
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical heterogeneity observed in the development of photo-oxidized PET micro- and nanoparticle weathered controls†
John
Pettibone
 *a,
Song-Syun
Jhang
b,
Eric
Windsor
a,
Tae Joon
Cho
a,
Thomas P.
Forbes
a,
Ying Tung
Kuo
b,
Lipiin
Sung
b and
Justin M.
Gorham
a
*a,
Song-Syun
Jhang
b,
Eric
Windsor
a,
Tae Joon
Cho
a,
Thomas P.
Forbes
a,
Ying Tung
Kuo
b,
Lipiin
Sung
b and
Justin M.
Gorham
a
aMaterials Measurement Science Division, National Institute of Standards and Technology, Gaithersburg, Maryland, USA. E-mail: john.pettibone@nist.gov
bInfrastructure Materials Group, National Institute of Standards and Technology, Gaithersburg, Maryland, USA
First published on 19th February 2025
Abstract
Determining unknown secondary micro and nanoplastic (MNP) composition remains a persistent analytical challenge for field collected samples. The availability of material standards should accelerate method development for improved identification. Here, ultraviolet irradiated polyethylene terephthalate (PET) films and particles were used as models for investigating the production of weathered controls. We investigated the weathered products' chemical signatures, their stability during sampling and examination, and reproducible production of the chemical distributions using commonly reported analytical approaches for small plastic particles. We found that both conditions during irradiation and the processing procedure used for MNP production significantly contribute to changes in product distribution and the persistence of the oxidized products within the particles. Measurements were also conducted directly on MNP powders after UV-exposure to minimize any possible perturbations in product distribution from sample handling and processing. Using the model PET system, differences in sensitivity for commonly used techniques and methods were compared and discussion on relative performance for detection sensitivity was provided. Together, these findings revealed unreported pitfalls affecting accurate identification of chemically modified MNP materials.
Environmental significanceDetection and identification of nanoplastics in environmental matrices continue to be persistent challenges with large associated uncertainties for nearly all methods employed. Furthermore, international regulatory agencies have outlined that the uncertainties associated with current analytical methods limit confidence in the assessment and use of published micro and nanoplastic research data from laboratory and field studies. The current work provides a basis to begin addressing the uncertainty for chemical identification by identifying previously unreported challenges associated with using and implementing weathered controls and their corresponding spectral signatures without understanding the contributions from extraction methods, sample preparation, and particle evaluation tools. Using the well-studied model system, UV irradiated polyethylene terephthalate, resulted in the ability to identify areas where weathering products, particle stability, and analytical method limitations (mass spectrometry and spectroscopy) could result in misleading conclusions regarding chemical transformation pathways, and ultimately fate. |
Introduction
Secondary micro and nanoplastics (MNP) are the degradation products resulting from production, storage, use, and disposal of commercial and industrial plastics. Secondary MNP, produced through combinations of chemical transformations and mechanical stresses, are complex.1,2 Furthermore, these processes are known to be both compositionally and production-process (i.e., manufacturing) dependent under similar weathering conditions.3,4 Combining the complexity resulting from composition and manufacturing with the heterogeneity that results from chemical modifications and mechanical stresses in natural and applied systems, the analytical challenges associated with detection, sampling, chemical identification, and quantification of MNP have proven difficult,5,6 even for monodisperse, well-studied and spherical particles.7 To help unify and benchmark fundamental and applied research efforts to understand the contributing factors leading to MNP formation, the need to develop and broadly adopt well-characterized reference materials has been widely recognized.8–10 Although reference materials are not required for validated method development, broad use of the same material to benchmark performance would provide internal controls for comparisons across research efforts.Our goal was to identify a small subset of polymeric materials that can be produced with nonspherical morphology, reproducible size distributions, and measurable modified chemical compositions. The small subset of materials ideally would be narrowed from the large pool of candidate materials representative of the major classes of plastics produced and chosen based on practical considerations for their generation and stability after chemical alteration, i.e., weathering. Weathered MNP controls of any kind are currently unavailable. Because weathering represents a broad range of modifications that can be initiated by irradiation, heat, reactive chemical species, and biotic (enzymatic) processing, the inherent complexity makes identifying relevant and reproducible weathering processes for control materials difficult.1 However, the complexity also reinforces the need for controls to improve chemical identification when evaluating MNP from different sources. Single batches of weathered MNP can be produced, but demonstrated reproducibility of the size distribution, morphology, and composition are limited by the outlined analytical challenges to adequately discern differences when present. This challenge becomes more difficult for nanoplastics, which we defined here as particles with the largest dimension, DP, less than 1000 nm, because generally these particles push the sensitivity limits for techniques and methods of detection and identification used in microplastics research. Previous efforts to generate the largest mass fraction of the smallest MNP, DP < 10 μm, have been reported from pristine and field collected samples, which can provide context on possible benefits from both approaches.11,12 Together, generating irregularly-shaped MNP under reproducible weathering procedures should provide the framework for improved assessment of analytical methods to detect and characterize changing chemical compositions for increasingly smaller secondary MNP.
