DOI:
10.1039/D5DT00106D
(Paper)
Dalton Trans., 2025,
54, 4484-4492
Synthesis of 2,3-dihydroquinazolinimines and 1,3-diiminoisoindolines catalyzed by rare earth metal alkyl complexes through tandem addition/cyclization of nitriles with amines†
Received
14th January 2025
, Accepted 10th February 2025
First published on 11th February 2025
Abstract
Mononuclear rare earth metal alkyl complexes with an anilido-imino ligand were synthesized and characterized, and their catalytic ability for the formation of dihydroquinazolines and isoindolines from nitriles and amines was investigated. Reactions of rare earth-metal trialkyl complexes RE(CH2SiMe3)3(THF)2 with proligand HL (L = (o-(NHDipp)C6H4CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(CH2)2NC4H8, Dipp = 2,6-iPr2C6H3)) in toluene afforded tridentate anilido-imino rare earth metal dialkyl complexes LRE(CH2SiMe3)2 (RE = Gd (1a), Y (1b), Yb (1c), Lu (1d)) in good yields. Further studies showed that these rare earth metal catalysts could achieve tandem addition/cyclization between ortho imine-functionalized benzonitriles and aromatic amines to afford 2,3-dihydroquinazolinimines for the first time. In addition, the catalysts are well applied to the reaction of substituted phthalonitriles with primary aromatic and aliphatic amines, producing various 1,3-diiminoisoindolines in moderate to excellent yields. During the process of the mechanism study, two anilido yttrium complexes LY(NHPh)2(THF) (7b) and LY(NHDipp)2 (8b) were isolated by the reaction of 1b with aniline and 2,6-diisopropylaniline, respectively, as well as their catalytic performance.
N(CH2)2NC4H8, Dipp = 2,6-iPr2C6H3)) in toluene afforded tridentate anilido-imino rare earth metal dialkyl complexes LRE(CH2SiMe3)2 (RE = Gd (1a), Y (1b), Yb (1c), Lu (1d)) in good yields. Further studies showed that these rare earth metal catalysts could achieve tandem addition/cyclization between ortho imine-functionalized benzonitriles and aromatic amines to afford 2,3-dihydroquinazolinimines for the first time. In addition, the catalysts are well applied to the reaction of substituted phthalonitriles with primary aromatic and aliphatic amines, producing various 1,3-diiminoisoindolines in moderate to excellent yields. During the process of the mechanism study, two anilido yttrium complexes LY(NHPh)2(THF) (7b) and LY(NHDipp)2 (8b) were isolated by the reaction of 1b with aniline and 2,6-diisopropylaniline, respectively, as well as their catalytic performance.
Introduction
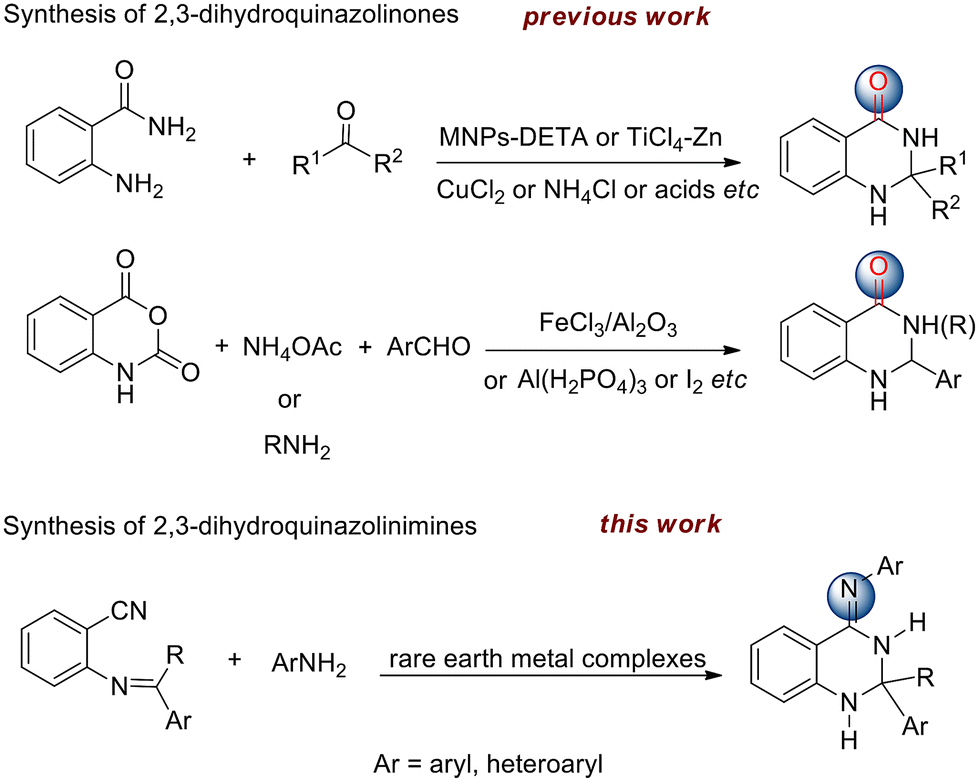
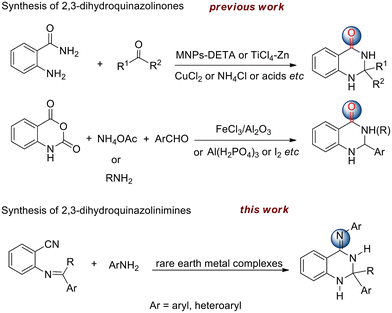
As one of the largest groups of organic compounds, heterocycles containing nitrogen have a considerable place in natural products, pharmaceuticals, and agrochemicals.1–3 Therefore, the development of convenient and straightforward synthetic approaches toward the construction of heterocycles is of great importance in organic chemistry. Recent research showed that quinazoline derivatives exhibited remarkable activity as antibacterial,4 antiviral,5 antitumor,6 and calcium channel antagonists.7 In contrast, the dihydroquinazoline scaffold features a saturated sp3 hybridized carbon at the 2 or 4 sites of the heterocyclic ring. In recent years, several synthetic protocols for the preparation of 2,3-dihydroquinazolinones have been reported. Among them, the most frequently used synthetic approaches are based on the reaction of 2-aminobenzamide with aldehydes or ketones. As a result, a variety of catalysts were employed to accomplish this transformation involving MNPs-DETA nanoparticles,8 CuCl2,9 TiCl4–Zn,10 ammonium chloride,11 acids12,13 and ionic liquids (Scheme 1).14 In addition, a more convenient strategy for the preparation of these compounds is a one-pot, three-component reaction of isatoic anhydride, aldehydes and ammonium acetate or primary amines in the presence of homo- or heterogeneous catalysts such as FeCl3/neutral Al2O3, [Al(H2PO4)3], and nanoparticles.15–21 However, in terms of 4-arylimino-functionalized dihydroquinazolines, namely 2,3-dihydroquinazolinimines, no report has described the synthesis and characterization to date (Scheme 1).
 |
| | Scheme 1 Synthesis of 2,3-dihydroquinazoline derivatives. | |
Isoindolines, being regarded as the hydrogenated analogues of isoindoles, are almost unknown in natural compounds but have important biological activities such as the commonly used nuevamine and lennoxamine alkaloids.22–24 In addition, bis(arylimino)isoindolines are regarded as an interesting class of pincer ligands due to their unique structural features and diverse coordination chemistry.25 A large number of complexes with varieties of transition metals derived from these ligands are known for their role as catalysts in organic transformations26–28 and as components of photoactive materials.29,30 With respect to the synthesis of bis(arylimino)isoindolines, the main methods of the direct reaction of 1,3-diiminoisoindoline with primary amines and the reaction of phthalonitrile with primary amines are involved. The condensation of 1,3-diiminoisoindoline with bulky amines or cyclic amines will lead to lower yields of products and often afford monosubstituted adducts. From 1997, several groups such as Siegl,31 Ziegler32 and Tang33 successively reported the reaction of phthalonitrile with aromatic amines or 2-pyridylamines to produce 1,3-diiminoisoindolines by using different catalysts. However, those catalysts suffer from one or more disadvantages such as lower yields, limited substrates, and difficult operation. Therefore, more efficient catalysts are expected. It is well known that rare earth metal complexes could be utilized to promote C–N bond formation through inter- and intramolecular hydroamination of various C–C unsaturated compounds such as alkenes, alkynes and allenes.34–38 Herein, we describe the first example of rare earth metal dialkyl complex-catalyzed tandem addition/cycloamination between ortho-imino-benzonitriles or substituted phthalonitriles and amines to provide a wide variety of 2,3-dihydroquinazolinimines and 1,3-diiminoisoindolines through the construction of two C–N bonds.
