
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast Li+ transport kinetics enabled by TiN nanofibers in hybrid polymer-based electrolytes for long-life Li metal batteries†
Yixin
Wu
a,
Zhen
Chen
 *a,
Kai
Shi
bc,
Yang
Wang
a,
Xian-Ao
Li
a,
Ziqi
Zhao
a,
Quan
Zhuang
*d,
Jian
Wang
*bc and
Minghua
Chen
*a
*a,
Kai
Shi
bc,
Yang
Wang
a,
Xian-Ao
Li
a,
Ziqi
Zhao
a,
Quan
Zhuang
*d,
Jian
Wang
*bc and
Minghua
Chen
*a
aKey Laboratory of Engineering Dielectric and Applications (Ministry of Education), School of Electrical and Electronic Engineering, Harbin University of Science and Technology, Harbin 150080, Heilongjiang, China. E-mail: chen.zhen@hrbust.edu.cn; mhchen@hrbust.edu.cn
bHelmholtz Institute Ulm (HIU), 89081 Ulm, Germany
cKarlsruhe Institute of Technology (KIT), 76021 Karlsruhe, Germany. E-mail: jian.wang@kit.edu
dInner Mongolia Key Laboratory of Solid State chemistry for Battery, Nano Innovation Institute (NII), College of Chemistry and Materials Science, Inner Mongolia Minzu University, Tongliao 028000, China. E-mail: zhuangquan21@outlook.com
First published on 19th February 2025
Abstract
Polymer-based solid-state electrolytes exhibit superior advantages in flexibility, light weight, and large-scale processability, rendering them promising for high-performance solid-state lithium metal batteries (SSLMBs) with enhanced safety. However, challenges like poor structural uniformity, sluggish Li+ transport kinetics, and inferior interface compatibility hinder their practical applications. Herein, a hybrid quasi-solid-state electrolyte (PHLT) composed of a titanium nitride (TiN) fibrous nanofiller and a poly(vinylidene fluoride-co-hexafluoropropylene)/lithium bis(trifluoromethanesulfonyl)imide (PVDF–HFP/LiTFSI) matrix was developed. The inorganic filler could decrease the crystallinity of PVDF–HFP, propel the polar transformation of the polymer, as well as adsorb and immobile the TFSI− anions, significantly enhancing Li-ion transport kinetics. Furthermore, the in situ generated fast Li-ion conductor, i.e., LixTiN, derived from lithiated TiN, along with a smooth but dense LiF interphase, effectively bridges the electrolyte|electrode interface and suppresses Li dendrite growth. Consequently, the as-fabricated Li|PHLT|LiFePO4 cells achieve exceptional cycling stability over 3000 cycles at 2 C with a superior average Coulombic efficiency of 99.8%. Notably, this strategy also enables great compatibility with matching high-loading cathodes (9.5 mg cm−2), moreover, it delivers impressive performance in large areal pouch cells as well as bilayer stacking cells. This work provides an innovative approach to constructing solid-state electrolytes with enhanced diffusion kinetics and interface compatibility, paving the way for practical SSLMB applications.
Broader contextIn the rapidly evolving fields of battery technology and energy storage, polymer-based electrolytes are gaining extensive attention for achieving high-energy-density lithium metal batteries (LMBs) with high safety owing to their exceptional flexibility and manufacturability. However, for the inorganic filler, most studies focus on a simple mixture with polymers to decrease the crystallinity of polymers to achieve a higher ion conductivity. Meanwhile, little attention has been paid to the interfacial ion transfer, which leads to higher barriers. In this work, as a proof-of-concept, an innovative conductive inorganic filler has been proposed to optimize the electrochemical performance of the quasi-solid-state polymer electrolytes of poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF–HFP). More importantly, the lithiated phase transformation of an inorganic filler into a lithiated LixTiN layer helps to accelerate the lithium salt dissociation, enhance the Li+ transport kinetics, and improve the interface compatibility, providing new insights for the design of advanced polymer electrolytes for high-performance LMBs. |
Introduction
The increasing prevalence of lithium-ion batteries (LIBs) in electric vehicles and large-scale energy storage systems has led to surging demand for high energy density, safety standards, and long life. Compared to the commercial graphite anode, the lithium metal anode exhibits an exceptionally high theoretical capacity (3860 mA h g−1) and the lowest reduction potential (−3.04 V vs. standard hydrogen electrode), rendering it the optimal choice for high-energy-density lithium metal batteries (LMBs).1–4 However, the failures of LMBs have shown that metallic Li cannot operate safely and stably in traditional liquid electrolyte and polyolefin separator systems. The main issue can be attributed to the growth of lithium dendrites that lead to short circuits and possible thermal runaway or even explosions. Furthermore, traditional organic liquid electrolytes are plagued by high flammability and serious side reactions with active metallic Li.5,6 As an alternative, solid-state electrolytes or quasi-solid-state electrolytes exhibit high mechanical strength and stability, inhibit the lithium dendrite growth, expand the operating temperature range of batteries, and fundamentally eliminate the risk of electrolyte leakage.7,8 Consequently, this advancement is expected to enable the realization of high energy density and high safety in LMBs.Compared to the hard and brittle inorganic sulfide or oxide-based electrolytes, polymer-based electrolytes such as polyethylene oxide,9 polyvinylidene fluoride (PVDF),10 poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF–HFP),11 and polyacrylonitrile,12 exhibit superior flexibility and toughness as well as the ease of large-scale fabrication, attracting extensive attention and investigations. Among them, PVDF–HFP stands out for its high dielectric constant (εr: ∼8–12), low crystallinity and superior thermal stability, making it highly suitable for a wide range of practical applications.13 Nevertheless, there are still some limitations to be overcome, such as low ionic conductivity at room temperature, a non-compact structure and interfacial parasitic reactions, which restrict their further development.
To address the above-mentioned challenges, numerous research approaches have been proposed including the utilization of electrolyte additives,14 the regulation of electrolyte composition,15 the grafting of polymers,6in situ polymerization strategies,16 and the incorporation of inorganic fillers.17 Among these strategies, the integration of inorganic fillers has demonstrated the potential to harmonize the electrochemical properties and mechanical strength of the electrolyte. Currently, the main inorganic fillers focus on oxides, while little attention has been paid to utilizing nitride materials such as BN, which shows exceptional insulating properties.18 Meanwhile, the research on conductive transition metal nitride fillers remains scarce, likely due to concerns about their high electronic conductivity potentially compromising the insulating properties of electrolytes. However, recent studies have reported that the inclusion of conductive fillers does not necessarily impair the electronic insulation of electrolytes.19 Instead, the surface polarization effect of conductive fillers can facilitate the dissociation of lithium salt in a manner analogous to high dielectric fillers.20 The polarization phenomenon redistributes the interfacial electric field, weakens the Coulombic interaction between cations and anions, and amplifies the local electric field strength within the polymer matrix. Therefore, incorporating metal-based nitride fillers can effectively enhance Li salt dissociation, increase carrier concentration, and improve the electrochemistry performance of electrolytes. Furthermore, the gradual conductive–dielectric transition significantly promotes fast ion transport, enabling superior performance of the LMBs.
