
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
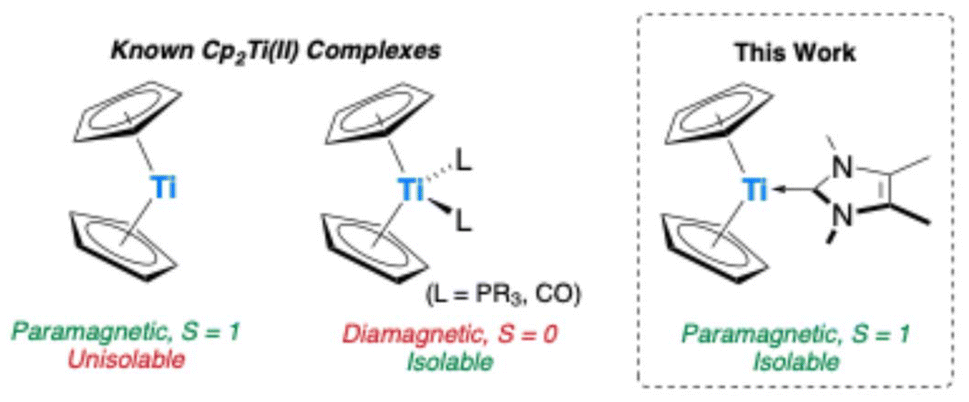
Isolation of the parent triplet titanocene via NHC stabilisation†
Felix Meyera,
Serhiy Demeshkoa,
Christopher Golzb,
Oliver P. E. Townrow *c and
Malte Fischer*a
*c and
Malte Fischer*a
aInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, D-37077 Göttingen, Germany. E-mail: malte.fischer@uni-goettingen.de
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, D-37077 Göttingen, Germany
cInstitute of Nanotechnology, Karlsruher Institut für Technologie, Hermann-von-Helmholtz-Platz 1, D-76344, Eggenstein-Leopoldshafen, Germany. E-mail: oliver.townrow@kit.edu
First published on 28th February 2025
Abstract
We present the synthesis and characterization of the parent isolable monomeric triplet titanocene complex, stabilized by the N-heterocyclic carbene (NHC) IMe4. Investigated by SQUID magnetometry and quantum-chemical calculations, reactivity studies of the titanium precursor [Cp2Ti(btmsa)] (2) with the NHC IiPr2Me2 and the zirconocene complex [Cp2Zr(py)(btmsa)] (1) revealed divergent reactivity patterns, highlighting the role of steric and electronic effects in stabilization.
Although the Group 4 elements titanium, zirconium, and hafnium show a strong thermodynamic preference for the +IV oxidation state, their complexes in the +II state are becoming increasingly significant in synthetic applications, ranging from catalysis to materials development.1 Combining Lewis acid reactivity in the +IV state and reductive transformations in the +II state, systems capable of reversible redox cycling between these have the potential to unlock exciting new possibilities for utilizing these Earth-abundant elements in efficient redox catalysis.1,2 Additionally, paramagnetic titanium(III) organometallics have also found recent attention as molecular spintronic components based on Earth-abundant metals, adding further relevance to finding novel examples.3
However, due to the thermodynamic stability of the +IV oxidation state, accessing low-valent species typically requires ‘masking’ through the incorporation of redox non-innocent ligands or ligands with pronounced backbonding capabilities (e.g. alkenes and alkynes). These ligands provide access to controlled sources of the low-valent form, serving as synthons for its generation.1,2,4 A key factor in advancing this field is a deep understanding of the electronic structure and nature of truly isolable low-valent Group 4 compounds and intermediates. Unlike their later transition metal homologues, titanocene and zirconocene [Cp2M] (Cp = η5-cyclopentadienyl; M = Ti, Zr) are not isolable and decompose through C–H activation processes.5 It must be noted that even the permethylated derivative [Cp*2Ti] (Cp* = η5-pentamethylcyclopentadienyl) is unstable and, for example, readily coordinates dinitrogen.6 However, its magnetic moment could be determined and was found to be in between 2.48 (129 K) and 2.60μB (298 K), slightly lower than the spin-only value of 2.83μB, and consistent with the presence of two unpaired electrons.6 Through the incorporation of bulky silyl groups at the cyclopentadienyl ligands, several triplet titanocene derivatives have been isolated and structurally characterized, such as [{C5Me4(SiMe2tBu)}2Ti], which has a magnetic moment of 2.40 μB.7 The reactive parent Group 4 metallocene