 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scanning electrochemical probe microscopy: towards the characterization of micro- and nanostructured photocatalytic materials†
Giada
Caniglia
 *,
Sarah
Horn
and
Christine
Kranz
*
*,
Sarah
Horn
and
Christine
Kranz
*
Institute of Analytical and Bioanalytical Chemistry, Ulm University, Albert-Einstein-Allee, 11 89081 Ulm, Germany. E-mail: christine.kranz@uni-ulm.de
First published on 15th July 2024
Abstract
Platinum-black (Pt-B) has been demonstrated to be an excellent electrocatalytic material for the electrochemical oxidation of hydrogen peroxide (H2O2). As Pt-B films can be deposited electrochemically, micro- and nano-sized conductive transducers can be modified with Pt-B. Here, we present the potential of Pt-B micro- and sub-micro-sized sensors for the detection and quantification of hydrogen (H2) in solution. Using these microsensors, no sampling step for H2 determination is required and e.g., in photocatalysis, the onset of H2 evolution can be monitored in situ. We present Pt-B-based H2 micro- and sub-micro-sized sensors based on different electrochemical transducers such as microelectrodes and atomic force microscopy (AFM)-scanning electrochemical microscopy (SECM) probes, which enable local measurements e.g., at heterogenized photocatalytically active samples. The microsensors are characterized in terms of limits of detection (LOD), which ranges from 4.0 μM to 30 μM depending on the size of the sensors and the experimental conditions such as type of electrolyte and pH. The sensors were tested for the in situ H2 evolution by light-driven water-splitting, i.e., using ascorbic acid or triethanolamine solutions, showing a wide linear concentration range, good reproducibility, and high sensitivity. Proof-of-principle experiments using Pt-B-modified cantilever-based sensors were performed using a model sample platinum substrate to map the electrochemical H2 evolution along with the topography using AFM-SECM.
1 Introduction
Monitoring of molecular hydrogen (H2) concentration is crucial in many fields, including in renewable energies such as light-driven water splitting, material science, and fuel production.1 Conventional methods for H2 quantification include head-space gas chromatography (GC), thermal conductivity, and mass spectroscopy,2 with head-space GC being the most used technique. However, even though these techniques provide sensitive and reliable determination of H2, they usually require a sampling step and longer irradiation times e.g., for heterogenized catalysts to produce sufficient H2, which reflects then an averaged measurement. Frequently photo(electro)catalytic materials are heterogeneous in nature and therefore, miniaturized sensors that allow in situ or operando measurements with high lateral and temporal resolution are required. Potentiometric and amperometric techniques for local H2 quantification are available and are characterized by several advantages in terms of sensitivity, selectivity, and the possibility of miniaturization.3Among the different techniques, scanning electrochemical probe microscopy such as scanning electrochemical microscopy (SECM)4–7 and scanning electrochemical cell microscopy (SECCM)8–11 have been widely used in the past years to locally monitor the H2 evolution at heterogenized photo(electro)catalytic systems.12
Even though H2 detection is mainly performed using platinum (Pt) electrodes due to the excellent electrocatalytic performance of Pt, a challenge remains when reducing the size of the electrodes due to the decreased electrochemically active surface area (EASA) and with that the number of available binding sites. To overcome this limitation, one strategy involves the fabrication of highly porous materials with significantly increased EASA.4,13–15 We recently demonstrated that highly porous platinum-black16 (Pt-B) microelectrodes and Pt-B hemispherical AFM-SECM probes are sensitive hydrogen peroxide (H2O2) microsensors, suitable for H2O2 detection in light-driven catalysis.17 These probes have been fabricated by the direct electrodeposition of Pt-B onto a recessed gold (Au) microelectrode obtained by focused ion beam (FIB) milling at the apex of a tipless cantilever.17 One of the main advantages of this method of fabrication is the possibility of tailoring the size of the Pt-B probe by controlling the size of the recessed Au electrode and the experimental conditions. For instance, using Pt-B probes obtained obtained by electrodepostion onto 5 μm-diameter recessed Au microelectrodes, the photocatalytic activity of a poly(heptazine imide)-based catalyst in terms of local H2O2 production could be monitored.17
Here, we explore the potential of Pt-B microsensors for local H2 sensing. Due to the increased surface area with a high number of active binding sites that favor the electron transfer, Pt-B films have been employed for decades as electrode materials (e.g., platinized Pt electrode) in the standard hydrogen reference electrode. Pt-B is also used as a catalyst in proton exchange membrane fuel cells (PEMFCs), along with Pt/C catalysts18,19 and as a sensing material for H2O2.17,20–23 Although, the EASA of Pt-B microelectrodes has been determined by using the H-desorption peak recorded in 0.5 M sulfuric acid, the electroanalytical detection/determination of H2 at photo(electro)catalysts using Pt-B microsensors has not been reported, and studies on the electrochemical behavior of Pt-B in terms of the hydrogen oxidation reaction (HOR) are still scarce.19,24,25
It is well-known that the HOR is a surface-dependent reaction related to the adsorption of H2, which takes place through the formation of underpotential-deposited hydrogen atoms (HUPD) at electrode materials that thermodynamically favor HUPD.26 At potentials more positive than the RHE, at noble metals such as Pt,27,28 and Pd,28 the HUPD formation is energetically favorable, whereas for other materials like Ni,29 the electrosorbed O-containing species (O2 and OH−) are thermodynamically more stable compared to the electrosorbed hydrogen species at the same applied potential.
The mechanism of HOR in aqueous solutions is well described and involves the formation of adsorbed hydrogen species on the Pt electrode followed by its dissociative oxidation, which in acidic media, generally proceeds following the combination of two steps.30,31 The first step is the dissociative adsorption of H2, which can follow two different pathways: the Tafel reaction (1) where a chemical dissociation with the formation of two hydrogen adsorbed species (or H adatoms) takes place, and the Heyrovsky reaction (2), where an electron transfer takes place from the H2 and the Pt electrode with the release of a proton and the formation of a single hydrogen adatom. The second and last step, the Volmer reaction (3), involves the discharge of adsorbed hydrogen. The general steps are summarized as follows:
| Tafel step: H2 + 2Pt*→2(Pt*Had) | (1) |
| Heyrovsky step: H2 + 2Pt* → Pt*Had + Pt* + H+ + e− | (2) |
| Volmer step: Pt*Had → Pt* + H+ + e− | (3) |
| Heyrovsky step: H2 + OH− + 2Pt* → Pt*Had + Pt* + H2O + e− | (4) |
| Tafel step: H2 + Pt*OHad → Pt*Had + H2O | (5) |
| Volmer step: Pt*Had + OH− → Pt* + H2O + e− | (6) |
2 Results and discussion
2.1. Preparation and electrochemical characterization of hemispherical Pt-B AFM-SECM probes
