
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of natural tissues and food waste into aerogels and their application in oleogelation†
Lara Gibowsky ae,
Lorenzo De Berardinisc,
Stella Plazzottac,
Erik Mankea,
Isabella Junga,
Daniel Alexander Méndezd,
Finnja Heidorna,
Gesine Lieseb,
Julia Husungb,
Andreas Liesebe,
Pavel Gurikovaf,
Irina Smirnovaae,
Lara Manzoccoc and
Baldur Schroeter*ae
ae,
Lorenzo De Berardinisc,
Stella Plazzottac,
Erik Mankea,
Isabella Junga,
Daniel Alexander Méndezd,
Finnja Heidorna,
Gesine Lieseb,
Julia Husungb,
Andreas Liesebe,
Pavel Gurikovaf,
Irina Smirnovaae,
Lara Manzoccoc and
Baldur Schroeter*ae
aInstitute of Thermal Separation Processes, Hamburg University of Technology, Eißendorfer Straße 38, 21073 Hamburg, Germany. E-mail: baldur.schroeter@tuhh.de
bInstitute of Technical Biocatalysis, Hamburg University of Technology, Denickestraße 15, 21073 Hamburg, Germany
cDepartment of Agricultural, Food, Environmental and Animal Sciences, University of Udine, Via Sondrio 2/A, 33100 Udine, Italy
dFood Safety and Preservation Department, Institute of Agrochemistry and Food Technology (IATA-CSIC), Valencia, Spain
eUnited Nations University Hub on Engineering to Face Climate Change at the Hamburg University of Technology, United Nations University Institute for Water, Environment and Health (UNU-INWEH), Hamburg, Germany
faerogel-it GmbH, Albert-Einstein-Str. 1, 49076 Osnabrück, Germany
First published on 4th April 2025
Abstract
In this work, various natural tissues were for the first time directly converted into nanostructured aerogels by utilizing their intrinsic (meso-)porosity. In contrast to common aerogel production, no use of pure biopolymers, their extraction, dissolution, gelation or use of additives (e.g. crosslinkers, acids and bases) was necessary. The production process required washing of the wet starting material with water, a solvent exchange with ethanol and drying with supercritical CO2. The resulting materials exhibited low bulk densities (0.01–0.12 g cm−3), significant specific surface areas (108–446 m2 g−1) and mesopore volumes (0.3–2.6 cm3 g−1). Assessment of 20 different tissues including fruit pulp and peel, vegetable pulp, and mushrooms showed the generality of the approach. A broad spectrum of different microstructures was identified, whereas especially textural properties of samples derived from water rich pulp were highly similar to those found in classical biopolymer aerogels, for instance based on pectin or cellulose. Furthermore, the capability of the materials to structure liquid sunflower oil was shown: the produced oleogels exhibited exceptionally high oil uptake (max. 99%) and rheological properties similar to those of solid fats. Results suggest that supercritical drying of tissues (e.g. based on food waste) is a suitable approach for their upcycling into value added materials by a complete green and sustainable process. This research also contributes to sustainable development by transforming food waste into valuable aerogels and promoting science education through accessible, open-source STEM resources.
Green foundation1. Our research enables the sustainable conversion of natural tissues into aerogels without biopolymer extraction or synthetic additives. Unlike conventional sol–gel methods, it eliminates strong acids, bases, and toxic solvents, reducing chemical waste and energy consumption.2. We directly transform food waste (e.g., fruit peels, vegetable fibers, mushrooms) into nanostructured aerogels using a green process involving water washing, ethanol exchange, and supercritical CO2 drying. These eco-friendly aerogels feature high surface areas and ∼99% oil absorption, making them ideal in application as sustainable oleogels. 3. Optimization could reduce ethanol and CO2 consumption via recovery systems. Low-energy vacuum drying and bio-based solvents could further enhance sustainability. Incorporating enzymatic treatments and expanding the use of agricultural by-products would strengthen the circular economy and broaden applications beyond food and pharmaceuticals. |
1 Introduction
Food loss and waste account for approx. 44% of global waste and around 30% of global food production: according to the United Nations Food Waste Index Report 2024, an estimated 931 million tons of food loss and waste are generated annually worldwide.1,2 In line with the UN sustainable development goal 12.3, the aim is to reduce this amount by 50% by 2030. To achieve this, both the avoidance and valorization of food waste have become crucial measures in recent years. The latter includes the upcycling of food and agricultural waste, reintegrating both edible and inedible parts (e.g. unutilized parts like residual pulp, peels, plant body leftovers) back into the human supply chain, but also its conversion into value added products which exceed the food-sector, for instance natural dyes, membranes, hydrogels and bio-fuels.2–5 An alternative/supplement to the direct conversion of food waste into high-value products is the extraction of bioactive compounds, including secondary metabolites, phytochemicals, oils, unsaturated fatty acids, and polysaccharides, as well as dietary fibers: such approaches require the development of green, cost-efficient, and selective extraction methods.3,6 Amongst various processes, supercritical fluid extraction (SFE) utilizing supercritical CO2 (scCO2) stands out as a widely used and green technique, which is comparably cheap, easily accessible, selective, and generally recognized as safe (GRAS).7,8Non-extractive valorization of food waste involves its direct use as a value added product, e.g. as a fertilizer or feed for livestock: hereby dry residues are normally used which contain approximately 65% carbohydrates, 12% crude proteins and a high fiber content of about 13%.3,9 Depending on the final solid shape (e.g. granules, chips, powders) and particular application, various different drying techniques are applied (for instance solar-, freeze-, and vacuum-drying, aeration, biological conversion, spray-, and drum processes).10 ScCO2-drying offers additional opportunities by enabling microbial inactivation and preservation of valuable properties and components.11–14 Furthermore, scCO2-drying can be used to avoid the occurrence of capillary forces during the drying process, effectively drying the material without causing microstructural damage.15,16 It is notable that the unique properties of supercritical fluids are utilized in both processes, SFE and scCO2-drying, but with different objectives. However, scCO2-drying of food products is still generally uncommon, particularly with the aim to preserve the intrinsic microstructure: pioneering studies have focused so far mostly on other aspects like microbial and enzymatic inactivation while providing nutrient retention and an increased shelf life as a result of the scCO2-drying process.12–14 However, in other fields, scCO2-drying is widely used for the fabrication of materials, whose primary value is rooted in their internal microstructure, namely aerogels.16
In a simple description, aerogels are highly porous solids (porosity ε of up to 99%) with low densities, whose microstructures offer a significant mesopore content (pore diameter dpore = 2–50 nm) and high mass specific surface areas (Sm) of up to >1000 m2 g−1.17,18 Even though no single definition of the term “aerogel” is so far provided in the literature, truly mesoporous aerogels are clearly distinguishable from other porous materials by their microstructural features. In particular, freeze-drying as opposed to supercritical drying leads to so called cryogels, which resemble coarse, solid foams due to their macroporous nature (pore diameter in the micrometer range) and lack therefore the distinguished properties (like a high Sm) of aerogels.16,19
This is especially demonstrated by works in which both drying techniques are directly compared.20,21 In this sense, the numerous works reporting the production of “aerogels” (from food waste and in general) via freeze-drying should critically be checked regarding the microstructural properties of the resulting materials (see e.g.22–26). Aerogels can principally be derived from any material (classically silica, but also various polymers and even metals). Amongst these, biopolymer aerogels show properties like biodegradability, biocompatibility and good viscoelasticity, which are desirable in food and pharma applications.17,27–29 The production of mesoporous biopolymer aerogels requires generally several steps, whereas the first step is in most cases the extraction and dissolution of a pure biopolymer in an appropriate solvent (Fig. 1).16 The subsequent build-up of three-dimensional pore networks (hydrogel formation, step 2) is achievable via sol–gel conversion: gelation of dissolved biopolymer molecules is hereby inducible via different methods (e.g. physical, ionic or covalent crosslinking).16 Follow-up steps are (3) the exchange of water by an organic solvent, in most cases ethanol and (4) removal of the liquid solvent by scCO2-drying, typically carried out at pressures in the range of 100–150 bar and slightly elevated temperatures >35 °C.30 Even though various biopolymer-to-aerogel routes have been established in recent years16 (including conversion of agricultural waste22 and biorefinery approaches31), it is crucial to minimize overall production costs of aerogels in order to enhance their competitiveness on the market. Besides optimization of solvent exchange32,33 and supercritical drying steps,34–36 the isolation of pure biopolymers from mixtures/composites and their dissolution should also be taken into account as cost driving and sustainability factors in the aerogel production line. For instance, cellulose is a not easily soluble biopolymer and the use of expensive or non-green solvents like aqueous alkali/urea mixtures,37 ionic liquids38 or molten salt hydrates39 is therefore required.15,40 Alternative approaches based on nanocellulose fibers avoid the sol–gel route and eliminate the dissolution step, but require the extraction of nanocellulose from biomass/cell walls (which is complicated) and demand typically further cost and energy intensive pretreatment, e.g. mechanical (such as grinding, separation), chemical, biological or combined methods.40 In the case of any biopolymer, the use of dissolution and crosslinking agents (like acids, bases, metal salts, etc.) increases the requirement for additional washing steps to remove chemical residuals from the pore matrix.37,41
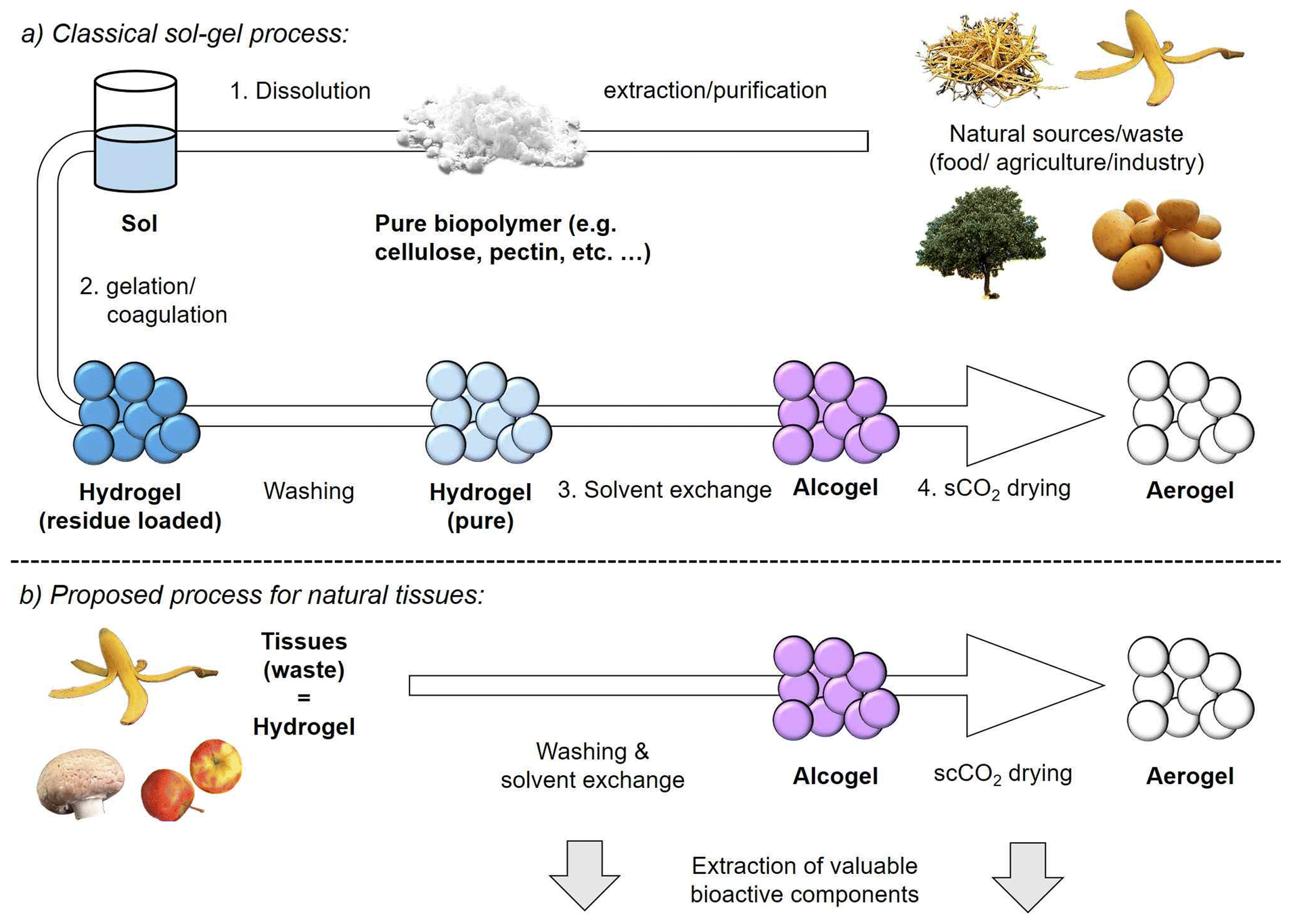 | ||
| Fig. 1 (a) Processing scheme for production of biopolymer aerogels from single components via the sol–gel route. (b) Proposed process for the direct conversion of natural tissues into aerogels. | ||
Recently, conversion of different orange parts into aerogels (peels, bagasse and pulp) by supercritical drying was reported.42 While this study highlights the possibility to transform a complete fruit-body into biopolymer aerogels, the intrinsic porosity of the orange-tissue was not used, since orange parts were first dried under ambient conditions (leading to mesopore-collapse). The pectin fraction was subsequently extracted and processed via the classical sol–gel route to form a porous matrix around non-dissolved orange parts.42 Generally, a question could be raised: why valuable biopolymers like pectin, cellulose, proteins and others should pass the steps of extraction, dissolution and gelation, rather than being directly converted to aerogels in their unaltered, intrinsically porous and natural tissue-based form. To address this matter, it is necessary to evaluate whether and to which extent supercritically dried natural tissues meet the criteria for being classified as “aerogels”, and how they compare to sol–gel-based aerogels in typical applications. So far, only a few pioneering works have dealt with scCO2-drying of natural tissues with respect to the resulting microstructures. Promising results were found for fresh cut salad waste, spent ground coffee and apple pomace tissues, whereas scCO2-dried products demonstrated aerogel-like behavior, e.g. low bulk densities, the presence of mesopores and significant specific surface areas (Sm = 112–229 m2 g−1).43,44 ScCO2-dried salad waste showed high water and oil sorption capability,45 indicating the potential use as a bulking agent or as a porous matrix for absorption of liquid, edible oils.
