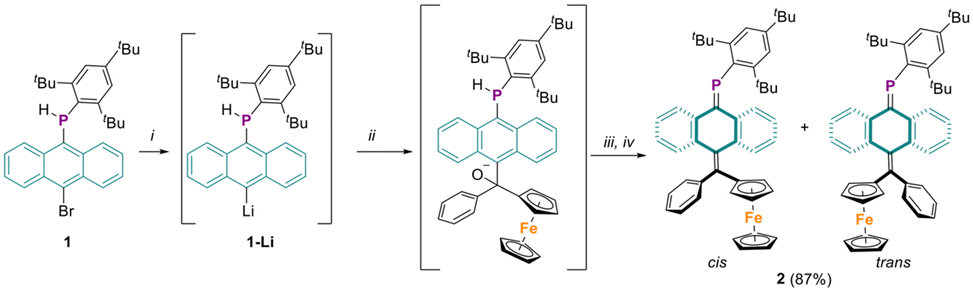 | ||
| Scheme 1 Synthesis of phosphaaquinoid 2. Conditions: (i) tBuLi, −78 °C; (ii) benzoylferrocene, −78 °C; (iii) DBN, r.t.; (iv) TMSCl, r.t. | ||
The molecular structures of both the cis (with Mes* and Fc units in cis orientation) and trans isomers of compound 2 were unambiguously elucidated through single-crystal X-ray diffraction analysis. Single crystals for both isomers, suitable for X-ray analysis, were obtained by the slow evaporation of benzene solutions at ambient temperature. The cis isomer crystallizes in a monoclinic space group, P21/n, forming orange-red block-shaped crystals, whereas crystals of the trans isomer are brownish red thin plates. Macroscopic separation of the two crystals was unfortunately impossible. The discussion here focuses on the cis isomer (denoted as 2cis), with detailed structural data for the trans isomer (2trans) provided in the ESI (Fig. S7†). Notably, the most distinguishing feature of the structures of compound 2cis is the highly contorted AQD core exhibiting a butterfly-like distortion (Fig. 2)—a novel characteristic for ferrocene-embedded quinoids. The central anthracene core folds to an angle of 136.71°, adopting a boat-like conformation with a puckering angle of 64.04(8)°. The PC bond distance is 1.701(2) Å, with a C–PC bond angle of 106.6(1)°, indicative of a regularly polarized phosphaalkene. The central CC bond linking the AQD core with the ferrocenyl unit measures 1.352(3) Å, consistent with similar AQD-based quinoidal molecules.4,6,22 The cyclopentadienyl (cp) rings exhibit a nearly staggered geometry, while the cp rings are essentially parallel, with a ring centroid–iron–ring centroid angle of 178.65°. The dihedral angle between the cp plane and the phenyl plane is 123.9°. The central CC bond deviates from the cp and phenyl rings with torsion angles of ∢C10–C11–C18–C22 = 132.1(3)° and ∢C10–C11–C12–C17 = −117.8(3)°, respectively.