Results and discussion
Synthesis of anilido-imino rare earth metal alkyl complexes
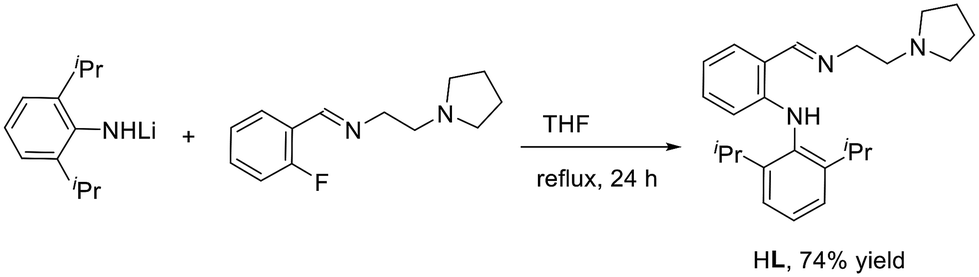
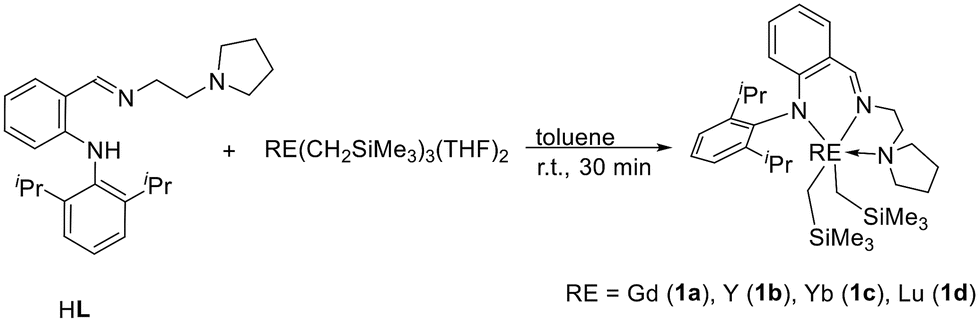
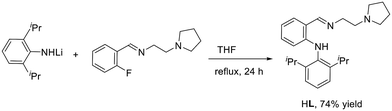
Treatment of lithium (2,6-diisopropylphenyl)amide with pyrrolidine-ligated fluorobenzene afforded an anilido-imino tridentate proligand HL (L = (o-(NHDipp)C6H4CHN(CH2)2NC4H8, Dipp = 2,6-iPr2C6H3) in 74% yield (Scheme 2). Subsequently, the reactions of rare earth metal trialkyl complexes RE(CH2SiMe3)3(THF)2 with an equimolar amount of HL in toluene at room temperature for 30 min afforded four distorted anilido-imino rare earth metal dialkyl complexes LRE(CH2SiMe3)2 (RE = Gd (1a), Y (1b), Yb (1c), Lu (1d)) (Scheme 3). Complexes 1a–1d have good solubility in toluene and THF.
 |
| | Scheme 2 Synthesis of the proligand. | |
 |
| | Scheme 3 Synthesis of tridentate anilido-imino rare earth metal dialkyl complexes. | |
The 1H NMR spectrum of 1b clearly shows the singlet of the CHN group at 7.80 ppm, the presence of the Y–CH2 species and the disappearance of the NH proton (at 10.54 ppm in HL). The diastereotopic methylene protons of the Y-alkyl groups are split into two doublets due to coupling to the 89Y metal with coupling constants of 2JH−H = 11.2 Hz and 2JY−H = 3.2 Hz (Fig. S4 and S6 in the ESI†). These values are consistent with the reported yttrium alkyl complexes,39,40 and similar results were observed in 1d (Fig. S8 in the ESI†).
These complexes were characterized by single-crystal X-ray crystallography and elemental analysis. The representative structural diagram of 1b is shown in Fig. 1, and those of other complexes are presented in the ESI.† Selected bond distances of 1a–1d are listed in Table 1. X-ray analysis revealed that the anilido-imino ligand is in a κ3N,N,N-coordinated fashion with the metal center, and the atoms N1, N2, N3, and C26 constitute a plane forming a distorted square pyramidal with one of the alkyl groups (labeled C30) taking the apical position (Fig. 1). The bond distance of Y1–N1 2.281(3) Å is obviously shorter than that of Y1–N2 2.390(5) Å in 1b. It is noteworthy that this difference of 0.1 Å for the Y–N bonds is greater than that of 0.02 Å in a tridentate β-diketiminato yttrium dialkyl complex (2.325(2) Å vs. 2.346(2) Å).39 Additionally, the bond distance of C13–N1 1.360(5) Å is clearly longer than that of C19–N2 1.298(7) Å in 1b, further demonstrating that the anilido/imino fragment exists in the rare earth metal alkyl complex due to phenyl ring embedding, resulting in electronic localization of the β-diketiminato unit, which is greatly different from the classical β-diketiminato analogues derived from acetylacetone with arylamines.41 Notably, the bond distance of Y1–N3 2.502(4) Å is significantly longer than that of Y1–N2 2.390(5) Å, suggesting that the steric effect has a great influence on the interaction between rare earth metal ions and adjacent atoms (Table 1). The same findings are also observed in gadolinium, ytterbium, and lutetium complexes (1a–1d). The RE–C bond distances ranging from 2.318(7) to 2.464(5) Å in 1a–1d are comparable to the values observed in reported five-coordinate rare earth alkyl complexes.42,43