To remove unnecessary uncertainty from material production reproducibility and ultimately provide a material that can be broadly used for method performance assessments, we used commercially available polyethylene terephthalate (PET) films and modified previously reported milling procedures to produce secondary MNP model systems.10,12,13 PET photooxidation (PO), primarily from ultraviolet (UV)-degradation, is a well-studied system,14–17 which provided known degradation products to monitor within the film and particle distributions. We monitored chemical composition after accelerated laboratory weathering16,17 with vibrational spectroscopy and mass spectrometry. An assessment of the MNP compositions was conducted as the PET underwent PO, milling, and sampling procedures to better understand the factors contributing to uncertainty associated with MNP characterization. Pristine and reproducibly weathered films and MNP were compared from two different sources and to previously reported surface chemical transformations examined spectroscopically.18 Multiple vibrational spectroscopy methods, attenuated total reflectance Fourier transform infrared (ATR-FTIR) and optical photothermal infrared (OPTIR) microscopy with Raman spectroscopy, were used to assess the changing chemical composition and were further supported by supplemental analytical methods where necessary. The IR-based methods were used to both compare previously reported degradation of PET films and powders, and investigate individual, small MNP particles by using increased spatial resolution and sensitivity afforded by OPTIR microscopy methods. Together, the work provides the basis to develop and implement well-characterized, reproducible research grade test materials that can used in other MNP workflows to evaluate relative method performance and provide data comparisons across research efforts.
Materials and methods
Reagents
Polyethylene terephthalate (PET) materials used were 1 mm thick amorphous films, (Goodfellow, Coraopolis, PA)‡ and a commercial water bottle used for comparing resulting products as function of irradiation and water content. Triton X-100 purchased from both Sigma-Aldrich (laboratory grade, St. Louis, US) and Arcos Organics (Thermo Scientific, Waltham, MA) were used as dispersants and reported to be DNAse, RNAse, protease, and peroxide free.Cryomilling procedure
As received and weathered PET films were ground using a Retsch CryoMill (Verder Scientific; Haan, Germany), a 50 mL stainless steel (SS) grinding jar and a single 25 mm diameter SS grinding ball. Prior to grinding, samples were cut into pieces (≤5 mm longest dimension). Approximately 4 grams of the cut material was used for grinding. The grinding jar was sealed, loaded into the cooling jacket of the CryoMill, and precooled with liquid nitrogen for 15 minutes. Grinding was performed in cycles. Each cycle consisted of 1 minute of grinding at a frequency of 30 Hz followed by 2 minutes of cooling. To increase the yield of sub-micrometer size particles, samples were ground for 3 hours (180 grinding cycles).SPHERE aging experiments
Accelerated laboratory exposures were carried out in the SPHERE (Simulated Photodegradation via High Energy Radiant Exposure) at 50 °C/dry condition in a sealed circular sample holder (photo) for 17 d and 35 d on both sides.16,17 Irradiance was measured at each location with a spectroradiometer at the conclusion of each experiment from which the irradiance through quartz window was calculated. Typical full irradiance at samples in each chamber was ≈150 W m−2 (295 nm to 400 nm). The daily UV radiation of SPHERE is around 13 MJ m−2, 17 d and 35 d in SPHERE is equivalent to outdoors in South Florida (280 MJ m−2 = 1 year) approximately 0.79 year to 1.63 years, respectively. Reaction cells were designed to allow samples to be exposed in dry, humid, and submerged in water during exposure. The uncertainty of the total irradiance is less than 3 percent (MJ m−2). Additional 33 d exposures of cryomilled powders directly irradiated in the cell without subsequent sample preparation were also conducted to eliminate possible perturbations from the cryomilling and extraction processes.Examination of films
ATR-FTIR spectra were collected on a Nicolet is50 spectrometer using a single-bounce diamond crystal (ThermoFisher Scientific) with a liquid nitrogen cooled MCAT detector. Spectra were averaged over 250 scans with 2 cm−1 resolution. Cryomilled (CM) spectra were also collected on multiple-reflection ATR-FTIR ZnSe (45-degree, Pike Technologies, Madison, WI) crystals to compare sensitivity of longer pathlength cells. Powders from the same CM stock source were measured in replicates (n ≥ 3) to determine reproducibility of integrated peak areas for the C–O regions.Optical photothermal infrared (OPTIR) microscopy measurements were collected on a mIRage-LS system (Photothermal Spectroscopy Corp., Santa Barbara, CA) equipped with MIRcat tunable quantum cascade laser as the pump (>100 mW at 100 ns pulse) and 532 nm laser (<200 mW) as the probe. Raman measurements were conducted on the same film using the manufacturer's mIRage system equipped with a pinhole to improve removal from spurious signal and evaluate performance of the OPTIR imaging at different focal-plane heights (Photothermal Spectroscopy, Newark, DE). Raman and OPTIR spectra can be simultaneously collected on the microscope, where the inelastically (Raman) and elastically (OPTIR) scattered light from the probe laser are measured independently from the same spot using separate detectors. The spectral range for the QCL laser was (940–1850) cm−1 and (2690–3000) cm−1 and was generally run at 20% power for both film and particle data collection. Probe power was run on both the standard and ultra-low power detector (0.01–0.9%) probe power range for weathered films. Spectra were collected at 2 cm−1 resolution and averaged over (5 to 10) scans depending on noise. Both the IR and probe powers were optimized based on voltage range and stability for each spot investigated. All spectral data were collected in reflection mode using 40× Cassegrain objective (Pike Technologies, Madison, WI). Manual adjustments to spatial response (xy plane) and focal plane height (z-plane) for individual particles were done during acquisition to ensure reproducibility in signal response and sample stability. Distances measured with the optical objectives were calibrated and have uncertainties generally less than ±1 μm. The reproducibility in the spectral response was evaluated by examining (n ≥ 4) spots on the film within a film feature using the optimized laser and detector settings for each sample. Polarization measurements were conducted to evaluate anisotropy in the previous amorphous PET particle populations after milling. The polarization dependence of a 5 μm particle was presented in Fig. S5† to demonstrate the effect anisotropy has on spectral features, which can affect identification using spectral matching algorithms.