The AFM-SECM probes were fabricated following an already published protocol17 in which the Pt-B is electrodeposited directly onto a FIB-milled recessed Au electrode to form a hemispherical Pt-B electrode (Fig. 1A and B) whose size can be tailored as a function of the recessed Au electrode diameter and the electrodeposition parameters. It should be noted that for Pt-B radii smaller than 300 nm, for imaging experiments, the position at the cantilever has to be considered. For most commercial AFMs, the AFM cantilever is mounted at an angle of 9–11°, therefore hemispherical sensors have to be positioned close to the free end of the cantilever. This distance is critical to ensure that the hemispherical probe touches the sample surface and not the leading edge of the cantilever (see Fig. S1A and B†). To determine the position at which the Au recessed electrode had to be milled to deposit the Pt-B, we calculated the limit ratio between the distance from the free end of the cantilever and the radius of the probe to prevent the edge of the cantilever from touching the surface. As shown in Fig. S1B,† the minimum radius (r0) corresponds to r0 = a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ, where a is the distance from the edge of the cantilever and the center of the probe with an angle θ of 9°. This leads to a a/r ratio of approx. 6, i.e., for a probe with radius r, the maximum distance from the free end of the cantilever at which the hemisphere can be deposited is 6 times r. For the fabrication of 250 nm-diameter nanoelectrodes, the recessed Au electrode was milled choosing an a/r ratio of 2 by 3, which is at approx. 500–700 nm from the free end of the cantilever. When considering a distance of 500 nm, the smallest hemispherical probe that can be produced without the risk that the edge of the cantilever touches the surface must have a radius of 78 nm. If the distance is decreased to 50 nm, probes with radii below 10 nm could be theoretically fabricated. Fig. 1 shows three exemplary Pt-B AFM-SECM probes obtained by using different diameters of the Au recessed electrode. The Pt-B probes displayed in the SEM images (Fig. 1C and D) were obtained from a recessed Au electrode with a diameter of 1 μm and 500 nm, respectively, via chronoamperometry by applying a potential of −0.06 V vs. Ag/AgCl, KCl (sat.) for 10 s in 31 mM H2PtCl6/0.67 mM Pb(NO3)2).23 The final radii of the hemispherical Pt-B electrodes are 1.5 μm and 800 nm, respectively. For diameters of the Au electrode smaller than 500 nm, we observed an overgrowth of the Pt-B probe with a final diameter similar to the depositions on a 500 nm-diameter recessed Au electrode. For better control of the Pt-B deposition, pulsed deposition was chosen as an alternative electrodeposition method.17 As shown in Fig. 1E, the application of pulse deposition (80 pulse cycles; cycle: −0.06 V for 0.5 s and 0 V for 0.5 s, potential vs. Ag/AgCl, KCl (sat.)) generates a Pt-B probe with a radius of approx. 500 nm. Interestingly, the porosity of the Pt-B decreases significantly when using pulse deposition, as shown in the FIB cross-section of the Pt-B probe depicted in Fig. S2.† We associate this denser deposition with the enhanced concentration of the active species during deposition at the electrode surface. Fig. 1F–H show exemplary cyclic voltammograms (CVs) performed in 5 mM FcMeOH/0.1 M KCl at each modification step of the cantilever. The CVs in black color correspond to the bare recessed Au electrodes and the CVs in red color to the corresponding Pt-B probes. The increased size and porosity of the Pt-B probe in comparison to the bare Au electrode can be observed in the difference in the shape and steady-state current (iss) of the CVs. Indeed, the iss increases by a factor of approx. 3 for 1 μm and 500 nm-diameters probes and it achieves an increment factor of 35 when the original diameter of the Au electrode is 250 nm. Furthermore, while in the 1 μm and 500 nm-diameter bare Au, the typical sigmoidal CVs with small capacitive current and steady-state current were observed, which are characteristic for micro- and sub-micro disk electrodes, the corresponding Pt-B modified probes exhibit a change in the shape of the voltammogram correlated to a change in the diffusional behavior and increased capacitive current due to the high porosity of the Pt-B probes. Significantly less pronounced changes were observed for a recessed Au electrode with a diameter of 250 nm and a pulsed deposition of Pt-B. The difference in porosity was also evaluated by calculating the EASA of each probe, by integrating the hydrogen desorption peak detected between approx. −0.25 V and 0.1 V vs. Ag/AgCl, KCl (sat.) in the CV performed in 0.5 M H2SO4 (Fig. S3†). Considering that the formation of an adsorbed hydrogen monolayer corresponds to a charge of Q = 210 μC cm−1,33 the calculated EASAs are 3111 μm2 (1 μm-diameter-probe), 2357 μm2 (500 nm-diameter probe), but only 150 μm2 for the pulsed deposition of Pt-B onto an Au electrode with a diameter 250 nm.
sinθ, where a is the distance from the edge of the cantilever and the center of the probe with an angle θ of 9°. This leads to a a/r ratio of approx. 6, i.e., for a probe with radius r, the maximum distance from the free end of the cantilever at which the hemisphere can be deposited is 6 times r. For the fabrication of 250 nm-diameter nanoelectrodes, the recessed Au electrode was milled choosing an a/r ratio of 2 by 3, which is at approx. 500–700 nm from the free end of the cantilever. When considering a distance of 500 nm, the smallest hemispherical probe that can be produced without the risk that the edge of the cantilever touches the surface must have a radius of 78 nm. If the distance is decreased to 50 nm, probes with radii below 10 nm could be theoretically fabricated. Fig. 1 shows three exemplary Pt-B AFM-SECM probes obtained by using different diameters of the Au recessed electrode. The Pt-B probes displayed in the SEM images (Fig. 1C and D) were obtained from a recessed Au electrode with a diameter of 1 μm and 500 nm, respectively, via chronoamperometry by applying a potential of −0.06 V vs. Ag/AgCl, KCl (sat.) for 10 s in 31 mM H2PtCl6/0.67 mM Pb(NO3)2).23 The final radii of the hemispherical Pt-B electrodes are 1.5 μm and 800 nm, respectively. For diameters of the Au electrode smaller than 500 nm, we observed an overgrowth of the Pt-B probe with a final diameter similar to the depositions on a 500 nm-diameter recessed Au electrode. For better control of the Pt-B deposition, pulsed deposition was chosen as an alternative electrodeposition method.17 As shown in Fig. 1E, the application of pulse deposition (80 pulse cycles; cycle: −0.06 V for 0.5 s and 0 V for 0.5 s, potential vs. Ag/AgCl, KCl (sat.)) generates a Pt-B probe with a radius of approx. 500 nm. Interestingly, the porosity of the Pt-B decreases significantly when using pulse deposition, as shown in the FIB cross-section of the Pt-B probe depicted in Fig. S2.† We associate this denser deposition with the enhanced concentration of the active species during deposition at the electrode surface. Fig. 1F–H show exemplary cyclic voltammograms (CVs) performed in 5 mM FcMeOH/0.1 M KCl at each modification step of the cantilever. The CVs in black color correspond to the bare recessed Au electrodes and the CVs in red color to the corresponding Pt-B probes. The increased size and porosity of the Pt-B probe in comparison to the bare Au electrode can be observed in the difference in the shape and steady-state current (iss) of the CVs. Indeed, the iss increases by a factor of approx. 3 for 1 μm and 500 nm-diameters probes and it achieves an increment factor of 35 when the original diameter of the Au electrode is 250 nm. Furthermore, while in the 1 μm and 500 nm-diameter bare Au, the typical sigmoidal CVs with small capacitive current and steady-state current were observed, which are characteristic for micro- and sub-micro disk electrodes, the corresponding Pt-B modified probes exhibit a change in the shape of the voltammogram correlated to a change in the diffusional behavior and increased capacitive current due to the high porosity of the Pt-B probes. Significantly less pronounced changes were observed for a recessed Au electrode with a diameter of 250 nm and a pulsed deposition of Pt-B. The difference in porosity was also evaluated by calculating the EASA of each probe, by integrating the hydrogen desorption peak detected between approx. −0.25 V and 0.1 V vs. Ag/AgCl, KCl (sat.) in the CV performed in 0.5 M H2SO4 (Fig. S3†). Considering that the formation of an adsorbed hydrogen monolayer corresponds to a charge of Q = 210 μC cm−1,33 the calculated EASAs are 3111 μm2 (1 μm-diameter-probe), 2357 μm2 (500 nm-diameter probe), but only 150 μm2 for the pulsed deposition of Pt-B onto an Au electrode with a diameter 250 nm.