fragment [Cp2M] is typically generated in situ by use of soluble, closed-shell-singlet synthons through the release of CO, PMe3, alkenes, alkynes, or by mixing [Cp2MCl2] with strong reducing agents.1c,4,8,9 Notable examples include ([Cp2ZrCl2]/nBuLi),10 [Cp2Ti(PMe3)2],11 [Cp2Ti(CO)2] (Scheme 1),12 and btmsa (bis(trimethylsilyl)acetylene) complexes [Cp2Zr(py)(btmsa)] (1) (py = pyridine), and [Cp2Ti(btmsa)] (2).1c,4,8,13 Their chemistry has been extensively reviewed on many occasions.1c,4,8,14 In contrast to these commonly employed diamagnetic Group 4 metallocenes, we present the synthesis and isolation of a crystalline paramagnetic parent titanocene, stabilised by an NHC which better mimics the electronic state of free [Cp2Ti].5 We further demonstrate how the choice of the metal precursor and NHC is crucial to metallocene formation, as varying these caused either ligand exchange, imidazole tautomerisation, or C–H activation. The reaction of 1 with IMe4 (1,3,4,5-tetramethylimidazol-2-ylidene) in C6D6 at room temperature resulted in an expected clean ligand exchange reaction in which the NHC replaces pyridine to yield [Cp2Zr(IMe4)(btmsa)] (3) (Scheme 2). The ligand exchange is visibly evident by an immediate colour change of the reaction mixture from intense purple to yellow–brown upon NHC addition. Inspection of the 1H NMR spectrum revealed the chemical shifts of free pyridine, in addition to divergence of the Me3Si signals of the btmsa ligand, indicating that free rotation is now sterically prohibited (Fig. S2†). The 13C chemical shift of the NHC carbene centre is located at δ13C{1H} = 197.8 ppm, confirmed by 1H/13C HMBC NMR (Fig. S4†), shifted when compared to that of the free carbene (δ13C{1H} = 213.3 ppm).15 It is worth noting that neither the addition of a second equivalent of IMe4 to 3 nor the direct addition of two equivalents of IMe4 to 1 led to any exchange of btmsa for IMe4, even at elevated temperatures (Fig. S5†). Yellow crystals of 3 suitable for single-crystal X-ray diffraction (SCXRD) were obtained from a saturated n-hexane solution at −30 °C, which allowed for structural confirmation. The coordination environment at zirconium is best described as distorted square pyramidal according to the τ5 structural parameter (τ5 = 0.11) (Fig. S6†).16 The CNHC–Zr bond length of 2.466(3) Å is in good agreement to other IMe4 ligated zirconium complexes, e.g. [(η7-C7H7)(η5-C5H5)Zr(IMe4)] (2.4452(17) Å).17,18 Interestingly, no reaction readily occurs between 1 and the slightly larger NHC IiPr2Me2 (1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). However, upon prolonged heating of this mixture up to 90 °C, 1 and IiPr2Me2 are completely consumed, and the characteristic signals of free pyridine are observed by 1H NMR spectroscopy. In addition, three signals in the typical range of cyclopentadienyl ligands and two new signals typical of hydride species are found, indicative of the formation of multiple products (Fig. S7†). After several attempts, a few dark yellow crystals were obtained from a saturated n-hexane solution at −30 °C, and the structure of the κ1-imidazole complex 4 was confirmed by SCXRD (Fig. S8†), with structural parameters in good agreement to those of 3.19 The formation of 4 suggests that propene is formed as a by-product. However, the expected ABX coupling pattern in the 1H NMR spectrum could not