
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inverse vulcanisation: a new Starter's guide to an emerging field
Liam James
Dodd

University of Liverpool, Department of Chemistry, Donnan and Robert Robinson Laboratories, Crown Street, Liverpool, L697ZD, UK. E-mail: sgldodd@liverpool.ac.uk
First published on 18th November 2024
Abstract
Inverse vulcanisation is a rapidly developing field of chemistry and materials science with the potential to afford low cost, green chemistry adherent, next generation polymeric materials from the industrial waste product: elemental sulfur. With tuneable properties, recyclability, as well as convenient and adaptable syntheses and processing, inverse vulcanised polymers may be used in several desirable applications, such as batteries, water purification, and advanced optical components. In the ten years since the field's conception, inverse vulcanisation has garnered growing research interest and popularity, and has even seen some recent commercial uptake. This review article is focused on supporting the growth of the inverse vulcanisation field by providing a resource for new researchers to have the most efficient possible start in the field. In that regard, this review article is designed to act as an ideal starting point for researchers looking to become invested in the field. This review first outlines the origin of inverse vulcanisation, before giving a small account of the applications of inverse vulcanisation and pointing to other useful reviews on these applications, thus making a case for research interest and providing sources of potential inspiration for new ideas. Most importantly, this review goes on to provide an effective resource for lab based researchers to establish themselves with foundational knowledge of the field, while offering a guide to practical skills in performing inverse vulcanisation. In doing so, this review offers a guide to standardising methods in inverse vulcanisation whilst also allowing new lab workers to avoid some of the pitfalls that are not obvious, and not common to other fields of chemistry. Then, this review examines methods of analysing inverse vulcanised polymers, which can be challenging, and sometimes needs careful consideration. Finally, this review looks at some mechanistic considerations of inverse vulcanisation before proposing directions for future research in the field.
1. Introduction
Inverse vulcanisation is an emerging field of polymer chemistry, founded in 2013 with a publication that as of August 2024, has more than a thousand citations.1 Inverse vulcanisation is a polymerisation reaction that utilises an industrial waste product, elemental sulfur, as a low-cost reagent alongside an organic comonomer, to produce polymers of high sulfur content by mass percentage. The reaction is diverse and versatile in that it can accept a wide variety of organic comonomers, with different loadings of sulfur, in many possible polymer architectures, all of which allows polymers of wide ranges of physical properties. The details of the polymerisation reaction will be discussed later in this review, but in short, inverse vulcanisation is usually a low cost, simple method, bulk copolymerisation of elemental sulfur with a species that usually contains two or more alkene bonds, resulting in crosslinked polymers with a vast array of attractive potential applications such as water purification sorbents, advanced optical materials, and cathode materials for next generation lithium–sulfur batteries to name but a few.2–7 Although inverse vulcanised polymers are yet to be applied outside of an academic context, recently inverse vulcanised polymers have seen commercial uptake by several companies, such as ThioTech, Outside the Box Materials, Uberbinder, and Clean Earth Technology, exemplifying that these polymers have desirable properties that could be exploited in mainstream applications with commercial benefit and societal impact, if some issues such as the scalability of the reaction and the cases of instability in the polymers can be addressed.8–11It is because of these properties and the low cost of their production that inverse vulcanised polymers have seen steadily growing research interest, however inverse vulcanisation suffers from a poorly understood mechanism and little regimentation or standardisation in how the syntheses are performed. Furthermore, the field of inverse vulcanisation is deceptively simple, both in its theory and practice, resulting in several non-obvious pitfalls and nuances that are not common to other fields of chemistry, and this combines with a severe challenge in characterising the polymers to give a research field that is less accessible than it seems at first glance.