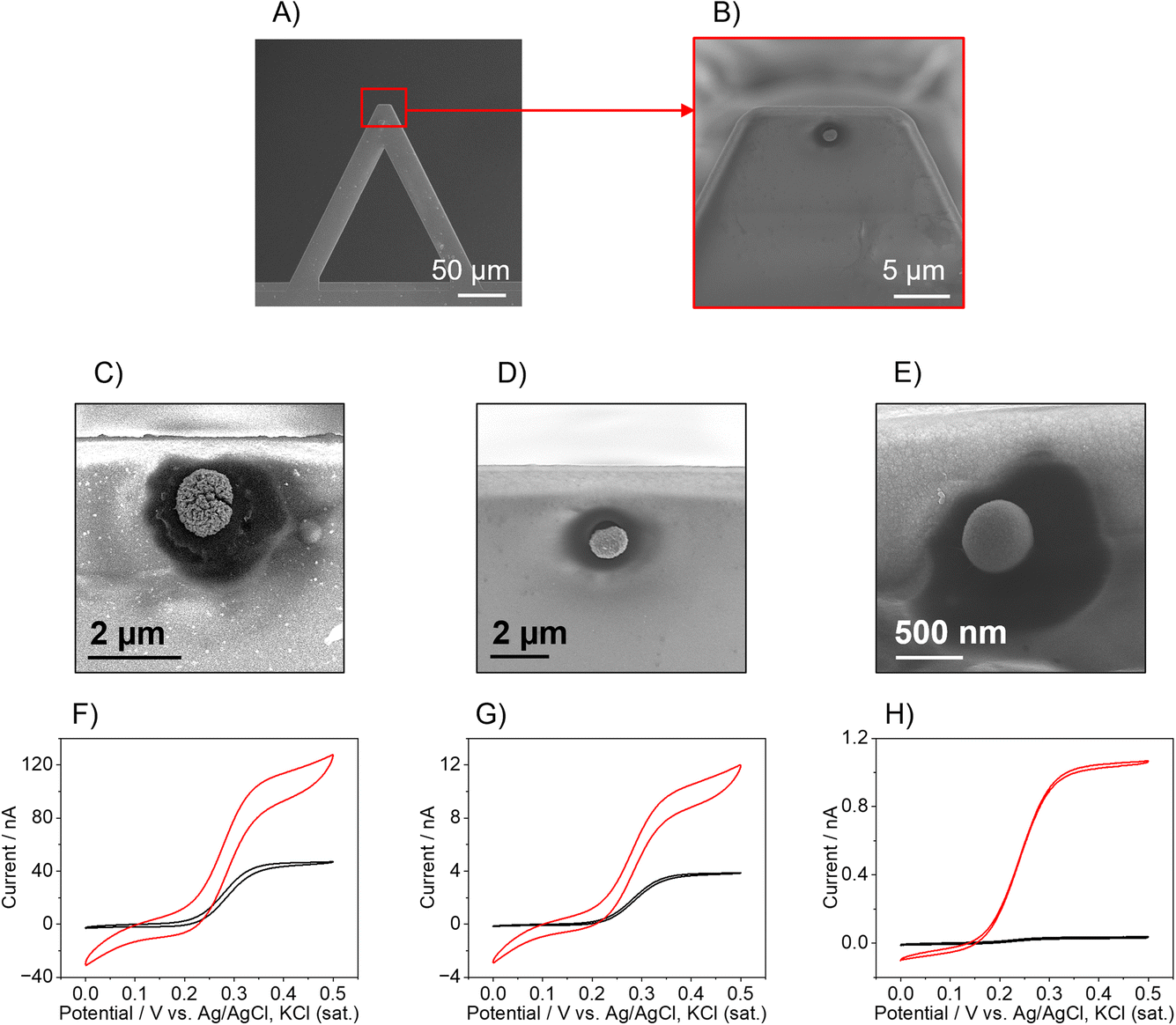 | ||
| Fig. 1 (A and B) Exemplary SEM images of the AFM cantilever before (A) and after (B) the deposition of Pt-B on a recessed disk Au electrode with a diameter equal to 500 nm. (C–E) SEM images of Pt-B probe electrodeposited on a recessed Au electrode with a diameter equal to (C) 1 μm, (D) 500 nm, and (E) 250 nm, respectively. (F–H) CVs of bare Au electrode (black curves) and after the deposition of Pt-B (red curves) recorded in 5 mM FcMeOH/0.1 KCl (sweep rate 50 mV s−1). The diameter of the bare Au electrode was, respectively, (F) 1 μm, (G) 500 nm, and (H) 250 nm. | ||
Further characterization for H2 quantification was performed in two different solutions using Pt-B probes produced from 1 μm-diameter recessed Au electrodes. Phosphate buffer saline (PBS) solution was chosen as a model medium, while a solution containing ascorbic acid was used to evaluate the compatibility of the Pt-B microsensors for photocatalytic H2 measurements since ascorbic acid is one of the most used sacrificial electron donors in e.g., molecular light-driven water splitting systems for H2 evolution.34,35 The pH was adjusted for both solutions to a value of pH 4 using either hydrochloric acid (HCl) or sodium hydroxide (NaOH). Further experiments at different pH values and using also triethanolamine (TEOA) solutions – another important sacrificial electron donor – were performed using Pt-B-modified microelectrodes (with diameters of approx. 25 μm), which will be discussed later. The quantification of H2 was performed via chronoamperometry. The applied potential for the experiments performed in PBS was −0.15 V vs. Ag/AgCl, KCl (sat.) since it was the highest anodic peak in the HUPD region of the CV obtained in PBS at pH 4.
For the quantification of H2 in ascorbic acid, the adsorption of ascorbic acid in the potential range from −0.4 V to approx. −0.06 V has to be considered.36,37 By applying a potential of −0.15 V like in the PBS solution at pH 4, the current response was not stable in ascorbic acid and the H2 calibration was affected by the presence of the organic species. Therefore, the H2 calibration in ascorbic acid was performed at −0.05 V, as the current response was stable at this potential.
Fig. 2 shows the calibrations of three individual sensors performed in 0.1 M PBS (Fig. 2A–C) and ascorbic acid (Fig. 2D–F). In both cases, calibrations exhibit linearity up to an H2 concentration of 300 μM, with a linearity range of 50 μM to 300 μM in PBS and 0 μM to 300 μM in ascorbic acid. As highlighted in Table 1, the LOD and LOQ, which were determined, are 10.7 ± 0.5 μM and 33 ± 2 μM in PBS and 10 ± 7 μM and 36 ± 10 μM in ascorbic acid, respectively. Also, the Pt-B probes showed excellent recovery as the sensor responses returned to the baseline current value upon the removal of H2 from the solution by purging with nitrogen after the completion of the calibration (Fig. 2B and E). The response time of the hemispherical H2 sensor was determined by the time required for the current to reach 90% of the steady-state current when the H2 concentration was increased.