be unambiguously assigned due to the formation of additional byproducts and overlapping signals. Therefore, the headspace of the reaction, performed in toluene as the solvent, was analysed via GC-MS, confirming the presence of propene (Fig. S19 and S20†). Furthermore, trimethylsilane, ethyltrimethylsilane, vinyltrimethylsilane, pyridine and the employed solvents were detected, with the presence of the various silanes underscoring the complexity of the reaction's outcome.19 It is worth noting that although the NHC-to-imidazole transformation is unusual, the reverse imidazole to NHC reactivity pattern has precedence in the literature.20 We then explored the reactions of these NHCs with the lighter, titanium, analogue [Cp2Ti(btmsa)] (2). Contrary to the formation of 4, the reaction of 2 with one equivalent of IiPr2Me2 in aromatic solvents occurs at room temperature, resulting in the immediate release of btmsa, as confirmed by its 1H NMR chemical shift at 0.16 ppm (Fig. S9†). Although the reaction onset occurred rapidly, full conversion of 2 proceeds slowly and plateaus at around 50% after 72 hours. Heating the mixture to 90 °C for an extended period eventually drives the reaction toward near completion; however, approximately half an equivalent of the NHC remains unreacted, and signs of a paramagnetic product begin to emerge.19 Upon work-up at this stage, dark violet crystals suitable for SCXRD are consistently obtained, revealing the formation of the hydride-bridged dititanium complex 5 (Fig. 1). Additionally, formal loss of dihydrogen and C–H activation of one cyclopentadienyl ligand led to the formation of bridging fulvalene and cyclopentadienyl ligands to both titanium atoms and only one NHC ligation, which explains why approximately half an equivalent of the NHC remains unreacted. The Ti1–C1 bond length is 2.318(2) Å and clearly exceeds the respective single bond covalent radii of the respective elements (Σcov(Ti–C) = 2.11 Å),21 indicating expected donor–acceptor interactions. This bond length aligns well with other NHC-ligated titanium hydrides, such as [(C8H4(1,4-SiiPr3)2)2{(IMe4)Ti(μ-H)TiH}] (I) reported by Tsoureas et al., in which also only one of the titanium(III) centres is coordinated by an NHC (2.300(2) Å).22 The Ti–H bond lengths (Ti1–H1 1.92(4) Å; Ti2–H1 1.84(4) Å) also compare favourably to I, as well as to [(η5-C5H3-1,2-Me2)2Ti(μ-H)Ti(η5-C5H3-1,2-Me2)(η5,η1-C5H2-1,2-Me2)] (II) (1.79(2) Å and 1.91(2) Å).22,23 Both titanium atoms exhibit a distorted tetrahedral coordination environment according to the τ4 and τ4′ structural parameters (Ti1: 0.83, 0.73; Ti2: 0.79, 0.73).24 The Ti2–C13 bond length of 2.166(2) Å is typical of a single bond, falling between the single-bond covalent radii of titanium and carbon (vide supra) and matching the analogous bond length in II (2.204(2) Å).21,23 The folding of the fulvalene ligand is evident from a dihedral angle of 17.6(2)° between the two planes defined by both five-membered rings, as well as the inter-ring carbon–carbon bond length of 1.452(4) Å, both of which are diagnostic of fulvalene dititanium complexes (cf. [{Cp2Ti(μ-F)}2(μ-η5:η5-C10H8)] (1.464(8) Å and 15.3(3)°)).25 Inspection of the 1H NMR spectrum from crystalline 5 in C6D6 at room temperature revealed several broad signals between δ1H = −25 and +35 ppm, indicative of a paramagnetic compound (Fig. S14†).