There are several excellent review articles on the field of inverse vulcanisation, including articles that cover the field in general, such as “polymerizations with elemental sulfur: a novel route to high sulfur content polymers for sustainability, energy and defense”, “sulfur chemistry in polymer and materials science”, and “recent advances in the polymerization of elemental sulphur, inverse vulcanization and methods to obtain functional Chalcogenide Hybrid Inorganic/Organic Polymers (CHIPs)”.12–14 Meanwhile, other review articles concentrate on one or more of the potential applications of the polymers; such reviews will receive due mention in the later parts of this review. This review article aims to address the aforementioned issues whilst also providing a convenient starting point for researchers wishing to become invested in the field of inverse vulcanisation. As such, this review will cover the origin of inverse vulcanisation and its chemical concepts in order to provide foundational knowledge that can act as a springboard to access the rest of the field. This review will then give a brief discussion of some of the core potential applications of inverse vulcanised polymers, outlining why the field of inverse vulcanisation deserves such research attention and simultaneously pointing to other reviews that comprehensively cover these applications, and can serve as inspiration for new research directions. Most importantly, this review will then tackle how inverse vulcanisation can be performed, what can be achieved with the syntheses, and what can be modified to alter the synthetic outcome toward a desired goal. This will also extend to post synthetic modification of the polymers, and look at alternate routes of synthesis. Next, this review will look at the commonplace analysis techniques used in inverse vulcanisation, informing new researchers what equipment they need access to, and what they stand to gain from such analyses. Penultimately, this review will give an overview of the poorly understood mechanisms that may be behind inverse vulcanisation, and what these may mean for the polymerisation. Finally, this review will give an outlook of where the field of inverse vulcanisation may proceed in terms of research direction, pointing to several current issues in the field and hopefully inspiring the focus of new starting researchers. Overall, this review article is intended to be an ideal starting point for new researchers to the field of inverse vulcanisation, teaching the prerequisite foundational knowledge to access the field, whilst also giving clear, easy to apply, standardised methods for practically performing inverse vulcanisation, which is something less addressed by prior reviews.
2. Origin of inverse vulcanisation
2.1. Sulfur
More than 60 million tonnes of elemental sulfur is produced annually by the refinement of petrochemical feedstocks.15 In order to prevent acid rain, fuels must be purified of sulfurous contaminant compounds, which would combust to form sulfur dioxide, then going on to dissolve in atmospheric rainwater to form sulfuric acid. As such crude fuels are subjected to hydrodesulfurisation and the Claus process, which convert sulfurous contaminant compounds in petrochemicals, to elemental sulfur.15–17 Although this sulfur has several applications, such as being used to create fertilisers and sulfuric acid, the supply greatly outweighs the demand, resulting in the excess sulfur being stored on the megaton scale in open to air stockpiles, with unexplored environmental consequences (Fig. 1).12,18 Elemental sulfur is known to have some antibacterial properties, which could mean that megaton scale sulfur stockpiles could be harmful to surrounding microbiological ecosystems which could have cumulative effects on food chains that rely on them.19 There is also the consideration that elemental sulfur can be ignited (Fig. 1), and burns to form sulfur dioxide, so if a sulfur stockpile were to be struck by lightning, there is a possibility of a large, hazardous fire that liberates toxic sulfur dioxide into the atmosphere, thereby undoing the efforts to mitigate acid rain.20 These stockpiles are expected to expand more rapidly in coming years as the depletion of fossil fuels forces the usage of previously avoided petrochemical resources of higher levels of sulfur contamination. | ||
| Fig. 1 Top: a sulfur stockpile at Syncrude near fort McMurray, 2007, Alberta, Canada. Bottom: the characteristic blue fire of burning molten sulfur, Kawah Ijen volcano, Indonesia. Top image source: https://metro.co.uk/2015/03/17/no-computer-trickery-here-just-real-bright-blue-lava-5108300/. Bottom image source: https://www.enersul.com/operations/. | ||
Because of the excess production of elemental sulfur, it is an abundant resource that is widely available, and often purchasable for close to the cost of shipping. One potential way to utilise the excess of elemental sulfur, is in the field of materials chemistry. At room temperature, elemental sulfur exists primarily as the α-sulfur allotrope, a crown shaped eight membered ring in an orthorhombic crystal structure, with traces of the less stable cyclo-heptasulfur allotrope present.18,21,22 At 95.3 °C α-sulfur converts to β-sulfur, another eight membered ring of sulfur atoms, this time in a monoclinic crystal structure.22,23 Continued heating results in the melting of elemental in the range of 114 °C to 120 °C, which is broad due to the interconversion of β-sulfur into various other forms of sulfur, all with different melting points.23,24
Through the molten phase to 159 °C or above, sulfur–sulfur bonds enter into an equilibrium where they homolytically cleave into thiyl radicals (Fig. 2), though the exact temperature where this process becomes non-negligible is debated.18,22,25 These thiyl radicals formed from the opening of elemental sulfur rings, are radical capped chains of sulfur atoms, and can attack upon one another and other sulfur rings to extend their chains forming sulfur oligomers, polymers, and large rings which are the predominant species when elemental sulfur forms a clear red solid at temperatures of 190 °C or above (Fig. 2).18 This polymeric solid is not stable to depolymerisation, and upon cooling, depolymerises back to cyclo-sulfur, prohibiting its use in any long term application.18,22,25 Note that the floor temperature refers to the temperature where elemental sulfur can begin to homopolymerise, indicated by changes in its properties, like viscosity. The temperature where elemental sulfur can form thiyl radicals may be lower than the floor temperature.