Imaging measurements were conducted at multiple focal plane heights and individual wavenumbers representing the C–O functionalities. Image analysis was conducted to search for oxidation products using individual wavelengths, 1790 cm−1 and 1680 cm−1, and ratios of those wavelengths to the νas(CO) peak maximum near 1710 cm−1 to search and visualize the presence of oxidation products over millimeters of substrate.
Pyrolysis-GC-MS
Chemical composition of PET samples was determined by pyrolysis-GC-MS and mass spectral comparison (i.e., MS Search) with the NIST mass spectral library. A single-step pyrolysis method heating up to 700 °C with a 30 s hold was employed with a CDS 6150 Pyroprobe (CDS Analytical LLC, Oxford, PA, USA). Dry powder PET samples were aliquoted into sample tubes and weighed on a microbalance on the order of (10s to 100) μg. The pyrolyzed samples were transferred (325 °C) to the GC-MS (Thermo Trace 1310 Gas Chromatograph/TSQ 8000evo Triple Quadrupole mass spectrometer, Thermo Fischer Scientific, Waltham, MA, USA). A 5% diphenyl dimethyl polysiloxane phase column, 30 m long, 0.25 mm ID, 0.25 μm film thickness (RXI-5 15HT, Agilent, Santa Clara, CA, USA) was used for chromatography with a 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 split ratio and helium carrier gas. A temperature program with a 1 min hold at 50 °C, followed by a 15 °C min−1 ramp up to 290 °C, and a 18 min hold. A standard 70 eV EI (electron ionization) source at 200 °C ionized samples for mass analysis.
:1 split ratio and helium carrier gas. A temperature program with a 1 min hold at 50 °C, followed by a 15 °C min−1 ramp up to 290 °C, and a 18 min hold. A standard 70 eV EI (electron ionization) source at 200 °C ionized samples for mass analysis.
Results and discussion
Infrared spectra for pristine and weathered (35 d exposures represented approximately 455 MJ m−2) amorphous PET films are shown along with the optical images of the films in the presence and absence of saturated relative humidity during exposure (Fig. 1). Representative normalized ATR-FTIR spectra (A/A0) for the PET film as a function of exposure are shown in Fig. S1 (ESI†). All films exhibited visible changes to physical and optical properties, which was consistent with previous work. The spectral features for both ATR-FTIR and OPTIR demonstrated similar spectral changes from the pristine to the highly PO films, indicating similar sensitivity to the weathering process. A discussion addressing the use of both infrared spectroscopy methods and comparisons of the spectra collected from the PET films can be found in the ESI.† In both methods, broadening of the νas(CO) region from (1650–1750) cm−1 was observed, representing the formation of terephthalic acid and benzoic acids at lower wavenumbers than 1710 cm−1 through Norrish reactions14,19 and secondary photochemical reactions to more highly oxidized species, e.g., anhydrides, at higher wavenumber bands near 1800 cm−1.20 In the ATR-FTIR spectra, increasing intensity of secondary photochemical products were observed in the presence of higher relative humidity and adsorbed water (Fig. S1B†). Furthermore, the water (vapor, adsorbed, and liquid phase) present that increased the production of photochemical products observed with IR measurements was also associated with observed film instability and mass loss from the films (Fig. 1 panels B and C).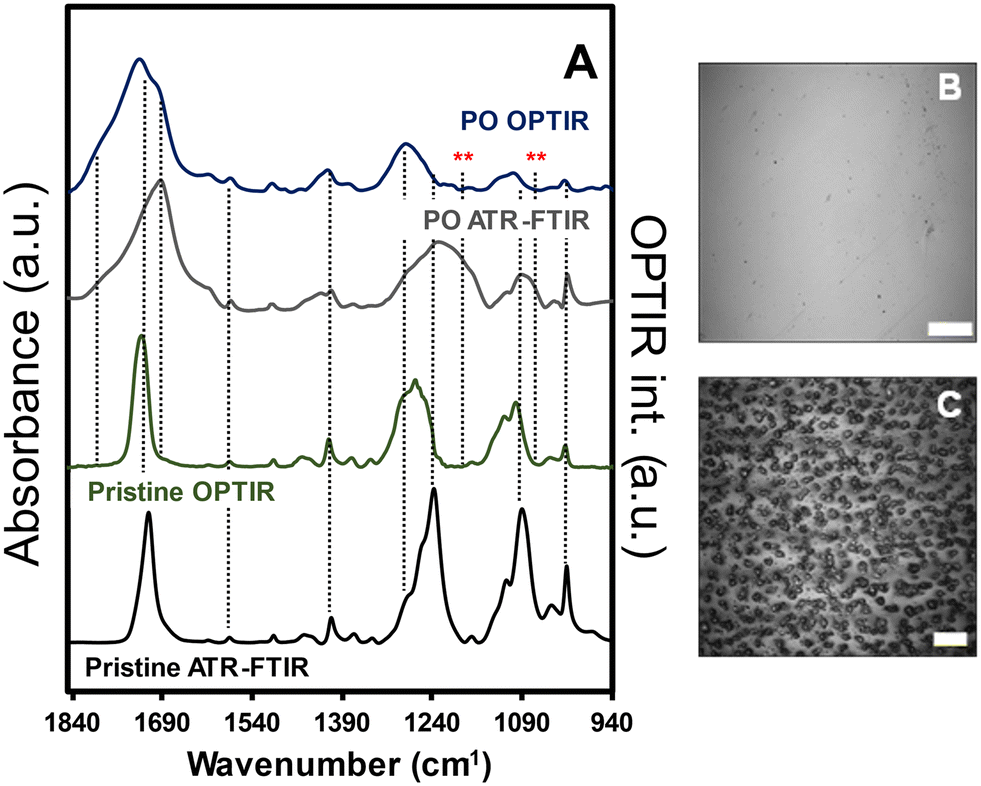 | ||