 | ||
| Scheme 1 Examples of bis(cyclopentadienyl)titanium(II) complexes. | ||
 | ||
| Scheme 2 Reactions of [Cp2Ti(btmsa)] (1) and [Cp2Zr(py)(btmsa)] (2) towards IMe4 and IiPr2Me2 to give complexes 3, 4, 5, and 6. | ||
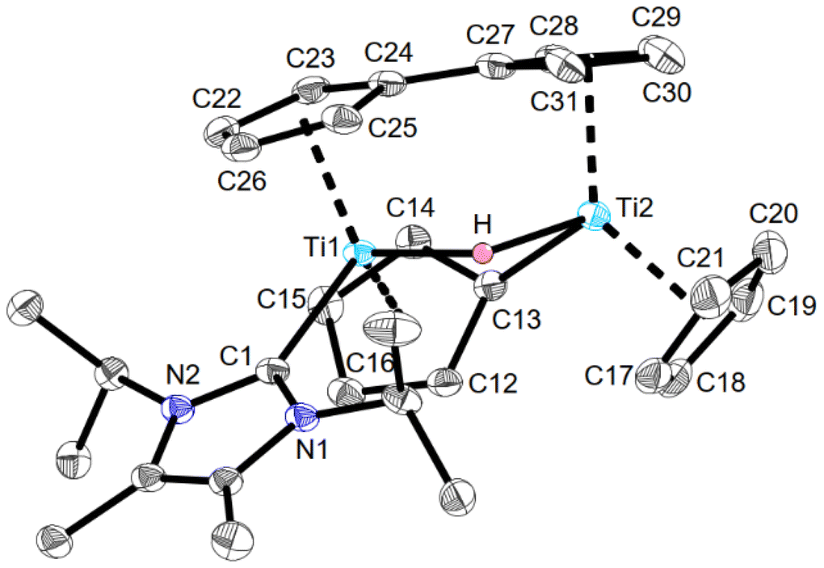 | ||
| Fig. 1 Molecular structure of 5 in the crystal. Anisotropic displacement parameters are drawn at the 50% probability level, with data collected at 100 K (hydrogen atoms attached to carbon omitted for clarity). Selected bond lengths (Å) and angles (deg): Ti1⋯Ti2 3.1183(6), Ti1–C1 2.318(2), Ti1–H1 1.92(4), Ti2–C13 2.166(2), Ti2–H1 1.84(4), Ti1–H1–Ti2 112.6, Ct1–Ti1–Ct2 136.0, Ct3–Ti2–Ct4 133.5 (Ct1 = C12–C15, Ct2 = C22–C26, Ct3 = C17–C20, Ct4 = C27–C31). | ||
Finally, we turned our attention to the remaining combination: [Cp2Ti(btmsa)] (2) and IMe4. Upon adding one equivalent of IMe4 to a C6D6 solution of 2, three new broad signals at −29.48, 9.92, and 93.46 ppm were detected alongside the characteristic signal of free btmsa in the 1H NMR spectrum, indicating a rapid ligand exchange reaction that occurs within a few minutes, yielding [Cp2Ti(IMe4)] (6) (Scheme 2 and Fig. S16, S17). Slow diffusion of n-pentane into a benzene solution of 6 produced dark yellow crystals, allowing for the unambiguous elucidation of the molecular structure by SCXRD (Fig. 2, top). Notably, complex 6 crystallises as dark yellow plates in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with three independent molecules in the unit cell, mainly due to different orientations of the Cp ligands (eclipsed and staggered conformers observed); the other structural parameters of all three molecules are in very good agreement. The average titanium–CNHC bond length is 2.27 Å, which is slightly shorter compared to the herein reported complex 5. In relation to a related formal three-coordinate and NHC-ligated titanium complex, such as [(C6H6)2Ti(IMe2H2)] (2.323(3) Å), this bond length is reduced and corresponds to the different titanium oxidation states in the two complexes.26 The titanium centre is in a trigonal planar coordination environment with a pronounced widening of the Ct1–Ti1–Ct2 angle (144.5°). The IMe4 ligand is aligned almost perpendicular to the Ct1–Ti1–Ct2 axis, exhibiting a dihedral angle of 81.8°. The UV-vis spectrum of Cp2Ti(IMe4) reveals an absorption maximum at 389 nm (c = 1 g L−1), in accordance to its (dark) yellow colour (Fig. S18†).