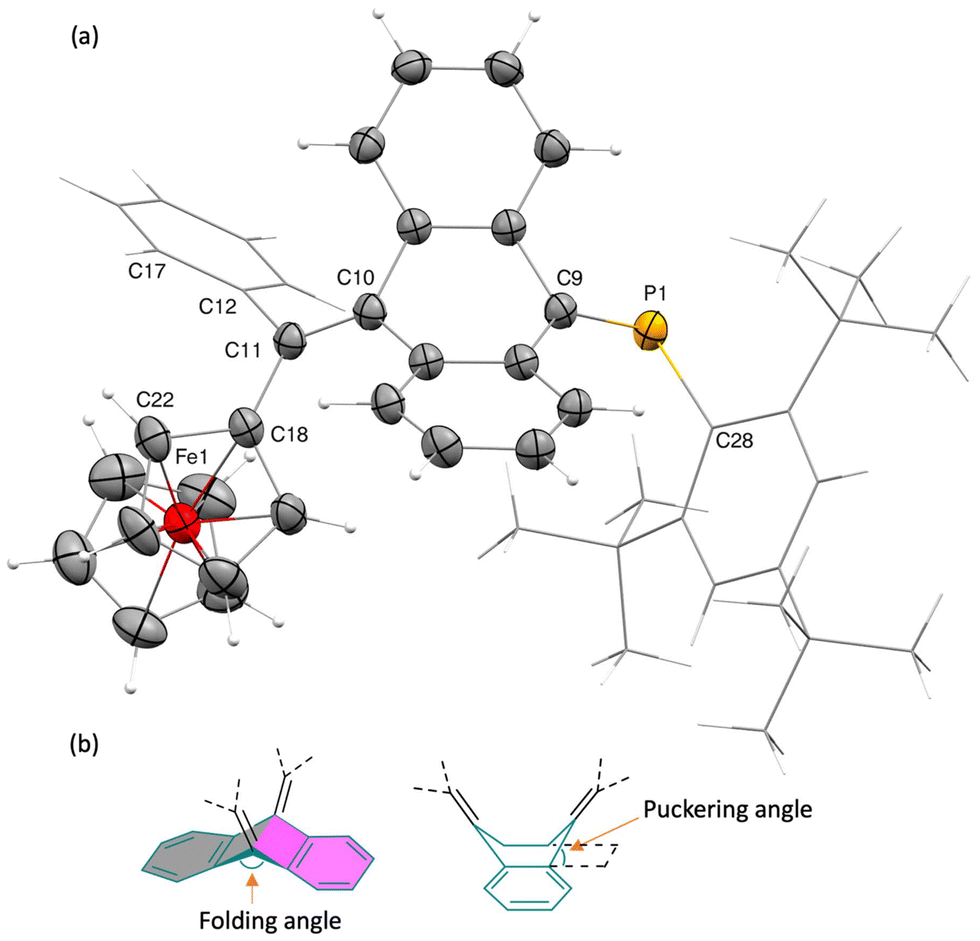 | ||
| Fig. 2 (a) Solid-state structure of 2cis. Thermal displacement ellipsoids are set at 50% probability levels, hydrogen atoms, benzene solvate are omitted, and Mes* and Ph units are shown as capped stick for clarity; (b) schematic representation defining the folding and puckering angles. | ||
In order to elucidate the impact of ferrocenyl substitution on the electronic structure of the molecular motif, we have performed a combined electrochemical and theoretical investigation. Cathodic scans revealed no reduction events within the solvent window, consistent with the distorted geometry of the phospha-anthraquinone framework. Anodic scans revealed a reversible oxidation at +0.051 V (vs. Fc/Fc+), attributed to ferrocene oxidation, followed by irreversible processes at +1.00 V and +1.36 V, with the former being characteristic of phosphaalkene-based oxidation (Fig. 3). Despite the presence of two isomers there is no distinction of the reduction potentials, highlighting a minimal stereoelectronic impact on the redox behavior. A scan rate-dependent analysis of the Fc/Fc+ redox event confirmed a diffusion-controlled electrochemical process (Fig. S11, ESI†). The reversible nature of the Fc/Fc+ redox event over the timescale of cyclic voltammetry, particularly at slower scan rates, prompted further investigation of its effect on the conformation of 2 through spectroelectrochemical techniques. The incorporation of the ferrocene moiety at one terminus of the quinoidal core induced a red shift in the absorption maximum (λmax) to 489 nm, as compared to the (thio)xanthene-capped AQD-based phosphaalkene.22 The involvement of ferrocene based electronic transitions was confirmed by TD-DFT calculations. Indeed, the cis and trans isomer, being almost isoenergetic (2cis: 0.0 kJ mol−1vs.2trans +0.3 kJ mol−1) in line with the 1:1 ratio of the 31P-NMR signals, allowing us to approximate the UV/vis spectra as simple unweighted average of both contributions (see figure and ESI† for further information). Controlled bulk electrolysis at 0.06 V (vs. Fc/Fc+) resulted in the emergence of a new band at 610 nm, attributed to the ferrocenium species, and a concurrent bleaching of the 489 nm band, associated with the pristine molecule. Additionally, a broad absorption feature centered around 805 nm emerged, suggesting additional electronic transitions in the oxidized state.