| Fig. 1 A) Normalized ATR-FTIR and OPTIR spectra of PET films prior to (pristine) and after exposure at <10% relative humidity. Optical images of film surface in the B) absence and C) presence of saturated relative humidity during exposure. The scale bars for panels B and C are 40 μm and 10 μm, respectively. The red asterisks (**) represent the vibrational modes observed with ATR-FTIR that were not present in the OPTIR spectra. | ||
Because both IR methods produced reproducible (albeit different) spectra for the pristine and weathered PET, OPTIR data was presented in the proceeding sections to characterize and compare the films and particle populations. First, we examined the cross-sectional composition of the PO films prior to milling to evaluate contributions from the milling process, sample handling, and exposure methods on the final measured chemical compositions of the weathered MNP population. In Fig. 2, the optical image of the PET film after 35 d exposure was shown with the corresponding OPTIR spectra. A gradient of optical properties from dark green to colorless was observed in the optical image from the surface to the center of the film (top panel). In addition to the optical gradients, clear delineations between specific regions were observed. From the film surface to approximately a 25 μm depth, the film exhibited a darker green region to a fainter green region, consistent with the previously reported penetration depth of the solar irradiation spectrum.14 At depths ≥85 μm (purple diamond), the green color was not observed, and the film was colorless.
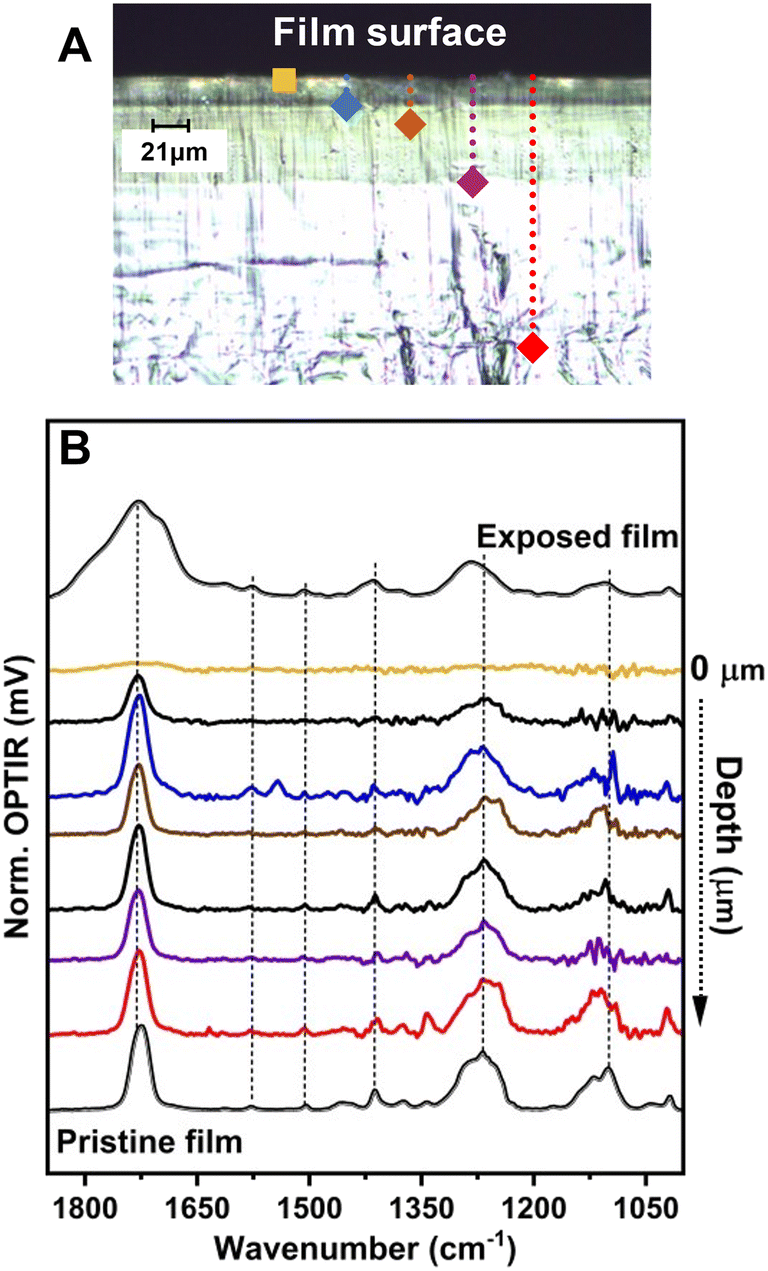 | ||
| Fig. 2 A) Optical cross-sectional image of the weathered PET film with color-coded markings for spectral acquisition. B) Representative OPTIR spectra collected across the film cross-section. The presented spectra were acquired at the surface (gold), 25 μm (blue), 35 μm (brown), 85 μm (purple), and 215 μm (red) from the surface. The black traces were representative spectra collected at intermediate depths between the regions identified with distinct color markings. | ||
Line scans were taken at approximately 3 μm intervals across >20 μm lateral spacings to collect multiple representative spectra from each region, including the film surface (bottom panel). Even at probe powers <1%, collecting spectra with characteristic C–O vibrational modes above the instrumental noise was difficult from the approximately top (1–2) μm. The OPTIR spectra demonstrated changing intensity of the PET characteristic vibrational modes as function of depth from the surface in the optically green regions (Fig. 2B). In the region near the interface from 5 μm to approximately 25 μm, characteristic PET vibrational modes emerge and the sample stability under the probe beam increases, characterized by a constant voltage of the signal during acquisition. The vibrational modes for the νas(CO) were consistent from near 25 μm to the center of the film. Additionally, the increased intensity of C–O–C stretching bands centered near 1115 cm−1 in the PO PET were characteristic of more crystalline films,21 which has been reported to occur through UV-induced cross-linking.14 Raman data provided qualitative evidence for changing chemical composition. Raman spectra collected simultaneously with OPTIR exhibited large fluorescence signals that saturated the detector in the top 20 μm of the film over the entire wavenumber range (Fig. S2†), consistent with previous studies.1 The changing fluorescence signals observed were not present in the pristine films and did not persist deeper into the film (e.g., red diamond), which provided further support for the presence of a changing chemical composition gradient within optically similar regions.