Considering their origin, it seems natural to use such materials in the food-sector: microstructural properties and composition render biopolymer aerogels indeed promising in food related applications, e.g. in food packaging or use as encapsulants and smart, functional ingredients.27,46 Open porosity and high specific surface area allow for quick, capillary force-driven uptake of oil, which is then held in the aerogel matrix via surface–oil interactions.27 This so called templating approach in oleogelation enables accordingly structuring of liquid, edible oils into semi-solid systems (oleogels), which represent a healthier alternative to solid, saturated fats and is e.g. applicable as a food ingredient in spreads.47,48 In previous studies, mesoporous whey- and potato protein isolate based aerogels have already been successfully converted into firm oleogels, showing hereby a high overall oil uptake >75% and sufficient oil holding capacities OHC (OHC up to 96%).47,49,50 It was found that these performance related criteria are connected to the microstructural properties of the aerogel template (e.g. specific surface area, meso- and macropore volumes) and to the polarity differences between oil and aerogel surfaces.20,27,49,50
The aim of this work is to propose a new way for food waste valorization by the direct conversion of natural tissues into biopolymer aerogels via scCO2-drying. So far, it has not been shown that a direct tissue-to-aerogel route (which avoids biopolymer and nanofiber isolation as well as sol–gel chemistry) is generally feasible and leads to competitive products. In order to assess the generality of the approach, a screening of twenty different tissues derived from fruit pulp, peels, mushrooms, and vegetables (including roots) is carried out. Samples are categorized based on their microstructure and compared with “conventional” sol–gel based biopolymer aerogels from single components. Finally, the application of the materials as oleogel templates is assessed by testing the capability of selected samples to adsorb edible liquid sunflower oil and to structure it into semi-solid oleogels. This research supports directly several UN Sustainable Development Goals (SDGs) as being a member of the United Nations University Hub at the Hamburg University of Technology.51 (1) By providing innovative methods to convert food waste into functional materials, advancing SDG 12 (responsible consumption and production) and SDG 9 (sustainable industrialization). (2) By upcycling natural food fabrics, we reduce environmental impact, promote resource efficiency and provide bio-based, sustainable alternatives to synthetic materials. (3) In addition, we address SDG 4 (quality education) through our open-access STEM platform, aerogel.kniffelix.de, which enables educators, students and the public to engage in science-based learning about aerogels, hands-on experimentation and sustainability-oriented innovation.52,53 This initiative not only advances sustainable technologies, but also empowers future generations to adopt environmentally friendly practices.
2 Materials and methods
2.1 Preparation of supercritically dried tissues
In total, 20 different tissue-based samples from fresh fruits, vegetables, and mushrooms have been prepared (for a complete sample list, see Table S1,† all starting materials were purchased at a local market). In the case of fruits and vegetables, the pulp has been separated from the remaining components like seeds and peels. The mushroom sample was processed without further separation. The desired parts (∼500 g) of the samples were subjected to 500 ml distilled water and shredded in a kitchen mixer (Bosch Stand Blender MMB6174S VitaPower). Shredded tissues were then washed in 10 L distilled water at room temperature for 12 h under stirring (overhead stirrer 750 rpm). The water was replaced, and the washing process was repeated three times with fresh distilled water to extract water-soluble components from the tissue-matrix. Subsequently, a direct solvent exchange from water to ethanol was carried out at room temperature, by placing samples in 99.9 wt% ethanol (EtOH, denatured, Carl Roth GmbH & Co. KG). This procedure was repeated with fresh EtOH until a minimum final concentration of 97.0 wt% EtOH inside the samples was reached (controlled by density measurements, Anton Paar, DMA 4500 M). In the case of samples which were later used in oleogelation, an additional wet milling step was applied after the solvent exchange was completed. This was done to achieve small particle sizes in the range of 50–100 μm which are optimal in terms of sensory effects during consumption.54 The procedure was carried out according to former works,47,55 using a colloid mill (IKA Magic LAB 1508) and applying a rotational speed of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and a gap width of 900 µm in the rotor-stator system. All solvent exchanged samples (alcogels) were sealed in filter paper bags and transferred into an autoclave (volume = 3.9 L). Drying with supercritical CO2 was then performed at a pressure of 120 bar and a temperature of 40 °C under a continuous CO2 flow (120–140 g min−1) until complete extraction of EtOH was achieved (approx. 3 hours drying time). Samples were collected after slow depressurization (2 bar min−1) of the autoclave and stored in 250 ml vessels sealed with parafilm in a desiccator over calcined silica gel until further use and analysis.
000 rpm and a gap width of 900 µm in the rotor-stator system. All solvent exchanged samples (alcogels) were sealed in filter paper bags and transferred into an autoclave (volume = 3.9 L). Drying with supercritical CO2 was then performed at a pressure of 120 bar and a temperature of 40 °C under a continuous CO2 flow (120–140 g min−1) until complete extraction of EtOH was achieved (approx. 3 hours drying time). Samples were collected after slow depressurization (2 bar min−1) of the autoclave and stored in 250 ml vessels sealed with parafilm in a desiccator over calcined silica gel until further use and analysis.
2.2 Characterization of supercritically dried tissues
Low temperature Nitrogen adsorption–desorption physisorption was carried out to investigate the microstructural properties of supercritically dried samples (Nova 3000e Surface Area Analyzer, Quantachrome Instruments, Boynton Beach, USA). Prior to each measurement, the corresponding sample was degassed in a measurement cell under vacuum at 60 °C for at least 6 h. A sample amount of 20–30 mg was used in each run. The Brunauer–Emmett–Teller (BET) method was used in the p/p0 range of 0.027–0.27 to estimate the mass specific surface area (Sm). The mesopore volume Vmeso and the mean mesopore diameter (dpore, mean) were estimated via the Barrett–Joyner–Halenda (BJH) method. Standard errors for determination of Sm and Vmeso were estimated based on a threefold determination (Sm ± 5 m2 g−1, Vmeso ± 0.14 cm3 g−1). The skeletal density ρS was determined via helium pycnometry (Multivolume Micromeritics AccuPyc II 1340, Norcross; GA USA) at room temperature. A 4-fold determination was performed for each sample. For estimation of the bulk density, a defined volume of sample (11.5 cm3) was filled in a container without tapping and the weight was determined on an analytical scale (Excellence Plus XP, Mettler Toledo). The solid bulk densities ρB were calculated as the ratio of the sample mass (m) and the sample volume (V) according to eqn (1):
 | (1) |
The porosity ε was estimated based on bulk- and skeletal densities:
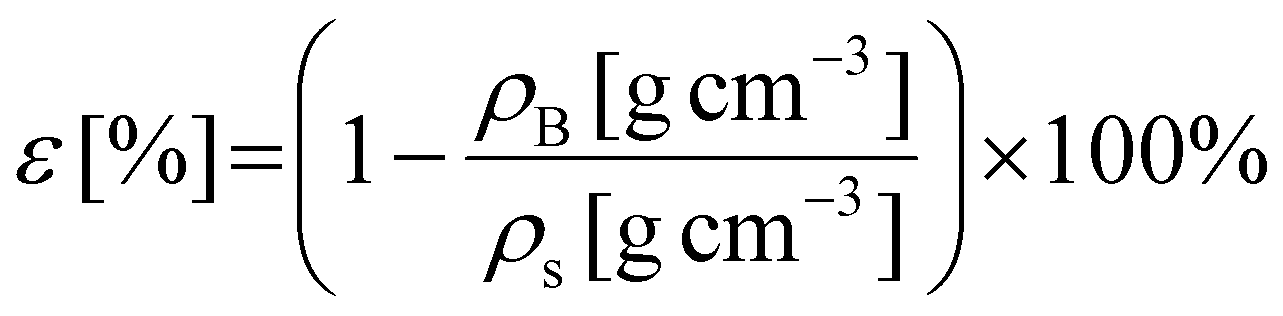 | (2) |
The inner pore structure of samples was characterized via scanning electron microscopy (SEM, Zeiss Supra VP55, Jena, Germany). Substrates were sputtered with a conductive, thin (approx. 6 nm) layer of gold (Sputter Coater SCD 050, BAL-TEC) prior to analysis. Measurements were carried out under high vacuum at an accelerating voltage of 3.00 kV, a working distance of 3.3–4.5 mm using an in-lens detector. Images of substrate powders were taken in a fotobox. The brightness and contrast of the SEM and photographic images were optimized using PowerPoint 2016. The neutral sugar and uronic acid composition of supercritically dried samples was analyzed following a modified procedure after acidic methanolysis.56 In brief, 1 mg of each sample was first freeze-dried and then subjected to methanolysis by incubation with 1 mL of 2 M HCl in anhydrous methanol at 100 °C for 5 hours. The reaction was then neutralized using pyridine, followed by drying under a stream of air. To further hydrolyze the samples, they were treated with 2 M trifluoroacetic acid at 100 °C for one hour. After repeated drying under an air stream, the samples were resuspended in MilliQ water, filtered (0.22 µm pore size), and injected for analysis. Monosaccharide analysis was carried out using high-performance anion exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD) on an ICS-6000 system (Thermo Fisher Scientific) fitted with a CarboPac PA1 column (4 × 250 mm, Dionex). Calibration was performed using control samples with known concentrations of monosaccharides, including glucose, fucose, rhamnose, galactose, arabinose, xylose, mannose, galacturonic acid, and glucuronic acid (Merck, Germany). The cellulose content was estimated as the difference of glucose between methanolysis and Saeman hydrolysis.57 In brief, 1 mg of each sample previously freeze dried was incubated with 0.125 mL of sulfuric acid for 3 hours at room temperature, then 1.375 mL of milliQ water was added and the samples were sealed and heated for 3 hours at 100 °C. The sample was filtered and diluted 1:10 with milliQ water, and then injected using the same equipment of the methanolysis method described previously. Calibration was performed using control cellulose with known concentrations (Merck, Germany). The protein content was quantified by the Dumas combustion method, employing a nitrogen conversion factor of 6.25, in accordance with ISO/TS 16634–2 (2016). All composition analyses were carried out in triplicate. The thermal stability was determined via thermogravimetric analysis (Linseis TGA PT 1600). An amount of 20–40 mg of sample was placed in an aluminum oxide crucible, heated with a heating rate of 10 °C min−1 to a final temperature of 850 °C under an oxidized atmosphere, followed by a dwelling phase of 120 min. A single bounce diamond ATR insert (MIRacle, Pike Technologies) and a liquid-cooled MCT detector were utilized for obtaining the ATR MIR spectra using a Vertex 70 spectrometer (Bruker Optik GmbH, Ettlingen, Germany). The Vertex 70 spectrometer was purged with dry and carbon dioxide-free air. The spectra were recorded from 4000 cm−1 to 600 cm−1 with 64 scans per spectrum using the OPUS 7.0 software package (Bruker Optik GmbH, Ettlingen, Germany).