, with three independent molecules in the unit cell, mainly due to different orientations of the Cp ligands (eclipsed and staggered conformers observed); the other structural parameters of all three molecules are in very good agreement. The average titanium–CNHC bond length is 2.27 Å, which is slightly shorter compared to the herein reported complex 5. In relation to a related formal three-coordinate and NHC-ligated titanium complex, such as [(C6H6)2Ti(IMe2H2)] (2.323(3) Å), this bond length is reduced and corresponds to the different titanium oxidation states in the two complexes.26 The titanium centre is in a trigonal planar coordination environment with a pronounced widening of the Ct1–Ti1–Ct2 angle (144.5°). The IMe4 ligand is aligned almost perpendicular to the Ct1–Ti1–Ct2 axis, exhibiting a dihedral angle of 81.8°. The UV-vis spectrum of Cp2Ti(IMe4) reveals an absorption maximum at 389 nm (c = 1 g L−1), in accordance to its (dark) yellow colour (Fig. S18†).
 | ||
| Fig. 2 Top: Molecular structure of [Cp2Ti(IMe4)] (6) in the crystal. Anisotropic displacement parameters are drawn at the 50% probability level, with data collected at 100 K (hydrogen atoms omitted for clarity). Selected bond lengths (Å) and angles (deg): Ti1–C1 2.2712(9), C1–N1 1.3624(10), C1–N2 1.3603(10), C4–C5 1.3543(13), Ct1–Ti1–Ct2 144.5, Ct1–Ti1–C1 106.8, Ct2–Ti1–C1 108.6; Bottom: variable-temperature χMT product for 6 (open circles) and best fit curve (red line); Insert: calculated spin density (Triplet; UPBE0-D3BJ/Def2-TZVP; isovalue = 0.15 Å−3). | ||
To gain further insight into the electronic ground state of complex 6, it was investigated using SQUID magnetometry (see ESI† for details). The χMT value of 0.85 cm3 mol−1 K at 295 K for 6 clearly indicates S = 1 spin state (expected spin-only value of 1.0 cm3 mol−1 K). When the temperature was lowered, the χMT value decreased below ∼70 K due to the sizeable zero-field splitting of −43.2 cm−1 (Fig. 2, bottom).19 To evaluate this computationally, the crystallographically derived geometries of 5 and 6 were optimised by density functional theory (DFT) with singlet and triplet wavefunctions, at the (U)PBE0-D3BJ/def2-TZVP level of theory, finding that the triplet states are energetically favoured by ΔE = 9.5 and 13.5 kcal mol−1, respectively.19 Analysis of the Kohn–Sham molecular orbitals of compound 6 found two occupied quasi-degenerate α-SOMOs with A1 and B1 symmetry (assuming a C2v point group), localised at the dz2 and dxz orbitals of the Ti centre. The spin density (Fig. 2, bottom) is also localised around the Ti centre and shows exclusively α-electron density. This is reflected in further analysis by natural bond orbital (NBO) theory, finding two (α) lone particles (two unpaired electrons) at Ti, accounting for 0.91 and 0.84 electrons, respectively. The latter (dz2) is lower than the former, accounting for delocalisation of electron density onto the metallocene rings, this was located by natural localised molecular orbital (NLMO) theory. Natural population analysis (NPA) gave a low natural charge, and a natural spin close to two (0.63 and 1.79, respectively), also indicative of a Ti(II) triplet system. The spin density on the carbene carbon is negligible. Analysis of dititanium complex 5 by NBO and NPA showed that both titanium centres have comparable NPA charges and are more positive than 6 (0.74, 0.78). Their natural spin densities (0.9, 0.97) place an unpaired electron on each Ti centre, indicating two Ti(III) centres (rather than a mixed valence species). There is no NBO describing a Ti–Ti interaction, thus, each Ti centre can be described as Ti(III) with 17 valence electrons. Inspection of the spin density reveals localisation at the titanium centres, with qualitatively the same d-orbitals which combine on one metal in the case of 6 (dz2 and dxz) (Fig. S30†).
In conclusion, we have synthesised and characterised a series of Zr and Ti metallocenes, produced by the reaction of small NHCs with metallocene sources [Cp2Zr(py)(btmsa)] (1) and [Cp2Ti(btmsa)] (2) which showed varied outcomes. Amongst these, we have developed a highly selective protocol for the synthesis of the parent isolable monomeric triplet Ti(II) metallocene and present our initial analysis of its electronic and magnetic properties. This communication acts as the first step of an in-depth study on Ti(II) triplet organometallics, which will be further explored as novel reagents and molecular spintronic components.
Data availability
The data supporting this article have been included as part of the ESI.† Additional references have been cited within the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27–47 Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2419251–2419254 for (3–6)† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.