 |
| | Fig. 1 Molecular structure of 1b. All the hydrogen atoms are omitted for clarity. | |
Table 1 Selected bond lengths (Å) of 1a–1d
| |
1a (Gd) |
1b (Y) |
1c (Yb) |
1d (Lu) |
| RE1–N1 |
2.311(3) |
2.281(3) |
2.256(5) |
2.245(5) |
| RE1–N2 |
2.443(5) |
2.390(5) |
2.331(8) |
2.337(7) |
| RE1–N3 |
2.552(4) |
2.502(4) |
2.463(7) |
2.462(6) |
| RE1–C26 |
2.464(5) |
2.430(4) |
2.326(7) |
2.380(6) |
| RE1–C30 |
2.410(5) |
2.377(4) |
2.372(7) |
2.318(7) |
| C13–N1 |
1.360(5) |
1.360(5) |
1.354(8) |
1.369(7) |
| C19–N2 |
1.279(7) |
1.298(7) |
1.278(12) |
1.283(10) |
| C13–C18 |
1.434(6) |
1.431(6) |
1.427(10) |
1.434(8) |
| C18–C19 |
1.437(12) |
1.406(7) |
1.437(12) |
1.421(11) |
Catalytic activity
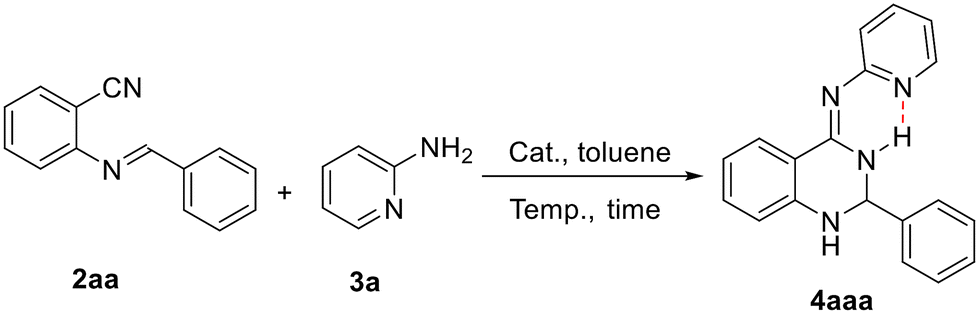
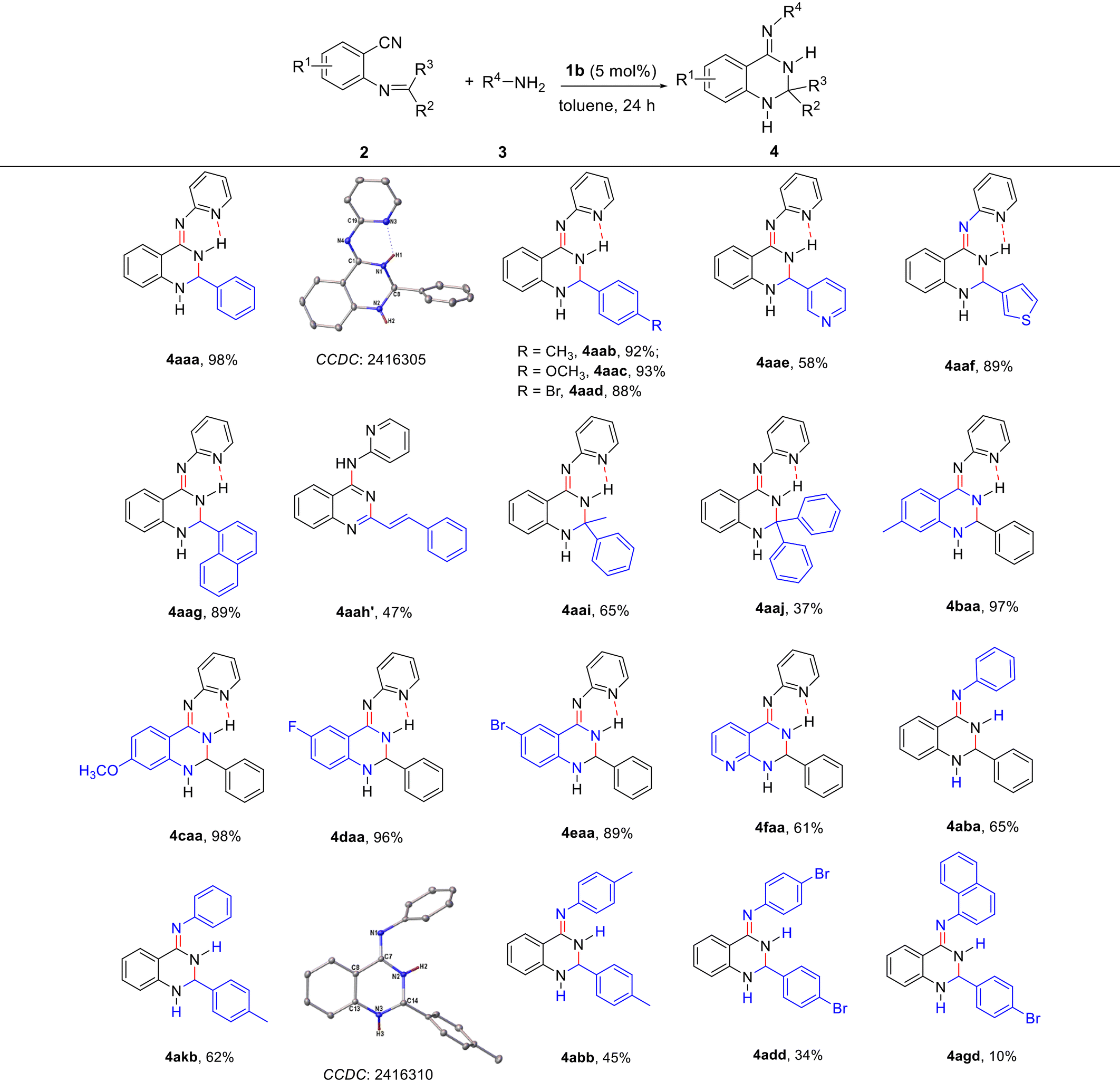
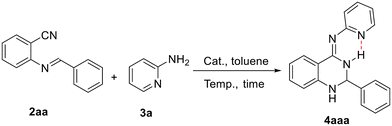
The catalytic activities of 1b were first evaluated by using 2-(benzylideneamino)benzonitrile and 2-aminopyridine as the model substrates for the preparation of 4-arylimino-substituted 2,3-dihydroquinazoline under different conditions. The representative results are shown in Table 2. The reaction of 2aa with 3a cannot take place in the absence of a catalyst even under heating condition; however, the rare earth metal complex 1b exhibits high activity in the catalytic formation of (2,3-dihydroquinazolin-4(1H)-ylidene)pyridin-2-amine 4aaa (CCDC 2416305†) (entries 1 and 2). Further tests showed that the yield of the catalytic product can be improved to 98% by adjusting reaction temperatures and substrate ratios (entries 3–7). It was noticed that no further conversion was observed even after prolonging the reaction time to 36 h (entry 8). The reaction proceeds well in the presence of 4 mol% 1b with 95% good yield (entry 9). Rare earth catalysts 1a–1d all showed high efficiencies where larger radius ions of Gd and Y performed better than those of Yb and Lu, probably due to a more favorable and open environment provided by larger metal ions (entries 10–12). The precursor Y(CH2SiMe3)3(THF)2 of 1b gave the catalytic product in 70% yield under the same conditions, suggesting the positive role of the N,N,N-tridentate anilido-imino ancillary ligand (entry 13).
Table 2 Optimization of the reaction conditionsa
|
|
| Entry |
Cat. (mol%) |
Temp. (°C) |
Amine (equiv.) |
Time (h) |
Yieldb (%) |
|
Reaction conditions: into a Schlenk glass reactor were successively placed 2a (1.0 mmol), 3a and the catalyst in toluene (1.5 mL).
Isolated yields were based on o-(benzylideneamino)benzonitrile.
Y[C]3 refers to the catalyst of Y(CH2SiMe3)3(THF)2.