2.3 Oleogel preparation and characterization
Oleogels were prepared by gradually adding sunflower oil (purchased in a local market) to an amount of 0.2 g aerogel, starting with an aerogel-oil weight ratio of 1:5. After each addition of oil, the sample was manually mixed, and the appearance was evaluated. Images were obtained after each oil addition using an image acquisition chamber (Immagini & Computer, Bareggio, Italy) equipped with a digital camera (EOS 550D, Canon, Milano, Italy).
The sample was placed on a spatula and positioned approx. 50 cm in front of the camera, with a black cardboard backdrop serving as the background. Light was provided by four 100 W photographic floodlights to optimize the pictures regarding shadow and gloss.
Viscoelastic properties (G′ and G″ moduli, and tanδ) were determined with a Rheometer RS6000 (Thermo Scientific RheoStress, Haake, Germany) equipped with a Peltier system for temperature control. All measurements were performed with a parallel plate geometry at 20 ± 0.5 °C and a gap width of 2 mm. First, the amplitude sweep test was carried out with increasing stresses from 1 to 6000 Pa at 1 Hz frequency to identify the linear viscoelastic region (LVR) of all oleogel samples. Afterward, the frequency sweep test was performed in duplicate at a stress of 10 Pa for all samples, which was determined in the LVR, and an increasing frequency of 0.1 to 15 Hz.
For confocal laser scanning microscopic measurements, oleogel samples were first stained with a 0.001% oil solution of Nile red and a 0.001% ethanolic solution of Fluorescent Brightener 28 (Sigma Aldrich, Milan, Italy), used to stain oil and polysaccharides, respectively. After staining, the samples were placed on a microscope slide, covered with a cover slide, and observed by using a confocal laser scanning microscope at 25 times magnification (Leica TCS SP8 X confocal system, Leica Microsystems, Wetzlar, Germany). Images were imported in tif.format using the software LasX 3.5.5 (Leica Microsystems, Wetzlar, Germany).
The oil holding capacity (OHC) was determined according to previous works.49 Eppendorf tubes were first weighed empty and then filled with approx. 0.5 g of the respective sample. Subsequently, samples were subjected to centrifugation (15 min at 15000g and 4 °C) (Mikro 120, Hettich Zentrifugen, Andreas Hettich GmbH and Co, Tuttlingen, Germany). After centrifugation, the released oil was removed, and the tubes were weighed again. The OHC of all oleogel samples was calculated according to eqn (3):
 | (3) |
All measurements were carried out in triplicate.
3 Results & discussion
To explore whether naturally grown tissues can be generally converted into aerogel-like materials, a screening of 20 different tissues, including pulps and peels sourced from fruits, vegetables, and mushrooms, was conducted. The names of supercritically dried samples correspond in the following discussion directly to the educts used in the production. If not further specified, samples were produced from the pulp; peel-based samples are pointed out in the sample name. In the first part of this study, different physical and textural properties of the dry products are described; in the second part, the applicability of selected samples as oil structuring templates for oleogelation is assessed.3.1 Properties of samples dried with supercritical CO2
Processing of natural tissues via shredding, washing, solvent exchange and subsequent supercritical drying yielded light weight, fine and odorless powders (Fig. 2). With some exceptions (plum, apple, mushroom, strawberry), colorless products were obtained, showing that pigments such as carotenoids were in most cases completely removed during solvent exchange and supercritical drying steps. The banana peel sample showed a quick color change after transfer into ethanol for solvent exchange due to enzymatic browning,58 and the coloration was preserved throughout the process (Fig. 2). Other non-polar components (e.g. chlorophylls,59 phytochemicals like polyphenols,60 fatty acids,61 and essential oils62) are easily dissolvable in supercritical CO2: therefore their extraction during the supercritical drying (with a high and continuous CO2-flow) is anticipated.43 A detailed composition analysis revealed as expected that dry samples were mainly composed of (poly-)saccharides and proteins (for a detailed single component overview for each sample, we refer to Table S2†). While component ratios varied to some extent in between samples of different origin (Table S2†), the average compositions represented typical vegetable/fruit cell wall fractions, with pectin being the main component (∼55 wt%), followed by cellulose (∼20 wt%), proteins (13 wt%), monosaccharides/sugar acids (∼11 wt%) and other components which included most likely fructose, hemicellulose and lignin (overall ∼11 wt%).63,64 As single exception, the mushroom sample (proteins ∼43 wt% and chitin ∼33 wt%) was not included in the calculation of the aforementioned averaged values. The overall significant monosaccharide fraction found in most samples indicated that some residual monosaccharides remained in the tissue matrix after washing and solvent exchange steps. Alternatively, these fractions could also be breakdown products of various biopolymers, which were obtained during the pre-processing (acidic methanolysis) of samples for the composition analysis. The biopolymer decomposition behaviour might – however – vary throughout the samples: for instance the amorphous regions of cellulose are more accessible and can therefore be broken down into glucose monomers more easily and quickly during acidic hydrolysis, whereas the crystalline regions require harsher conditions for degradation. Furthermore, also hemicelluloses such as xyloglucans and glucomannans contain glucose units that can be released under certain acidic hydrolysis conditions.65 All identified biopolymer fractions belong to the most common starting materials in biopolymer aerogel production, with cellulose and pectin making up alone for a significant part in all works.16 Skeletal densities of samples varied slightly (average ρs = 1.73 ± 0.33 g cm−3) (Table S1†) and fell in most cases within the usual range reported for the main components.15,19 Additional FT-IR spectroscopic analysis was performed on all samples, which were subsequently used for oil structuring. The spectra of all samples were largely similar, with the exception of the mushroom based tissue. Although the resulting spectra represented the mixed (partly overlapping) signals of all components, specific bands corresponding to the main biopolymeric fractions (pectin, cellulose, and chitin in the case of the mushroom sample) were clearly identifiable. For a more detailed interpretation of the spectra, we refer to the description in the ESI and to Fig. S1.† Special attention was given to determine the degree of esterification of the pectin fraction, as this is a crucial factor influencing pectin behavior within cell walls, its gelation properties, hydrogen bonding capability and its interaction with oil.66 The degree of pectin methoxylation (%DM) was estimated based on the areas of bands at ∼1740 cm−1 (A1740) and at ∼1600–1630 cm−1 (A1630) according to eqn (4).67,68
 | (4) |
 | ||
| Fig. 2 Images of powders obtained after solvent exchange and sCO2-drying of various natural tissues. Respective sample names are given in the individual images. | ||
Pectins present in the variety of samples in this work were expected to show pronounced differences at the structural and molecular levels, which could result in different chemical stabilities and decomposition behaviors.69 Nonetheless, all samples fell within the range of low-methoxyl (LM) pectins, with a %DM of approximately 20% for pectins in banana peel, kiwi, and nectarine and around 40% in those from orange pulp, orange peel, and radish samples. LM pectins are prone to crosslinking in the presence of divalent ions (e.g., Ca2+), generally soluble in water at pH 7 (in the absence of ions) and their dissolution behaviour is generally sensitive to various conditions during the washing step (beyond pH, e.g. also temperature and the presence of ions and other biopolymers). While the washing conditions (pH-neutral, demineralized water, 20 °C, ion-free) used in this work were generally suitable for dissolving LM pectin, our results showed that the final products still contained significant amounts. This could be due to a variety of factors, as pectins can interact in complex ways within multi-component food systems, which might affect their dissolution behavior. Consequently, a more in-depth understanding of each individual system would be required to fully explain the particular pectin dissolution behavior in each case. Regarding their use as food ingredients, LM pectins are useful for low-calorie or sugar-free products and are, for instance, used in low-fat desserts, reduced-calorie jams, puddings, and plant-based gelatin alternatives. In terms of the given use case (oleogelation) weaker interactions with oil, compared to high %DM (>50%) pectins, are expected, due to the increased number of free carboxyl groups which lead to a higher hydrophilicity. Insights into the thermal stability of samples were provided via TGA analysis under an oxidizing atmosphere. As anticipated, the individual decomposition patterns exhibited some variation (Fig. S2a†). However, all curves followed a general trend typical of (bio)polymer decomposition (Fig. S2b†).70 Three distinct phases were identified: (1) loss of residual water (dehydration), causing a slight mass decrease (T = 20–100 °C), (2) decomposition of organic materials (e.g., cellulose, chitin, pectin, proteins), leading to significant mass loss (T = 100–465 °C). This phase includes decomposition mechanisms like main- and side-chain scission, elimination, depolymerization, cyclation and crosslinking.70 While the most pronounced biopolymer decomposition occurred at temperatures above 165 °C, slow decomposition occurred already at lower temperatures. (3) Continued decomposition of the remaining organic matter under formation of inorganic residues (e.g. ash), with stable and lower mass loss (T = 465–850 °C).