27–47 Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers 2419251–2419254 for (3–6)† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support (VCI Liebig fellowships for O.P.E.T. and M.F.; YIG Prep Pro fellowship for O.P.E.T.) is gratefully acknowledged. The NMR, MS, and EA services at the Faculty of Chemistry of the Georg-August-Universität Göttingen are kindly acknowledged for measurements and technical assistance. Support from the DFG (INST 186/1237-1 and INST 186/1324-1) is also gratefully acknowledged. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG for access to the JUSTUS 2 cluster.References
- Selected recent review articles: (a) M. Manßen and L. L. Schafer, Chem. Soc. Rev., 2020, 49, 6947–6994 RSC; (b) S. Fortier and A. Gomez-Torres, Chem. Commun., 2021, 57, 10292–10316 RSC; (c) U. Rosenthal, ChemistryOpen, 2021, 10, 1234–1243 CrossRef CAS PubMed.
- E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem., 2019, 3, 15–34 CrossRef PubMed.
- (a) L. C. de Camargo, M. Briganti, F. S. Santana, D. Stinghen, R. R. Ribeiro, G. G. Nunes, J. F. Soares, E. Salvadori, M. Chiesa, S. Benci, R. Torre, L. Sorace, F. Totti and R. Sessoli, Angew. Chem., Int. Ed., 2020, 60, 2588–2593 CrossRef PubMed; (b) M. Briganti, G. Serrano, L. Poggini, A. L. Sorrentino, B. Cortigiani, L. C. de Camargo, J. F. Soares, A. Motta, A. Caneschi, M. Mannini, F. Totti and R. Sessoli, Nano Lett., 2022, 22, 8626–8632 CrossRef CAS PubMed; (c) S. Wisbeck, A. L. Sorrentino, F. S. Santana, L. C. de Camargo, R. R. Ribeiro, E. Salvadori, M. Chiesa, N. Giaconi, A. Caneschi, M. Mannini, L. Poggini, M. Briganti, G. Serrano, J. F. Soares and R. Sessoli, Chem. Sci., 2024, 15, 14390–14398 RSC.
- U. Rosenthal, Chem. Soc. Rev., 2020, 49, 2119–2139 RSC.
- (a) M. Polasek and J. Kubista, J. Organomet. Chem., 2007, 692, 4073–4083 CrossRef CAS; (b) P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS.
- J. E. Bercaw, J. Am. Chem. Soc., 1974, 96, 5087–5095 CrossRef CAS.
- (a) P. B. Hitchcock, F. M. Kerton and G. A. Lawless, J. Am. Chem. Soc., 1998, 120, 10264–10265 CrossRef CAS; (b) L. Lukesova, M. Horacek, P. Stepnicka, K. Fejfarova, R. Gyepes, I. Cisarova, J. Kubista and K. Mach, J. Organomet. Chem., 2002, 663, 134–144 CrossRef CAS; (c) L. Lukesova, J. Pinkas, M. Horacek, R. Gyepes, J. Kubista and K. Mach, J. Organomet. Chem., 2006, 691, 748–758 CrossRef CAS.
- (a) U. Rosenthal, Organometallics, 2020, 39, 4403–4414 CrossRef CAS; (b) U. Rosenthal, ChemistryOpen, 2019, 8, 1036–1047 CrossRef CAS PubMed.
- See for example: (a) D. W. Stephan, Organometallics, 1992, 11, 996–999 CrossRef CAS; (b) G. Bousrez, I. Dechamps, J.-L. Vasse and F. Jaroschik, Dalton Trans., 2015, 44, 9359–9362 RSC; (c) G. Bousrez, D. Harakat, S. Chevreux, I. Déchamps-Olivier and F. Jaroschik, Dalton Trans., 2024, 53, 15595–15601 RSC.
- (a) E. Negishi, F. E. Cederbaum and T. Takahashi, Tetrahedron Lett., 1986, 27, 2829–2832 CrossRef CAS; (b) E. Negishi and S. Huo, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH, Weinheim, 2002, pp. 1–49 Search PubMed.