|
| 1 |
— |
110 |
1 |
24 |
NR |
| 2 |
1b (5) |
110 |
1 |
24 |
85 |
| 3 |
1b (5) |
120 |
1 |
24 |
75 |
| 4 |
1b (5) |
100 |
1 |
24 |
80 |
| 5 |
1b (5) |
110 |
1.2 |
24 |
87 |
| 6 |
1b (5) |
110 |
1.5 |
24 |
90 |
| 7 |
1b (5) |
110 |
2 |
24 |
98 |
| 8 |
1b (5) |
110 |
2 |
36 |
98 |
| 9 |
1b (4) |
110 |
2 |
24 |
95 |
| 10 |
1a (5) |
110 |
2 |
24 |
97 |
| 11 |
1c (5) |
110 |
2 |
24 |
95 |
| 12 |
1d (5) |
110 |
2 |
24 |
92 |
| 13 |
Y[C]3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (5) c (5) |
110 |
2 |
24 |
70 |
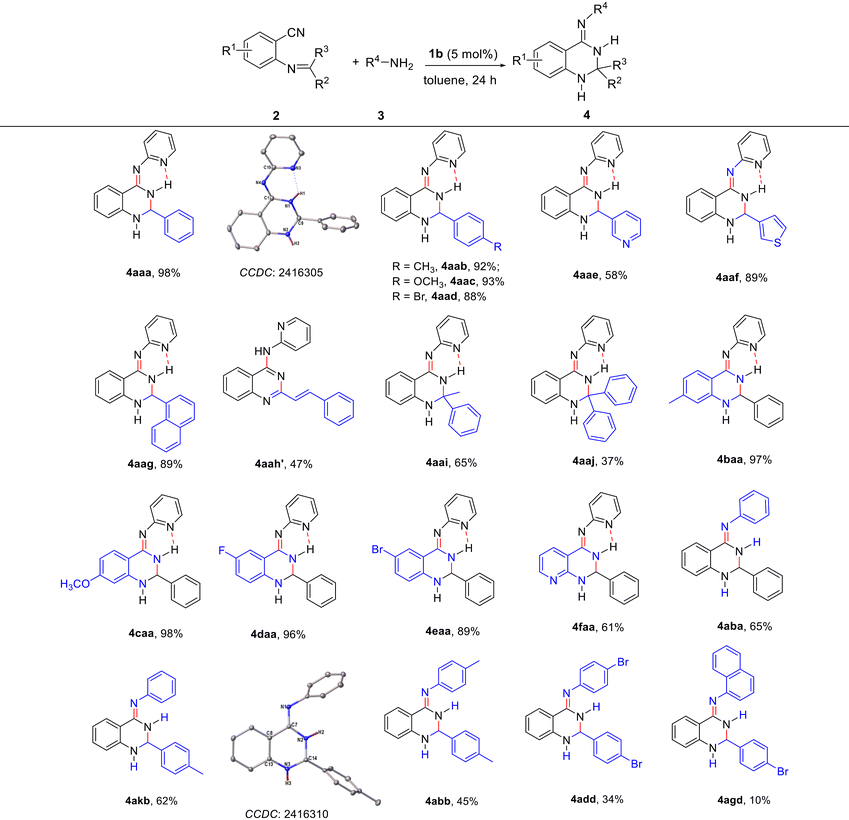
To evaluate the scope of the synthesis of 2,3-dihydroquinazolinimines, various 2-imine-functionalized benzonitriles and amines were examined under the optimized conditions and the results are represented in Table 3. The results indicated that some substituents such as methyl, methoxyl and bromine on the phenyl ring of the imines can smoothly react with 2-aminopyridine, affording the corresponding products 4aab–4aad in good yields (88–93%). In addition, pyridine and thiophene-functionalized imino nitriles could be used in this reaction, producing 2,3-dihydroquinazolinimine derivatives 4aae and 4aaf, respectively. Notably, (Z)-N-(2-(naphthalen-1-yl)-2,3-dihydroquinazolin-4(1H)-ylidene)pyridin-2-amine 4aag was obtained in 89% yield regardless of the use of the bulky naphthyl group. It is remarkable to find that an aromatic compound of the quinazoline 4aah′ was generated from the reaction of 2-aminopyridine with a conjugated α,β-unsaturated imine. Subsequently, the reactions of ketimino-functionalized nitriles with the primary amine 2aa were tested, affording 2,2-bis(substituted) dihydroquinazolinimine products 4aai and 4aaj in moderate yields. Further studies showed that not only electron-donating (Me and OMe) but also electron-withdrawing (F and Br) substituents on the phenyl ring of benzonitrile were tolerated with a limited effect on the reaction yields. Even o-(benzylideneamino) nicotinonitrile can work with 2aa to deliver product 4faa in 61% isolated yield. The scope of this methodology for the preparation of 2,3-dihydroquinazolinimines was demonstrated by the synthesis of 4aba, 4akb (CCDC 2416310†) and 4abb in moderate yields by the reactions of 2-imine-functionalized benzonitriles with aniline or p-toluidine. Two electron-withdrawing bromine atoms or a sterically-bulky naphthalenyl group were used in this reaction, resulting in lower yields of the products of 2-aryl-4-arylimino-2,3-dihydroquinazolines 4add and 4agd. Generally, the rare earth metal alkyl catalyst provides direct and flexible access to diverse 2,3-dihydroquinazolinimines substituted at the C2, N4-positions.
Table 3 Scope of 2-iminobenzonitriles and aminesa
|
Reaction conditions: into a Schlenk glass reactor were successively placed 2 (1.0 mmol), 3 (2.0 mmol) and 1b in toluene (1.5 mL), 110 °C.
|
|
After the successful formation of dihydroquinazolines, we want to continue to expand the variety of reactions available. The initial studies were carried out by the reaction of phthalonitrile with aniline under similar conditions, and the detailed results are presented in Table S1 in the ESI.† To our delight, 1,3-bis(benzylideneamino)isoindoline 6aa was synthesized and isolated. The ratio of Z,Z to Z,E-isomers was about 2:1, as determined by NMR spectroscopy. After screening, the reaction of phthalonitrile with aniline to furnish 1,3-bis(benzylideneamino)isoindoline was performed in the presence of 5 mol% of 1b in toluene at 130 °C for 24 h with an excellent 98% isolated yield. Next, a variety of substituted phthalonitriles and amines were examined and the results are shown in Table 4. Under the same conditions, treatment of phthalonitrile with ortho-, meta- and para-substituted toluidine gave products 6ab–6ad in 85%, 89%, and 98% yields, respectively. The results implied that a great steric effect of the substituents on the reactivity was observed in the reaction, which was further reflected by the substrate 2,6-dimethylaniline, affording the desired product 6ab in 47% yield. Similarly, satisfactory yields of 6af and 6ag were obtained when 4-(tert-butyl) and 4-methoxy aniline were employed in this reaction. Unfortunately, 4-chloroaniline or 4-bromoaniline failed to afford the corresponding isoindoline derivatives. However, naphthylamine can work well with phthalonitrile affording 6ah in 79% yield. More significantly, benzophenone hydrazine and 1-aminoisoquinoline could undergo tandem addition and cyclization, affording the corresponding products 6ai and 6aj in high yields with exclusive Z,Z-configuration, probably due to larger steric hindrance or intramolecular hydrogen bond formation. As for the heteroaromatic amines, 2-aminopyridine exhibited excellent activity, producing 1,3-bis(2-pyridylimino)isoindoline 6ak in 95% isolated yield, which was widely used as a chelating ligand in organometallic chemistry.44 In contrast to 2-aminopyridine, 2-aminopyrimidine showed an inert feature in this reaction. Catalyst 1b is also applicable for aliphatic amines, such as cyclohexanamine, tert-butylamine and tetrahydrofurfurylamine, delivering 1,3-bis(alphatic imino)-functionalized isoindolines 6al, 6am and 6an in 54%–67% yields under the optimized conditions. Further studies showed that some substituents (Me, nBuO and Br) on the phenyl ring of phthalonitrile were tolerated, giving 1,3-diiminoisoindolines with little influence on the activity, and the result is completely different from the reaction of phthalonitrile with 4-bromoaniline. In addition, other types of o-dicarbonitriles, such as pyridine-, pyrazine- and naphthalene-2,3-dicarbonitriles, were reacted with 2-aminopyrimidine to produce the corresponding products 6qk–6sk in good yields. Remarkably, treatment of phthalonitrile with an equimolar amount of meta-phenylenediamine produced a macrocyclic compound 6ao in 58% yield.