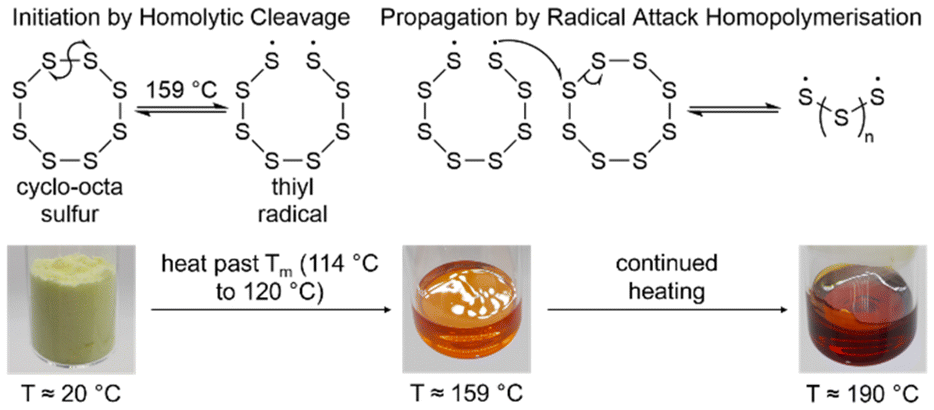 | ||
| Fig. 2 The homolytic cleavage of cyclo-octasulfur to form thiyl radicals and subsequently, their homopolymerisation to form a sulfur polymer, as well as photographs of elemental sulfur at various stages of heating.18,22,23 | ||
2.2. Inverse vulcanisation
For nearly two centuries, elemental sulfur has been used in conventional vulcanisation for the modification of polymer properties by crosslinking organic polymer chains with sulfur bridges, though the mechanism of this has long been debated.25 It is presumed that the thiyl radicals formed by homolytic cleavage of cyclo-sulfur utilise α-allyl hydrogen abstraction to form carbon–sulfur bonds, as suggested by Coleman et al., of which Fig. 3a shows a schematic adapted from their work.26 It should however be noted that it has not been proven beyond reasonable doubt that thiyl radicals are responsible for this crosslinking, and that an alternate theorem is that elemental sulfur cleaves heterolytically to form a zwitterion, though there is less evidence of this.26 Other theories suggest mechanisms closer to that of thiol–ene click chemistry.27 Regardless, conventional vulcanisation does not use large amounts of sulfur, and so cannot be the solution to the ever-increasing excess sulfur problem.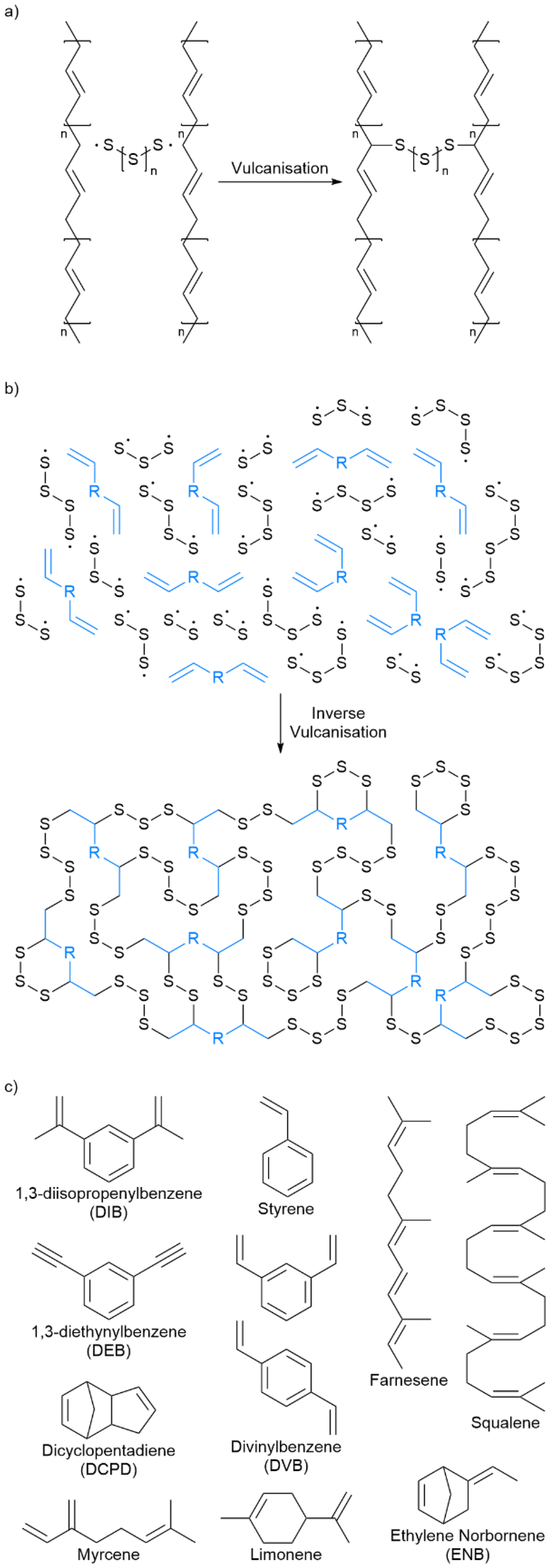 | ||
| Fig. 3 (a) A simplified reaction scheme for conventional vulcanisation. Note that the carbon–carbon double bonds may be incorporated into the macromonomer chains as part of the backbone or as pendant groups. (b) A simplified reaction scheme for inverse vulcanisation. (c) Names, abbreviations and structures of representative examples of small organic molecules that are active in inverse vulcanisation.32–37 Note that divinylbenzene is usually supplied as a mixture of the ortho and para isomers as shown. | ||
Inspired by conventional vulcanisation, some anionic copolymerisations with elemental sulfur, and some earlier works on the copolymerisation of sulfur with styrene or dicyclopentadiene (DCPD), Pyun and coworkers made the landmark discovery of inverse vulcanisation in 2013: a bulk polymerisation of molten sulfur with an organic small molecule crosslinker that contains at least two carbon–carbon double bonds (Fig. 3b).1,28–31 This reaction is capable of forming inverse vulcanised polymers of high sulfur content that can sometimes be stable to depolymerisation, and the variety of reactive crosslinkers gives rise to a plethora of unique materials with differing properties. The reaction itself requires no solvent (though one can be applied if desired to