- (a) L. B. Kool, M. D. Rausch, H. G. Alt, M. Herberhold, U. Thewalt and B. Wolf, Angew. Chem., Int. Ed. Engl., 1985, 24, 394–401 CrossRef; (b) L. B. Kool, M. D. Rausch, H. G. Alt, M. Herberhold, B. Honold and U. Thewalt, J. Organomet. Chem., 1987, 320, 37–45 CrossRef CAS.
- (a) J. G. Murray, J. Am. Chem. Soc., 1959, 81, 752–753 CrossRef CAS; (b) J. L. Atwood, K. E. Stone, H. G. Alt, D. C. Hrncir and M. D. Rausch, J. Organomet. Chem., 1975, 96, C4–C6 CrossRef CAS.
- (a) V. V. Burlakov, A. V. Polyakov, A. I. Yanovsky, Y. T. Struchkov, V. B. Shur, M. E. Vol'pin, Z. Rosenthal and H. Görls, J. Organomet. Chem., 1994, 476, 197–206 CrossRef CAS; (b) U. Rosenthal, A. Ohff, W. Baumann, A. Z. Tillack, H. Görls, V. V. Burlakov and V. B. Shur, Z. Anorg. Allg. Chem., 1995, 621, 77–83 CrossRef CAS; (c) J. R. Nitschke, S. Zürcher and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 10345–10352 CrossRef CAS; (d) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884–900 CrossRef CAS; (e) M. Fischer, L. Vincent-Heldt, M. Hillje, M. Schmidtmann and R. Beckhaus, Dalton Trans., 2020, 49, 2068–2072 RSC.
- See for example: (a) P. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS; (b) Z. Xi and Z. Li, Top. Organomet. Chem., 2004, 8, 27–56 CrossRef.
- J. Klaucke, N. Sinthathurai, C. Golz, O. P. E. Townrow and M. Fischer, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-4lzc5.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- R. J. Baker, T. Bannenberg, A. Kunst, S. Randoll and M. Tamm, Inorg. Chim. Acta, 2006, 359, 4797–4801 CrossRef CAS.
- For a summary of Group 14 NHC complexes and structural parameters see: C. D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC.
- For details see the ESI.†.
- See for example: (a) J. Ruiz and B. F. Perandones, J. Am. Chem. Soc., 2007, 129, 9298–9299 CrossRef CAS PubMed; (b) M. Brill, J. Diaz, M. A. Huertos, R. Lopez, J. Perez and L. Riera, Chem. – Eur. J., 2011, 17, 8584–8595 CrossRef CAS PubMed.
- (a) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed; (b) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- N. Tsoureas, J. C. Green and F. G. N. Cloke, Chem. Commun., 2017, 53, 13117–13120 RSC.
- S. S. Semproni, C. Milsmann and P. J. Chirik, Organometallics, 2012, 31, 3672–3682 CrossRef CAS.
- (a) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC; (b) A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- P. Yu, E. F. Murphy, H. W. Roesky, P. Lubini, H.-G. Schmidt and M. Noltemeyer, Organometallics, 1997, 16, 313–316 CrossRef CAS.
- H. Braunschweig, C. Brückner, M. A. Celik, K. Dück, F. Hupp, T. Kramer, J. Krebs and I. Krummenacher, Chem. – Eur. J., 2015, 21, 11056–11064 CrossRef CAS PubMed.
- C. Barnett, M. L. Cole and J. B. Harper, Eur. J. Inorg. Chem., 2021, 4954–4958 CrossRef CAS.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- E. Bill, Simulation of the experimental magnetic data was performed with the julX_2S program, Max-Planck Institute for Chemical Energy Conversion, Mülheim/Ruhr, Germany Search PubMed.
- Bruker, SAINT, V8.40B, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, B72, 171–179 Search PubMed.
- D. Kratzert, FinalCif, V132, https://dkratzert.de/finalcif.html.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, Phys. Rev. A:At., Mol., Opt. Phys., 1998, 38, 3098–3100 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B:Condens. Matter Mater. Phys, 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Eznzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- NBO 6.0, E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013 Search PubMed.
- A. Gilbert, IQMol 3.1.4, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of the reported compounds, crystallographic and computational details. CCDC 2419251–2419254. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00480b |
| This journal is © The Royal Society of Chemistry 2025 |