Table 4 Scope of substituted phthalonitriles and aminesa
|
Reaction conditions: into a Schlenk glass reactor were successively placed 5 (1.0 mmol), 3 (4.0 mmol) and the catalyst in toluene (1.5 mL), isolated yields. The ratios in brackets refer to isomers of Z,Z to Z,E-products as determined by 1H NMR spectroscopic analysis.
|

|
Reaction of 1b with arylamines
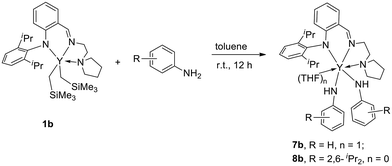
In order to gain more insights into the catalytic mechanism, several control experiments were carried out. The reaction of 1b with an equimolar amount of aniline at room temperature for 12 h afforded a solvated yttrium bis(anilido) complex LY(NHPh)2(THF) 7b. During the process, the deprotonation of the NH2 group and the release of two molecules of SiMe4 took place (Scheme 4). When increasing the amount of aniline to 2 equiv., complex 7b was obtained in 68% isolated yield. Similarly, treatment of 1b with a bulky substrate 2,6-diisopropylaniline under the same conditions produced an unsolvated yttrium bis(anilido) complex LY(NHDipp)28b. Next, upon treatment of 7b with 2-(benzylideneamino)benzonitrile or phthalonitrile in a 1:1 molar ratio at 120 °C for 24 h, no desired rare earth metal complex was isolated except for the catalytic products detected. As for the role of the anilido complex in the catalytic reaction, 1H NMR spectral monitoring of 7b with 2aa was performed in toluene-d8 (Fig. S96 in the ESI†). The insertion reaction between the Y–N bond and the cyano group occurred quickly to form the yttrium complex bearing an amidinate functional group (ab at 5.79 ppm in Fig. S96 in the ESI†), and it disappeared completely after 24 h reaction time. At the same time, the signal of 2,3-dihydroquinazolinimine 4aba was found in this reaction, indicating that the anilido complex could be regarded as an active intermediate in this catalytic reaction.
 |
| | Scheme 4 Reaction of 1b with arylamines. | |
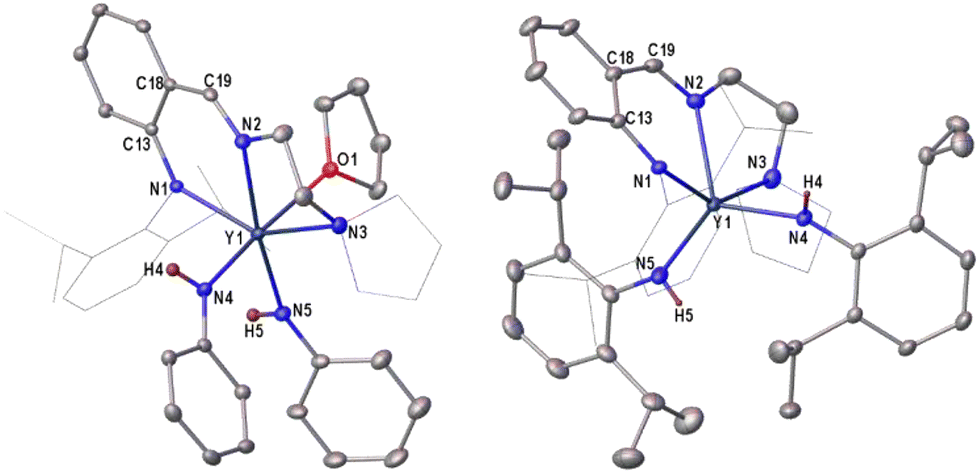
In the 1H NMR (C6D6, 500 MHz) spectra, signals at 4.16 ppm for 7b and 4.58 ppm for 8b are associated with the NHAr group (Fig. S10 and S12†). The values are similar to that of 4.83 ppm in a β-diketiminato yttrium anilido complex.45 X-ray analysis showed that the geometry of the six-coordinate 7b is different from that of the five-coordinate 1b. These atoms (N1, N2, N3 and N5) in 7b constitute a plane and other atoms of N4 and O1 occupy two apical positions, respectively. In contrast, the structure of 8b is similar to that of 1b, and two alkyl groups in 1b were replaced by two anilido groups, forming a five-coordinate yttrium complex (Fig. 2). The bond distances of 2.238(2) Å and 2.271(3) Å of Y–Nanilide are obviously shorter than those of 2.335(2) and 2.435(2) Å of Y–Nanilido-imino in 7b, probably due to the bulky steric hindrance of the anilido-imino ligand, and this finding was also observed in 8b.
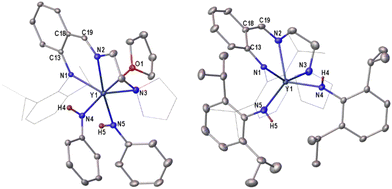 |
| | Fig. 2 Molecular structures of 7b (left) and 8b (right). All the hydrogen atoms are omitted for clarity except for H4 and H5. Selected bond lengths (Å) and angles (deg.). 7b: Y1–N1 2.335(2), Y1–N2 2.435(2), Y1–N3 2.599(3), Y1–N4 2.238(2), Y1–N5 2.271(3), Y1–O1 2.4655(19), C13–N1 1.373(3), C19–N2 1.283(4), C13–C18 1.441(3), C18–C19 1.426(4), N4–Y1–O1 169.55(8). 8b: Y1–N1 2.311(3), Y1–N2 2.367(3), Y1–N3 2.569(3), Y1–N4 2.210(3), Y1–N5 2.218(4), C13–N1 1.360(5), C19–N2 1.294(5), C13–C18 1.425(5), C18–C19 1.417(6), N5–Y1–N4 125.53(14). | |
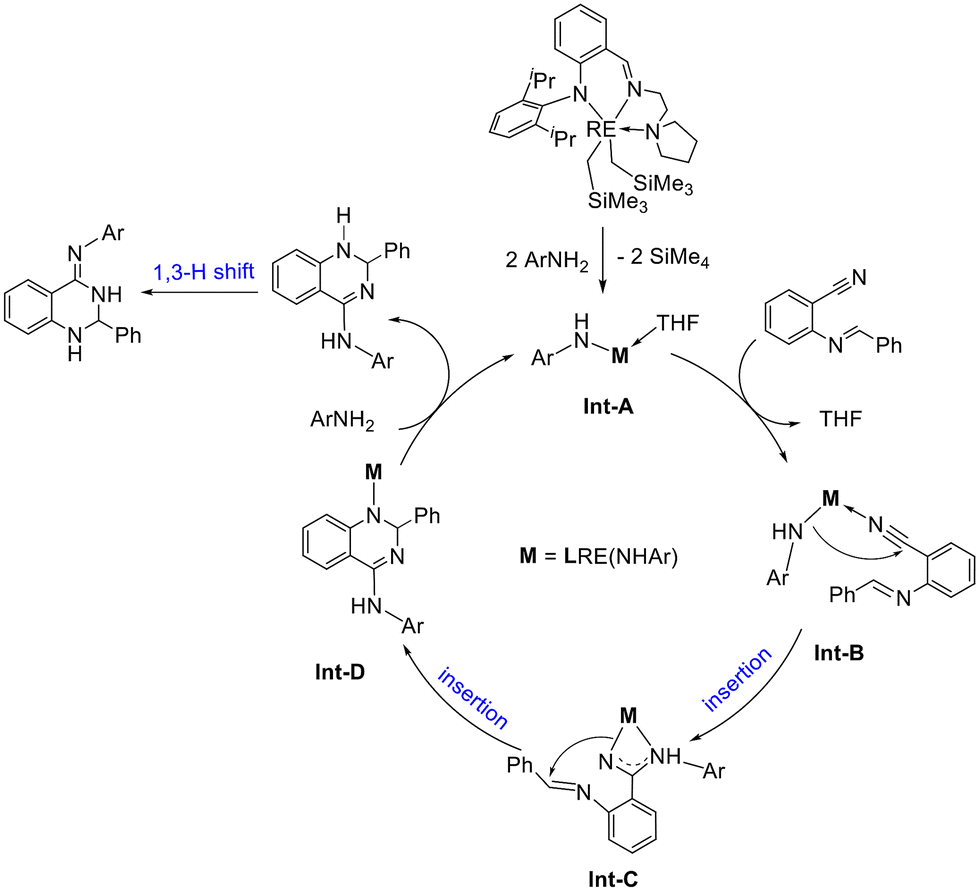
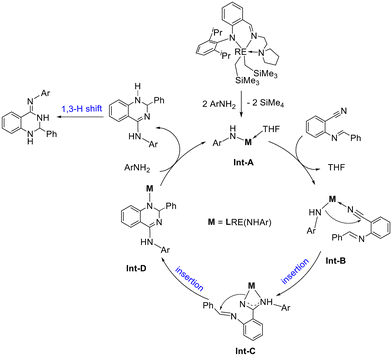
Based on the control experiments, a plausible reaction mechanism was proposed by using the synthesis of 2,3-dihydroquinazolimines as an example (Scheme 5). The deprotonation of arylamines with the anilido-imino rare earth metal dialkyl complexes produced Int-A by the release of SiMe4 (as 7b and 8b). Then the coordination of ortho imine-functionalized benzonitriles with the metal center generated Int-B, which underwent the first insertion of RE–N to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N unit to produce Int-C. Subsequently, the second insertion led to the formation of a six-membered heterocyclic intermediate Int-D. Treatment of Int-D with an amine resulted in the release of dihydroquinazolines following a 1,3-H shift to form the catalytic product 2,3-dihydroquinazolinimines and, meanwhile, regenerated the amido species Int-A to fulfill a catalytic cycle. The formation of 1,3-diiminoisoindolines is given in the ESI† as it is similar to the reaction described in Scheme 5.