Herein, as a “proof-of-concept”, a dielectric PVDF–HFP based quasi-solid-state polymer electrolyte (QPE) integrating optimized TiN nanofiber fillers was proposed. The TiN fillers possess superior mechanical properties, thermal conductivity, exceptional chemical/electrochemical stability, and metallic-like conductivity, effectively enhancing the comprehensive properties of the electrolyte. In this design, as exhibited in Fig. 1A, the incorporation of TiN nanofibers greatly promotes the dissociation of LiN(SO2CF3)2 (LiTFSI) and accelerates Li-ion kinetics through several aspects: (1) the stronger adsorption of TFSI− anions on the TiN (111) surface provides an “anchor effect” to restrict anion mobility; (2) TiN facilitates the phase transformation of PVDF into its β-phase, which features with a higher dielectric constant enabling reduced dissociation barrier; and (3) the surface of TiN is in situ lithiated to form the LixTiN layer during cycling, further enhancing Li-ion transport dynamics alongside the heterogeneous interfaces. Additionally, the high aspect ratio of fibrous TiN increases the proportion of organic–inorganic interfaces. Combined with the contribution from favored β-phase transformation, continuous and shortened Li-ion transport pathways are created for uniform and fast Li-ion flux, yielding substantially enhanced Li+ transport kinetics. Consequently, these advantages enable stable operation of the Li‖Li symmetric cells and Li‖LiFePO4 full cells. More impressively, the bipolar stacking cells and pouch cells also demonstrate excellent safety and stability, showcasing the practical feasibility of this design.
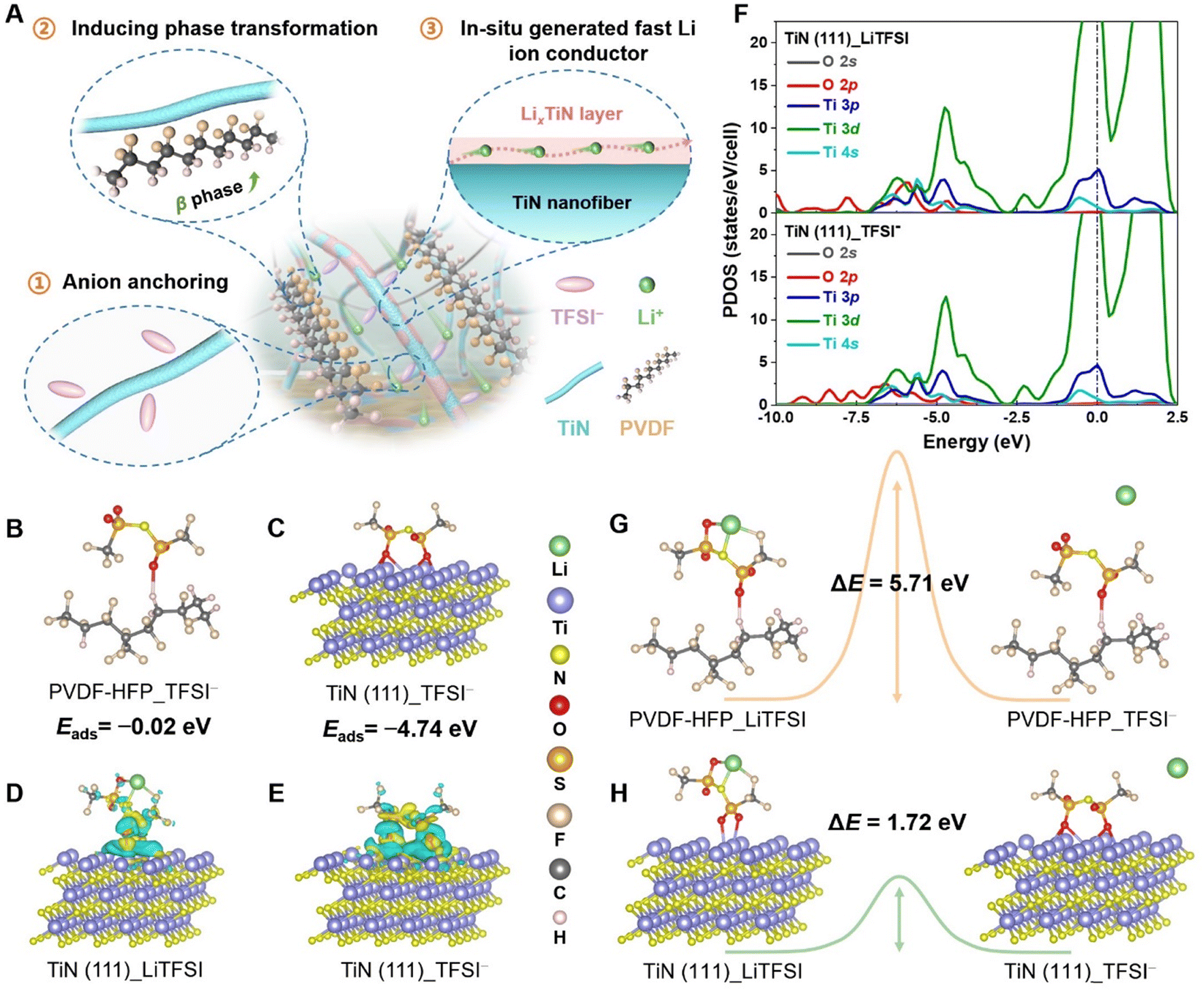 | ||
| Fig. 1 (A) Schematic diagram of the functions of TiN nanofibers in the electrolyte. The adsorption configurations of TFSI− anion on the (B) PVDF–HFP and (C) TiN (111). The calculated charge density difference for (D) LiTFSI and (E) TFSI− adsorption on TiN (111); cyan and yellow iso-surfaces represent electrons corresponding to charge density contours of −0.0025 and +0.0025 eÅ−3, respectively. (F) PDOS plots of LiTFSI and TFSI− on TiN (111). The dissociation energy barrier of LiTFSI on surfaces of (G) PVDF–HFP and (H) TiN (111), respectively. | ||
Results and discussion
Theoretical simulations of TiN fillers on the dissociation behavior of LiTFSI
Firstly, density functional theory (DFT) calculations were conducted to gain insights into the interactions and dissociation ability of LiTFSI on both the pristine PVDF–HFP chain segments and the TiN (111) crystal plane. Fig. 1B illustrates that the adsorption energy of TFSI− anions on PVDF–HFP is minimal at only 0.02 eV. Conversely, TiN exhibits significantly stronger adsorption energy as high as 4.74 eV (Fig. 1C). This adsorption is conducive to limiting the free movement of anions, thereby increasing the Li+ transference number and reducing the polarization of the electrolyte. The charge density difference plots between LiTFSI/TFSI− and Ti atoms further reveal the interaction between TiN and TFSI− anions (Fig. 1D and E), evidenced by the charge accumulations between TFSI− and the O atoms. The interactions can also be corroborated by the projected density of states (PDOS) and crystal orbital Hamilton population (COHP) analyses. The hybridization between the electronic orbitals of O and Ti atoms is evident, aligning well with the COHP results, which show fully occupied bonding states for the Ti–O bonds. This suggests strong anion confining by TiN (Fig. 1F and Fig. S1, ESI†). The bonding strength can be quantified by the integral COHP (ICOHP), which measures the energy-weighted population of bonding wave functions between two atomic orbitals. The larger absolute value of ICOHP reflects stronger bonding. For LiTFSI/TFSI− adsorption on the TiN (111) surface, two and five Ti–O bonds are formed with average lengths of 2.26 Å and 2.38 Å, corresponding well with the absolute ICOHP values of 1.67 and 1.34.Due to the strong attraction exerted by TiN, the dissociation barrier for lithium salts is significantly reduced to 1.72 eV (Fig. 1G), which is considerably lower than that of the PVDF–HFP (5.71 eV, Fig. 1H)—approximately one-third of the barrier in the pristine polymer matrix. The above investigations and findings reveal that TiN is more conducive to decoupling LiTFSI and acting as an “anchor” for TFSI− as well, thereby reducing the interaction between Li+ and TFSI−. Consequently, the utilization of TiN as a modified filler is anticipated to facilitate the dissociation of lithium salts, liberating a substantial amount of Li+, which in turn enhances the electrochemical properties of the electrolyte. Simultaneously, the confinement of TFSI− anions further contributes to improved ionic transport and stability, making TiN an effective additive for advancing solid-state lithium-ion battery performance.