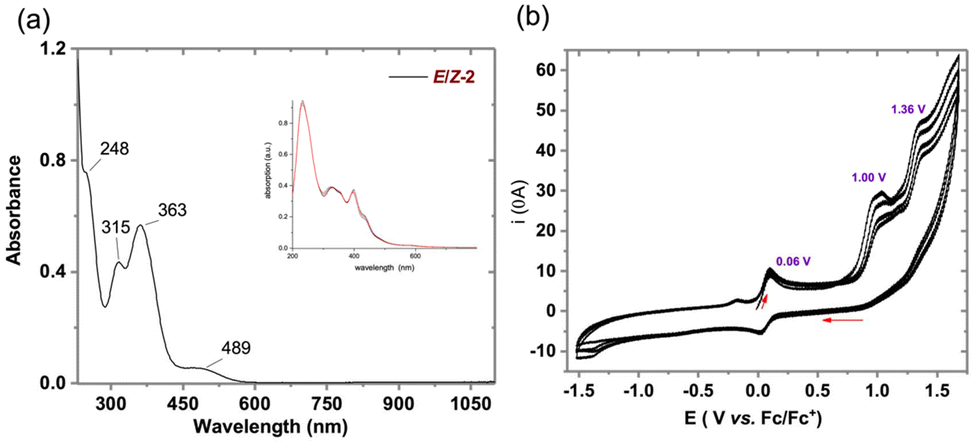 | ||
| Fig. 3 Opto-electronic characterization of compound 2. (a) Experimental electronic absorption spectra of compound 2; inset: calculated spectra (average of E and Z isomers, for details sees ESI†); (b) cyclic voltammograms of compound 2 (1 mM) in 5:1 acetonitrile-DCM. Full scans at 100 mV s−1 (further details see ESI†). | ||
To get deeper insight into the electronic properties of the oxidised species, TD-DFT calculations of cationic species were performed, considering both folded and twisted conformations of the central anthrylene core. The folded cationic species reproduces the absorption band at approximately 600 nm, whereas the twisted conformation accounts for the longer-wavelength absorption features in the 600–800 nm region, partially aligning with the experimental spectro-electrochemical data. However, additional absorption bands observed experimentally could not be attributed to either the folded or twisted cationic species, suggesting that oxidation at the ferrocene unit is accompanied by subsequent processes on the spectroelectrochemistry timescale. Despite extensive experimental efforts, and theoretical studies we were unable to elucidate the precise reactivity of the oxidized intermediate [2+]. This suggests that, despite the fully reversible one-electron oxidations observed by means of cyclic voltammetry (CV) and differential pulse voltammetry (DPV), the intricate nature of the electronic and conformational dynamics inherent to this system lead to a more complex electrochemistry at longer timescale. The computed energy difference of a twisted and folded cation (2+twistedvs.2+folded) suggests a slight preference for the twisted conformation (5.5 kJ mol−1), further supporting possible dynamic redox behavior of this system. Such complexity is consistent with literature reports on P-containing conjugated systems functionalized with ferrocenyl units, where oxidation often triggers subsequent reactivity at the P-center due to its oxidizable lone pair.31,32 The initial formation of folded and twisted cationic species is likely followed by subsequent chemical transformations, potentially involving, among other processes, insertion of a phosphenium center into the tBu groups of the Mes* substituent or more complex intermolecular interactions.33 Unfortunately, attempts to isolate or characterize the oxidized species through both electrochemical and chemical oxidation consistently resulted in intractable mixtures of products, further underscoring the complexity of this system.