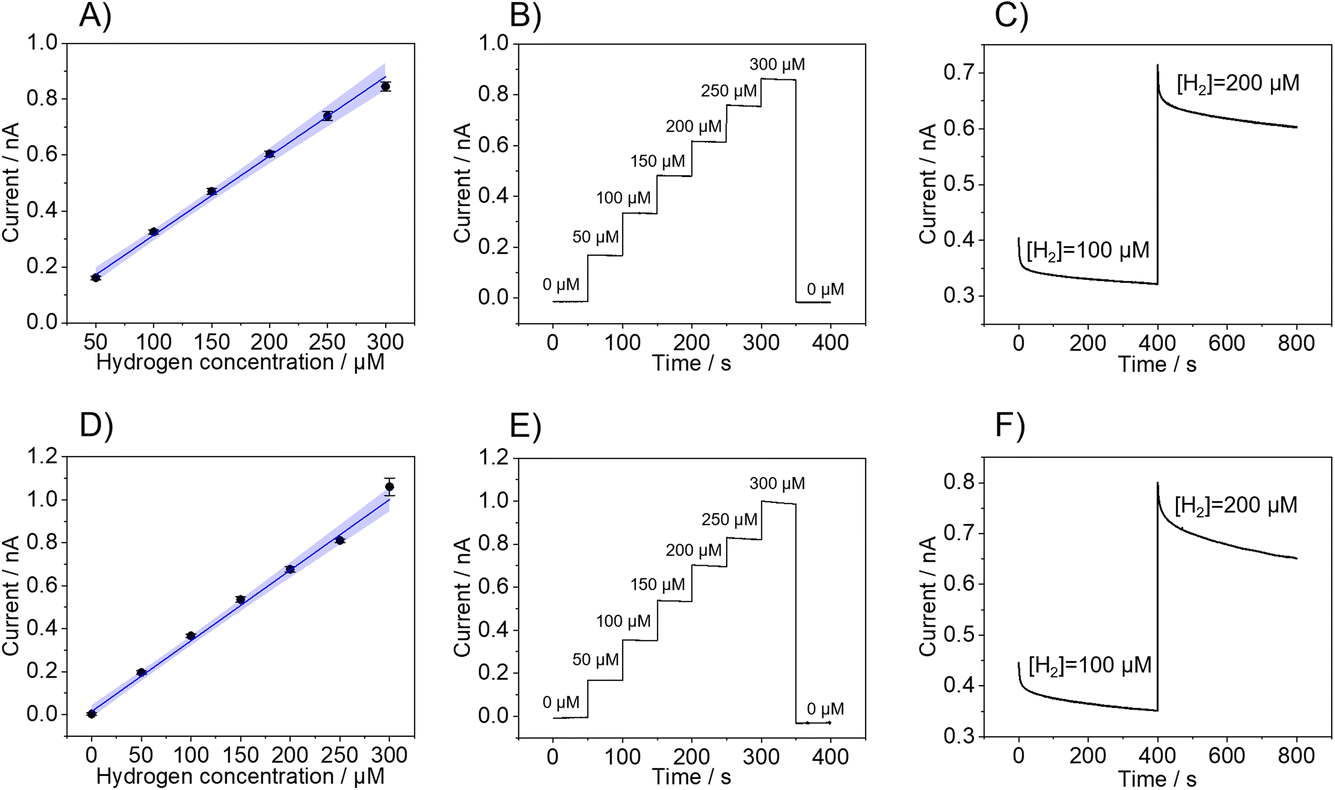 | ||
| Fig. 2 (A and D) Calibration curves (blue line) and (B and E) corresponding chronoamperometry recorded at a Pt-B modified AFM-SECM probe (d = 1 μm) with increased H2 concentrations immersed in (A and B) 0.1 M PBS at pH 4 (Eprobe = −0.15 V vs. Ag/AgCl) and (D and E) 0.1 M ascorbic acid at pH 4 (Eprobe = −0.05 V vs. Ag/AgCl). The blue area denotes the uncertainty range, and the error bars reflect three repetitive measurements. (C and F) Temporal response of the probes changing the H2 concentration from 100 μM to 200 μM for the Pt-B sensor in (C) 0.1 M PBS at pH 4 (Eprobe = −0.15 V vs. Ag/AgCl) and (F) 0.1 M ascorbic acid at pH 4 (Eprobe = −0.05 V vs. Ag/AgCl). | ||
| Solution | E vs. Ag/AgCl, KCl (sat.), V | LOD μM | LOQ, μM | Dynamic range, μM | Sensitivity, μA M−1 |
|---|---|---|---|---|---|
| PBS (pH 4) | −0.15 | 10.7 ± 0.5 | 33 ± 2 | 33.3–300.00 | 2.8 ± 0.6 |
| Ascorbic acid 0.1 mol L−1 (pH 4) | −0.05 | 10 ± 7 | 36 ± 10 | 36–300.00 | 3.3 ± 0.2 |
Fig. 2C and F show the current change after increasing the H2 concentration from 100 μM to 200 μM in PBS (Fig. 2C) and ascorbic acid (Fig. 2F), which reveal a time response of 7.7 s (PBS) and 50 s (ascorbic acid), respectively. Even though the current response is not influenced by any redox reaction involving the ascorbic acid present in the solution, there might be interactions with the Pt-B surface that produce a delay in the time response of the sensor but does not significantly affect the overall analytical performance of the sensor. Indeed, it has been shown that ascorbic acid can adsorb at Pt electrodes via the carbon atom linking with the first hydroxyl group of the molecule and that this interaction may affect the hydrogen adsorption–desorption region of the Pt electrode.36,37
2.2. In situ H2 mapping
As proof-of-principle experiments, the hemispherical Pt-B probes were tested using two different model samples. The first experiment was performed using TiO2 powder (particle size > 100 nm) as photocatalytically active material. As shown in Fig. 3A, the setup consisted of two compartments separated by a porous (track-etched) polycarbonate membrane (pore sizes: 50 nm). The bottom compartment contained a TiO2 suspension in ascorbic acid, which was illuminated at λ = 365 nm to trigger the photocatalytic production of H2, following the reactions (7)–(9):38,39| TiO2 + hv → TiO2 (eCB− + hVB+) | (7) |
| 2eCB− + 2H+ → H2 | (8) |
| 2hVB+ + H2A → H+ + DHA | (9) |
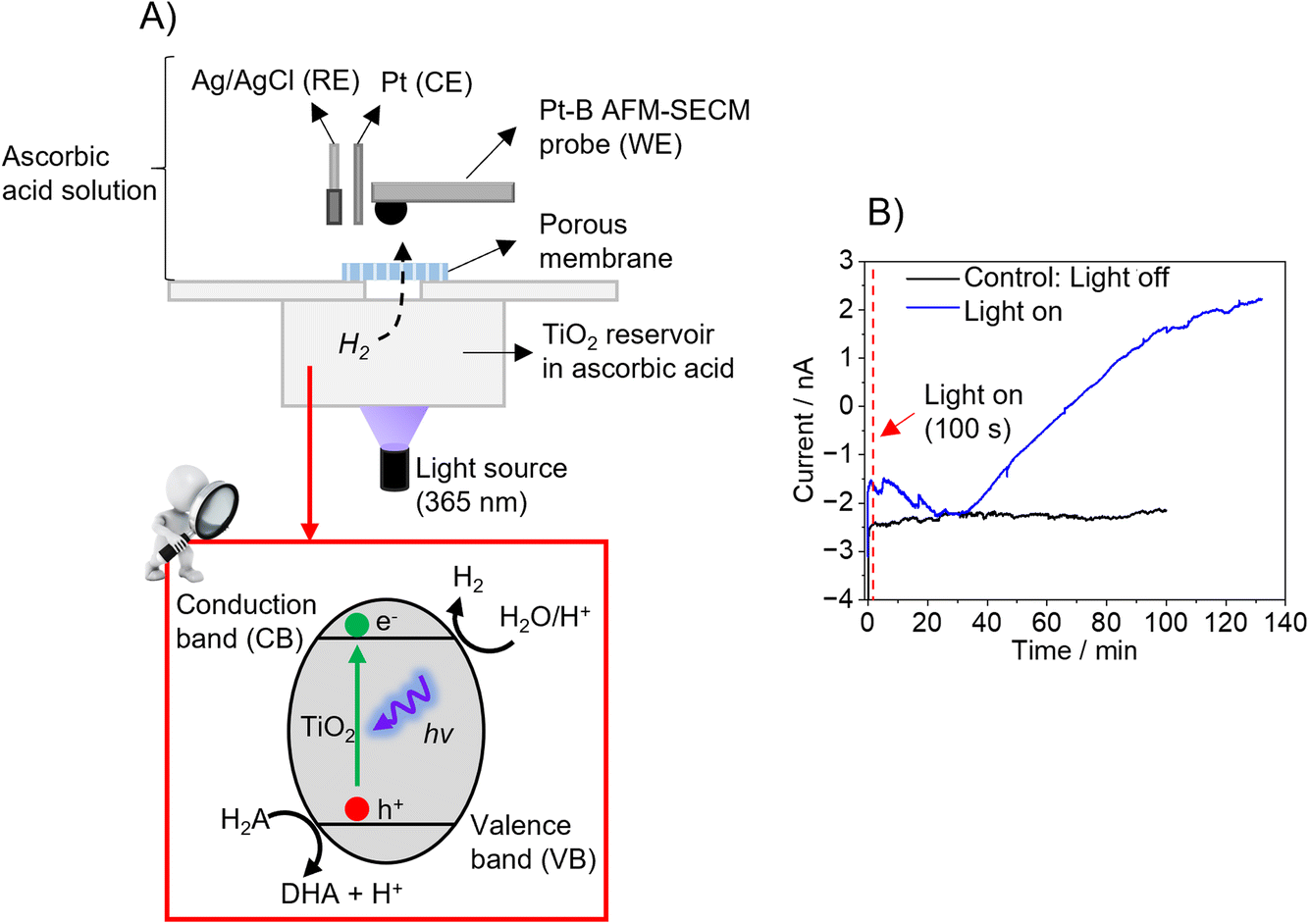 | ||
| Fig. 3 (A) Schematic of the two-compartment setup used for the proof-of-principle H2 measurement using the Pt-B AFM-SECM probe. (B) Chronoamperometry (i–t curve) recorded at the Pt-B AFM-SECM probe (Eprobe = −0.05 V vs. Ag/AgCl) under illumination (λ = 365 nm, blue line) and dark conditions (black line). | ||
During illumination, H2 evolves, which diffuses through the track-etched membrane to the upper compartment, in which the hemispherical Pt-B probe was positioned close to the membrane in a three-electrode configuration. To detect the H2, the hemispherical Pt-B probe was first moved at its open circuit potential to the membrane under dark conditions using the electronic feedback loop of AFM until it contacted the membrane and was then retracted up to 20 μm (false engagement). Then, a potential of −0.05 V vs. Ag/AgCl was applied to the probe. Fig. 3B (blue line) shows the current response recorded over time under illumination conditions. When comparing the current–time curve recorded in dark conditions as a control experiment, it is evident that illumination leads to an increase in current due to the evolution of H2, which diffuses through the tracked-etched membrane to the upper compartment. A decrease in current was observed during the first 30 minutes of illumination and the subsequent increase in current after this time until reaching a plateau after approx. 130 minutes. This plateau might be due to the system having reached an equilibrium since the saturation of the sensor is not expected, as shown in the H2 calibrations (Fig. 2D and E).