To evaluate if MNP test materials would retain weathered products detected in the film after processing, we used a modified cryomilling procedure that increased the mass fraction of nanoplastic particles previously reported.10,12,13 Single particle spectra from the CM batch are presented in Fig. 3. The representative optical images for the sample contained color coordinated locations with the representative spectra to demonstrate size (Fig. 3 inset). Overall, more than 500 different particles were evaluated from the CM batch, which were all in the size range from ≥800 nm to 10 μm. Some differences in the averaged νas(CO) widths were observed when compared to additional samples prepared from the same batch (not shown). Therefore, we showed representative spectra of particles, without the removal of outliers, to make comparisons to both intact films and unexposed CM MNP. The peak shape for the CM particles narrowed and the peak maximum shifted up to 10 cm−1 from the pristine PET film. Peaks that represented highly oxidized species centered near 1790 cm−1 were not observed. A shoulder present near 1677 cm−1 may represent the presence of terephthalic acid, but those peaks were also observed in the unexposed CM samples (Fig. S5†). Overall, the spectral changes observed in the collected single particle spectra ostensibly represented changes to particle bulk properties (crystallinity) more than chemical transformation observed in the PO reference films. Additional imaging measurements were conducted to detect vibrational signatures for either the 1685 cm−1 or 1790 cm−1 bands and ratioed values of those peaks with 1720 cm−1. However, this resulted in no detectable particles with signatures representing the chemical composition changes observed in the PO films. Imaging was conducted on CM samples both drop casted as well as dry powder. Neither sample preparation method resulted in areas in the field of view where increased intensity of the 1685 cm−1 nor the 1790 cm−1 bands were detected when compared to the pristine CM sample, consistent with the trend for weathered films in water (Fig. S1B†).
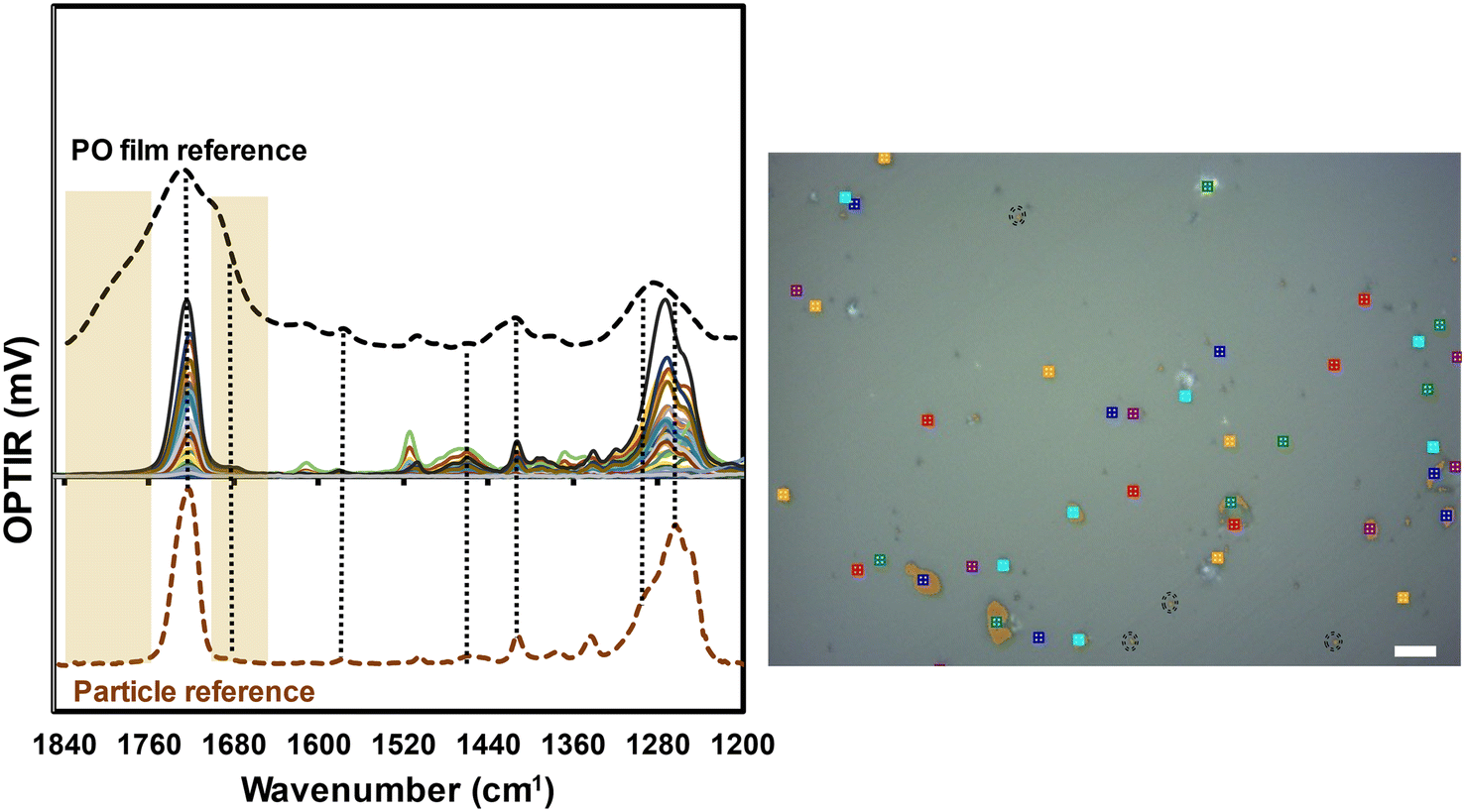 | ||
| Fig. 3 Comparison of OPTIR spectra from the photooxidized film (black dashed line), a subset of individual particles evaluated (overlapping colored spectra), and the cryomilled pristine particle reference (orange dashed line). The PO PET particles here were produced from cryomilling PO films. All the particle spectra were collected from particles in the size range of ≥800 nm to 10 μm. The OPTIR film spectra (black dashed line) was provided for reference to demonstrate the oxidized products were detected in the exposed film. The marked image was a representative optical image used to denote locations and general particle sizes for collected spectra. All spectra presented are from particles less than 10 μm for the largest dimension. The black dashed circles are locations where spectra came from particles ≤2 μm. The scale bar in the optical image is 10 μm. | ||