modify the properties of the product) and has a theoretical atom economy of 100% (note that side reactions occur in practice that reduce the atom economy).1Fig. 3c shows the structures of some representative small organic molecules that can be used in inverse vulcanisation reactions as comonomers. Note that there are several examples of crosslinker molecules derived from renewable sources, for example limonene, and that the scope of inverse vulcanisation has been expanded to include molecules with one double bond, more than two double bonds, and even triple bonds.32–37 It is also important to remember that the structure shown in Fig. 3b as a representative of an inverse vulcanised polymer structure has not been proven conclusively, and is a presumed structure. Side reactions and newer research that will be discussed later, all point to the structure shown in Fig. 3b being a drastic simplification.
Previously, there has been some lack of clarity regarding the definition of an inverse vulcanised polymer. The definition that shall be used in this article, is that a conventionally vulcanised polymer is one where sulfur chains crosslink a macromonomer, and that an inverse vulcanised polymer is one where sulfur chains crosslink small organic molecules. Both of these definitions refer to a direct thermal polymerisation of sulfur and comonomer, and discussed later will be alternate routes of synthesis that do not use heat to generate derivatives of inverse vulcanised polymers.
In their seminal publication, Pyun and coworkers reacted 1,3-diisopropenylbenzene (DIB) with molten elemental sulfur in a bulk polymerisation method, at 185 °C using between 50 and 90% by mass in the reaction feed.1 Within five minutes a glassy red solid formed, which was then cooled to room temperature, revealing that it was stable to depolymerisation over the course of months, shape-persistent, and showed no signs of crystalline elemental sulfur contained within its structure. Powder X-ray diffraction (PXRD) revealed only amorphous scattering with no peaks corresponding to crystalline elemental sulfur, and differential scanning calorimetry (DSC) indicated glass transition temperatures (Tg's) which are diagnostic of a polymer structure, and showed no peaks corresponding to the melting of either α-sulfur or β-sulfur (95 °C and 115 °C respectively) when the sulfur feed ratio was 80% by mass or less.1,21,24 It is worth noting that more recent research has pointed out that both PXRD and DSC rely on elemental sulfur being in a crystalline state in order to detect it, though this will be discussed later in detail.