The second model sample consists of a partially Pt-covered silicon (Si) substrate. A 40 × 40 μm area at the border between the Pt and Si area was scanned using a 1 μm-diameter Pt-B AFM-SECM probe. Prior to the AFM experiments, the sample (area of Pt/Si) was first imaged by SEM-EDX. Fig. S4† shows the SEM and EDX data which reveals that there is no sharp border between the two areas due to the evaporation not masking the border effectively. For the Pt-coated area, H2 evolution is expected when applying a potential of −1.0 V vs. Ag/AgCl. The topography of the sample obtained by AFM using the hemispherical Pt-B probe (Fig. 4A) shows a change in height of around 60 nm between the Pt and the Si region. As the sample is fairly flat, the AFM topography does not reveal any obvious artifacts. In fact, an area with variations in morphology (upper part of the AFM image) can be clearly revealed with the hemispherical probe. Of course, as previously shown and discussed,17 samples with features such as narrow trenches or steep features would certainly generate topographical artifacts due to the shape and size of the hemispherical probe. The topography image was obtained in a 0.1 M PBS (pH 4) solution by keeping both the AFM-SECM probe and Pt@Si substrate at their open circuit potential. Once the probe was retracted 20 μm from the substrate, the probe was biased at −0.15 V vs. Ag/AgCl in order to detect the presence of H2 in SECM generation/collection mode,40 while the Pt@Si substrate was polarized at −1.0 V vs. Ag/AgCl, a sufficiently cathodic potential for the evolution of H2 from the Pt modified area. The current change detected when scanning the probe from the Si substrate (no H2 evolution) to the Pt-covered area was approx. 200 nA, with a clear increase over the Pt area (Fig. 4B). The change in current at the Pt–Si border was not pronounced, which is expected as there is no sharp border between the two areas (as shown in the SEM/EDX and AFM topography). However, as shown in the line scan in Fig. 4C, the current decreased gradually from the Pt area to the Si region, which fits also the topographical information. This gradual decrease in current might be due to a fast diffusion of the H2 produced at the Pt area.
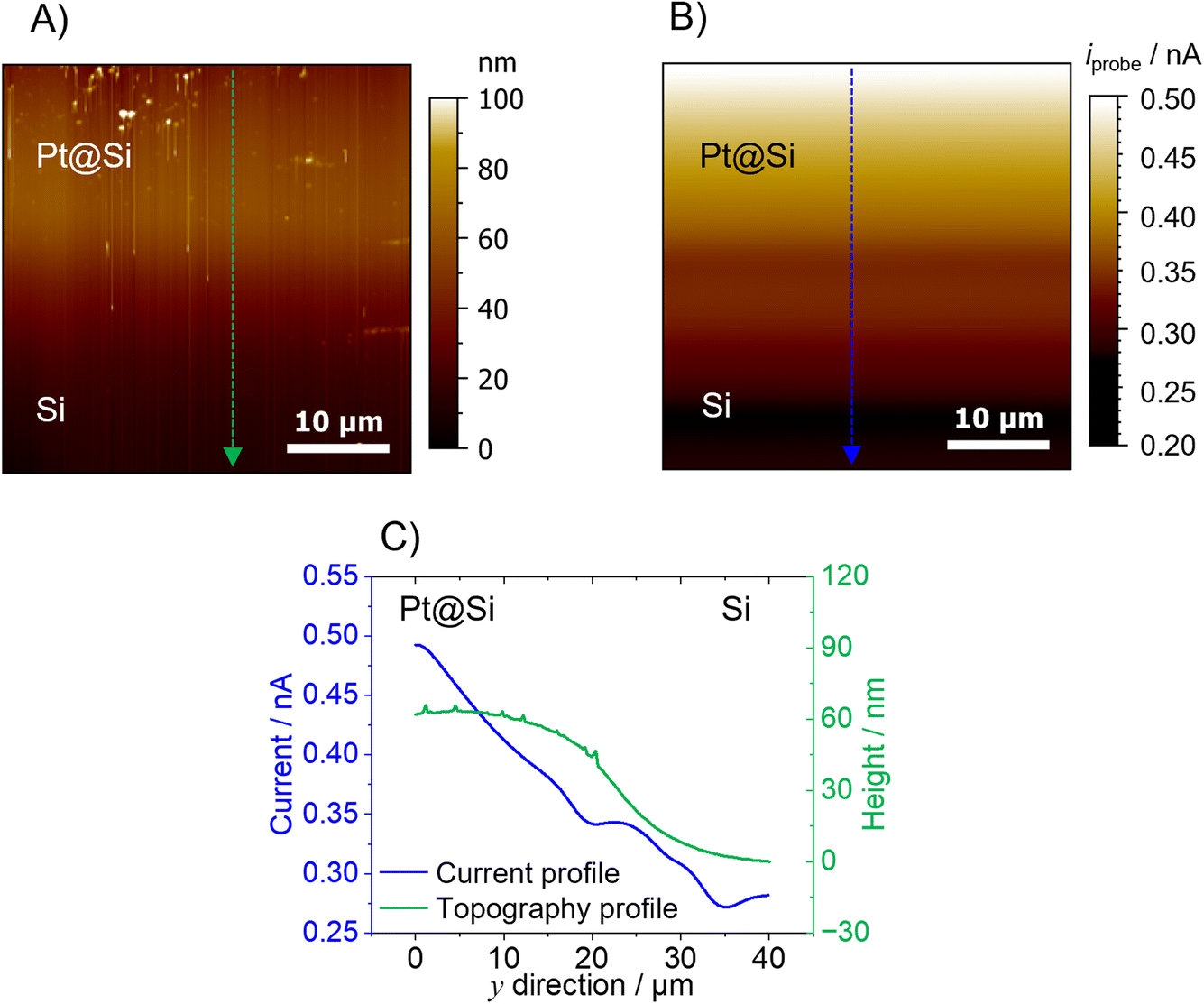 | ||
| Fig. 4 (A) Contact mode AFM topography map performed using a Pt-B probe immersed in 0.1 M PBS (pH 4). The probe and the substrate were kept in their open circuit potential. (B) Current map of the H2 evolution at the Pt electrode of the Pt@Si substrate after the Pt-B probe was retracted by 20 μm (Eprobe = −0.15 V vs. Ag/AgCl; Esubstrate = −1.0 V vs. Ag/AgCl). (C) Corresponding topography profile (green line) and current profile (blue line). All images were recorded with a scan speed of 0.2ln s−1. | ||
The current recorded at the Pt-B probe during the imaging can be used to calculate the concentration using the H2 calibration performed in bulk conditions (Fig. 2A). According to this correlation, the highest concentration of H2 evolved from the Pt was approx. 150 μM, while the lowest concentration detected over the Si substrate was approx. 84 μM. As with conventional SECM measurements in constant height mode, it is important to consider that the current map (and hence the concentration calculations) displays an averaged diffusion profile and that its resolution, among other factors, is a function of the distance between the substrate and the (AFM)-SECM probe. Taking into account the absence of a sharp border between the two areas, the size of the scanned area, and the fast diffusion of H2, the concentration of H2 over Si can be explained. Fig. S5† shows the control experiment performed by keeping the Pt substrate in its open circuit potential, so no H2 evolution should be observed. Indeed, no current change was detected between the Pt and Si regions.