In order to assess the question, if samples can be classified as aerogels, the presence of internal mesoporosity was evaluated via nitrogen physisorption and SEM measurements. Physisorption data were evaluated for 17 out of 20 samples (general criteria: R2 of the BET-plot ≥0.999, positive C constants and regular isotherms, see Table S1 and Fig. S3 S4†). Significant specific surface areas in the range of Sm = 108–354 m2 g−1 were estimated for all samples (Table 1). Hereby, lower values <230 m2 g−1 were in the same range of those previously reported for supercritically dried salad leaf, apple and coffee tissues, whereas the majority of samples exhibited higher specific surface areas.43,44 Adsorption–desorption isotherms of the corresponding measurements showed in all cases a hysteresis (type IV), which is related to mesoporous adsorbents (for normalized isotherms averaged by shape see Fig. 3a, for single sample isotherms we refer to Fig. S3†).71–73
 | ||
| Fig. 3 (a) Normalized nitrogen adsorption–desorption isotherms, averaged by sample category (see Table 1). (b) Normalized cumulative specific surface area plotted against the pore diameter. (c) Normalized pore size distributions, estimated via the BJH method. Lines in (a)–(c) are drawn to guide the eye, hatched areas represent the standard deviation of the average and n equals the number of samples taken into account. (d) Relationship between mesopore volume and specific surface area for samples of different categories. Error bars represent the absolute error of the methods. Linear trend lines are drawn to guide the eye. | ||
| Sample | Smb overall [m2 g−1] |
Vmesoc overall [cm3 g−1] |
Ratio Sma (≤6.5 nm) [%] |
Ratio Sma (6.5–10 nm) [%] |
Ratio Sma (10–50 nm) [%] |
||
|---|---|---|---|---|---|---|---|
| a Contribution of pores in the given range of pore diameters (nm) to the overall specific surface area in %.b Standard error of method ± 5 m2 g−1.c Standard error of method ± 0.14 cm3 g−1. | |||||||
| Sample category | 1 | Radish | 328 | 0.65 | 61 | 30 | 9 |
| Apple | 202 | 0.55 | 63 | 30 | 7 | ||
| Bell pepper | 190 | 0.31 | 63 | 31 | 6 | ||
| Cucumber | 238 | 0.59 | 60 | 30 | 10 | ||
| Carrot | 125 | 0.27 | 62 | 26 | 12 | ||
| 2 | Mushroom | 281 | 0.78 | 43 | 31 | 26 | |
| Banana peel | 240 | 0.43 | 53 | 29 | 18 | ||
| Pear | 168 | 0.53 | 48 | 37 | 15 | ||
| Plum | 259 | 0.93 | 47 | 26 | 27 | ||
| Tomato | 251 | 0.74 | 42 | 28 | 30 | ||
| Orange peel | 285 | 1.15 | 36 | 37 | 27 | ||
| Onion | 108 | 0.30 | 53 | 27 | 20 | ||
| 3 | Kiwi | 354 | 2.56 | 3 | 14 | 83 | |
| Orange | 322 | 1.36 | 26 | 40 | 34 | ||
| Honey Pom. | 305 | 1.25 | 30 | 39 | 31 | ||
| Strawberry | 277 | 0.94 | 25 | 38 | 37 | ||
| Nectarine | 248 | 1.34 | 9 | 24 | 67 | ||
While the shape of the hysteresis loops did not necessarily match the idealized IUPAC classifications (H1–H6 shape), three distinct isotherm shapes were identified (Fig. 3a), which suggested the presence of separate sample categories with different microstructural properties. Differences at the microstructural level were evaluated in detail by plotting the normalized, cumulative internal surface areas against the mesopore diameter (Fig. 3b). This allowed us to assess up to which extent mesopores of different size-regimes contributed to the overall specific surface area of a given sample (Table 1).
Based on the results, samples were assignable to three distinct categories which represent different microstructures. Samples with small mesopores (dpore ≤ 6.5 nm) contributing to a large extent to the overall specific surface area (≥60%) were assigned to category 1. Larger mesopores (dpore = 10–50 nm) presented only a small amount of surface area in these cases. This picture was reversed in category 3 samples, where large mesopores contributed most significantly to Sm (up to 83%) and small mesopores in the range of dpore = 6.5–10 nm accounted for max. 30% of the Sm (with some exceptions where these pores were almost negligible, e.g. in kiwi and nectarine samples). Category 2 samples showed a significant contribution of all mesopore size-regimes to the Sm, with the slightly highest contribution of the smallest mesopores on average. The aforementioned relations consequently also reflected in the shapes of the corresponding pore size distributions (PSDs) (for normalized PSDs averaged by category see Fig. 3c, single PSDs are shown in ESI, Fig. S5†). Category 1 samples contained mainly mesopores with diameters below 18 nm (dpore,mean ∼ 6–10 nm, Table S1†). The provided pore volume increased towards lower mesopore diameters and the presence of even smaller pores (dpore < 4 nm) with sizes below the detection limit of the BJH-method is proposed. PSDs of category 2 samples showed in addition to a narrow peak with a maximum at dpore ∼ 10 nm a broad flank, which suggested the presence of various sized, larger pores with pore diameters dpore ≥ 20 nm up to the range of small macropores. Category 3 samples exhibited mesopore size distributions which covered the whole mesoporous range with the mean pore diameter falling in the range of medium sized mesopores (dpore,mean ∼ 18–30 nm, Table S1†). The corresponding isotherms showed H3-shaped hysteresis loops with a steep increase towards higher p/p0 values (Fig. 3a and Fig. S3†), which is consistent with the additional presence of macropores.71 Such isotherms/PSDs are also typical of various sol–gel based biopolymer aerogels with fibrous, multiscale pore networks as e.g. reported in recent works on pectins,73–75 cellulose41,76 or various proteins47,77 and pointed towards a high similarity between nanostructures of many “classical” biopolymer aerogels and category 3 samples. Samples of all different categories showed an increasing specific surface area, as the mesopore volume was higher: the overall mesopore volume follows the trend Vmeso, category 1 < Vmeso, category 2 < Vmeso, category 3, whereas values were generally in the typical range reported for biopolymer aerogels (Fig. 3d).15 It is notable that the relationship between Vmeso and Sm varied in between samples of different categories. While variation of Vmeso was not as large throughout category 1 samples, even a slight increase in Vmeso resulted in a significant increase of the Sm. This is because these samples presented mainly small mesopores, which provided a comparably high surface to volume ratio. For instance, the radish sample showed a high Sm equal to the range of category 3 samples, even though it presented a significantly smaller mesopore volume in comparison. With increasing presence of larger mesopores (and decreasing contribution of smaller ones), more mesopore volume was required to create additional Sm, which was reflected by the trend for category 3 samples (larger variation of Vmeso, narrow range of Sm). More insights into different microstructures were available by taking results obtained via scanning electron microscopy (SEM) into account. In the following, we discuss results with respect to the overall organization of plant tissues and with special emphasis on the question, which parts of the samples are mainly contributing to the mesoporous structure. Due to the diversity of samples and the corresponding cellular and microstructural features, we emphasize however, that the discussion in this study is limited to most fundamental aspects. In general, plant cells can contain various different types of porous tissue: for instance, fruit cells are separated by thin, rather unlignified and nanostructured cell walls which contain pectin/cellulose nanofibers in their subunits (primary and secondary cell walls).64 These fibers stabilize the cell wall by creating pore networks composed of heterogeneous and variably branched/entangled micro- to nanofribrils, whereas the biopolymer chain length can vary widely in a range from a few tenths of up to several thousand nanometers.78 Cytoplasmic bridges (plasmodesmata) of various sizes form channels through these walls and enable mass transport (e.g. water, nutrients, other materials) between individual cells.64,79 While the initial definition of these bridges has changed with the development of advanced detection techniques, dimensions and accurate determination of the plasmodesmata size are currently still under discussion: generally, plant biologists acknowledge plasmodesmata nowadays as nanoscaled pores.79 In the SEM images of lower magnification (250–5000×, Fig. 4 and Fig. S6† for all other tissue-based samples of the screening) cells with sizes of ∼20–100 µm were indeed visible in most samples (exceptions: peel & mushroom samples). Folding/deformation of cell walls occurred in many cases; this might be a result of diffusive forces which occurred during processing (liquids/CO2 entering and leaving the inner cell body) or caused by the occurrence of mechanical stress during grinding steps.
 | ||
| Fig. 4 SEM images of selected samples (radish, mushroom, and kiwi) from different sample categories obtained at different magnifications: (a)–(c) 250×),( d)–(f) 5000×, (g)–(i) 150000×. | ||
The stability of the cellular-structure depended on the individual tissue and should generally also be influenced by several, source-related factors, such as the status of the ripening process, the particular microstructure and thickness of the cell wall and the cell size.64 The flesh of softer fruits (e.g. kiwi and orange) consists often of looser tissue with larger intercellular spaces which leads to higher flexibility in contrast to the smaller and denser cell structures of harder fruits and vegetables (e.g. radish and carrot, Fig. 4 and Fig. S6†). Consequently, intact/non-deformed cells were not visible in the SEM images of comparably soft category 3 samples (Fig. 4a and Fig. S6†). Images obtained at higher magnifications (50000–150000×) were used to identify the most representative mesopore structures, which contributed to Vmeso and Sm. Hereby, clear differences between mesopore structures across sample categories 1–3 were detected. In category 1 samples, small mesopores were homogenously distributed throughout the cell walls (Fig. 4g and Fig. S6†). In this case, SEM imaging was limited to the outer cell-wall surface and no other mesopores e.g. in intercellular spaces or fibrils were determined. No macropores were visible throughout category 1 samples, which is consistent with the N2-isotherm shape, showing a rather gradual increase in the amount of absorbed nitrogen in the higher p/p0-range. The small size of pores and the narrow range of the PSD distribution indicated that the mesoporous volume in category 1 samples was partly provided by confined, organized structures. These could include, for example, internal plasmodesmal channels in the cell walls or subunits (e.g. the desmotubule, presumed transport channels, cytosolic sleeves), whose dimensions have been reported to be within the range of pore sizes estimated via the BJH method.79,80 With respect to the overall low volume of such channels, it is nevertheless not clear to which extent they could have possibly contributed to the overall surface area. In contrast, fibrillar and multiscale pore networks with nanometer sized fibers were visible in samples categorized as type 3 (Fig. 4i, 5d–f and Fig. S6†). These fibrillar pore networks were most likely part of the diverse pectin- and cellulose based fibrous cell wall fractions and contained both mesopores of various sizes as well as small macropores in the range of pore diameters from several hundred nanometers of up to a few micrometers. Noteworthy is the high similarity of these microstructures with those of common sol–gel based biopolymer aerogels. Pectin aerogels with comparable macro- to mesoporous networks are e.g. producible from apple-, citrus- and watermelon extracted pectin via calcium-ion induced and physical crosslinking.56,75,81,82
 | ||
| Fig. 5 Pore structures of calcium-ion crosslinked, pectin aerogels produced via the sol route (top, Reprinted from81 with permission from Elsevier, Copyright 2018) and various tissue based samples produced in this work (bottom). Sol–gel based samples were produced with different pectin-contents in the sol: (a) 2 wt%, (b) 4 wt%, and (c) 6 wt%. Represented pore structures from tissue-based samples are: (d) nectarine (e) strawberry and (f) orange. | ||
In the case of cellulose aerogels, comparable pore structures are for instance obtainable from microcrystalline cellulose using different cellulose-solvents (e.g. NaOH/water, ZnCl2/water, Ca(SCN)2/water, ionic liquids).20,39,40 An exemplary, direct comparison between various category 3 samples produced in this work and pectin aerogels obtained from citrus extracted pectin is provided in Fig. 5. While the morphology and density of the pore network can be controlled by the amount of dissolved pectin or by the presence of cations in the sol–gel process (see Fig. 5a–c), such kind of control is reduced in the case of tissue-based samples, where the particular morphology depends also on the inherent microstructure of the used tissue (see Fig. 5d–f). Presented examples highlight although the huge variety of mesopore-morphologies present within samples of category 3 in this work. Category 2 samples were generally highly inhomogeneous and included the fungal as well as peel samples, which were differently organized at the cellular level as compared to fruit and vegetable pulp. Besides cell wall residues, small mesopores (equal to category 1) and fibrillar pore networks (equal to category 3) were both visible (Fig. 4b and Fig. S6†). Nitrogen physisorption data indicated that both types of pores contributed in these cases significantly to the overall mesoporous texture (see Fig. 3c). With respect to the overall inhomogeneity of samples (as compared to “pure” aerogels), it is to be mentioned that high resolution SEM imaging of pore structures provided only qualitative insights of selected sample areas. Particularly category 2 and 3 samples contained in many cases also larger nano- (diameter of a few hundred nm) and microfibers or other amorphous parts.