The lack of compositional changes to MNP particle distributions that occurred during production to the best of our knowledge have not been reported for any weathered test or control MNP development. We used a zeroth order approximation to estimate the amount of oxidation that should be detected in the CM sample based on film thickness (1 mm). The outer 50 μm (2 sides of 25 μm depths) represented 5% of the processed mass and the entire optically green film represented approximately 17% of the sample mass that should be represented in the CM particles. The general assumption was weathering products would persist and could be detected with single particle spectroscopy or mass spectrometry.
However, the persistence of these photochemical degradation products was not observed after processing of the films into CM particles and subsequent washing steps. The summarized comparison spectra from OPTIR for the pristine film, unexposed CM MNP, and averaged weathered MNP spectra are presented in Fig. S3.† In general, exposure to the PET film resulted in a narrowing of the carboxylic stretching region from (1660–1800) cm−1 and differences in crystallinity may have been present based on the ratioed intensities near (1100 and 1120) cm−1.22 However, the broad chemical heterogeneity observed for intact weathered films were not present for any particle spectra observed. Comparison of pyrolysis gas chromatography mass spectrometry (py-GC-MS) measurements, which represent the current gold standard for identification and quantification in the field, also demonstrated difficulty in detecting chemical differences. Fig. S4† shows the spectral comparison of a pristine CM film, an oxidized film that was CM (panel A), and CM particles that were directly UV-exposed without further perturbation prior to analysis (Panel B). Based on the simple approximation of the film thickness above, 25% conversion of films and likely greater percentage for the directly CM samples should result in detectable changes to the weathered spectrometric signatures. However, neither comparison provided readily distinguishable differences between the pristine and oxidized samples. These results indicate that quantifying chemical changes to the PET products in film or particle form are difficult with py-GC-MS using the simple method employed. Furthermore, the inability to distinguish differences identified another source of uncertainty in the quantification of chemically modified plastics with current analytical methods.23,24
The analytical challenge of determining chemical composition across broad size ranges of MNP has been recognized, especially as the DP decreases below 1000 nm.2,9,25 Our current study used measured PET PO as a reproducible degradation pathway that resulted in previously reported, heterogeneous product distributions that could be verified in the exposed films with spectroscopic methods. However, trying to follow and detect the persistence of degradation products on the model system for CM particles demonstrated challenges for detection, sample handling, and technique and method evaluation. These challenges only become more complex for MNP with unknown origins and that undergo multiple weathering processes in environmental and applied systems. Thus, to begin making meaningful progress towards bridging the gap between controls and MNP that pose potential risks to human and environmental health, approaches that can be implemented broadly and promote cross comparisons of datasets with some levels of confidence are required. To promote progress, the development of an accessible, broadly adopted control material would be useful if implemented to assess relative method performance in multiple systems and would provide an expanding dataset for the plastics community to build further measurement capabilities.