2.3. Electrochemical quantification of H2 using Pt-B microelectrodes
Further calibration experiments using Pt-B sensors were also performed using a 25 μm-diameter microelectrode. The Pt-B microelectrodes were fabricated by electrodepositing Pt-B directly onto Pt disk microelectrodes via chronoamperometry (E = −0.06 V vs. Ag/AgCl) for 40 s in 31 mM H2PtCl6/0.67 mM Pb(NO3)2.23 To calculate the EASA of the Pt-B modified microelectrodes, the hydrogen desorption region, detected between approx. −0.25 V and 0.1 V in the CV recorded in 0.5 M H2SO4, was integrated (Fig. S6†).41 Considering again that the formation of an adsorbed hydrogen monolayer corresponds to a charge of Q = 210 μC cm−1, the calculated EASA is 0.20 ± 0.01 mm2 (n = 3). The Pt-B microsensors were tested in terms of H2 quantification in different solutions and at different pH values. PBS solution was again chosen as a model electrolyte testing the sensors at three different pH levels: 4, 7, and 10. Solutions containing ascorbic acid at pH 4 and triethanolamine (TEOA) at pH 10 were also used to evaluate the compatibility of the microsensors for photocatalytic H2 measurements.34Like in the experiments performed using Pt-B AFM-SECM probes, the highest anodic peak in the HUPD region of the CV performed in the corresponding solution was chosen for the chronoamperometric experiments and H2 calibration. Worth mentioning is the shift of approx. 0.13 V, when the pH of the solution in PBS changes from 4 to 10, and the even larger peak shift (approx. 0.3 V) when the CV was performed in the presence of 10% v/v TEOA (pH 10). The peaks for the calibrations and the analytical performance of the sensor are summarized in Table 2. All calibration curves are shown in Fig. S7.†
| Solution | E vs. Ag/AgCl, KCl (sat.), V | LOD, μM | LOQ, μM | Dynamic range, μM | Sensitivity, μA M−1 |
|---|---|---|---|---|---|
| PBS (pH 4) | −0.15 | 12.5 ± 6 | 41.7 ± 19 | 41.7–700.00 | 50.3 ± 3 |
| PBS (pH 7) | −0.20 | 5.25 ± 2 | 17.5 ± 9 | 17.5–700.00 | 47.8 ± 3 |
| PBS (pH 10) | −0.28 | 30.0 ± 1 | 100.0 ± 4 | 100.0–700.00 | 47.3 ± 3 |
| Ascorbic acid 0.1 mol L−1 (pH 4) | −0.05 | 24.0 ± 2 | 80.0 ± 8 | 80.0–700.00 | 50.9 ± 4 |
| TEOA 10 v/v in K2HPO4 (pH 10) | −0.47 | 4.0 ± 3 | 13.3 ± 3 | 13.3–200.00 | 29.0 ± 2 |
On average, the best performance of the Pt-B microsensors was achieved in PBS solution at pH 7 with a wide dynamic range and a sensitivity close to 50 μA M−1. Considering the results just in PBS (without the presence of organic compounds) in alkaline pH, the sensor exhibits the lowest efficiency in terms of dynamic range but still maintains a rather good sensitivity. This might be due to the slower HOR kinetics of Pt-B, and in general of Pt electrodes, at alkaline pH values.27,42
When comparing the calibrations performed at alkaline pH in PBS at pH 10, the highest H2 concentration that could be quantified is 700 μM (approx. the saturation of H2 in water), whereas in the presence of TEOA, this drops down to 200 μM. Even though TEAO exhibits only one redox potential at potentials higher than +0.6 V vs. Ag/AgCl, KCl (sat.), there might be possible interactions with the Pt-B electrode that affect the H2 quantification. A comprehensive study of the interactions between TEOA and Pt-B was not within the scope of this work.
Like for the characterization of the hemispherical Pt-B probes, to obtain information on the recovery of the Pt-B microelectrode, after each calibration, the H2 was removed from the solution by purging with pure nitrogen for at least 10 minutes. After purging, again H2 measurements at the corresponding potential were performed, and the current response dropped to or close to the baseline of 0 μM concentration at all investigated pH values. Fig. S7F† shows an exemplary chronoamperometry experiment performed with consecutive increments of H2 at the Pt-B microelectrode and the drop of current close to the baseline after completing the calibration and purging the electrochemical cell with nitrogen. When comparing the performance of the hemispherical Pt-B probes and the Pt-B microelectrodes, the hemispherical Pt-B probes exhibit a linearity up to an H2 concentration of 300 μM, being lower than the linearity achieved with the microelectrodes in the same conditions (700 mM, see Tables 1 and 2). This might be due to the smaller EASA of the hemispherical Pt-B probes in comparison to the microelectrodes, being approx. 7 times smaller. However, even though the working range is reduced, the LOD and LOQ in the AFM-SECM probes are improved, as shown in Table 1.
The Pt-B modified microelectrode was also compared to bare Pt microelectrode (diam. 25 μm) in PBS at pH 4 in the presence of H2 in solution. The higher current response of the Pt-B modified microelectrode is evident in Fig. S8† in which the CVs performed in PBS (pH 4) using the Pt-B sensor (black line) and the disk Pt electrode (red line) are shown. The lower current response in the CVs is also reflected in the H2 calibration as evidenced in Fig. S7A.† The disk Pt electrode not only does not show a good linearity but also a current response of approx. 95% lower than the response achieved with Pt-B at the same concentration (example shown at 100 μM).
In previous works,4,43 we presented a Pd-modified microelectrode for H2 quantification in acidic conditions (pH 4–5.5) that was successfully used for the detection of light-driven H2 evolution from micro-spot arrays of cobaloxime catalysts using SECM. The H2 absorption using the Pd-modified microelectrodes biased at −600 mV vs. Ag/AgCl was used for these measurements. A linearity from 0 μM up to 96.4 μM, with LOD and LOQ in the range of, respectively, 0.34–0.95 μM and 1.14–3.17 μM was obtained for these H2 microsensors.4 Although the Pt-B microsensors presented here exhibit less favorable LODs and LOQs, the Pt-B microsensors show a larger dynamic range compared to the Pd-modified microsensors, reaching the quantification of H2 up to its saturation concentration in water. Moreover, the Pt-B microsensors do not exhibit saturation and therefore must not be regenerated and can be used for longer measurement times. Pd-modified microelectrodes, instead, exhibit saturation due to the H2 absorption which requires a cleaning step in which a potential of 0.2 V vs. Ag/AgCl is applied.