Overall, our results suggest that the amount of free accessible nanofibrous structures was the largest in the pulp of relatively soft and aqueous fruit bodies, leading on average to higher mesopore volumes and specific surface areas. It is however important to note the composite nature of samples from all categories, with amorphous components being present alongside open-mesoporous, and nanostructured networks. Given the differences between natural tissue-based samples and “sol–gel” based biopolymer aerogels, it is interesting to compare their typical characteristics, such as density and specific surface area (Fig. 6). Parameters were compared with recent works (2008–2023) on aerogels from single components (pectin, cellulose and proteins), which represent ∼75% of the composition in tissue-based samples. Studies in which freeze-drying was used as a drying-technique were not included, due to the microstructural differences between cryogels and aerogels. A comparison of the densities (Fig. 6a) highlights the overall light-weight nature of tissue-based samples with bulk densities ranging from ∼0.01–0.12 g cm−3 (Table S3†). While this points towards a high porosity ≥97% of the powdered samples, we emphasize that ρb is a rough estimate for the internal pores. Since the bulk density was calculated from the non-compressed bed of irregular particles ε encompassed both the internal porosity of particles as well as interparticle voids. Because compared works include studies on different aerogel shapes (e.g. also monoliths), such interparticle voids were not necessarily present in these cases (leading to higher median values of the density). In the case of Sm, the median of tissue-based samples falls within the same order of magnitude with compared works (Fig. 6b): the overall accessible surface areas in the sol–gel route are nonetheless principally higher due to the homogeneous nature of the samples. In summary, tissue-based samples fulfill the key criteria for materials to be assigned as “aerogels”: namely high porosities, low densities, the presence of a significant specific surface area >100 m2 g−1 and open, three-dimensional mesoporous solid networks.18,31,40 Differences in tissue-based samples as compared to “sol–gel” based aerogels lie in their heterogeneous nature, leading to a composite-like character. Our results suggest that scCO2-dried natural tissues are principally applicable in many directions, where low densities, a high specific surface area and the presence of mesoporosity play a role. This accounts for leftover pulp and peels as well and highlights the potential use of food waste (e.g. generated in juice, jam and essential oil production or in processing for ready meals) as a mesoporous matrix.
 | ||
| Fig. 6 Comparison of densities (a) and specific surface areas (b) of supercritically dried natural tissues with cellulose, pectin and protein aerogels obtained via sol–gel processing of pure biopolymers. The category “tissues” includes combined results from this work as well as available data from former reports.41,42 The violin plots represent the density of the data along the y-axis, the width of the plot represents the frequency density for different values. The black box shows the range between the 1st quartile (25th percentile) and the 3rd quartile (75th percentile). Whiskers represent the range of 1.5 IQR; the white dot corresponds to the median. n = number of samples taken into account. All references are listed in Table S3.† | ||
3.2 Optimization and ageing
From an application point of view, it is important to evaluate how long food waste can be stored before being converted to an aerogel without compromising the resulting microstructure and composition. A showcase experiment was carried out with orange leftovers, which were directly obtained as a kitchen waste mixture (peel and pulp) after juice production in a fruit press. The samples were stored under ambient conditions (∼20 °C) under open air for different times (up to three weeks), prior to further processing. As compared to samples described in section 3.1, significantly less sample (50 g) was used in relation to water during the washing process. This was done in order to (1) ensure the complete removal of monosaccharidic fractions or decomposition products being formed during storage; (2) evaluate if intensified washing influences the final aerogel properties.In the case of the fresh sample, a significantly increased max. Sm of 446 m2 g−1 was found (Fig. 7a), which is most likely attributed to the intensified washing. This value exceeds so far reported values for tissue-based samples (compare Table 1 and former studies43,44) and is competitive to many sol–gel/nanofiber based aerogels. No pronounced changes of Sm, Vmeso, and pore size distributions were determined for samples produced after different storage times, and aerogel properties were still obtained after three weeks (Fig. 7). This result showed that no significant evaporative drying of liquid fractions in the inner pores occurred in the investigated time frame. Minor differences between samples were probably related to (1) slight variations in the peel to pulp ratio of individual samples, which was not exactly controlled; (2) changes induced by microbiological/enzymatic conversion, particularly of the pectin fraction. During enzymatic degradation, pectins are broken down by pectinases, leading to depolymerization, solubilization and de-esterification by pectin methylesterases, reducing its %DM. While a corresponding decrease of the %DM over time was expected, our results showed a different trend. Only slight changes in the %DM occurred up to one week storage time and a severe decrease was observed after two weeks (Fig. 7b, for the corresponding IR spectra we refer to Fig. S7†). This was consistent with significant browning of the sample, which occurred after one week (Fig. 7a) and indicated the presence of oxidized phenols (e.g., through polyphenol oxidases) and/or degradation products from pectin. However, after three weeks, a significant increase in the %DM was observed. We surmise that further conversion (including depolymerization and desertification) of some pectin fractions between two and three weeks storage time led to an increased water solubility and loss of these fractions due to out washing. The remaining pectin in the solid aerogel backbone (detected after three weeks) was then a high %DM fraction. In summary, our results suggested that no severe changes of the aerogel microstructure happened due to storage, but the composition (and therefore chemical functionality) was altered. Microbial and enzymatic induced processes led to changes in chemical functionality, particularly affecting the pectin fraction. We'd like to emphasize that the showcased example represents a single case and cannot be directly applied to other samples, as each requires individual analysis.
 | ||
| Fig. 7 (a) Specific surface areas (black) and mesopore volumes (red) of aerogels made from waste generated during orange juice production, shown as a function of storage time. Error bars correspond to the estimated standard error of the methods; dashed lines highlight the average values. The images (inset) show the sample appearances after different storage times (top) and after the conversion to the corresponding aerogel powder (bottom). (b) Pore size distributions and %DM values of the samples. | ||
It must also be noted that the upcycling of food residues as raw materials for the production of bioaerogels intended as food ingredients requires the waste material to comply with stringent hygienic and quality requisites. In particular, food raw materials cannot be deteriorated but freshly transformed through the application of food-grade procedures. This would of course require a change in the actual food waste management practices, involving the set-up of dedicated lines for waste collection, pretreatment and final bioaerogel production. It can be affirmed that the main hazard found in fresh fruit and vegetable waste is microbial growth, due to the perishable nature of this biomass. It is – however – worth mentioning that solvent exchange with ethanol, which is necessary for bioaerogel production, offers an easy step of stabilization of the waste from a microbiological point of view, whose antimicrobial effects are further supported by the subsequent supercritical-CO2-drying.83 Additionally, the extraction of decomposition products during aerogel-processing is expected. This was evident in our case, as all final aerogel samples were nearly color and odorless, despite significant microbiological activity (including mold growth) occurring after approximately two weeks storage time (Fig. 7a).
3.3 Environmental assessment
In this section, we compare the process presented in this work with a conventional sol–gel aerogel fabrication method, focusing on their environmental friendliness. So far, numerous methods for environmental assessment have been developed. In this work, we adopt the environmental assessment methodology and the approach proposed by Biwer and Heinzle84,85 to identify the environmental “hot spots” of the process. It is important to emphasize that our analysis focuses solely on the process itself and does not evaluate whether producing a given product is reasonable or sustainable. To benchmark our approach, we compare our process with an archetypal sol–gel cellulose aerogel fabrication process reported by Cai et al.86In the conventional process, microcrystalline cellulose is dissolved in an aqueous solution of sodium hydroxide and urea. The cellulose is then regenerated into a gel form by casting it into a regeneration bath containing diluted sulfuric acid. The resulting hydrogel is washed with deionized water, followed by solvent exchange with ethanol and drying using supercritical CO2. The process parameters and assumptions for both the conventional and our novel processes are detailed in the ESI (Tables S4–S8†). Briefly, we first establish a material balance for both the conventional and novel processes. Based on this material balance, we calculate the Mass Index (MI) for all input and output components.87 For input materials, the MI represents the amount of a given component consumed to produce a unit mass of the final product (all calculations are standardized to 1 g of aerogel). For output components, the MI indicates the quantity of a component generated and subsequently disposed of per unit mass of the final product.
The environmental impact of each compound involved in the process is assessed across 15 impact categories (ICs), including land use, acute toxicity, and global warming potential, among others. Each component is assigned to one of three relevance classes (A, B, or C) within each category, corresponding to high, moderate, and low relevance, respectively (see assignment in ESI, Tables S4–8†). These ICs are further grouped into six impact groups, each representing key environmental, health, or safety concerns. In the next step, numerical values are assigned to classes A, B, and C, allowing us to derive an environmental factor (EF) for each component based on its classification within the impact groups. The final environmental impact is then determined by multiplying each component's MI by its EF, yielding the Environmental Index (EI).
This index helps identify the most environmentally significant components in the process. We computed the EI for both the conventional (Fig. 8a) and novel (Fig. 8b) processes, considering all input and output components. The analysis clearly demonstrates that ethanol and carbon dioxide are the two most environmentally impactful substances in both processes, primarily due to their use in the final solvent exchange and supercritical drying steps. These compounds contribute the most to the overall environmental indices. Notably, integrated process designs have been proposed to reduce their consumption.88,89 Moreover, their environmental impact is significantly lower at the industrial scale, where both ethanol and CO2 can be recovered and reused in a closed-loop system (annotated with “rec.” in Fig. 8). This highlights the importance of process optimization and scale-up strategies in aerogel production in order to minimize environmental burdens. The total sum of all EIs (both input and output) represents the EI of the entire process and provides an overall measure of its environmental impact. Our results indicate that the process developed in this work (EI = 28 index points per gram of aerogel) is significantly more environmentally favorable than the conventional sol–gel regeneration process (EI = 57). Notably, the material intensity of both processes is comparable (669.8 g g−1 aerogel vs. 740.1 g g−1 aerogel, respectively). Furthermore, our process does not involve any chemicals beyond ethanol and carbon dioxide, whereas the conventional sol–gel process relies on NaOH/urea solutions and an acidic regeneration bath. In conclusion, the tissue-based approach provides a more environmentally sustainable alternative to conventional biopolymer aerogel fabrication while maintaining comparable material efficiency. Our process eliminates the need for strong acids and bases and significantly reduces its overall environmental impact. While the comparison in this section was based on a specific example, the findings are likely relevant to other sol–gel or nanofiber-based methods, which generally require additional chemicals beyond ethanol and CO2. This highlights the potential of the tissue-based approach as a promising and sustainable alternative for aerogel production.