Structural characterization of TiN nanofibers and electrolyte membranes
The TiN nanofibers retain the fibrous morphology, similar to the precursor (Fig. S2, ESI†), with a diameter of approximately 500 nm (Fig. S3A, ESI†). The structure is consistent with the X-ray diffraction (XRD) pattern of the Osbornite TiN phase (PDF #06-0642) (Fig. S3B, ESI†). Ionic conductivity testing demonstrates that the quasi-solid-state electrolyte incorporating 5 wt% TiN (denoted as PHLT) exhibits the highest ionic conductivity, ca., 1.29 mS cm−1 (Fig. S4, ESI†), which is four times higher than that of the PVDF–HFP electrolyte without fillers (PHL, 0.29 mS cm−1). As exhibited in Fig. 2A, the as-synthesized PHLT membrane appears much smoother and more compact, indicating the potential for tight electrolyte/electrode contact. Cross-sectional scanning electron microscope (SEM) images show that the thicknesses of PHL and PHLT are approximately 55 and 53 μm, respectively (Fig. 2B and Fig. S5, ESI†). Notably, the long fibrous structure facilitates superior dispersibility of filler within the polymer matrix (Fig. S6, ESI†).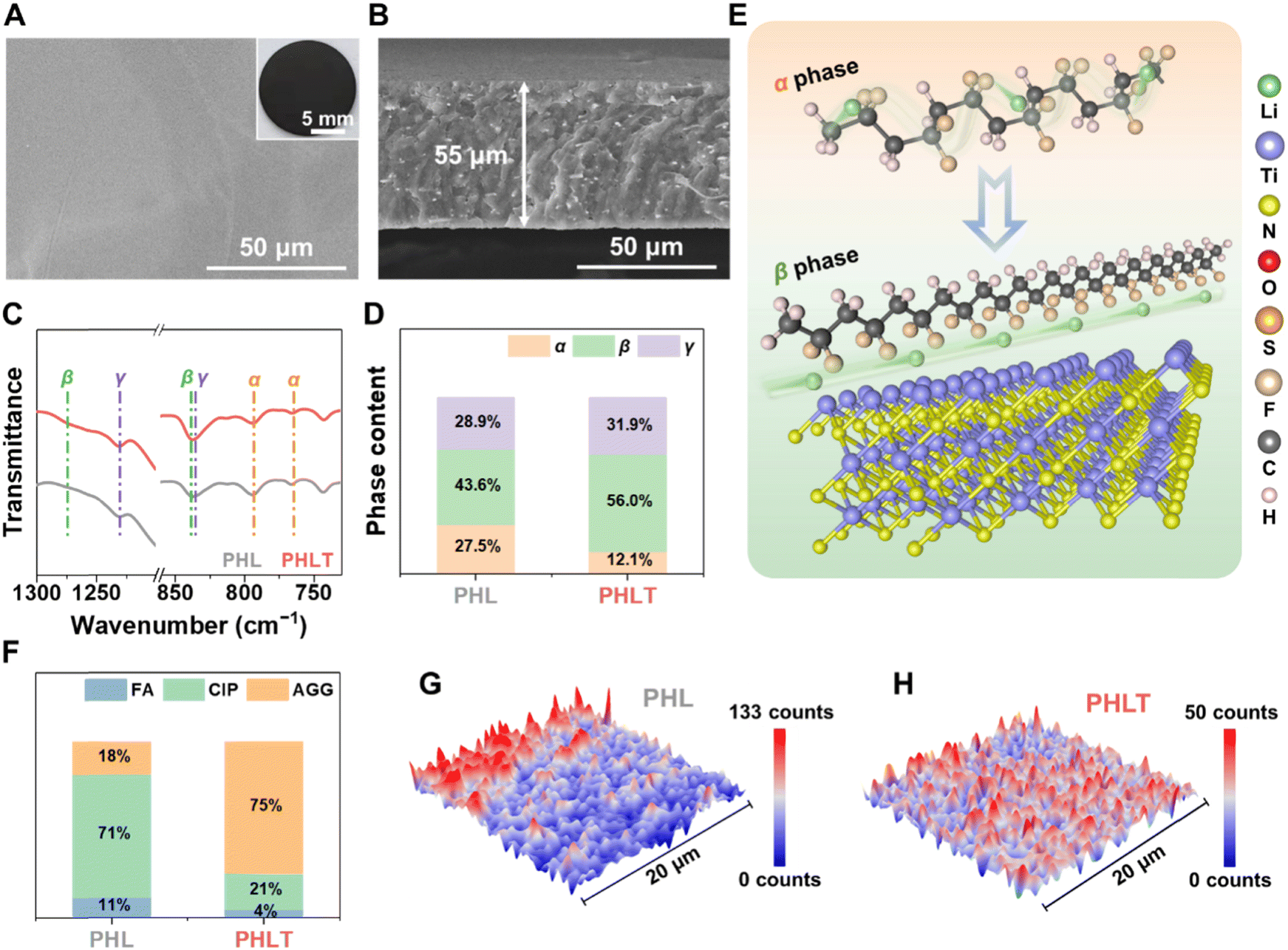 | ||
| Fig. 2 (A) The top-view and (B) cross-sectional SEM images of PHLT, respectively; the inset is an optical photograph. (C) Fourier transform infrared spectrometer (FT-IR) spectra and (D) the crystal phase content of PVDF in PHL and PHLT. (E) Schematic diagram of the conformational transformation of the polymer matrix and the facilitated Li+ transport pathway. (F) The corresponding Raman quantification results of the TFSI− state in the electrolytes. Raman mapping plots of the TFSI− signal at 730–760 cm−1 of (G) PHL and (H) PHLT. | ||
For practical applications, the mechanical properties of electrolytes are crucial. Fig. S7 (ESI†) displays an optical photograph of PHLT possessing a smooth and dense morphology with excellent flexibility and foldability, which is beneficial for future flexible smart devices. The PHLT achieves a significantly higher mechanical toughness and strain of 5.78 MPa and 197.9% (Fig. S8, ESI†), remarkably outperforming PHL (2.86 MPa and 17.1%). Even after wetting with liquid electrolyte, the PHLT still retains superior mechanical strength and improved ductility, ensuring effective dendrite suppression while maintaining excellent interfacial compatibility. Furthermore, the PHLT exhibits superior thermal conductivity, evidenced by uniform heat distribution and a minor temperature difference (Fig. S9, ESI†), which is anticipated to balance the uneven electrochemical kinetics in space caused by thermal contrast. The decomposition temperature of PHLT is about 20 °C higher than that of the PHL, and the dimethyl formamide (DMF) contents in PHL and PHLT are less than 3 wt% (Fig. S10, ESI†).21 FT-IR results confirm that all DMF molecules are coordinated with Li+ ions, ensuring their complete interaction with the electrolyte matrix (Fig. S11, ESI†).