Summary and implications
The current work describes the first thorough examination of particle specific chemical signatures for weathered, test MNP produced through the commonly reported cryomilling procedures. Because of the current analytical challenges associated with characterizing chemical composition of smaller polymeric and plastic particles, assumptions were generally made in previous studies about the distribution of particle composition based on bulk or ensemble measurements that may not be representative. Here, the chemical transformations observed in the PO PET film were not detected in the final MNP particle distributions, suggesting either the stability or solubility of the weathered products and terephthalic acid moieties resulted in their removal during processing and preparation steps. The removal of the products from the weathered PET particles provides both i) insight into signatures likely present in environmental samples resulting from solely solar irradiation and ii) demonstrated the importance of testing control material behavior in specific applications. The removal of the weathered products during production was consistent with the observed distribution for the highly oxidized products of weathered films submerged in water (Fig. S1†), where the removal of the most highly oxidized products near 1800 cm−1 were observed. However, additional contributions from processing other than water must have contributed to the removal of the terephthalic and benzoic acid products that persisted in all PO film experiments and should be examined further. The implications of the current study provide a material to develop insights into chemical distributions during sample preparation and handling. Implementing weathered controls in established methods provides an opportunity to evaluate observed changes to chemical distributions for PET in environmental or other media, such as food matrices. The ability to compare and examine chemical composition between control materials and other PET MNP should result in improved understanding of contributions from media components to sorb, bind, and stabilize chemically transformed products, which currently represents a difficult analytical challenge.To develop and produce MNP particles with specific properties, the methods for generating said materials (e.g., powder processing, solvent control, secondary reaction pathway development) need to be further evaluated to better tailor the production process for specific applications. However, these results only represent half of the characterization necessary (chemical heterogeneity) for fully describing the particle properties of the material to evaluate toxicity. More work is required to identify size-dependent chemical stability of weathered products during examination using IR microscopy, Raman-based methods, or other single particle methods. Additionally, to investigate the stability and fate of real secondary MNP, controls will have to be implemented with a suite of measurement methods for characterization,2 oxidative extent,26 and appropriate sample handling and preparation methods for examination.27
In our effort to develop reproducible methods for generating weathered PET particle populations, we identified primary contributors to their formation and stability. The promising outcome from the current research was that cryomilling of weathered films resulted in reproducible chemical distributions within the films and the processed MNP particles characterized with both ensemble and single particle methods used (albeit different between each). Having a representative, well-characterized, and readily available control for different classes of plastics should be sufficient to benchmark data sets across experiments and research laboratory efforts when wide adoption occurs. Our ongoing efforts, which will be presented in contemporaneous works, are also focused on evaluating particle size distributions with some measured uncertainty and to provide dimensional properties (size and shape) across the entire CM particle distribution. Both efforts are non-trivial and represent challenges that require cross-disciplinary efforts to make progress. However, the ability to evaluate both the chemical composition and physical dimensions across the entire particle size distribution would provide the tools for tailoring weathered controls that are more fit for purpose. Overall, well-characterized test materials as benchmarks in applied studies and methods to monitor weathered product persistence during sample preparation and handling should help the broad plastic community quantify uncertainty associated with chemical composition, size, and morphology in more complex systems. The understanding of measurement capabilities and chemical stability should also enable more mechanistic studies of fragmentation, MNP formation, and fate as well as contributions from additives and adsorbed contaminants by differentiating particle-specific properties from other factors.
Data availability
All vibrational spectra and chromatograms will be available on the NIST Public Data Repository at https://data.nist.gov/sdp/#/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Frank Weston for his help on the Raman measurements. Certain commercial equipment, instruments, or materials are identified in this article to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by NIST nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.References
- R. Tian, K. Li, Y. Lin, C. Lu and X. Duan, Characterization Techniques of Polymer Aging: From Beginning to End, Chem. Rev., 2023, 123, 3007–3088 CrossRef CAS PubMed.
- N. P. Ivleva, Chemical Analysis of Microplastics and Nanoplastics: Challenges, Advanced Methods, and Perspectives, Chem. Rev., 2021, 121, 11886–11936 CrossRef CAS PubMed.
- J. G. Speight, in Handbook of Industrial Hydrocarbon Processes, ed. J. G. Speight, Gulf Professional Publishing, Boston, 2011, ch. 14 – Monomers, Polymers, and Plastics, pp. 499–537 Search PubMed.
- B. Pinlova and B. Nowack, From cracks to secondary microplastics - surface characterization of polyethylene terephthalate (PET) during weathering, Chemosphere, 2024, 352, 141305 Search PubMed.
- G. Binda, G. Kalčíková, I. J. Allan, R. Hurley, E. Rødland, D. Spanu and L. Nizzetto, Microplastic aging processes: Environmental relevance and analytical implications, TrAC, Trends Anal. Chem., 2024, 172, 117566 CrossRef CAS.
- O. S. Alimi, D. Claveau-Mallet, R. S. Kurusu, M. Lapointe, S. Bayen and N. Tufenkji, Weathering pathways and protocols for environmentally relevant microplastics and nanoplastics: What are we missing?, J. Hazard. Mater., 2022, 423, 126955 CrossRef CAS PubMed.
- T. Gouin, R. Ellis-Hutchings, M. Pemberton and B. Wilhelmus, Addressing the relevance of polystyrene nano- and microplastic particles used to support exposure, toxicity and risk assessment: implications and recommendations, Part. Fibre Toxicol., 2024, 21, 39 CrossRef CAS PubMed.