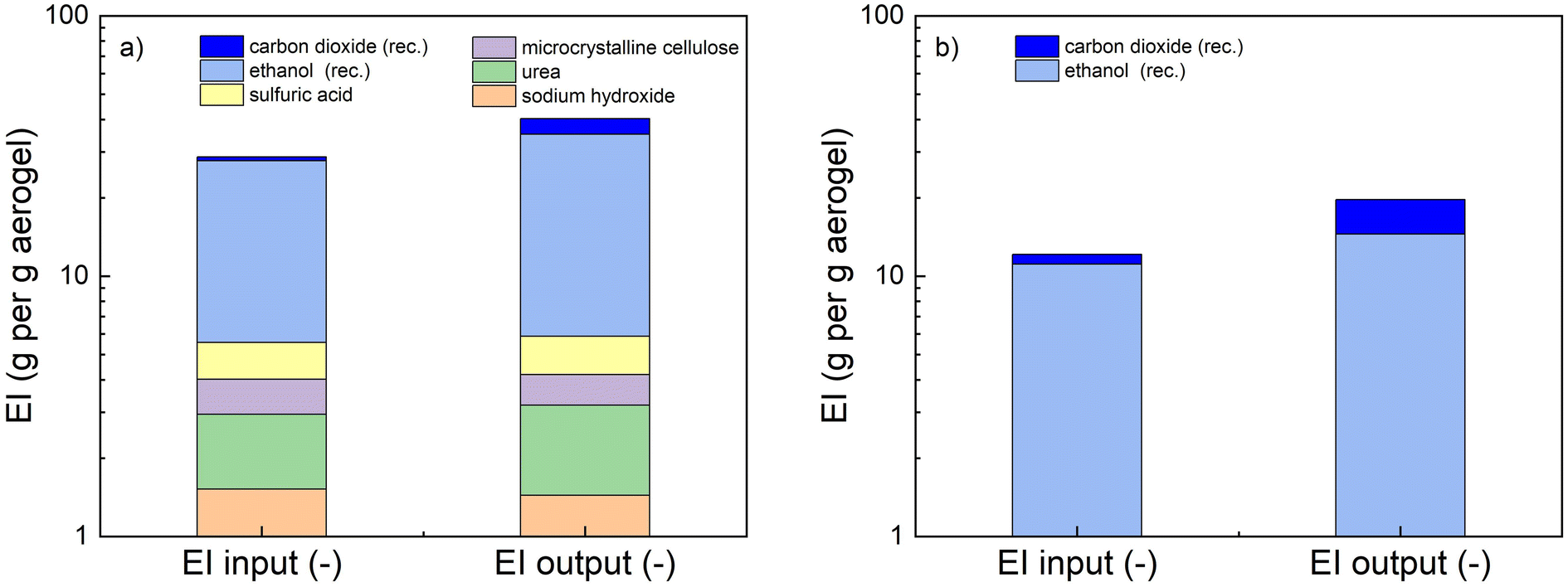 | ||
| Fig. 8 Comparison of Environmental Indices (EIs) of the input and output for a conventional sol–gel (a) and the process reported in this work (b). Note log-scale on the y-axis. | ||
3.4 Use of supercritically dried tissues in oleogelation
The oil structuring capacity and use of various tissue-derived aerogels as oleogel templates are evaluated in this section. To this aim, among aerogels shown in Fig. 2, those obtained from orange (pulp and peel), banana peel, kiwi, radish, nectarine and mushroom were selected. This selection was based on (a) microstructural, (b) compositional and (c) source related aspects. Samples with max. specific surface area (kiwi and radish) were chosen from different microstructural categories with mainly small (category 1) and large (category 3) mesopores. Additional examples from category 3 (nectarine and orange samples) represent samples with lower mesopore volume and specific surface area than the kiwi aerogels. Peels were included to assess the use of classical food waste/inedible parts as functional food ingredients. The mushroom sample is representative for a different biopolymer composition (highest protein content and presence of chitin). The selected aerogels were added with increasing amounts of sunflower oil: corresponding gradual changes in appearance are exemplary shown for the orange pulp sample in the main text (Table 2), for all other samples we refer to Fig. S8 and S9.†| Oil uptake [%] | 83.3% | 90.9% | 93.9% = oilgel | 95.2% | 96.2% |
|---|---|---|---|---|---|
| Appearance |  |
 |
 |
 |
 |
Upon the initial addition of oil, all samples displayed a granular morphology, characterized by the formation of small aggregates. In these aerogel–oil mixtures, capillary forces promoted oil absorption into the aerogel pores, with minimal surface interaction among the aerogel particles.90 As the oil content increased, it progressively interacted with the hydrophobic groups present on the surface of the aerogel particles, forming oil bridges between them (Table 2 and Fig. S8, S9†). Concomitantly, hydrophilic interactions among the aerogel particles in a lipophilic environment contributed to the formation of a continuous particle network.91 By further increasing the oil amount, particle dilution in the oil reduced surface particle–particle interactions, thereby weakening the particle network and resulting in a fluid system. This behavior aligns with observations reported in previous studies relevant to oil structuring using sol–gel-based protein aerogels.47 Based on the results, a minimum oil content oilgel could be identified, at which the transition from a granular solid to a continuous self-standing oleogel occurred (Table 2 and Fig. S8 and S9†) and was chosen for further comparison of oleogel performances. The appearance of the obtained oleogels, along with their physical and rheological properties, is summarized in Table 3. The minimum oil content necessary to form oleogels depended on the nature of the aerogel-matrix, varying in the range of high values from oilgel = 89–99% (Table 3). These values are significantly higher than those formerly reported for sol–gel-based aerogels derived from both whey and potato proteins (70–84%).47,50,55
δ), and microstructure of oleogels prepared from different tissue-derived aerogels and containing the minimum oil amount (%) leading to a continuous structure
| Tissue | oilgel (%) | Appearance | OHC (%) | G′ (104 Pa) | Tan δ |
|---|---|---|---|---|---|
| Orange peel | 88.9 |  |
97.1 ± 0.7 | 25.4 ± 4.3 | 0.043 ± 0.010 |
| Orange | 93.8 |  |
90.5 ± 1.8 | 6.1 ± 0.5 | 0.076 ± 0.007 |
| Banana peel | 93.8 |  |
91.6 ± 0.8 | 6.5 ± 0.5 | 0.054 ± 0.001 |
| Kiwi | 96.1 |  |
91.7 ± 3.2 | 5.1 ± 0.2 | 0.057 ± 0.002 |
| Radish | 98.7 |  |
91.6 ± 0.8 | 1.2 ± 0.1 | 0.057 ± 0.002 |
| Nectarine | 95.2 |  |
93.8 ± 1.3 | 5.8 ± 3.1 | 0.073 ± 0.005 |
| Mushroom | 96.0 |  |
97.2 ± 1.1 | 2.6 ± 0.3 | 0.066 ± 0.003 |
By contrast, the high oil absorption capacities are in agreement with those relevant to tissue-aerogels prepared from lettuce leaves without previous grinding.45 In that case, the high oil absorption capacity was attributed to the peculiar response of plant tissues to the aerogel production process, which was demonstrated to lead to an expansion of the cellular structure, possibly enhancing the volume available for oil uptake.43 It is noteworthy that in the present work, plant tissues were finely ground, causing a loss of cell integrity with the release of compartmentalized cellular content (Fig. 4). Upon oil absorption, fractions of cell walls and thylakoids (in fluorescence) dispersed in oil (in yellow) were clearly evident, with morphologies depending on the initial vegetable material, in the confocal micrographs of oleogels (Fig. 9 and Fig. S10†). It can be observed that these cellular fractions, which appeared tightly packed in the mesoporous aerogel structure (see Fig. 4 and Fig. S6†), tended to expand, detaching one from the other and begetting large spaces holding oil among them. In other words, the aerogel particles likely swelled upon absorbing oil, which probably expanded the fibrous matrix and exposed functional groups.
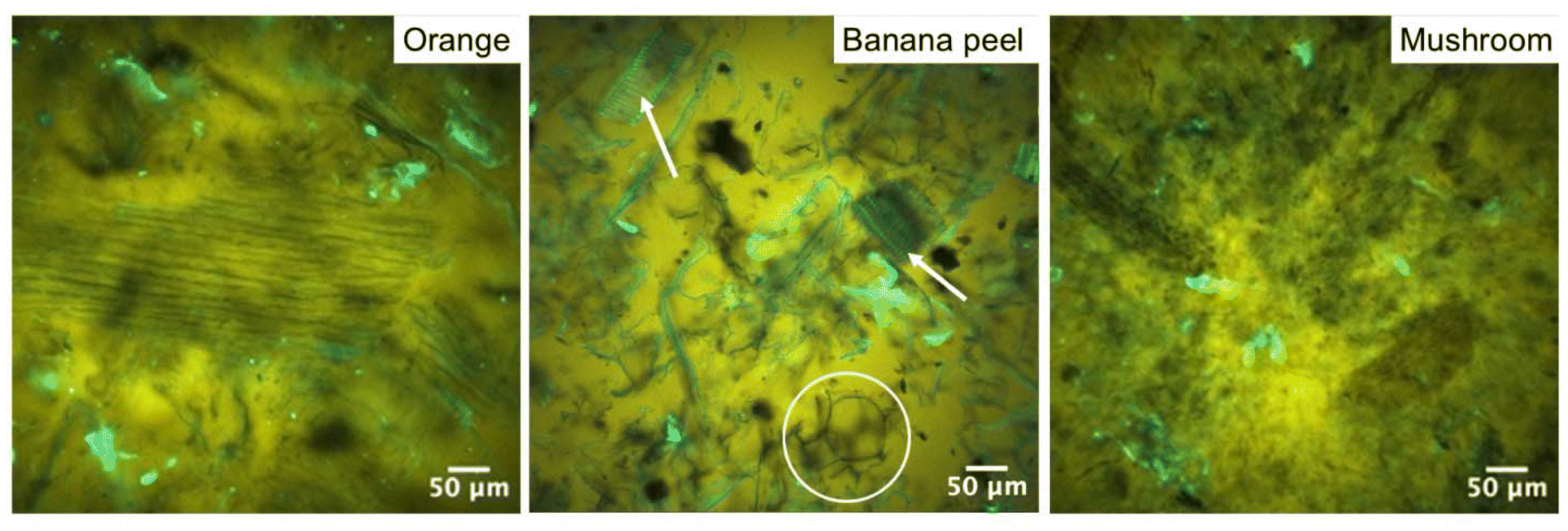 | ||
| Fig. 9 Confocal micrographs of oleogels based on different tissue-based aerogels. Highlighted areas in the middle image: white circle = detached cell walls, white arrows = closely spaced xylem fibers. | ||
Among these, hydrophobic groups present in proteins, which were shown to be still present in the aerogel matrix (see Table S2†), could play a not negligible role in driving interactions of aerogels with oil. Aerogel swelling was also observed for the mushroom based sample: however, since mushrooms do not present a cellular structure analogous to fruits and vegetables, they beget oleogels formed by fine stranded fibers homogeneously distributed in the oleogel network. As shown in Table 3, the oleogel oil content depended on the original vegetable material. In detail, radish led to the oleogel with the highest oil content, followed by mushroom, kiwi, nectarine, orange, banana peel, and orange peel. Unlike sol–gel-based, pure protein aerogels,47,50,55 no direct significant correlation between oil absorption and microstructural features (surface area and mesoporous volume) was observed (p > 0.05), suggesting that oil absorption could be driven not only by microstructural properties but also by compositional factors in our case. No relationship with the pectin content was identified, which aligns with the low %DM values found in our samples, which indicated a rather weak interaction of the pectin fraction with oil. An interesting correlation (R = 0.92, p < 0.05) was found between oil absorption and the reciprocal of the product of the protein content and surface area of the aerogels (Fig. 10).