The lower the crystallization of the polymer, the higher the Li-ion transport kinetics. The XRD (Fig. S12 and S13, ESI†) and differential scanning calorimetry (DSC) analyses (Fig. S14, ESI†) reveal that the crystallinity degree and crystallization temperature are simultaneously reduced in PHLT, suggesting that the TiN fillers facilitate the formation of amorphous regions to enhance the transport of Li+. Additionally, a shift from non-polar α-phase (766 cm−1) to polar β/γ-phases (840 cm−1) is also witnessed by FT-IR, which is primarily attributed to the interactions between TiN and the dipole moment of the polymer (Fig. 2C). The proportion of the β-phase and γ-phase can be determined by the absorbance peaks at 1276 and 1232 cm−1, respectively.22 As estimated, the incorporation of TiN leads to a decreased α-phase and increased β-phase by 15.4% and 12.4%, respectively (Fig. 2D). Thus, the incorporation of TiN fillers in PHLT has profound effects on reducing the polymer crystallization and promoting the favored composition of the β-phase, enhancing Li-ion transport kinetics. Furthermore, the polarity phase could enhance the dissociation of LiTFSI and shorten the Li+ migration path, thereby reducing the diffusion barrier of Li+ (Fig. 2E).23 Raman analysis further reveals that the TFSI− anion presents a higher proportion of contact ion pairs (CIP, ∼71%) and a lower proportion of free anion (FA) in PHL, indicating the weak dissociation of salt. The aggregate (AGG, ∼75%) dominates in the PHLT, corresponding to the coupling between TFSI− and TiN (Fig. 2F and Fig. S15, ESI†).
Furthermore, the Raman mapping of PHL reveals the presence of a lithium salt-enriched area, confirming that PHL has an uneven distribution of anions, leading to a heterogeneous solid electrolyte interphase (SEI) structure and electrochemical performance (Fig. 2G). In contrast, PHLT shows a uniform distribution of anions (Fig. 2H), indicating consistent composition and electrochemical properties at the micrometer scale owing to the even dispersion of TiN fillers and their anchoring effect on TFSI− anions.
Evaluation of electrochemical properties and stability against the Li metal anode
As expected, the PHLT not only demonstrates a higher ionic conductivity but also a reduced activation energy of 0.14 eV (Fig. 3A). The Li+ transference number (tLi+) increases to 0.46 (Fig. 3B and Fig. S16, ESI†) due to the “anchor” effect of TiN on anions. Notably, TiN fillers exhibit a negligible effect on the electronic conductivity of PHLT, ensuring it is sufficient to meet the requirement of batteries (Fig. S17 and Table S1, ESI†). Furthermore, the PHLT shows a higher exchange current density of 0.142 mA cm−2 and a limiting current density of up to 4.3 mA cm−2, surpassing those of PHL (Fig. 3C and Fig. S18, ESI†). The electrochemical stability window of the PHLT is broadened to 0–4.89 V (Fig. 3D), and the flat cyclic voltammetry (CV) curves of Li|PHLT|stainless steel cells in the voltage range of 0–3 V and 3–5 V (Fig. S19, ESI†) further confirm its superior electrochemical stability both towards oxidation and reduction reactions. The enhanced ionic conductivity facilitates efficient charge transfer at the electrode–electrolyte interface, reducing Li+ accumulation and minimizing localized overpotentials, thereby reducing the risk of localized oxidation. Additionally, TiN fillers