N unit to produce Int-C. Subsequently, the second insertion led to the formation of a six-membered heterocyclic intermediate Int-D. Treatment of Int-D with an amine resulted in the release of dihydroquinazolines following a 1,3-H shift to form the catalytic product 2,3-dihydroquinazolinimines and, meanwhile, regenerated the amido species Int-A to fulfill a catalytic cycle. The formation of 1,3-diiminoisoindolines is given in the ESI† as it is similar to the reaction described in Scheme 5.
 |
| | Scheme 5 A proposed mechanism for the synthesis of 2,3-dihydroquinazolinimines. | |
Conclusions
In summary, novel N,N,N-tridentate anilido-imino rare earth metal dialkyl complexes were synthesized and characterized. Further investigation showed that these dialkyl complexes provided a convenient and atom-economical approach for the construction of 5- and 6-membered heterocyclic architectures including quinazolines and isoindolines via tandem addition and cyclization. 4-Arylimino-2,3-dihydroquinazoline derivatives were readily achieved by the reaction of 2-imine-functionalized benzonitriles with (hetero)aromatic primary amines for the first time. Moreover, a series of 1,3-diiminoisoindolines were synthesized in moderate to excellent yields from the reaction of o-dicarbonitriles with aromatic or aliphatic amines. The rare earth metal catalysts featured some advantages such as step economy, high activity and good tolerance to functional groups. During the process of determining the catalytic reaction mechanism, two yttrium anilido complexes were isolated and fully characterized.
Experimental
General remarks
All experiments were performed under an argon atmosphere using standard Schlenk techniques or in a glovebox. Solvents (toluene, hexane, and THF) were dried by refluxing over sodium/benzophenone ketyl and distilled before use. Amines and nitriles were purchased from commercial vendors and distilled before use. Pyrrolidine-ligated fluorobenzene46 and 2-arylimino-benzonitriles were prepared according to the literature.47 Elemental analysis data were obtained on a PerkinElmer Model 2400 Series II elemental analyzer. The 1H NMR and 13C {1H} NMR spectra were recorded on a Bruker Model spectrometer. Chemical shifts (δ) were reported in ppm. J values were reported in Hz. HRMS was obtained on a Thermo Scientific LTQ Orbitrap XL mass spectrometer.
Crystal structure determination
Single crystals for X-ray diffraction were sealed in a thin-walled capillary. Diffraction was carried out on a Bruker SMART CCD area detector diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). An empirical absorption correction was applied using the SADABS program.48 All structures were solved by direct methods, completed by subsequent difference Fourier syntheses, and refined anisotropically for all non-hydrogen atoms by full-matrix least-squares calculations on F2 using the SHELXTL program package. The program SQUEEZE in PLATON was used to remove the disordered solvents.49
Synthesis of proligand HL
18.0 mL of n-BuLi in hexane (1.6 M, 28.8 mmol) was added to a solution of 2,6-diisopropylaniline (4.5 mL, 24 mmol) in THF (40 mL) at −78 °C, and the mixture was warmed to room temperature for 8 h. Then N-(2-fluorobenzylidene)-2-(pyrrolidin-1-yl)ethanamine (4.4 g, 20 mmol) was added and the mixture was refluxed for 24 h. After filtration, the solvent was evaporated under reduced pressure. Colorless crystals were obtained by recrystallization from methanol (5.6 g, 74% yield). M.P.: 134–136 °C. 1H NMR (400 MHz, CDCl3): δ 10.54 (s, NH, 1H), 8.49 (s, CHN, 1H), 7.34–7.24 (m, ArH, 4H), 7.10–7.05 (m, ArH, 1H), 6.66 (t, J = 7.6 Hz, ArH, 1H), 6.21 (d, J = 8.0 Hz, ArH, 1H), 3.80 (t, J = 6.8 Hz, NCH2, 2H), 3.12 (sept, J = 6.8 Hz, CH, 2H), 2.79 (t, J = 7.2 Hz, NCH2, 2H), 2.60–2.57 (m, NCH2, 4H), 1.80–1.76 (m, CH2, 4H), 1.16 (d, J = 6.8 Hz, CH3, 6H), 1.14 (d, J = 7.2 Hz, CH3, 6H). 13C{1H} NMR (100 MHz, CDCl3, ppm): δ 164.8 (CN), 149.4, 147.5, 135.1, 133.4, 130.9, 127.1, 123.6, 116.6, 114.9, 111.6 (ArC), 61.1 (NCH2), 57.4 (NCH2), 54.7 (NCH2), 28.4, 24.9, 23.4, 22.8. HRMS (ESI): calcd for C25H35N3 [M + H]+: 378.2904, found: 378.2914.
Synthesis of complex 1
Under an argon atmosphere, proligand HL (0.37 g, 1.0 mmol) and RE(CH2SiMe3)3(THF)2 (1.0 mmol) were mixed in toluene (1.5 mL) and stirred at room temperature for 5 min. A suitable amount of hexane (1–3 mL) was added and then filtered. The crystals were obtained by standing the solution at −30 °C for 6–24 h.
Complex 1a.
Orange crystals. (0.46 g, 65% yield). Anal. calcd for C33H56N3Si2Gd: C, 55.96; H, 7.97; N, 5.93. Found: C, 55.59; H, 7.88; N, 5.79.
Complex 1b.
Yellow crystals. (0.38 g, 60% yield). 1H NMR (400 MHz, C6D6, ppm): δ 7.80 (s, HCN, 1H), 7.36–7.29 (m, ArH, 3H), 7.07 (dd, J = 1.2, 8.0 Hz, ArH, 1H), 6.91–6.87 (m, ArH, 1H), 6.43–6.40 (m, ArH, 1H), 6.32 (d, J = 8.8 Hz, ArH, 1H), 3.36 (sept, J = 6.8 Hz, CHMe2, 2H), 3.25–3.19 (m, NCH2, 2H), 2.94 (t, J = 6.0 Hz, NCH2, 2H), 2.26 (t, J = 6.0 Hz, NCH2, 4H), 1.60–1.52 (m, CH2, 2H), 1.43 (d, J = 6.8 Hz, CH3, 6H), 1.29–1.25 (m, CH2, 2H), 1.15 (d, J = 6.8 Hz, CH3, 6H), 0.14 (s, SiCH3, 18H), −0.78 (dd, J = 3.2, 11.2 Hz, YCH2, 2H), −0.91 (dd, J = 3.2, 11.2 Hz, YCH2, 2H). 13C{1H} NMR (100 MHz, C6D6, ppm): δ 169.4 (HCN), 156.9, 146.0, 141.2, 137.6, 134.5, 126.7, 125.4, 118.3, 118.0, 113.7 (ArC), 57.4 (NCH2), 55.2 (NCH2), 53.5 (NCH2), 35.8 (d, JY–C = 41.0 Hz, YCH2), 28.5, 25.8, 25.1, 22.7, 4.5 (SiMe3). Anal. calcd for C33H56N3Si2Y: C, 61.94; H, 8.82; N, 6.57. Found: C, 61.70; H, 8.94; N, 6.54.