Conclusions
In summary, we have reported a high yielding and straight forward synthetic strategy for AQD-based ferrocenyl phosphaquinoid by reacting anthracene based secondary phosphine Mes*AnPHBr with benzoyl ferrocene. X-ray crystallographic analysis of both cis and trans isomers revealed a pronounced butterfly-like distortion in the central AQD core, marking this molecule as rare example of non-planar, ferrocene-based quinoidal main-group compounds. Notably, the synthesized phosphaalkenes demonstrate significant stability under ambient conditions, in contrast to the high reactivity of most main-group quinoidal compounds. The 31P NMR resonance observed at ∼233 ppm supports the presence of a regularly polarized phosphaalkene unit in cis-/trans-2, which is further corroborated by the PC bond distance derived from crystallographic data. Electrochemical analysis demonstrated a reversible ferrocene-centered oxidation process within the cyclic voltammetry timescale. However, the resulting cationic species undergoes subsequent chemical transformations on the spectroelectrochemistry timescale or upon chemical oxidation.
The introduction of ferrocene, a well-known redox-active unit, into the quinoidal framework opens up new possibilities for tuning the electronic properties of quinoidal molecules through redox manipulation. Further research is underway to investigate the reactivity of this molecule in the context of coordination chemistry and small molecule activation, which could provide further insight into its potential applications in catalysis and material science.
Data availability
Crystallographic data for compound 2cis and 2trans have been deposited with the CCDC under deposition number 2412505 (for 2cis), and 2412506 (for 2trans).†Experimental details, spectral data and Cartesian coordinates for the DFT-optimised geometries are available in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Swedish Research council for their continuous support (A. O. project 202017-03727, 21-03658). The Wenner Gren Foundation and Olle Engkvists Stiftelse are acknowledged for their financial support. The computations were enabled by resources in project NAISuybmissioS 2023/5-355 provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at UPPMAX, funded by the Swedish Research Council through grant agreement no. 2022-06725.References
- M. B. S. Wonink, B. P. Corbet, A. A. Kulago, G. B. Boursalian, B. de Bruin, E. Otten, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 18020–18028 CrossRef CAS PubMed.
- C.-H. Liu, Z. He, C. Ruchlin, Y. Che, K. Somers and D. F. Perepichka, J. Am. Chem. Soc., 2023, 145, 15702–15707 CrossRef CAS PubMed.
- Y. Ishigaki, Y. Hayashi and T. Suzuki, J. Am. Chem. Soc., 2019, 141, 18293–18300 CrossRef CAS PubMed.
- T. Nishiuchi, S. Aibara, H. Sato and T. Kubo, J. Am. Chem. Soc., 2022, 144, 7479–7488 CrossRef CAS PubMed.
- Y. Ishigaki, T. Harimoto, T. Shimajiri and T. Suzuki, Chem. Rev., 2023, 123, 13952–13965 CrossRef CAS PubMed.
- X. Yin, J. Z. Low, K. J. Fallon, D. W. Paley and L. M. Campos, Chem. Sci., 2019, 10, 10733–10739 RSC.
- D. Rottschäfer, N. K. T. Ho, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef PubMed.
- A. Maiti, S. Chandra, B. Sarkar and A. Jana, Chem. Sci., 2020, 11, 11827–11833 RSC.
- G. E. Rudebusch, A. G. Fix, H. A. Henthorn, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 3627–3633 RSC.
- H. Hwang, D. Khim, J.-M. Yun, E. Jung, S.-Y. Jang, Y. H. Jang, Y.-Y. Noh and D.-Y. Kim, Adv. Funct. Mater., 2015, 25, 1146–1156 CrossRef CAS.