promote anion anchoring, which inhibits their deposition and decomposition at the cathode side to some extent, further improving the oxidation stability of the electrolyte.24 Subsequently, the critical current density (CCD) values are determined in Li‖Li cells, as illustrated in Fig. 3E and F and Fig. S20 (ESI†). In the time-constant mode, the CCD value increases from 0.6 mA cm−2 to 0.85 mA cm−2. In the capacity-constant mode, PHLT exhibits CCD values of 3.1, 1.6, and 0.7 mA cm−2 when the capacity is set at 0.2, 0.5, and 1 mA h cm−2, respectively, significantly higher than those of the PHL (2.1, 1.0, and 0.3 mA cm−2). These enhanced CCD values highlight that TiN nanofibers reinforce the electrolyte's capability of inhibiting dendritic lithium growth. The PHLT-based Li symmetric cells operate stably over 1400 h at 0.1 mA cm−2 (Fig. 4A), significantly outlasting the Li|PHL|Li symmetric cell (376 h, Fig. S21, ESI†). Increasing the current density to 0.2 mA cm−2, the Li|PHLT|Li symmetric cell still enables a longer life (Fig. S22, ESI†) with smaller resistances (Fig. S23, ESI†). To investigate the complex interfacial evolution and kinetics characteristic, electrochemical impedance spectroscopy (EIS) was further analyzed by the distribution of relaxation times (DRT), which is a model-free, time-domain analysis technique (Fig. 4B, C and Fig. S24, ESI†). Three typical electrochemical processes are identified at 10−6 s, 10−4–10−3 s, and 10−2–10−1 s, corresponding to electrolyte resistance (Rb), SEI resistance (RSEI), and charge-transfer resistance (Rct), respectively.25 The Li|PHL|Li cell exhibits a high RSEI value initially, which rapidly decreases due to lithium dendrite growth. Meanwhile, dead lithium and parasitic reactions lead to the deterioration of PHL, resulting in a rapid rise in Rb. In stark contrast, the Li|PHLT|Li cell displays much more stable cycling behavior with minimal fluctuations. Interestingly, a unique new peak emerges at 10−5 s after initial cycling, indicative of the formation of a novel Li+ conductive phase, which differs from the pristine PHLT (Fig. 4C). Correspondingly, the Li–N bond has been detected on the cycled TiN surface by ex situ X-ray photoelectron spectroscopy (XPS) analysis (Fig. 4D and E), supporting the formation of the lithiated TiN phase, i.e. LixTiN. Previous studies have demonstrated the feasibility of TiN lithiation, with the lithiation behavior occurring on its surface and grain boundary.26,27 In addition, even if there exists the possible dendrite formation on the Li metal surface, the TiN fillers help mitigate lithium dendrite growth by reacting with dendrites, thereby preventing short-circuiting and prolonging the lifespan of lithium metal batteries. These findings suggest that TiN filler not only enhances the ion transport kinetics of the electrolyte but also stabilizes the interface between the electrolyte and Li metal, thereby improving cycling stability. Furthermore, the presence of the LiTFSI signal on the cycled TiN surface confirms that LiTFSI molecules are strongly bound to the filler surface and cannot be completely removed even after repeated washing and centrifugation, providing additional evidence for the strong adsorption between LiTFSI and TiN.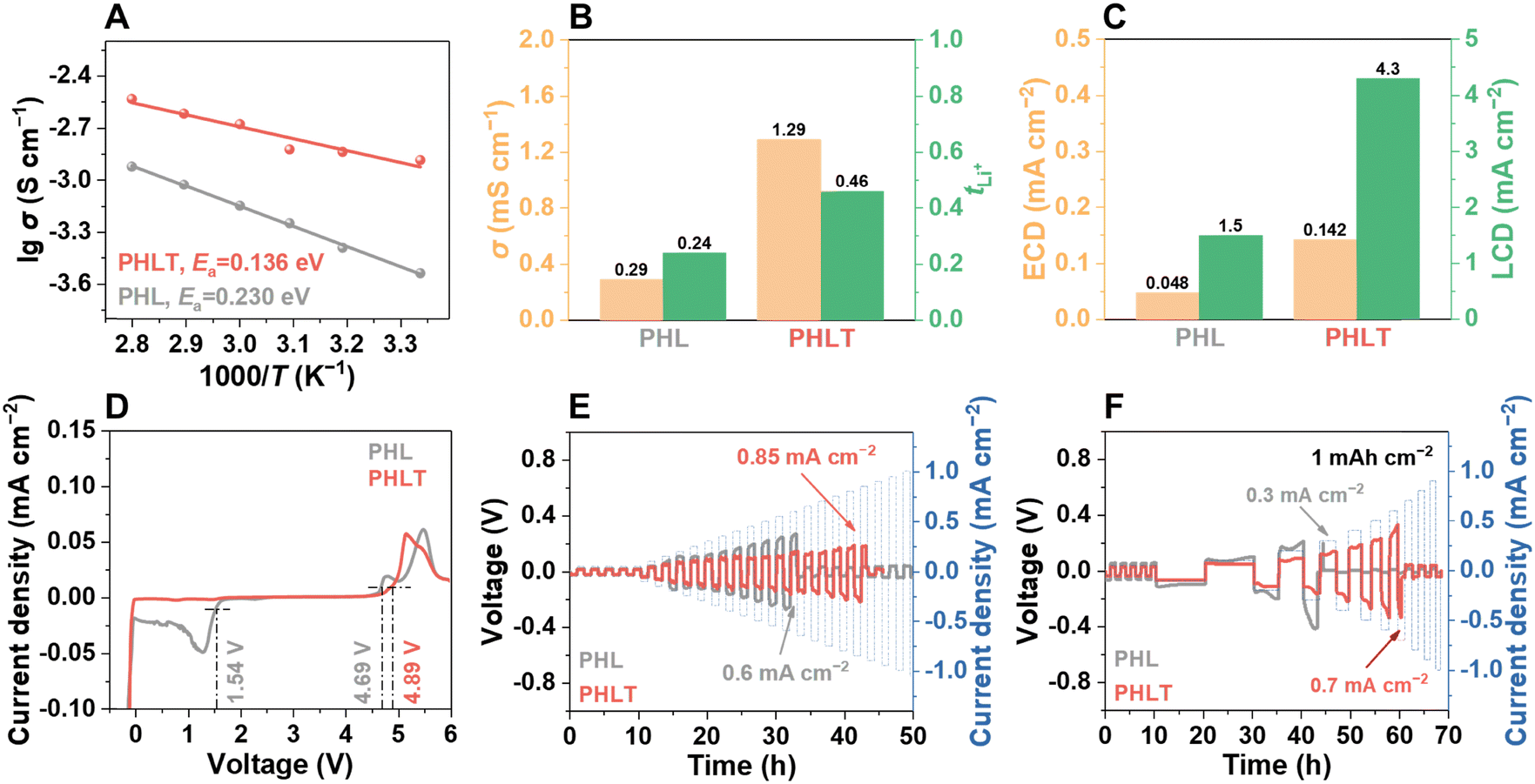 | ||