Pyun and coworkers also found that the polymers’ Tg's were tuneable with sulfur content; highest with more DIB, and lowest with more sulfur.1 The reasons for this trend will be explained later, as they are multifaceted, and recent findings regarding DIB add an extra element to the discussion. When the prepolymer was poured into a hot mould towards the end of the five-minute reaction time, it was found that the prepolymer would take on the shape of the mould before forming into a shape persistent red glass, thereby allowing for melt processability.1 To conclude their publication, Pyun and coworkers examined the electrochemical behaviour of their DIB inverse vulcanised polymers as the cathode material of lithium ion batteries. They found that the cyclic voltammogram of their fabricated batteries were quite similar to those of elemental sulfur, but in contrast to other lithium sulfur batteries, their materials demonstrated good capacity retention with extensive discharge and recharge cycles.1
3. Applications of inverse vulcanised polymers
Pyun and coworkers demonstrated in their initial publication that inverse vulcanised polymers could find application in the field of energy storage materials, however in the years since, numerous potential applications for inverse vulcanised polymers have become apparent as their properties have been explored.1 At the time of publication, inverse vulcanised polymers have not been applied outside of an academic context, and have not transitioned to industrial chemistry scales, but with further incubation of the technology, inverse vulcanised polymers may see full commercialisation in the potential applications that will soon be discussed, possibly through the aforementioned companies or new start-ups that could arise in the future.8–11 In addition to their low-cost reagents, simple and facile synthesis, and potential for recyclability, inverse vulcanised polymers can sometimes display self-healing properties. These self-healing properties, as well as their combination of thermoplastic and thermoset properties, and recyclability and remoulding potential, are all resultant from the reversible breakage and formation of the sulfur–sulfur bonds within their structures.12 It is worth bearing in mind during the following discussion of the potential applications of inverse vulcanised polymers, that many inverse vulcanised polymers possess these properties innately, making them attractive candidates for next generation materials. The fact that inverse vulcanised polymers adhere well to the principles of green chemistry on the following grounds: their solventless synthesis; the reagent sulfur is an abundant industrial waste product; the organic comonomer can come from a renewable source; and the polymers can be recycled, all furthers how attractive inverse vulcanised polymers are as next generation materials in a society that is pushing towards being more sustainable.1,19,24,25 It is not the aim of this review to discuss at length, the applications of inverse vulcanised polymers, but it would be remiss not to mention the core reasons why inverse vulcanised polymers receive such research attention. As such, a brief mention of the most popular potential applications will be given, as well as directing the reader towards more comprehensive articles on these topics.3.1. Lithium–sulfur batteries
Lithium–sulfur batteries are an attractive class of next generation energy storage devices due to the low cost of the sulfur cathode and the relatively high specific capacities they can provide.38 However, the poor processability of elemental sulfur can make fabrication of effective cathodes difficult, and the discrete molecular nature of sulfur promotes the loss of electrochemical intermediates into the electrolyte, and away from the cathode where they are useful, (referred to as the shuttling effect) which limits the retention of specific capacity and ultimately decreases the cycle life of the battery.39 Inverse vulcanised polymers may be able to remedy these drawbacks as they can provide sulfur source in the form of a melt or solution processable solid, that contains sulfur bonded into and therefore trapped as part of, a crosslinked matrix, which may be able to prevent the shuttling effect.1,38,39As a result, lithium–sulfur battery cathodes have become one of the most extensively researched topics regarding inverse vulcanised polymers. This application requires a detailed understanding of battery electrochemistry which cannot be included here, however there are several commendable reviews that already exist on this topic: “engineering strategies for suppressing the shuttle effect in lithium–sulfur batteries”, “revisiting the role of polysulfides in lithium–sulfur batteries”, and “recent advances in applying vulcanization/inverse vulcanization methods to achieve high-performance sulfur-containing polymer cathode materials for Li–S batteries”.40–42