- K. Drzewinska and S. Belz, Stakeholder survey on microplstics materials needs for European Commission, J. R. C, Luxembourg, 2024 Search PubMed.
- T. V. Duncan, S. A. Khan, A. K. Patri and S. Wiggins, Regulatory Science Perspective on the Analysis of Microplastics and Nanoplastics in Human Food, Anal. Chem., 2024, 96, 4343–4358 CrossRef CAS PubMed.
- L. Sørensen, M. H. Gerace and A. M. Booth, Small micro- and nanoplastic test and reference materials for research: Current status and future needs, Cambridge Prisms: Plastics, 2024, 2, e13 CrossRef.
- F. Blancho, M. Davranche, F. Fumagalli, G. Ceccone and J. Gigault, A reliable procedure to obtain environmentally relevant nanoplastic proxies, Environ. Sci.: Nano, 2021, 8, 3211 RSC.
- C. J. McColley, J. A. Nason, B. J. Harper and S. L. Harper, An assessment of methods used for the generation and characterization of cryomilled polystyrene micro- and nanoplastic particles, Microplast. Nanoplast., 2023, 3, 20 CrossRef CAS.
- T. Gardon, I. Paul-Pont, G. Le Moullac, C. Soyez, F. Lagarde and A. Huvet, Cryogrinding and sieving techniques as challenges towards producing controlled size range microplastics for relevant ecotoxicological tests, Environ. Pollut., 2022, 315, 120383 CrossRef CAS PubMed.
- T. Sang, C. J. Wallis, G. Hill and G. J. P. Britovsek, Polyethylene terephthalate degradation under natural and accelerated weathering conditions, Eur. Polym. J., 2020, 136, 109873 CrossRef CAS.
- M. Djebara, J. P. Stoquert, M. Abdesselam, D. Muller and A. C. Chami, FTIR analysis of polyethylene terephthalate irradiated by MeV He+, Nucl. Instrum. Methods Phys. Res. B: Beam Interact. Mater. At., 2012, 274, 70 CrossRef CAS.
- J. Chin, T. Nguyen, E. Byrd and J. Martin, Validation of the reciprocity law for coating photodegradation, JCT Res., 2005, 2, 499 CAS.
- J. Chin, E. Byrd, N. Embree, J. Garver, B. Dickens, T. Finn and J. Martin, Accelerated UV weathering device based on integrating sphere technology, Rev. Sci. Instrum., 2004, 75, 4951 CrossRef CAS.
- J.-L. Gardette, A. Colin, S. Trivis, S. German and S. Therias, Impact of photooxidative degradation on the oxygen permeability of poly(ethyleneterephthalate), Polym. Degrad. Stab., 2014, 103, 35 CrossRef CAS.
- F. B. Marcotte, D. Campbell, J. A. Cleaveland and D. T. Turner, Photolysis of poly(ethylene terephthalate), J. Polym. Sci., 1967, 5, 481 CrossRef CAS.
- A. Rivaton, Photochemistry of poly(butyleneterephthalate): 2—Identification of the IR-absorbing photooxidation products, Polym. Degrad. Stab., 1993, 41, 297 CrossRef CAS.
- K. Aljoumaa and M. Abboudi, Physical ageing of polyethylene terephthalate under natural sunlight: correlation study between crystallinity and mechanical properties, Appl. Phys. A: Mater. Sci. Process., 2015, 122, 6 CrossRef.
- I. Donelli, G. Freddi, V. A. Nierstrasz and P. Taddei, Surface structure and properties of poly-(ethylene terephthalate) hydrolyzed by alkali and cutinase, Polym. Degrad. Stab., 2010, 95, 1542 CrossRef CAS.
- T. P. Forbes, J. M. Pettibone, E. Windsor, J. M. Conny and R. A. Fletcher, Rapid Chemical Screening of Microplastics and Nanoplastics by Thermal Desorption and Pyrolysis Mass Spectrometry with Unsupervised Fuzzy Clustering, Anal. Chem., 2023, 95, 12373 CrossRef CAS PubMed.
- M. E. Seeley and J. M. Lynch, Previous successes and untapped potential of pyrolysis–GC/MS for the analysis of plastic pollution, Anal. Bioanal. Chem., 2023, 415, 2873 Search PubMed.
- J. Gigault, H. El Hadri, B. Nguyen, B. Grassl, L. Rowenczyk, N. Tufenkji, S. Feng and M. Wiesner, Nanoplastics are neither microplastics nor engineered nanoparticles, Nat. Nanotechnol., 2021, 16, 501 CrossRef CAS PubMed.
- C. di Luca, J. Garcia, M. Munoz, M. Hernando-Pérez, Z. M. de Pedro and J. A. Casas, Strategies for the quantification and characterization of nanoplastics in AOPs research, Chem. Eng. J., 2024, 493, 152490 CrossRef CAS.
- N. M. Ainali, D. Kalaronis, A. Kontogiannis, E. Evgenidou, G. Z. Kyzas, X. Yang, D. N. Bikiaris and D. A. Lambropoulou, Microplastics in the environment: Sampling, pretreatment, analysis and occurrence based on current and newly-exploited chromatographic approaches, Sci. Total Environ., 2021, 794, 148725 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4en00841c |
| ‡ Certain commercial equipment, instruments, or materials are identified in this article to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by NIST nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose. |
| This journal is © The Royal Society of Chemistry 2025 |