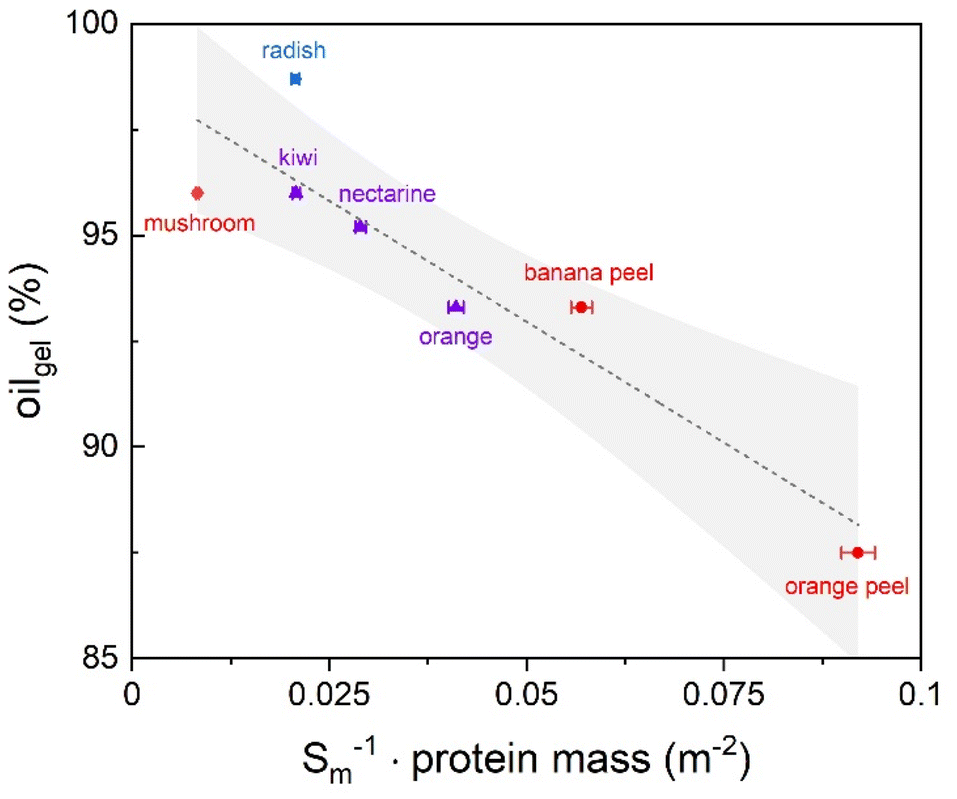 | ||
| Fig. 10 Correlation of oil absorption of tissue-based aerogels with the reciprocal of the product of protein content and surface area. X-errors represent the standard error of BET measurements; the straight line corresponds to linear fitting and the hatched area to the 95% confidence interval. The color/symbol code is according to aerogel categories as defined in section 3.1 (see Fig. 3). | ||
This relationship might suggest that oil absorption increases synergically with the availability of surfaces exposing hydrophobic groups, reasonably derived from proteins. Throughout the samples with the highest oil absorption capability, the mushroom aerogel contains the by far highest protein content (∼43%), while the radish aerogel presents an elevated internal surface area combined with a still significant protein amount (∼15%) as compared to most other samples, leading most probably to the maximum oil uptake observed in this work. The combination of proteins, acting as surface active materials, with the representative high-content pectin polysaccharides, can provide stable steric repulsions: therefore, the high amount of protein covalently bound to a highly branched polysaccharide structure represented by the unique content of rhamnose from the radish sample may result in particularly beneficial surface–oil interactions.92 As regards the OHC, all samples exceeded 90%, demonstrating that the oil was strongly retained in the aerogel structure in all cases (Table 3). From a rheological point of view, the obtained oleogels exhibited typical gel-like rheological profiles, characterized by a storage modulus (G′) higher than the loss modulus (G″) in the linear viscoelastic region (LVR) (Table 3). The yield resistance and elastic properties of the oleogels were attributed to the network formed by aerogel particles in the oil, resulting in a plastic and deformable structure. Moreover, for all the oleogels, the frequency sweep test showed the independence of G′ and G″ in the frequency range considered, suggesting the strong gel behavior of the samples. Likewise, the low loss tangent (tanδ) value, approaching zero, indicated that the elastic (solid-like) behavior of the material dominated over its viscous (liquid-like) modulus. These results are in agreement with the rheological properties of oleogels obtained from sol–gel-based aerogel particles.50,55 It must be underlined how the rheological parameters of the oleogels were comparable to those of commercial hard fat (e.g., butter, palm oil, margarine, laminating shortening)93 making these materials ideal fat-replacers in real-food matrixes, while requiring only small amounts of aerogel regarding the overall oil mass (down to 1% in the case of radish aerogels).
4 Conclusion
In this study, we demonstrated for the first time that the inherent porosity of various natural tissues – which have not yet been utilized as aerogels – can be directly harnessed to convert them into biopolymer aerogels. Unlike sol–gel or nanofiber-based aerogel production, our approach eliminates the need for biopolymer extraction, dissolution, gelation, and additional additives. This streamlines the production process, making aerogel manufacturing more environmentally friendly and sustainable. Our key finding is that tissue based aerogels exhibit textural properties comparable to many single-component materials obtained via sol–gel processes. The materials presented high specific surface areas, with a maximum of up to ∼446 m2 g−1 (in the case of orange residues obtained after juice production). These results underscore the potential of food waste as a valuable porous high-performance material. The specific use case in this study was the application of tissue based aerogels as oleogel-templates: our results showed that our materials can transform liquid oils into oleogels with rheological properties similar to solid fats, achieving exceptionally high efficiency (oil uptake of up to ∼99%) and excellent oil retention (91–97%).For dissemination, our project can leverage open public science via the aerogel.kniffelix.de platform: for access to the interactive learning platform, we refer to aerogel.kniffelix.de and for download of learning materials to aerogel-download-material.kniffelix.de, where experiments are accessible to the public and can be carried out in part using common household materials. This open access STEM resource will support engagement through educational videos and hands-on aerogel experiments, promoting both scientific literacy and community engagement in sustainable materials science. Overall, this study highlights the feasibility of utilizing food industry by-products, such as fruit and vegetable tissues, as sources of valuable aerogel products, aligning with sustainable development goals by contributing to the reduction and valorization of food waste through innovative, environmentally friendly technological applications. We suggest that the fundamental idea behind this study – seeing all kinds of organic tissues as naturally grown hydrogels – is extendable to a huge variety of other sources and hope our results promote the further exploration of the direct tissue-to-aerogel route.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Human and animal rights
This manuscript does not contain any studies with human participants or animals performed by any of the authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
All authors declare that they have no conflict of interest.Acknowledgements
The authors wish to acknowledge support for this research received from the Bundesministerium für Bildung und Forschung (BMBF) within the framework of the “Dahrendorf-project for the EU project NanoHybrids”, funding code 01DT22001.This work was carried out in the frame of the COST Action CA18125 “Advanced Engineering and Research of aeroGels for Environment and Life Sciences” (AERoGELS) and funded by the European Commission.
The Accreditation as Center of Excellence Severo Ochoa IATA-CSIC, CEX2021–001189-S funded by MCIU/AEI/10.13039/501100011033 is also fully acknowledged.
References
- S. Kaza, L. Yao, P. Bhada-Tata and F. Van Woerden, What a Waste 2.0 - A Global Snapshot of Solid Waste Management to 2050, World Bank Publications, Washington, DC, 2018 Search PubMed.
- United Nations Environment Programme, Food Waste Index Report 2024. Think Eat Save: Tracking Progress to Halve Global Food Waste, UNEP, Nairobi, 2024.
- S. P. Sha, D. Modak, S. Sarkar, S. K. Roy, S. P. Sah, K. Ghatani and S. Bhattacharjee, Front. Microbiol., 2023, 14, 1260071 Search PubMed.
- A. Alammar, R. Hardian and G. Szekely, Green Chem., 2022, 24(1), 365–374 CAS.
- H. Liu, T. Wang, J. Wang, G. Gao, Y. Li and D. Ren, Green Chem., 2025, 27, 1125–1144 CAS.
- C. Y. Cheok, N. Mohd Adzahan, R. Abdul Rahman, N. H. Zainal Abedin, N. Hussain, R. Sulaiman and G. H. Chong, Crit. Rev. Food Sci. Nutr., 2018, 58, 335–361 Search PubMed.
- T. Ahmad, F. A. Masoodi, S. A. Rather, S. M. Wani and A. Gull, J. Biol Chem. Chron., 2019, 5(1), 114–122 Search PubMed.
- S. R. R. Comim, K. Madella, J. V. Oliveira and S. R. S. Ferreira, J. Supercrit. Fluids, 2010, 54(1), 30–37 CAS.
- D. San Martin, S. Ramos and J. Zufía, Valorisation of Food Waste to Produce New Raw Materials for Animal Feed, Food Chem., 2016, 198, 68–74 CAS.
- C. H. Tan, C. L. Hii, C. Borompichaichartkul, P. Phumsombat, I. Kong and L. P. Pui, Drying Technol., 2022, 40(8), 1514–1538 Search PubMed.
- M. Vetralla, G. Ferrentino, A. Zambon and S. Spilimbergo, ETP Int. J. Food Eng., 2018, 4(3), 186–190 Search PubMed.
- A. Zambon, S. Bourdoux, M. F. Pantano, N. M. Pugno, F. Boldrin, G. Hofland, A. Rajkovic, F. Devlieghere and S. Spilimbergo, Drying Technol., 2021, 39(2), 259–267 CrossRef CAS.
- A. Zambon, P. Facco, G. Morbiato, M. Toffoletto, G. Poloniato, S. Sut, P. Andrigo, S. Dall'Acqua, M. De Bernard and S. Spilimbergo, Food Chem., 2022, 397, 133789 CrossRef CAS PubMed.
- K. Pravallika, S. Chakraborty and R. S. Singhal, J. Food Eng., 2023, 343, 111375 CrossRef CAS.
- T. Budtova, Cellulose, 2019, 26(1), 81–121 CrossRef CAS.
- S. Zhao, W. J. Malfait, N. Guerrero-Alburquerque, M. M. Koebel and G. Nyström, Angew. Chem., Int. Ed., 2018, 57(26), 7580–7608 CrossRef CAS PubMed.
- R. Subrahmanyam, P. Gurikov, P. Dieringer, M. Sun and I. Smirnova, Gels, 2015, 1(2), 291–313 CrossRef PubMed.
- Springer Handbook of Aerogels, ed. M. A. Aegerter, N. Leventis, M. Koebel and S. A. Steiner III, Springer Handbooks; Springer International Publishing, Cham, 2023 Search PubMed.
- K. Ganesan, T. Budtova, L. Ratke, P. Gurikov, V. Baudron, I. Preibisch, P. Niemeyer, I. Smirnova and B. Milow, Materials, 2018, 11(11), 2144 CrossRef PubMed.
- F. Ciuffarin, M. Négrier, S. Plazzotta, M. Libralato, S. Calligaris, T. Budtova and L. Manzocco, Food Hydrocolloids, 2023, 140, 108631 CrossRef CAS.
- V. Baudron, P. Gurikov, I. Smirnova and S. Whitehouse, Gels, 2019, 5(1), 12 CrossRef CAS PubMed.
- P. T. T. Nguyen, N. H. N. Do, X. Y. Goh, C. J. Goh, R. H. Ong, P. K. Le, N. Phan-Thien and H. M. Duong, Waste Biomass Valorization, 2022, 13(4), 1825–1847 CrossRef PubMed.
- N. H. N. Do, T. P. Luu, Q. B. Thai, D. K. Le, N. D. Q. Chau, S. T. Nguyen, P. K. Le, N. Phan-Thien and H. M. Duong, Mater. Technol., 2020, 35(11–12), 807–814 CrossRef CAS.
- S. Chaudhary and D. T. K. Dora, Biomass Convers. Biorefinery, 2023 Search PubMed.
- P. X. T. Nguyen, K. H. Ho, C. T. X. Nguyen, N. H. N. Do, A. P. N. Pham, T. C. Do, K. A. Le and P. K. Le, IOP Conf. Ser. Earth Environ. Sci., 2021, 947(1), 012011 CrossRef.
- Y. Fan, B. Lu, W. Chen, X. Zhang and X. Li, Elastic white radish-based cellulose aerogel for efficient oil-water separation, Research Square, 2023, preprint, DOI:10.21203/rs.3.rs-2478285/v1.
- L. Manzocco, K. S. Mikkonen and C. A. García-González, Food Struct., 2021, 28, 100188 CrossRef CAS.
- Y. Jeong, R. Patel and M. Patel, Biomimetics, 2024, 9(7), 397 CrossRef CAS PubMed.
- C. A. García-González, A. Iglesias-Mejuto, C. López-Iglesias, V. Santos-Rosales, I. Ardao and C. Alvarez-Lorenzo, in Engineering of Natural Polymeric Gels and Aerogels for Multifunctional Applications, ed. S. Thomas, B. Seantier and B. Joseph, Elsevier, 2024, ch. 5, pp 125–157 Search PubMed.