Complex 1c.
Red crystals. (0.51 g, 70% yield). Anal. calcd for C33H56N3Si2Yb: C, 54.74; H, 7.80; N, 5.80. Found: C, 54.45; H, 7.80; N, 5.67.
Complex 1d.
Yellow crystals. (0.47 g, 65% yield). 1H NMR (500 MHz, C6D6, ppm): δ 7.79 (s, HCN, 1H), 7.35–7.30 (m, ArH, 3H), 7.06 (dd, J = 1.5, 8.0 Hz, ArH, 1H), 6.91–6.88 (m, ArH, 1H), 6.42–6.37 (m, ArH, 2H), 3.42 (sept, J = 7.0 Hz, CHMe2, 2H), 3.22–3.17 (m, NCH2, 2H), 2.92 (t, J = 6.0 Hz, NCH2, 2H), 2.29–2.26 (m, NCH2, 2H), 2.21 (t, J = 6.0 Hz, NCH2, 2H), 1.56–1.50 (m, CH2, 2H), 1.46 (d, J = 7.0 Hz, CH3, 6H), 1.25–1.22 (m, CH2, 2H), 1.16 (d, J = 6.5 Hz, CH3, 6H), 0.13 (s, SiCH3, 18H), −0.93 (d, J = 12.0 Hz, LuCH2, 2H), −1.05 (d, J = 12.0 Hz, LuCH2, 2H). 13C{1H} NMR (125 MHz, C6D6, ppm): δ 169.6 (HCN), 157.8, 145.5, 144.1, 137.3, 134.5, 126.4, 125.2, 119.1, 118.2, 113.9 (ArC), 57.3 (NCH2), 54.6 (NCH2), 53.1 (NCH2), 43.4, 28.4, 25.9, 25.1, 22.5, 4.7 (SiMe3). Anal. calcd for C33H56N3Si2Lu: C, 54.60; H, 7.78; N, 5.79. Found: C, 54.68; H, 7.43; N, 5.49.
Synthesis of complexes 7b and 8b
Under an argon atmosphere, complex 1b (96 mg, 0.15 mmol) and ArNH2 (0.30 mmol) were mixed in tetrahydrofuran (4.0 mL) and stirred at room temperature for 5 min. A suitable amount of hexane (1–2 mL) was added and then filtered. The crystals were obtained by standing the solution at room temperature for 48 h.
Complex 7b.
Yellow crystals. (73 mg, 68% yield). 1H NMR (500 MHz, C6D6, ppm): δ 7.82 (s, HCN, 1H), 7.28–7.22 (m, ArH, 3H), 7.13–7.10 (m, ArH, 5H), 6.88–6.85 (m, ArH, 1H), 6.57 (t, J = 7.5 Hz, ArH, 2H), 6.44–6.38 (m, ArH, 5H), 6.23 (d, J = 8.5 Hz, ArH, 1H), 4.16 (s, NH, 2H), 3.48–3.41 (m, CHMe2 and OCH2, 6H), 3.04 (t, J = 5.5 Hz, NCH2, 2H), 2.77–2.30 (m, NCH2, 4H), 2.21 (t, J = 5.5 Hz, NCH2, 2H), 1.36 (s, OCH2CH2, 4H), 1.28–1.26 (m, NCH2CH2, 4H), 1.24 (d, J = 6.5 Hz, CH3, 6H), 1.07 (d, J = 6.5 Hz, CH3, 6H). 13C{1H} NMR (125 MHz, C6D6, ppm): δ 169.8 (HCN), 158.0, 157.5, 147.5, 142.0, 137.9, 134.1, 129.5, 126.6, 125.2, 119.2, 117.6, 116.3, 113.5, 113.2 (ArC), 69.5 (OCH2), 58.5 (NCH2), 56.9 (NCH2), 55.6 (NCH2), 28.6, 26.2, 25.6, 24.2, 23.0. Anal. calcd for C41H54N5OY: C, 68.22; H, 7.54; N, 9.70. Found: C, 68.53; H, 7.48; N, 9.94.
Complex 8b.
Yellow crystals. (88 mg, 72% yield). 1H NMR (500 MHz, C6D6, ppm): δ 7.67 (s, HCN, 1H), 7.12 (d, J = 8.0 Hz, ArH, 1H), 7.07 (d, J = 8.0 Hz, ArH, 4H), 7.01 (d, J = 7.0 Hz, ArH, 3H), 6.78 (t, J = 7.5 Hz, ArH, 2H), 6.75–6.72 (m, ArH, 1H), 6.39 (d, J = 7.0 Hz, ArH, 1H), 6.19 (d, J = 9.0 Hz, ArH, 1H), 4.58 (s, NH, 2H), 3.47 (sept, J = 7.0 Hz, CHMe2, 2H), 3.11–3.03 (m, CHMe2, 2H), 2.97–2.87 (m, CHMe2 and NCH2, 6H), 2.36 (t, J = 6.0 Hz, NCH2, 2H), 2.10 (s, NCH2, 2H), 1.74–1.70 (m, NCH2CH2, 2H), 1.41–1.36 (m, NCH2CH2, 2H), 1.28 (d, J = 7.0 Hz, CH3, 12H), 1.17 (d, J = 7.0 Hz, CH3, 6H), 1.15 (d, J = 6.5 Hz, CH3, 12H), 1.03 (d, J = 6.5 Hz, CH3, 6H). 13C{1H} NMR (125 MHz, C6D6, ppm): δ 171.2 (HCN), 158.3, 151.6, 151.6, 146.9, 142.0, 137.9, 137.6, 134.2, 133.2, 129.3, 128.6, 126.8, 125.7, 125.4, 123.3, 120.4, 117.8, 115.8, 113.5 (ArC), 57.2 (NCH2), 56.4 (NCH2), 55.8 (NCH2), 30.2, 28.8, 27.0, 24.8, 24.3, 24.0, 23.0, 21.4. Anal. calcd for C49H70N5Y: C, 71.95; H, 8.63; N, 8.56. Found: C, 71.62; H, 8.61; N, 8.83.
General procedure for the synthesis of 2,3-dihydroquinolinimines
Catalyst 1b (32 mg, 5 mol%) was dissolved in 1.5 mL of toluene. Then amines (2.0 mmol) and ortho-imino-functionalized benzonitriles (1.0 mmol) were added. The reaction was stirred at 110 °C for 24 h. After that, the volatiles were removed under reduced pressure, and the catalytic products were purified by silica gel column chromatography (ethyl acetate:petroleum ether = 1:1–1:30).
General procedure for the synthesis of 1,3-diiminoisoindolines
Catalyst 1b (32 mg, 5 mol%) was dissolved in 1.5 mL of toluene. Then primary amines (4.0 mmol) and phthalonitrile (1.0 mmol) were added. The reaction was stirred at 130 °C for 24 h. After that, the volatiles were removed under reduced pressure, and the catalytic products were purified by silica gel column chromatography (ethyl acetate:petroleum ether = 1:2–1:50).
Data availability
The data used to support the findings of this study are included within the article.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22171003, 22171004 and 22301005) and the Anhui Teaching and Research Program (2022xsxx069).
References
- H. Jin, H.-G. Dan and G.-W. Rao, Heterocycl. Commun., 2018, 24, 1–10 CrossRef CAS.
- E. Cohen, B. Klarberg, J. Vaughan and R. James, J. Am. Chem. Soc., 1959, 81, 5508–5509 CrossRef CAS.
- A. M. Starosotnikov and M. A. Bastrakov, Chem. Heterocycl. Compd., 2017, 53, 1181–1183 CrossRef CAS.
- E. L. Ellsworth, T. P. Tran, H. D. H. Showalter, J. P. Sanchez, B. M. Watson, M. A. Stier, J. M. Domagala, S. J. Gracheck, E. T. Joannides, M. A. Shapiro, S. A. Dunham, D. L. Hanna, M. D. Huband, J. W. Gage, J. C. Bronstein, J. Y. Liu, D. Q. Nguyen and R. Singh, J. Med. Chem., 2006, 49, 6435–6438 CrossRef CAS PubMed.