| Fig. 3 (A) Arrhenius plots of the QPEs. (B) Ionic conductivity and Li+ transference number, (C) exchange current density (ECD) and limiting current density (LCD) results, (D) LSV curves and CCD tests using (E) time-constant mode (1 h for platting and 1 h for stripping), and (F) the capacity-constant mode (1 mA h cm−2) at 25 °C, respectively. | ||
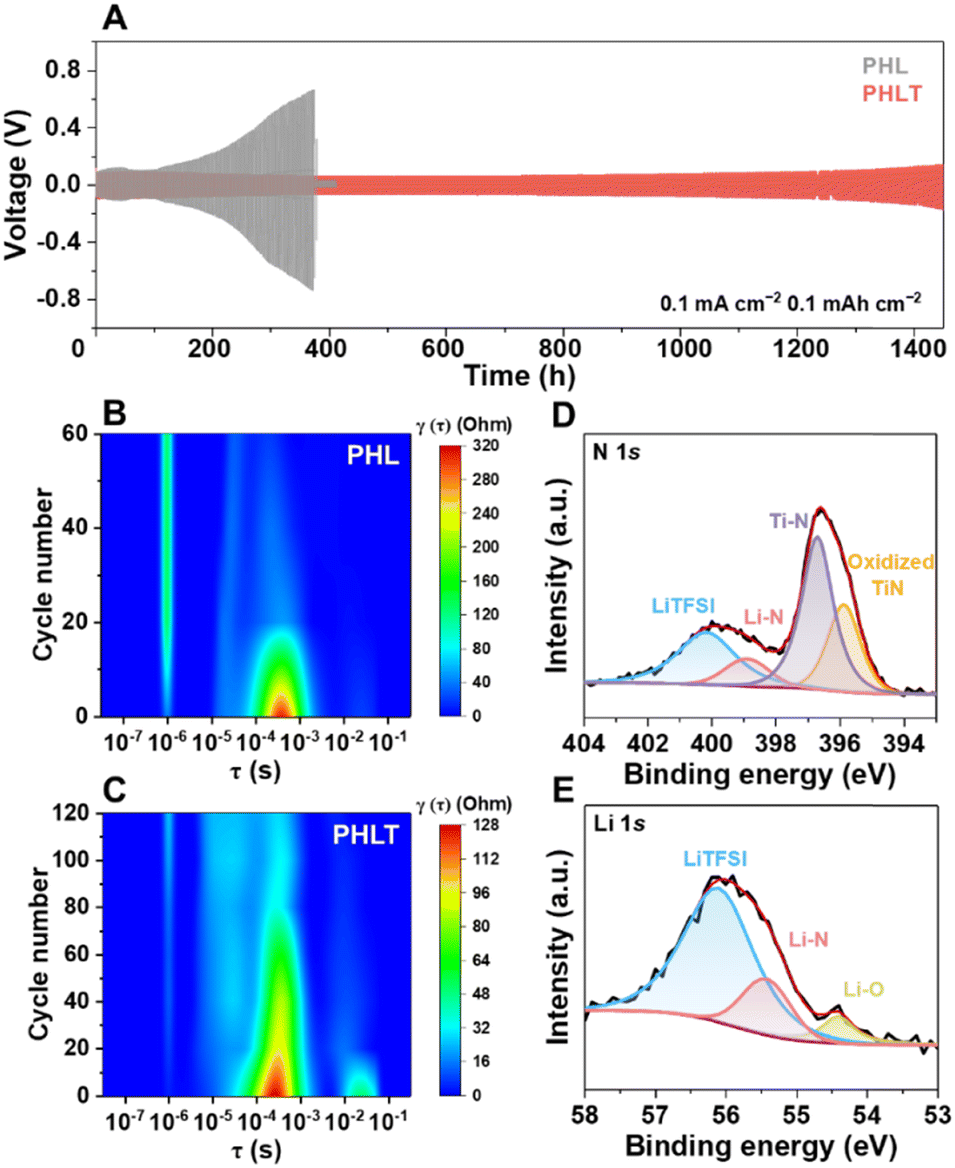 | ||
| Fig. 4 (A) Lithium stripping-plating profiles at 0.1 mA cm−2 and 0.1 mA h cm−2. DRT results of the (B) Li|PHL|Li and (C) Li|PHLT|Li cells cycled at 0.2 mA cm−2 and 0.2 mA h cm−2. Ex situ XPS analysis of (D) N 1s and (E) Li 1s spectra of recovered TiN nanofibers. | ||
Investigation of the composition and structure of the solid electrolyte interphase
The Li‖Li cells (300 h, 0.1 mA cm−2, and 0.1 mA h cm−2) were disassembled to analyze the morphologies of the cycled lithium metal electrodes and the SEI components. Fig. S25 (ESI†) shows the SEM image of fresh Li, which displays a relatively smooth and flat surface. While in the case of using PHL as the electrolyte, the Li metal shows a mossy and porous surface with an 80–130 μm thick reaction layer (Fig. 5A, B and Fig. S26A, B, ESI†). In sharp contrast, a significantly flat and denser morphology is observed by using PHLT electrolyte (Fig. 5C, D and Fig. S26C, D, ESI†), indicating suitable contact and uniform Li+ distribution. Ex situ XPS reveals significant differences in the SEI composition of the two systems (Fig. S27 and Table S2, ESI†). In the PHL system, the SEI contains massive organic components and Li2CO3 is produced due to the decomposition of solvents, while LiF and Li3N are found only in minor quantities (Fig. 5E and Fig. S28, ESI†). However, in the case of PHLT, the organic components are minimal (∼4%), instead, with an F-rich SEI layer as high as ∼53% (Fig. 5F). The robust and stable LiF exceeds over 90% in the F 1s spectrum, and a higher proportion of Li3N is also observed (Fig. S29, ESI†). Besides, the ex situ time of flight secondary ion mass spectrometry (ToF-SIMS) analysis was also performed to provide the spatial distribution of SEI components.28,29 The depth profiling of secondary ion fragments (Fig. 5G and Fig. S30, ESI†) demonstrates that in the PHL system, the concentration of the F species is relatively low, whereas the proportions of organic species (CH2−), anionic fragments (SO2N−), and CO3− are higher. | ||
| Fig. 5 Ex situ SEM images of the (A) and (C) surface and (B) and (D) cross-sectional view of the cycled Li electrodes recovered from Li‖Li cells. Ex situ XPS spectra of (E) C 1s and (F) F 1s of the cycled Li electrodes. (G) ToF-SIMS depth profiling, (H) surface mapping, and (I) 3D reconstruction images of the F−, CH2−, and SO2N− fragments of the Li electrode operated with PHLT. Schematic representation of lithium deposition morphology on the Li anode using (J) PHL or (K) PHLT as the electrolyte. | ||
In sharp contrast, PHLT promotes an abundant and homogeneous distribution of F species within the SEI, with a reduction of CH2− and SO2N− species (Fig. 5H, I and Fig. S31, S32, ESI†). Consequently, TiN fillers are instrumental in promoting the formation of a robust, LiF-dominated dense SEI, which substantially inhibits the lithium dendrite growth, and minimizes the interfacial parasitic reactions (Fig. 5K). In contrast, in the PHL system, persistent side reactions lead to the generation of dead lithium and lithium dendrites (Fig. 5J).