- I. Smirnova and P. Gurikov, J. Supercrit. Fluids, 2018, 134, 228–233 CrossRef CAS.
- T. Budtova, D. A. Aguilera, S. Beluns, L. Berglund, C. Chartier, E. Espinosa, S. Gaidukovs, A. Klimek-Kopyra, A. Kmita, D. Lachowicz, F. Liebner, O. Platnieks, A. Rodríguez, L. K. Tinoco Navarro, F. Zou and S. J. Buwalda, Polymers, 2020, 12(12), 2779 CrossRef CAS PubMed.
- C. S. A. Bento, B. Agostinho, A. Teixeira, M. S. Reis, H. C. De Sousa and M. E. M. Braga, J. Supercrit. Fluids, 2024, 211, 106321 CrossRef CAS.
- M. Schwan, S. Nefzger, B. Zoghi, C. Oligschleger and B. Milow, Front. Mater., 2021, 8, 662487 CrossRef.
- R. Subrahmanyam, I. Selmer, A. Bueno, D. Weinrich, W. Lölsberg, M. Fricke, S. Movahhed, P. Gurikov and I. Smirnova, in Springer Handbook of Aerogels, ed. M. A. Aegerter, N. Leventis, M. Koebel and S. A. Steiner III, Springer International Publishing, Cham, 2023, pp 93–120 Search PubMed.
- İ. Şahin, Y. Özbakır, Z. İnönü, Z. Ulker and C. Erkey, Gels, 2017, 4(1), 3 Search PubMed.
- I. I. Khudeev, A. E. Lebedev, M. S. Mochalova and N. V. Menshutina, Drying Technol., 2024, 42(5), 812–835 CAS.
- J. Cai, S. Kimura, M. Wada, S. Kuga and L. Zhang, ChemSusChem, 2008, 1(1–2), 149–154 CAS.
- O. Aaltonen and O. Jauhiainen, Carbohydr. Polym., 2009, 75(1), 125–129 CAS.
- A. Rege, M. Schestakow, I. Karadagli, L. Ratke and M. Itskov, Soft Matter, 2016, 12(34), 7079–7088 CAS.
- A. Zaman, F. Huang, M. Jiang, W. Wei and Z. Zhou, Energy Built Environ., 2020, 1(1), 60–76 Search PubMed.
- B. Schroeter, V. P. Yonkova, N. A. M. Niemeyer, I. Jung, I. Preibisch, P. Gurikov and I. Smirnova, Cellulose Aerogel Particles: Control of Particle and Textural Properties in Jet Cutting Process, Cellulose, 2021, 28(1), 223–239 CAS.
- L. T. Possari, C. G. Otoni, T. Budtova and S. H. P. Bettini, Upcycling orange waste biomass into bio-aerogels, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-q8gtn.
- S. Plazzotta, S. Calligaris and L. Manzocco, Innovative Food Sci. Emerging Technol., 2018, 47, 485–492 CAS.
- G. Gaggero, R. P. Subrahmanyam, B. Schroeter, P. Gurikov and M. Delucchi, Gels, 2022, 8(11), 691 CAS.
- S. Plazzotta, S. Calligaris and L. Manzocco, LWT, 2018, 89, 276–283 CAS.
- I. Selmer, C. Kleemann, U. Kulozik, S. Heinrich and I. Smirnova, Development of Egg White Protein Aerogels as New Matrix Material for Microencapsulation in Food, J. Supercrit. Fluids, 2015, 106, 42–49 CrossRef CAS.
- I. Jung, B. Schroeter, S. Plazzotta, L. De Berardinis, I. Smirnova, P. Gurikov and L. Manzocco, Food Hydrocolloids, 2023, 142, 108758 CAS.
- S. Plazzotta, S. Calligaris and L. Manzocco, J. Food Eng., 2023, 351, 111522 CAS.
- L. Manzocco, S. Plazzotta, J. Powell, A. De Vries, D. Rousseau and S. Calligaris, Food Hydrocolloids, 2022, 122, 107117 CAS.
- S. Plazzotta, I. Jung, B. Schroeter, R. P. Subrahmanyam, I. Smirnova, S. Calligaris, P. Gurikov and L. Manzocco, Polymers, 2021, 13(23), 4063 CAS.
- N. Shokri, B. Stevens, K. Madani, J. Grabe, M. Schlüter and I. Smirnova, ACS Eng. Au, 2023, 3(1), 3–6 CAS.
- L. Geuer, G. Liese, M. Roth, A. Liese, S. Perret, A. Schlindwein and R. Ulber, Chem. Unserer Zeit, 2024, 58(4), 216–223 CAS.
- G. Liese, F. Rohweder, J. Husung, L. Gibowsky, B. Schroeter, I. Smirnova and A. Liese, Chem. Ing. Tech., 2022, 94(9), 1379–1380 Search PubMed.
- C. Kleemann, I. Selmer, I. Smirnova and U. Kulozik, Food Hydrocolloids, 2018, 83, 365–374 CAS.
- S. Plazzotta, S. Calligaris and L. Manzocco, Food Res. Int., 2020, 132, 109099 CAS.
- D. A. Méndez, B. Schroeter, A. Martínez-Abad, M. J. Fabra, P. Gurikov and A. P. López-Rubio, Carbohydr. Polym., 2023, 306, 120604 CrossRef PubMed.
- J. F. Saeman, Ind. Eng. Chem., 1945, 37(1), 43–52 CrossRef CAS.
- M. A. Joslyn and J. D. Ponting, Advances in Food Research, Elsevier, 1951, 3, pp 1–44 Search PubMed.
- A. Hosikian, S. Lim, R. Halim and M. K. Danquah, Int. J. Chem. Eng., 2010, 2010, 1–11 CrossRef.
- T. Arumugham, K. Rambabu, S. W. Hasan, P. L. Show, J. Rinklebe and F. Banat, Chemosphere, 2021, 271, 129525 CrossRef CAS PubMed.
- J. Martínez and A. Aguiar, Curr. Anal. Chem., 2013, 10(1), 67–77 CrossRef.
- E. Reverchon, J. Supercrit. Fluids, 1997, 10(1), 1–37 CrossRef CAS.
- N. A. M. Eskin and E. Hoehn, in Biochemistry of Foods, Elsevier, 2013, ch. 2, pp 49–126 Search PubMed.
- D. A. Brummell, Funct. Plant Biol., 2006, 33(2), 103 CrossRef CAS.
- M. Becker, K. Ahn, M. Bacher, C. Xu, A. Sundberg, S. Willför, T. Rosenau and A. Potthast, Cellulose, 2021, 28(13), 8719–8734 CrossRef CAS PubMed.
- S. Levigne, M. Thomas, M.-C. Ralet, B. Quemener and J.-F. Thibault, Determination of the Degrees of Methylation and Acetylation of Pectins Using a C18 Column and Internal Stand-ards, Food Hydrocolloids, 2002, 16(6), 547–550, DOI:10.1016/S0268-005X(02)00015-2.
- J. S. J. Santiago, C. Kyomugasho, S. Maheshwari, Z. Jamsazzadeh Kermani, D. Van De Walle, A. M. Van Loey, K. Dewettinck and M. E. Hendrick, Food Hydrocolloids, 2018, 77, 482–493 CrossRef CAS.
- E. L. La Cava, E. Gerbino, S. C. Sgroppo and A. Gómez-Zavaglia, J. Food Sci., 2018, 83(6), 1613–1621 CrossRef CAS PubMed.
- A. G. J. Voragen, G.-J. Coenen, R. P. Verhoef and H. A. Schols, Struct. Chem., 2009, 20(2), 263–275 CrossRef CAS.
- H. M. Ng, N. M. Saidi, F. S. Omar, K. Ramesh, S. Ramesh and S. Bashir, Encyclopedia of Polymer Science and Technology, Wiley, 2018, pp. 1–29, DOI:10.1002/0471440264.pst667.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87(9–10), 1051–1069 CrossRef CAS.
- G. Horvat, M. Pantić, Ž. Knez and Z. Novak, Gels, 2022, 8(7), 438 CrossRef CAS PubMed.
- A. Veronovski, G. Tkalec, Ž. Knez and Z. Novak, Carbohydr. Polym., 2014, 113, 272–278 CrossRef CAS PubMed.
- M. Pantić, G. Horvat, Ž. Knez and Z. Novak, Molecules, 2020, 25(5), 1187 CrossRef PubMed.
- C. Rudaz, R. Courson, L. Bonnet, S. Calas-Etienne, H. Sallée and T. Budtova, Biomacromolecules, 2014, 15(6), 2188–2195 CrossRef CAS PubMed.
- B. Schroeter, I. Jung, K. Bauer, P. Gurikov and I. Smirnova, Polymers, 2021, 13(17), 3000 CrossRef CAS PubMed.
- D. J. Andlinger, A. C. Bornkeßel, I. Jung, B. Schroeter, I. Smirnova and U. Kulozik, Food Hydrocolloids, 2021, 112, 106305 CrossRef CAS.
- S. Posé, C. Paniagua, A. J. Matas, A. P. Gunning, V. J. Morris, M. A. Quesada and J. A. Mercado, Trends Food Sci. Technol., 2019, 87, 47–58 CrossRef.
- W. S. Peters, K. H. Jensen, H. A. Stone and M. Knoblauch, J. Plant Physiol., 2021, 257, 153341 CrossRef CAS PubMed.
- A. W. Robards, Nature, 1968, 218(5143), 784–784 CrossRef.
- S. Groult and T. Budtova, Carbohydr. Polym., 2018, 196, 73–81 CrossRef CAS PubMed.
- S. Groult and T. Budtova, Eur. Polym. J., 2018, 108, 250–261 CAS.
- A. Zambon, F. Michelino, S. Bourdoux, F. Devlieghere, S. Sut, S. Dall'Acqua, A. Rajkovic and S. Spilimbergo, Drying Technol., 2018, 36(16), 2016–2021 CrossRef CAS.
- E. Heinzle, A. P. Biwer and C. L. Cooney, Development of Sustainable Bioprocesses: Modeling and Assessment, Wiley, 2006, DOI:10.1002/9780470058916.
- A. Biwer and E. Heinzle, J. Chem. Technol. Biotechnol., 2004, 79(6), 597–609 CrossRef CAS.
- J. Cai, S. Kimura, M. Wada, S. Kuga and L. Zhang, ChemSusChem, 2008, 1(1–2), 149–154 CrossRef CAS PubMed.
- E. Heinzle, D. Weirich, F. Brogli, V. H. Hoffmann, G. Koller, M. A. Verduyn and K. Hungerbühler, Ind. Eng. Chem. Res., 1998, 37, 3395–3407 CrossRef CAS.
- R. Subrahmanyam, P. Gurikov, I. Meissner and I. Smirnova, J. Visualized Exp., 2016, 113 Search PubMed.
- A. Lebedev, E. Suslova, A. Troyankin and D. Lovskaya, Gels, 2021, 7(1), 4 CrossRef CAS PubMed.
- I. Selmer, J. Karnetzke, C. Kleemann, M. Lehtonen, K. S. Mikkonen, U. Kulozik and I. Smirnova, J. Food Eng., 2019, 260, 1–11 CrossRef CAS.
- A. De Vries, Y. L. Gomez, E. Van Der Linden and E. Scholten, RSC Adv., 2017, 7(19), 11803–11812 RSC.
- D. A. Mendez, M. J. Fabra, A. Martínez-Abad, M. Martínez-Sanz, M. Gorria and A. López-Rubio, Food Hydrocolloids, 2021, 120, 106957 CrossRef CAS.
- A. I. Blake and A. G. Marangoni, Factors Affecting the Rheological Properties of a Structured Cellular Solid Used as a Fat Mimetic, Food Res. Int., 2015, 74, 284–293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05703a |
| This journal is © The Royal Society of Chemistry 2025 |