To give a brief overview, Pyun and coworkers investigated the electrochemical properties of a DIB inverse vulcanised polymer, as part of a standardised testing cell (known as a coin cell).1 Cyclic voltammetry experiments found that coin cells of sulfur and coin cells of DIB inverse vulcanised polymers behaved relatively similarly. The reduction peaks between 2.3 V and 2.4 V in both cyclic voltammetry traces were assigned to the initial reduction of sulfur to linear lithium polysulfides (Li+ S−–Sn–S− Li+) and the reduction peaks between 2.0 V and 2.1 V were assigned to further reduction of these linear lithium polysulfides to shorter chain linear lithium polysulfides, though cyclic voltammetry experiments alone cannot confirm these assignments. More in depth studies of the battery cycling behaviour revealed that the DIB inverse vulcanised polymer coin cells had long cycling lives as well as specific capacities in the range of other cutting edge cathode composites for lithium sulfur batteries, which were at that time, the best for any polymer-based cathode in such batteries, outperforming those of a much more expensive and difficult fabrication.1 Besides review articles and the seminal publication of inverse vulcanisation, another noteworthy publication is that of Hoefling et al., who used a challenging solid state NMR approach to derive critical insights into the operation of inverse vulcanised polymer cathodes in lithium–sulfur cathodes, and is therefore recommended reading for those interested in the battery applications of inverse vulcanised polymers.39
3.2. Optical components
Since inverse vulcanised polymers are comprised of chemical bonds with very low polarity, mostly sulfur–sulfur bonds and carbon–sulfur bonds, (note that carbon and sulfur have the same Pauling electronegativity) they often have low infra-red absorbances, as their stretching modes either do not, or very weakly, satisfy the selection rule for infra-red spectroscopy, that being, infra-red vibrational modes must cause a change in dipole moment.43 In addition to this sulfur is a highly polarisable atom, and a high proportion of polarisable atoms within a material's structure gives rise to high refractive index.44 As such, inverse vulcanised polymers are potential next generation materials for infra-red optics, which are applied in thermal imaging devices.A high refractive index allows for lenses to be made thinner and still achieve the same lens power, which can be important to the application of the lens (lightweight devices), but also its lifetime, as it can be that heavier lenses are gradually deformed by their own weight, pulling them out of their precise shape which is vital to their function. Thinner lenses also require less material, inherently lowering the cost, though inverse vulcanised polymers are far cheaper than the current industry standard chalcogenide glasses and crystalline semiconductors.44 Crystalline semiconductors such as silicon, germanium, and zinc selenide give high performance components, but can be hard to refine to a sufficient purity, difficult to appropriately shape, and have poor scalability to their synthesis and purification raising the cost as a result.44 Meanwhile chalcogenide glasses, defined as a non-crystalline compound of a chalcogen (sulfur, selenium, or tellurium) with a group 14 or 15 element, are almost as infra-red transparent as crystalline semiconductors, with the additional advantage of solution processability under an inert atmosphere. Chalcogenide glasses are accepting of a wide variety of chalcogen to group 14 or 15 element, with heavier chalcogens tending to give chalcogenide glasses of poorer mechanical properties and shape persistence but greater transparency. However, the raw materials to make chalcogenide glasses must be very pure, and need multiple, expensive, high temperature purification processes for material fabrication. Worse, the end-of-life products of these materials can be toxic, and so require special disposal to avoid environmental and health consequences.44
Since inverse vulcanised polymers remedy many of the drawbacks of chalcogenide glasses and crystalline semiconductors, in that they can be mechanically robust, melt processable, and highly infra-red transparent and refractive, as well as being cheap, easy to make, scalable, and often have no need for purification, they could be ideal replacements for chalcogenide glasses and crystalline semi-conductors, from which several publications have been centred.44 The publication titled “100th anniversary of macromolecular science viewpoint: high refractive index polymers from elemental sulfur for infrared thermal imaging and optics” gives an excellent overview of these publications as well as setting out the field, and is thoroughly recommended reading.44 Here, a few noteworthy publications that exemplify important principles in inverse vulcanisation will be mentioned.