- T. Goldner, G. Hewlett, N. Ettischer, H. Ruebsamen-Schaeff, H. Zimmermann and P. Lischka, J. Virol., 2011, 85, 10884–10893 CrossRef CAS PubMed.
- J. H. Heo, H. N. Seo, Y. J. Choe, S. Kim, C. R. Oh, Y. D. Kim, H. Rhim, D. J. Choo, J. Kim and J. Y. Lee, Bioorg. Med. Chem. Lett., 2008, 18, 3899–3901 CrossRef CAS PubMed.
- J. A. Jeong, H. Cho, S. Y. Jung, H. B. Kang, J. Y. Park, J. Kim, D. J. Choo and J. Y. Lee, Bioorg. Med. Chem. Lett., 2010, 20, 38–41 CrossRef CAS PubMed.
- L. Shiri, S. Rahmati, Z. R. Nejad and M. Kazemi, Appl. Organomet. Chem., 2017, 31, e3687 CrossRef.
- R. J. Abdel-Jalil, W. Voelter and M. Saeed, Tetrahedron Lett., 2004, 45, 3475–3476 CrossRef CAS.
- D. Q. Shi, L. C. Rong, J. X. Wang, Q. Y. Zhuang, X. S. Wang and H. W. Hu, Tetrahedron Lett., 2003, 44, 3199–3201 CrossRef CAS.
- A. Shaabani, A. Maleki and H. Mofakham, Synth. Commun., 2008, 38, 3751–3759 CrossRef CAS.
- M. Rueping, A. P. Antonchick, E. Sugiono and K. Grenader, Angew. Chem., Int. Ed., 2009, 48, 908–910 CrossRef CAS PubMed.
- X. Cheng, S. K. Vellalath and R. Goddard, J. Am. Chem. Soc., 2008, 130, 15786–15787 CrossRef CAS PubMed.
- A. Davoodnia, S. Allameh, A. R. Fakhari and N. Tavakoli-Hoseini, Chin. Chem. Lett., 2010, 21, 550–553 CrossRef CAS.
- S. J. Wu, Z. Q. Zhao, J. S. Gao, B. H. Chen and G. F. Chen, Res. Chem. Intermed., 2019, 45, 2327–2339 CrossRef CAS.
- H. R. Shaterian, A. R. Oveisi and M. Honarmand, Synth. Commun., 2010, 40, 1231–1242 CrossRef CAS.
- L.-Y. Zeng and C. Cai, J. Heterocycl. Chem., 2010, 47, 1035–1039 CrossRef CAS.
- M. Z. Kassaee, S. Rostamizadeh, N. Shadjou, E. Motamedi and M. Esmaeelzadeh, J. Heterocycl. Chem., 2010, 47, 1421–1424 CrossRef CAS.
- J. Safari and S. Gandomi-Ravandi, J. Mol. Catal. A: Chem., 2013, 371, 135–140 CrossRef CAS.
- M. Alipour, Z. Hossaini, S. Khaksar and F. Rostami-Charati, Curr. Org. Synth., 2020, 17, 40–45 CrossRef CAS.
- K. Niknam, M. R. Mohammadizadeh and S. Mirzaee, Chin. J. Chem., 2011, 29, 1417–1422 CrossRef CAS.
- G. Albano and L. A. Aronica, Eur. J. Org. Chem., 2017, 7204–7221 CrossRef CAS.
- E. Andrade-Jorge, J. R. Bahena-Herrera, J. Garcia-Gamez, I. I. Padilla-Martínez and J. G. Trujillo-Ferrara, Med. Chem. Res., 2017, 26, 2410–2431 CrossRef.
- E. Valencia, A. J. Freyer, M. Shamma and V. Fajardo, Tetrahedron Lett., 1984, 25, 599–602 CrossRef CAS.
- D. C. Sauer, R. L. Melen, M. Kruck and L. H. Gade, Eur. J. Inorg. Chem., 2014, 28, 4715–4725 CrossRef.
- B. L. Dietrich, J. Egbert, A. M. Morris, M. Wicholas, O. P. Anderson and S. M. Miller, Inorg. Chem., 2005, 44, 6476–6481 CrossRef CAS PubMed.
- D. C. Sauer, M. Kruck, H. Wadepohl, M. Enders and L. H. Gade, Organometallics, 2013, 32, 885–892 CrossRef CAS.
- S. Gaire, B. R. Schrage, V. N. Nemykin and C. J. Ziegler, Dalton Trans., 2021, 50, 826–829 RSC.
- K. Hanson, L. Roskop, P. I. Djurovich, F. Zahariev, M. S. Gordon and M. E. Thompson, J. Am. Chem. Soc., 2010, 132, 16247–16255 CrossRef CAS.
- R. Csonka, G. Speier and J. Kaizer, RSC Adv., 2015, 5, 18401–18419 RSC.
- W. Siegl, J. Org. Chem., 1977, 144, 1872–1878 CrossRef.
- I.-S. Tamgho, J. T. Engle and C. J. Ziegler, Tetrahedron Lett., 2013, 54, 6114–6117 CrossRef CAS.
- X. Kong, H. Zhao and Y. Tang, Tetrahedron, 2023, 54, 133546 CrossRef.
- J. Escorihuela, A. Lledós and G. Ujaque, Chem. Rev., 2023, 123, 9139–9203 CrossRef CAS PubMed.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS.
- X. Zhu, J. Sun, Z. Wu, D. Guo, S. Zhou and S. Wang, Adv. Synth. Catal., 2024, 366, 1405–1411 CrossRef CAS.
- B. Feng, H.-Y. Zhang, H. Qin, Q. Peng, X. Leng and Y. Chen, CCS Chem., 2022, 4, 3309–3318 CrossRef CAS.
- K. Manna, M. L. Kruse and A. D. Sadow, ACS Catal., 2011, 1, 1637–1642 CrossRef CAS.
- X. Zhu, Y. Qi, Y. Yang, D. Guo, Z. Huang, L. Zhang, Y. Wei, S. Zhou and S. Wang, Inorg. Chem., 2022, 61, 3202–3211 CrossRef CAS.
- B. Wang, D. Wang, D. Cui, W. Gao, T. Tang, X. Chen and X. Jing, Organometallics, 2007, 26, 3167–3172 CrossRef CAS.
- P. G. Hayes, W. E. Piers, L. W. M. Lee, L. K. Knight, M. Parvez, M. R. J. Elsegood and W. Clegg, Organometallics, 2001, 20, 2533–2544 CrossRef CAS.
- X. Zhu, Y. Li, D. Guo, S. Wang and Y. Wei, Dalton Trans., 2018, 47, 3947–3957 RSC.
- X. Xu, X. Y. Xu, Y. F. Chen and J. Sun, Organometallics, 2008, 27, 758–763 CrossRef CAS.
- G. Reshma, V. Padmanabhan, A. R. Varma, M. S. Gouri, U. R. Nair, P. B. Parvathy, N. V. Kulkarni and D. Senthurpandi, J. Mol. Struct., 2021, 1226, 129344 CrossRef CAS.
- H. Zou, Z. Wang, D. Guo, L. Zhang, S. Wang and X. Zhu, Dalton Trans., 2024, 53, 267–275 RSC.
- P. G. Hayes, G. C. Welch, D. J. H. Emslie, C. L. Noack, W. E. Piers and M. Parvez, Organometallics, 2003, 22, 1577–1579 CrossRef CAS.
- L. H. Leijendekker, J. Weweler, T. M. Leuther and J. Streuff, Angew. Chem., Int. Ed., 2017, 56, 6103–6106 CrossRef CAS PubMed.
- O. V. L. Dolomanov, J. R. Bourhis, J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Xiancui
Zhu
and
Xiancui
Zhu