3 Conclusions
Photocatalytic and electrocatalytic materials for H2 evolution are often heterogeneous in nature. Therefore, it is highly important to investigate possible heterogeneities at the micro- and nanoscopic level under in situ and/or operando conditions. AFM-SECM-based sensors with sub-micron geometric dimensions are a suitable approach for studying morphological features along with mapping the activity or electrochemical properties. In this work, we demonstrate the feasibility of fabricating micro- and sub-mcro-sized probes based on Pt-B electrodeposition for the electrochemical detection of H2 evolution. The increased EASA of the hemispherical Pt-B probes allows effective quantification of H2 in a wide concentration range, up to an H2 concentration of 300 μM. These probes were successfully tested using two different model systems as examples for photocatalytic and electrocatalytic H2 evolution, demonstrating that the hemispherical Pt-B probes are suitable candidates for future in situ/operando investigations of heterogenized photo(electro)catalytic materials.Current research is focused on using AFM-SECM nanoprobes based on Pt-B to investigate the electrochemical activity and photo-degradation screening of immobilized catalysts, such as earth-abundant catalysts as recently shown for cobaloxime catalysts deposited on solid surfaces such as carbon nanomembranes.43
4 Experimental
4.1. Chemicals and materials
Potassium dihydrogen phosphate (KH2PO4), dipotassium hydrogen phosphate (K2HPO4), sodium chloride (KCl), sulfuric acid (85% v/v), sodium hydroxide (NaOH), chlorohydric acid (HCl), hydrogen hexachloroplatinate (H2[PtCl6]), lead(II) nitrate (Pb(NO3)2), triethanolamine (TEOA), titanium dioxide (TiO2), and ferrocene-methanol (FcMeOH) were obtained from Merck, Germany. All solutions were prepared with ultrapure water (Elga water system, UK, conductivity 18.0 MΩ cm).4.2. Preparation of the Pt-black modified probes and calibration procedure
Pt microelectrodes were fabricated following well-established procedures,44 by sealing a 12.5 μm radius Pt wire (Goodfellow, Bad Nauheim, Germany) in a borosilicate glass capillary (Hilgenberg GmbH, Malsfeld, Germany). The microelectrodes were then sequentially polished, cleaned in an ultrasonic bath with ultrapure water, and characterized via optical microscopy and cyclic voltammetry. AFM-SECM probes were produced as described elsewhere.17,45 Briefly, commercially available tipless AFM cantilevers (Bruker NP-O, k = 0.06 N m−1) were modified with a Au layer via evaporation and insulated with silicon oxide/silicon nitride layers using plasma-enhanced chemical vapor deposition. The insulation layer was locally removed through FIB milling (Quanta 3D FEG, ThermoFisher Scientific, USA) to expose a recessed, Au disk-shaped (sub)-microelectrode at the apex of the cantilever.The electrochemical preparation and characterization of the Pt-B modified probes were performed in a three-electrode setup using a CHI660C potentiostat (CH Instruments, USA). Pt-B deposition on Pt-microelectrodes and Au-milled AFM-SECM probes was performed in a three-electrode set-up consisting of a Pt wire serving as a counter electrode, an Ag/AgCl, KCl (sat.) reference electrode, and the corresponding AFM-SECM probe as the working electrode. The probe was immersed in a solution containing 31 mM H2PtCl6 and 0.67 mM Pb(NO3)2 in PBS (pH = 3.2) and a constant potential of −0.06 V vs. Ag/AgCl, KCl (sat.) was applied for 40 seconds (Pt-microelectrodes) or 10 s (AFM-SECM probes); for AFM-SECM probes with a disk Au electrode smaller than 500 nm pulsed deposition was used by applying 40 cycles with a potential pulse sequence of −0.06 V/0.5 s; 0.0 V/0.5 s vs. Ag/AgCl, KCl (sat.).
The H2 calibration of the Pt-B modified microelectrodes was performed using chronoamperometry in PBS at three different pH values (pH 4, 7, and 10), in 0.1 M ascorbic acid (pH 4) and 10% v/v TEOA (pH 10). H2 calibration of the Pt-B modified AFM-SECM probes was performed in PBS and 0.1 M ascorbic acid at pH 4. Before each calibration experiment, a CV from −0.7 V to 1.2 V vs. Ag/AgCl, KCl (sat.) in the corresponding oxygen-free solution was recorded. For the calibration, the electrochemical cell (a homemade closed glass chamber equipped with an inlet and outlet gas line) was purged with a mixture of H2 and N2. The H2 concentration (0 μM to 700 μM) was controlled by mixing nitrogen (N2) and H2 gas at different ratios using a mass flow controller (Bronkhorst GmbH, Kamen, Germany). Data was analyzed using OriginPro 2021 v. 9.8.0.200 (OriginLab Corporation, USA) software.
4.3. In situ hydrogen measurement via AFM-SECM
AFM-SECM measurements for H2 evolution were performed in either a three- or four-electrode cell using a 5500 AFM/SPM microscope (Keysight Technologies, USA) and a CHI 832A bipotentiostat (CH Instruments, USA).The photocatalytic sample consists of two compartments separated by a porous polycarbonate membrane (pore density approx. 4.5 × 1012 pores per m2 and pore size of 50 nm). The scheme of the setup is depicted in Fig. 3A. The lower compartment is filled with a 0.05 M TiO2 suspension in 0.1 M ascorbic acid, while in the upper compartment, the three-electrode configuration is placed with the Pt-B AFM-SECM probe serving as the working electrode, an Ag/AgCl as a pseudo-reference electrode and a Pt wire as the counter electrode. The upper compartment was filled with a solution of 0.1 M ascorbic acid. For H2 measurements the compartment with the TiO2 suspension was illuminated from the bottom using an optical fiber (diameter 1000 μm, M59-L01, Thorlabs GmbH, Bergkirchen, Germany) connected to a 141 mW blue LED (365 nm, M365FP1, Thorlabs GmbH). Prior to the illumination, the AFM-SECM probe was moved towards the membrane under dark conditions and after contact lifted to 20 μm, the distance at which the photocatalytic H2 detection was performed. Once the AFM-SECM probe was positioned, a potential of −0.06 V vs. Ag/AgCl was applied to the probe and after the current response stabilized, the light was turned on to trigger H2 evolution.
The electrocatalytic sample for H2 evolution consisted of a 60 nm Pt layer deposited via sputtering on a silicon substrate (Pt@Si). The electrochemical cell consisted of an Ag/AgCl pseudo-reference electrode, a Pt wire serving as a counter electrode, and the Pt-B probe and the Pt@Si substrate as the first and second working electrodes, respectively. Topographical images were performed in contact mode in solution maintaining both working electrodes at their open circuit potential, while images of H2 evolved from the substrates were carried out after retracting the Pt-B probe by a distance of 20 μm (false engagement). The probe was biased at −0.15 V vs. Ag/AgCl and the Pt@Si substrate was biased at −1.0 V vs. Ag/AgCl to favor the electrocatalytic evolution of H2 at the Pt area. For the control experiment, the same sample area was scanned leaving the Pt@Si substrate at open circuit potential. All images were recorded in 0.1 M PBS (pH 4) with a scan rate of 0.2ln s−1. Data evaluation was performed using MoutainSPIP v. 9 (Digital Surf, France) and OriginPro 2021 software, v 9.8.0.200 (OriginLab Corporation).
Data availability
Data for this article are available at the Open Access Repositorium der Universität Ulm und Technischen Hochschule Ulm (OPARU) at https://doi.org/10.18725/OPARU-53116.Author contributions
Giada Caniglia: conceptualization, data curation, writing. Sarah Horn: performed part of the experimentation with microelectrodes. Christine Kranz: supervision, conceptualization, review, and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the FIBCenter UUlm and Dr Gregor Neusser (Institute of Analytical and Bioanalytical Chemistry, Ulm University) for the FIB-SEM measurements and Dr Sven Daboss (Institute of Analytical and Bioanalytical Chemistry, Ulm University) for the gold coating and insulation of the tipless AFM cantilevers. Georgia Mandela is acknowledged for contributions to electrochemical measurements. The project is funded by the Deutsche Forschungsgemeinschaft (DFG – German Research Foundation) – project number 364549901 – TRR 234, subproject C4.References
- M. González Martínez, M. Elsaddik and A. Nzihou, Int. J. Hydrogen Energy, 2023, 48, 22113–22131 CrossRef.