Evaluation of full cell performance and safety property
The CV curves (Fig. S33, ESI†) confirm the superior reversibility and enhanced reaction kinetics of the PHLT electrolyte. The Li|PHLT|LiFePO4 (LFP) cell delivers specific discharge capacities of 158.7 (0.1 C), 153.3 (0.2 C), 143.3 (0.5 C), 131.6 (1 C), 117.2 (2 C), and 88.6 (5 C) mA h g−1, respectively (Fig. 6A), which are much higher than those of the Li|PHL|LFP cell, i.e., 141.2 (0.1 C), 122.3 (0.5 C) and 38.9 (5 C) mA h g−1. Furthermore, the Li|PHLT|LFP cell exhibits a flatter charge/discharge voltage and smaller polarization (Fig. S34, ESI†). The Li|PHLT|LFP cell achieves excellent long-term cycling stability, evidenced by a capacity retention of 62.2% after 3000 cycles at 2 C with an average Coulombic efficiency of 99.8% (Fig. 6B). | ||
| Fig. 6 (A) Rate capabilities and (B) cycling stability of the Li‖LFP cells. (C) A comparison of C-rate, cycling number, and capacity retention between this work and other reported PVDF–based electrolytes in recent literature. (D) Cycling stability and (E) charge/discharge voltage profiles of the bipolar stacking cell. (F) The cycling performance of a Li|PHLT|LFP pouch cell at 0.2 C. (G) Optical photographs illustrating the bending and cutting tests of the pouch cell. | ||
This performance contrasts sharply with the Li|PHL|LFP cell, which only retains a capacity of 19.7 mA h g−1 after 700 cycles, corresponding to a capacity retention ratio of 18.0%. Correspondingly, the impedance evolution of the Li|PHL|LFP cell demonstrates continuous growth over 400 cycles, while the Li|PHLT|LFP cell maintains remarkably stable impedance throughout cycling (Fig. S35, ESI†). The Li|PHLT|LiNi0.8Co0.1Mn0.1O2 cells also display superior rate capability and cycling stability (Fig. S36 and Table S3, ESI†). Compared with the recently reported PVDF–based electrolytes modified by inorganic fillers, the TiN nanofiber enhanced polymer-based electrolyte stands out with its impressive cycling stability and overall electrochemical performance (Fig. 6C and Table S4, ESI†).30–39
Thanks to the immobilization properties of the QPE, we successfully constructed bilayer bipolar stacking cells comprising two Li|PHLT|LFP units in series using stainless steel sheets as the bipolar plates (Fig. S37, ESI†). This design enables a high-voltage output in a single cell, reduces internal resistance, simplifies packaging, and improves the overall energy density of the cells, demonstrating its significant potential for practical applications. At 0.1 C, the cell enables outstanding capacity retention of 89.5% after 100 cycles and an average Coulombic efficiency of 99%, outputting a discharge voltage platform of 6.7 V (Fig. 6D and E). To the best of our knowledge, the bipolar stacking cells in this work exhibit superior cycling stability and capacity retention compared to others reported in the literature (Fig. S38, ESI†).40–45 Furthermore, coupling with high-mass-loading cathode electrodes of 9.5 mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2 (∼1.6mA hcm−2), the Li|PHLT|LFP cell still delivers a high specific discharge capacity of over 150 mA h g−1 (0.1 C) for more than 30 cycles (Fig. S39, ESI†). Additionally, a single-layer pouch cell (4.5 × 5.8 cm2) was assembled with an LFP cathode and thin lithium foil (80 μm). The pouch cell delivers a discharge capacity of 153 mA h g−1 at 0.2 C, and capacity retention of 89.2% after 300 cycles (Fig. 6F and Fig. S40, ESI†). The pouch cell shows stable operation even under severe abuse conditions such as bending or cutting (Fig. 6G and Fig. S41, ESI†), underscoring its superior mechanical flexibility and excellent safety. These results, collectively, highlight the PHLT as a promising candidate for practical diverse applications.
cm−2 (∼1.6mA hcm−2), the Li|PHLT|LFP cell still delivers a high specific discharge capacity of over 150 mA h g−1 (0.1 C) for more than 30 cycles (Fig. S39, ESI†). Additionally, a single-layer pouch cell (4.5 × 5.8 cm2) was assembled with an LFP cathode and thin lithium foil (80 μm). The pouch cell delivers a discharge capacity of 153 mA h g−1 at 0.2 C, and capacity retention of 89.2% after 300 cycles (Fig. 6F and Fig. S40, ESI†). The pouch cell shows stable operation even under severe abuse conditions such as bending or cutting (Fig. 6G and Fig. S41, ESI†), underscoring its superior mechanical flexibility and excellent safety. These results, collectively, highlight the PHLT as a promising candidate for practical diverse applications.
Conclusions
In summary, we propose a structurally compact, kinetically enhanced, and interface-stable QPE by incorporating multifunctional TiN nanofiber fillers into PVDF–HFP-based electrolyte. The interactions between the polymer matrix and TiN fillers promote conformational transformation and enhance the dissociation of lithium salt while constraining the movement of anions. Additionally, the introduction of TiN facilitates the formation of a LiF-rich and dense SEI, which ensures uniform lithium deposition, inhibits lithium dendrite growth, and effectively mitigates parasitic reactions. Consequently, the Li‖LiFePO4 cells can operate stably for over 3000 cycles at 2 C. The two-layer bipolar stacking cells (5–8 V), high mass-loading coin cells (9.5 mg cm−2), and pouch cells also exhibit excellent cycling and safety performance. Our work provides an efficient and valuable strategy for leveraging the multifunctional properties of fillers to enhance the performance of polymer electrolytes, facilitating their practical applications in SSLMBs.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (52277215 and 52377206), the China Postdoctoral Science Foundation (2023M730884), the Heilongjiang Provincial Postdoctoral Science Foundation (LBH. Z23024), and the opening funding from Key Laboratory of Engineering Dielectrics and Its Application (Harbin University of Science and Technology) (No. KFM202507, Ministry of Education). Dr. Jian Wang also thanks the fellowship support from the Alexander von Humboldt Foundation.References
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- X. Wang, Z. Chen, K. Jiang, M. Chen and S. Passerini, Adv. Energy Mater., 2024, 14, 2304229 CrossRef CAS.
- J. Wang, J. Zhang, Y. Zhang, H. Li, P. Chen, C. You, M. Liu, H. Lin and S. Passerini, Adv. Mater., 2024, 36, e2402792 CrossRef PubMed.
- J. Wang, L. Li, H. Hu, H. Hu, Q. Guan, M. Huang, L. Jia, H. Adenusi, K. V. Tian, J. Zhang, S. Passerini and H. Lin, ACS Nano, 2022, 16, 17729–17760 CrossRef CAS PubMed.
- Q. Wang, L. Jiang, Y. Yu and J. Sun, Nano Energy, 2019, 55, 93–114 CrossRef CAS.
- F. Ye, Z. Wang, M. Li, J. Zhang, D. Wang, M. Liu, A. Liu, H. Lin, H. T. Kim and J. Wang, Nano Lett., 2024, 24, 6850–6857 CrossRef CAS PubMed.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- Y. Wang, Z. Chen, K. Jiang, Z. Shen, S. Passerini and M. Chen, Small, 2024, 20, 2402035 CrossRef CAS PubMed.
- Z. Chen, G.-T. Kim, Z. Wang, D. Bresser, B. Qin, D. Geiger, U. Kaiser, X. Wang, Z. X. Shen and S. Passerini, Nano Energy, 2019, 64, 103986 CrossRef CAS.
- M. Chen, W. Liu, Z. Yue, Y. Wang, Y. Wu, Y. Li and Z. Chen, Batteries, 2023, 9, 270 CrossRef CAS.
- Y. Wu, Z. Chen, Y. Wang, Y. Li, C. Zhang, Y. Zhu, Z. Yue, X. Liu and M. Chen, J. Energy Chem., 2024, 89, 437–448 CrossRef CAS.
- Y. Wang, Z. Chen, Y. Wu, Y. Li, Z. Yue and M. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 21526–21536 CrossRef CAS PubMed.
- Y. Wu, Y. Li, Y. Wang, Q. Liu, Q. Chen and M. Chen, J. Energy Chem., 2022, 64, 62–84 CrossRef CAS.
- Y. Jiang, C. Xu, K. Xu, S. Li, J. Ni, Y. Wang, Y. Liu, J. Cai and C. Lai, Chem. Eng. J., 2022, 442, 136245 CrossRef CAS.