Pyun and coworkers recognised that if their previous organic crosslinker of DIB contained a third olefin moiety, then the crosslinking potential of a single molecule could be increased, thereby resulting in a higher crosslink density which would give higher Tg and mechanical robustness.45 It would also allow for more sulfur stabilisation, raising the percentage of sulfur in the polymer, which has a twofold effect: it raises the proportion of highly polarisable sulfur atoms for greater refractive index, and it reduces the percentage content of organic units, which are the main cause for infra-red absorption. As such 1,3,5-triisopropenylbenzene was synthesized, and inverse vulcanised to yield materials’ whose Tg was tuneable with sulfur content and showed self-healing when exposed to heat, allowing for it to be repaired when damaged, which is a large advantage over the current industry standard materials which must be discarded if damaged.45
A seemingly sensible advancement toward inverse vulcanised polymers as infra-red optical components, would be the substitution of sulfur for selenium, which as a heavier element, is more polarisable and therefore would instil a higher refractive index to the resultant material.46 However, as will be discussed later, there are numerous issues with substituting elemental selenium into inverse vulcanisation reactions. To circumvent these issues, two methods have been employed. In the first method, elemental sulfur was doped with selenium, to replace some sulfur atoms in cyclo-octasulfur with selenium, leading to a precursor material: S90Se10, and although the selenium loading was limited, it was successfully employed in inverse vulcanisation with DIB to generate a polymer with higher refractive index than the selenium free analogue.47 The second method achieved higher loadings of selenium and avoided the issues incurred by its many allotropic phase transitions, by heating elemental sulfur to the molten state, and then introducing grey selenium (an allotrope of elemental selenium) to the molten elemental sulfur. The thiyl radicals of the molten elemental sulfur cracked the chains of selenium atoms, creating an in situ mixed chalcogen, which was then used to crosslink DIB. The resultant material showed higher loadings of selenium than the products of the first method, as well as exhibiting refractive indices as high as 2.1.52
In another publication, it was recognised that because most of the infra-red absorbance of inverse vulcanised polymers stems from their organic comonomer units, choosing a comonomer of low infra-red absorbance would allow inverse vulcanised polymers of minimal infra-red absorbance could be prepared.49 DFT was used to predict which co-monomers might have low infra-red absorbance, directing to norbornadiene, though it was rationalised that norbornadiene had a boiling point too low to be successfully implemented in inverse vulcanisation, and so a dimer which was termed dinorbornadiene was employed in inverse vulcanisation, and the resultant polymer was subjected to a study of its optical properties.49 Another noteworthy publication in regard to inverse vulcanised polymers for infra-red optics is that of Tonkin et al., and is recommended reading.50 Tonkin et al. used an atypical synthetic method that will be discussed later to generate a cyclopentadiene inverse vulcanised polymer for infra-red optics, supported by DFT simulation of the infra-red spectra, to create a polymer of optimised infra-red transparency.50
3.3. Antimicrobial materials
Elemental sulfur itself has antimicrobial properties, but is limited in its applications due to its powdered form.19,51 Inverse vulcanised polymers are a potential means to provide sulfur as a continuous solid with sufficiently robust mechanical properties that they can be applied in a wider range of antimicrobial materials, for example, thin coatings.1 One publication by Smith et al. examined the antimicrobial properties of the inverse vulcanised polymers of two organic crosslinkers, DIB and DCPD, finding that inverse vulcanised polymers of both organic crosslinkers prevented exponential growth and biofilm (a surface film made up of adhered bacteria) formation of E. coli bacteria on their surfaces.52 Further, more controlled characterisation of the surface bactericidal effects of these inverse vulcanised polymers, showed that the DIB inverse vulcanised polymer caused a 99.9% reduction in the survival of two different species of bacteria on its surface, E. coli and S. aureus, while the DCPD inverse vulcanised polymer showed a lesser reduction in the number of surviving E. coli bacteria on its surface, and showed little reduction in the population of S. aureus surviving on its surface. From these results, it is clear that the choice of organic crosslinker has significant influence over the extent to which, and over the species of which an inverse vulcanised polymer can kill bacteria.52Another important publication in the field of antibacterial studies was that of Dop et al., who used more in-depth methods to characterise the bactericidal effects of a range of different inverse vulcanised polymers made from a wider range different crosslinkers.53 It was found that not only does the bactericidal effects depend on the identity of the organic crosslinker, but also upon the Tg of the polymer. Of exceptional importance, Dop et al. discovered that their inverse vulcanised polymers showed little or no bactericidal effects when tested at temperatures below their Tg, but showed significantly greater bactericidal effects when tested at temperatures exceeding their Tg. This not only informs future research, but also suggests that it could be the reversible sulfur–sulfur bond formation may have a role to play in the antibacterial properties of inverse vulcanised polymers, as it is above the Tg where homolytic bond cleavage becomes more significant, presenting a greater presence of thiyl radicals, and Dop et al. noted that other studies have shown that such sulfur radicals can be toxic to bacteria. Dop et al. also tested whether the hydrophobicity of the polymers had an effect on their bactericidal effect but found no correlation. Because Dop et al. observed a reduction in viable cells in solutions treated with inverse vulcanised polymers, a leaching study was conducted, despite the fact that Smith et al.'s worked had steered away from the conclusion of leaching effects.52 They found that some inverse vulcanised polymers showed more rapid reductions in viable cells when tested above their Tg which could be because when the polymer is above its Tg it has greater chain mobility, permitting easier diffusion of bactericidal species out of its structure and into the surroundings.53 Additionally, yellow precipitates formed in the test containers by the end of the tests, which DSC confirmed to be elemental sulfur. 