- A. Das, B. J. Elvers, N. Chrysochos, S. I. Uddin, T. Gangber, I. Krummenacher, D. Borah, A. Mishra, M. Shanmugam, C. B. Yildiz, H. Braunschweig, C. Schulzke and A. Jana, J. Am. Chem. Soc., 2024, 146, 9004–9011 CrossRef CAS PubMed.
- T. Nozawa, M. Nagata, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 5773–5775 CrossRef CAS PubMed.
- Y. K. Loh, P. Vasko, C. McManus, A. Heilmann, W. K. Myers and S. Aldridge, Nat. Commun., 2021, 12, 7052 CrossRef CAS PubMed.
- Y. Su, X. Wang, Y. Li, Y. Song, Y. Sui and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 1634–1637 CrossRef CAS PubMed.
- A. Maiti, J. Stubbe, N. I. Neuman, P. Kalita, P. Duari, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Angew. Chem., Int. Ed., 2020, 59, 6729–6734 CrossRef CAS PubMed.
- G. Kundu, S. De, S. Tothadi, A. Das, D. Koley and S. S. Sen, Chem. – Eur. J., 2019, 25, 16533–16537 CrossRef CAS PubMed.
- D. Rottschäfer, B. Neumann, H.-G. Stammler, R. Kishi, M. Nakano and R. S. Ghadwal, Chem. – Eur. J., 2019, 25, 3244–3247 CrossRef PubMed.
- G. Märkl, R. Hennig and K. M. Raab, Chem. Commun., 1996, 17, 2057–2058 RSC.
- S. Sasaki, F. Murakami and M. Yoshifuji, Angew. Chem., Int. Ed., 1999, 38, 340–343 CrossRef CAS PubMed.
- F. Murakami, S. Sasaki and M. Yoshifuji, Angew. Chem., Int. Ed., 2002, 41, 2574–2576 CrossRef CAS PubMed.
- F. Murakami, S. Sasaki and M. Yoshifuji, J. Am. Chem. Soc., 2005, 127, 8926–8927 CrossRef CAS PubMed.
- R. Deka, M. A. Ansari, S. Chattopadhyay, R. Lomoth, A. Thapper and A. Orthaber, Angew. Chem., Int. Ed., 2024, e202406076, DOI:10.1002/anie.202406076.
- P. Štěpnička, Dalton Trans., 2022, 51, 8085–8102 RSC.
- R. Pietschnig and E. Niecke, Organometallics, 1996, 15, 891–893 CrossRef CAS.
- R. Pietschnig, E. Niecke, M. Nieger and K. Airola, J. Organomet. Chem., 1997, 529, 127–133 CrossRef CAS.
- D. Miesel, M. Korb, A. Hildebrandt and H. Lang, Inorg. Chim. Acta, 2018, 471, 741–745 CrossRef CAS.
- D. Miesel, A. Hildebrandt, M. Korb, P. J. Low and H. Lang, Organometallics, 2013, 32, 2993–3002 CrossRef CAS.
- S.-i. Kato, M. Kivala, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht and F. Diederich, Chem. – Eur. J., 2009, 15, 8687–8691 CrossRef CAS PubMed.
- A. El Nahhas, M. A. Shameem, P. Chabera, J. Uhlig and A. Orthaber, Chem. – Eur. J., 2017, 23, 5673–5677 CrossRef CAS PubMed.
- Y. V. Svyaschenko, A. Orthaber and S. Ott, Chem. – Eur. J., 2016, 22, 4247–4255 CrossRef CAS PubMed.
- F. Barrière, R. U. Kirss and W. E. Geiger, Organometallics, 2005, 24, 48–52 CrossRef.
- A. J. Downard, N. J. Goodwin and W. Henderson, J. Organomet. Chem., 2003, 676, 62–72 CrossRef CAS.
- X. Pan, X. Wang, Z. Zhang and X. Wang, Dalton Trans., 2015, 44, 15099–15102 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2412505 and 2412506. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03542a |
| This journal is © The Royal Society of Chemistry 2025 |