- M. Li, H. An, Y. Song, Q. Liu, J. Wang, H. Huo, S. Lou and J. Wang, J. Am. Chem. Soc., 2023, 145, 25632–25642 CrossRef CAS PubMed.
- X. Song, R. Zhao, J. Zhu, J. Zhang, N. Xu, J. Liu, Y. Liu, H. Zhang, Y. Ma, C. Li and Y. Chen, Natl. Sci. Rev., 2025, nwaf016 CrossRef.
- Y. Zhu, Z. Lao, M. Zhang, T. Hou, X. Xiao, Z. Piao, G. Lu, Z. Han, R. Gao, L. Nie, X. Wu, Y. Song, C. Ji, J. Wang and G. Zhou, Nat. Commun., 2024, 15, 3914 CrossRef CAS PubMed.
- C. M. Thomas, W. J. Hyun, H. C. Huang, D. Zeng and M. C. Hersam, ACS Energy Lett., 2022, 7, 1558–1565 CrossRef CAS.
- Q. Zhou, X. Yang, X. Xiong, Q. Zhang, B. Peng, Y. Chen, Z. Wang, L. Fu and Y. Wu, Adv. Energy Mater., 2022, 12, 2201991 CrossRef CAS.
- X. Guo, Z. Ju, X. Qian, Y. Liu, X. Xu and G. Yu, Angew. Chem., Int. Ed., 2023, 62, e202217538 CrossRef CAS PubMed.
- Y. Yuan, L. Chen, Y. Li, X. An, J. Lv, S. Guo, X. Cheng, Y. Zhao, M. Liu, Y.-B. He and F. Kang, Energy Mater. Dev., 2023, 1, 9370004 Search PubMed.
- A. Ma, C. Fu, H. Chu, X. Ran and W. Nie, Chin. J. Appl. Chem., 2020, 37, 1411–1419 CAS.
- Y. Huang, J. Zeng, S. Li, C. Dai, J.-F. Liu, C. Liu and Y.-B. He, Adv. Energy Mater., 2023, 13, 2203888 CrossRef CAS.
- J. Zhang, J. Zhu, R. Zhao, J. Liu, X. Song, N. Xu, Y. Liu, H. Zhang, X. Wan, Y. Ma, C. Li and Y. Chen, Energy Environ. Sci., 2024, 17, 7119–7128 RSC.
- Q. Gao, D. Wu, Z. Wang, P. Lu, X. Zhu, T. Ma, M. Yang, L. Chen, H. Li and F. Wu, Energy Storage Mater., 2023, 63, 103007 CrossRef.
- K. H. T. Raman, T. R. Penki, N. Munichandraiah and G. M. Rao, Electrochim. Acta, 2014, 125, 282–287 CrossRef CAS.
- H.-S. Tsai, C.-H. Hsu, C.-C. Chi, Y.-C. Wang, F.-W. Liu, S.-Y. Tang, C.-J. Tsai, H. Ouyang, Y.-L. Chueh and J.-H. Liang, Inorg. Chem. Front., 2019, 6, 172–175 RSC.
- J. Wang, J. Zhang, J. Wu, M. Huang, L. Jia, L. Li, Y. Zhang, H. Hu, F. Liu, Q. Guan, M. Liu, H. Adenusi, H. Lin and S. Passerini, Adv. Mater., 2023, 35, e2302828 CrossRef PubMed.
- Z. Zhang, J. Wang, H. Qin, B. Zhang, H. Lin, W. Zheng, D. Wang, X. Ji and X. Ou, ACS Nano, 2024, 18, 2250–2260 CrossRef CAS PubMed.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y.-B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS PubMed.
- Q. Wu, M. Fang, S. Jiao, S. Li, S. Zhang, Z. Shen, S. Mao, J. Mao, J. Zhang, Y. Tan, K. Shen, J. Lv, W. Hu, Y. He and Y. Lu, Nat. Commun., 2023, 14, 6296 CrossRef CAS PubMed.
- W. Yang, Y. Liu, X. Sun, Z. He, P. He and H. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202401428 CrossRef CAS PubMed.
- X. An, Y. Liu, K. Yang, J. Mi, J. Ma, D. Zhang, L. Chen, X. Liu, S. Guo, Y. Li, Y. Ma, M. Liu, Y.-B. He and F. Kang, Adv. Mater., 2024, 36, 2311195 CrossRef CAS PubMed.
- W. Huang, S. Wang, X. Zhang, Y. Kang, H. Zhang, N. Deng, Y. Liang and H. Pang, Adv. Mater., 2023, 35, 2310147 CrossRef CAS PubMed.
- J. Pan, Y. Zhang, J. Wang, Z. Bai, R. Cao, N. Wang, S. Dou and F. Huang, Adv. Mater., 2022, 34, 2107183 CrossRef CAS PubMed.
- S. Lv, X. He, Z. Ji, S. Yang, L. Feng, X. Fu, W. Yang and Y. Wang, Adv. Energy Mater., 2023, 13, 2302711 CrossRef CAS.
- L. Bi, L. He, Y. Song, Y. Wang, Q. Xie, P. Dong, Y. Zhang, Y. Yao, J. Liao and S. Wang, Adv. Funct. Mater., 2024, 34, 2311848 CrossRef CAS.
- H. Cheng, D. Li, B. Xu, Y. Wei, H. Wang, B. Jiang, X. Liu, H. Xu and Y. Huang, Energy Storage Mater., 2022, 53, 305–314 CrossRef.
- Y. Jin, X. Zong, X. Zhang, Z. Jia, H. Xie and Y. Xiong, Energy Storage Mater., 2022, 49, 433–444 CrossRef.
- S.-H. Kim, K.-H. Choi, S.-J. Cho, J. Yoo, S.-S. Lee and S.-Y. Lee, Energy Environ. Sci., 2018, 11, 321–330 RSC.
- S.-H. Kim, J.-H. Kim, S.-J. Cho and S.-Y. Lee, Adv. Energy Mater., 2019, 9, 1901841 CrossRef CAS.
- Z. Chen, G.-T. Kim, J.-K. Kim, M. Zarrabeitia, M. Kuenzel, H. Liang, D. Geiger, U. Kaiser and S. Passerini, Adv. Energy Mater., 2021, 11, 2101339 CrossRef CAS.
- J. Lee, J. Song, H. Lee, H. Noh, Y.-J. Kim, S. H. Kwon, S. G. Lee and H.-T. Kim, ACS Energy Lett., 2017, 2, 1232–1239 CrossRef CAS.
- H. W. Kim, J. Kim, D. Kim, Y. Kim and W.-G. Lee, J. Mater. Chem. A, 2023, 11, 14655–14662 RSC.
- K.-S. Oh, J.-H. Kim, S.-H. Kim, D. Oh, S.-P. Han, K. Jung, Z. Wang, L. Shi, Y. Su, T. Yim, S. Yuan and S.-Y. Lee, Adv. Energy Mater., 2021, 11, 2101813 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee06035k |
| This journal is © The Royal Society of Chemistry 2025 |