- T. Hübert, L. Boon-Brett, G. Black and U. Banach, Sens. Actuators, B, 2011, 157, 329–352 CrossRef.
- G. Korotcenkov, S. Do Han and J. R. Stetter, Chem. Rev., 2009, 109, 1402–1433 CrossRef CAS PubMed.
- J. Kund, J. Romer, E. Oswald, A. Gaus, M. Küllmer, A. Turchanin, M. von Delius and C. Kranz, ChemElectroChem, 2022, 9, e202200071 CrossRef CAS.
- D. Koster, R. Gutkowski, J. Masa and W. Schuhmann, J. Electroanal. Chem., 2018, 812, 207–212 CrossRef CAS.
- H. Li, M. Du, M. J. Mleczko, A. L. Koh, Y. Nishi, E. Pop, A. J. Bard and X. Zheng, J. Am. Chem. Soc., 2016, 138, 5123–5129 CrossRef CAS PubMed.
- C. Iffelsberger, D. Rojas and M. Pumera, J. Phys. Chem. C, 2022, 126, 9016–9026 CrossRef.
- C. Iffelsberger, S. Ng and M. Pumera, Chem. Eng. J., 2022, 446, 136995 CrossRef CAS.
- J. W. Hill and C. M. Hill, Nano Lett., 2019, 19, 5710–5716 CrossRef CAS PubMed.
- Y. Liu, C. Jin, Y. Liu, K. H. Ruiz, H. Ren, Y. Fan, H. S. White and Q. Chen, ACS Sens., 2021, 6, 355–363 CrossRef CAS.
- Y. Wang, E. Gordon and H. Ren, J. Phys. Chem. Lett., 2019, 10, 3887–3892 CrossRef CAS.
- C. Santana Santos, B. N. Jaato, I. Sanjuán, W. Schuhmann and C. Andronescu, Chem. Rev., 2023, 123, 4972–5019 CrossRef CAS.
- B. J. Ostertag, M. T. Cryan, J. M. Serrano, G. Liu and A. E. Ross, ACS Appl. Nano Mater., 2022, 5, 2241–2249 CrossRef CAS PubMed.
- H. du Toit and M. Di Lorenzo, Sens. Actuators, B, 2014, 192, 725–729 CrossRef CAS.
- M. E. Sandison, N. Anicet, A. Glidle and J. M. Cooper, Anal. Chem., 2002, 74, 5717–5725 CrossRef CAS PubMed.
- A. M. Feltham and M. Spiro, Chem. Rev., 1971, 71, 177–193 CrossRef CAS.
- A. Hellmann, G. Neusser, S. Daboss, M. M. Elnagar, J. Liessem, D. Mitoraj, R. Beranek, S. Arbault and C. Kranz, Anal. Chem., 2024, 96, 3308–3317 CrossRef CAS.
- L. Fan, J. Zhao, X. Luo and Z. Tu, Int. J. Hydrogen Energy, 2022, 47, 5418–5428 CrossRef CAS.
- A. R. Kucernak and E. Toyoda, Electrochem. Commun., 2008, 10, 1728–1731 CrossRef CAS.
- S. Ben-Amor, E. Vanhove, F. Sékli Belaïdi, S. Charlot, D. Colin, M. Rigoulet, A. Devin, N. Sojic, J. Launay, P. Temple-Boyer and S. Arbault, Electrochim. Acta, 2014, 126, 171–178 CrossRef CAS.
- Y. Wang, J.-M. Noël, J. Velmurugan, W. Nogala, M. V. Mirkin, C. Lu, M. Guille Collignon, F. Lemaître and C. Amatore, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11534–11539 CrossRef CAS PubMed.
- Y. Li, K. Hu, Y. Yu, S. A. Rotenberg, C. Amatore and M. V. Mirkin, J. Am. Chem. Soc., 2017, 139, 13055–13062 CrossRef CAS.
- A. Hellmann, S. Daboss, F. Zink, C. Hartmann, P. Radermacher and C. Kranz, Electrochim. Acta, 2020, 353, 136458 CrossRef CAS.
- T. Kessler, A. M. Castro Luna, W. E. Triaca and A. J. Arvia, J. Appl. Electrochem., 1986, 16, 693–702 CrossRef CAS.
- T. C. Franklin and S. L. Cooke, J. Electrochem. Soc., 1960, 107, 556 CrossRef CAS.
- E. S. Davydova, S. Mukerjee, F. Jaouen and D. R. Dekel, ACS Catal., 2018, 8, 6665–6690 CrossRef CAS.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef.
- D. S. Hall, C. Bock and B. R. MacDougall, J. Electrochem. Soc., 2013, 160, F235–F243 CrossRef CAS.
- W. Vogel, L. Lundquist, P. Ross and P. Stonehart, Electrochim. Acta, 1975, 20, 79–93 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- H. H. Do, D. L. T. Nguyen, X. C. Nguyen, T.-H. Le, T. P. Nguyen, Q. T. Trinh, S. H. Ahn, D.-V. N. Vo, S. Y. Kim and Q. van Le, Arabian J. Chem., 2020, 13, 3653–3671 CrossRef CAS.
- Q.-S. Chen, J. Solla-Gullón, S.-G. Sun and J. M. Feliu, Electrochim. Acta, 2010, 55, 7982–7994 CrossRef CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- U. Pal, S. Ghosh and D. Chatterjee, Transition Met. Chem., 2012, 37, 93–96 CrossRef CAS.
- X. Xing, I. T. Bae, M. Shao and C.-C. Liu, J. Electroanal. Chem., 1993, 346, 309–321 CrossRef CAS.
- P. Karabinas and D. Jannakoudakis, J. Electroanal. Chem. Interfacial Electrochem., 1984, 160, 159–167 CrossRef CAS.
- M. Rafique, S. Hajra, M. Irshad, M. Usman, M. Imran, M. A. Assiri and W. M. Ashraf, ACS Omega, 2023, 8, 25640–25648 CrossRef CAS.
- V. Kumaravel, S. Mathew, J. Bartlett and S. C. Pillai, Appl. Catal., B, 2019, 244, 1021–1064 CrossRef CAS.
- D. Polcari, P. Dauphin-Ducharme and J. Mauzeroll, Chem. Rev., 2016, 116, 13234–13278 CrossRef CAS PubMed.
- S. Trasatti and O. A. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
- V. Briega-Martos, A. Ferre-Vilaplana, E. Herrero and J. M. Feliu, Electrochim. Acta, 2020, 354, 136620 CrossRef CAS.
- E. Oswald, A. L. Gaus, J. Kund, M. Küllmer, J. Romer, S. Weizenegger, T. Ullrich, A. K. Mengele, L. Petermann, R. Leiter, P. R. Unwin, U. Kaiser, S. Rau, A. Kahnt, A. Turchanin, M. von Delius and C. Kranz, Chem.–Eur. J., 2021, 27, 16896–16903 CrossRef CAS PubMed.
- F. R. F. Fan and D. Damaille, Preparation of tips for scanning electrochemical microscopy, in Scanning Electrochemical Microscopy, ed. A. J. Bard and M. V. Mirkin, 2nd edn, CRC Press, Boca Raton, 2012, pp. 25–51 Search PubMed.
- P. Knittel, H. Zhang, C. Kranz, G. G. Wallace and M. J. Higgins, Nanoscale, 2016, 8, 4475–4481 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00136b |
| This journal is © The Royal Society of Chemistry 2025 |