1H NMR was conducted on the solution that had been treated with the inverse vulcanised polymer, and no signals were detected, suggesting that the leachate contained no hydrogen containing species such as inverse vulcanised oligomers. However, inductively coupled plasma optical emission spectroscopy revealed that the solution did contain elemental sulfur. Dop et al. did note that further studies should be conducted to determine with greater certainty, what species leach out of the polymer, such as H2S. It is worth noting that in a following publication, Dale et al. showed that inverse vulcanised polymers generate hydrogen sulfide gas over long periods of contact with water.54 Regardless, Dop et al.'s study inadvertently highlighted that analysis of the presence of crystalline elemental sulfur alone in inverse vulcanised polymers is not sufficient, as none of their polymers contained crystalline elemental sulfur, yet either amorphous sulfur leached out and crystallised, or the polymers depolymerised to form it.53
3.4. Remediation of water
Elemental sulfur itself can be used to sequester chemically soft metals from water, which are a significant health concern in many parts of the world.4 However, sulfur has relatively low affinity for these metals in comparison to specialised metal uptake materials, and it is difficult to process into effective devices due to its high crystallinity and powdered morphology.4,13,17 Inverse vulcanised polymers are a potential alternative as they have been shown to have high affinities for soft metals like palladium, gold, and mercury, and are often melt processable into useful shape persistent devices.4,6 Inverse vulcanised polymers are also of lower cost in comparison to other technologies, making inverse vulcanised polymer sorbents more economically accessible in the poorer parts of the world, which is where this technology is most needed.4,6,8 Recommended reading on this topic includes “the mercury problem in artisanal and small-scale gold mining” and “inverse vulcanized polymers for sustainable metal remediation”, as well as “best practices for evaluating new materials as adsorbents for water treatment” which gives some much needed standardised methods for heavy metal sorption testing, which has been an issue in the field of inverse vulcanisation.4,6,55Chalker and coworkers were among the first to report inverse vulcanised polymers with application of water remediation as the target.36 They reported a material, produced by a modified inverse vulcanisation method, comprised of elemental sulfur and limonene. The reaction was performed with an active vacuum distillation attached to the reaction vessel, which drew off p-cymene, volatile thiols and sulfides as the distillate. Chalker and coworkers noted that the formation of aromatic p-cymene from limonene in the presence of sulfur had been reported in the literature previously, but it does hint to the conclusion that the mechanism of inverse vulcanisation is likely more complex than simple radical attack, and can have unexpected branches to its reaction pathways.56 The product of the reaction was too low molecular weight to be considered a true polymer, but rather an oligomer which was otherwise easy to synthesize, scalable to 100 g in its synthesis, and had an affinity for aqueous mercury(II) and palladium(II). The waxy material was capable of taking up 55% of mercury(II) from a HgCl2 solution (10 mL, 2000 parts per billion) in water, and the mercury(II) could not dissociate from the material back into water once bound. Though 55% uptake is relatively poor compared to some materials, sulfur–limonene polysulfide's remarkably low cost and ease of processing could make it an attractive material. What makes it more attractive is the fact that the material undergoes a distinct colour change upon absorbing mercury(II) which may provide a simple means of assessing the material's lifetime.36
Since Chalker and coworkers’ publication, several other inverse vulcanised polymers have been demonstrated to uptake mercury and other harmful metals from water, with greater efficiency than Chalker and coworkers’ sulfur–limonene material and better investigated absorption thermodynamics and kinetics. It has become apparent that the choice of the organic monomer strongly influences metal uptake, but also that the morphology and microscopic structure of inverse vulcanised polymers play critical roles in several factors regarding metal uptake. It has also been shown that modifications to the polymer structure, both during and post synthesis can have a large impact on the mercury uptake of inverse vulcanised polymers.57,58 These modification methods will be discussed later in more detail.
4. Performing inverse vulcanisation
As described above, inverse vulcanisation is a polymerisation that reacts elemental sulfur with an organic comonomer.1 There is wide variety in the comonomers that can be used, though it is generally accepted that inverse vulcanisation utilises comonomers with at least one carbon–carbon double bond. Use of comonomers with a single alkene generates linear inverse vulcanised polymers, with chains of sulfur atoms that are interrupted by organic units.59 Much more common is the use of comonomers with multiple alkenes, thereby leading to a crosslinked network, where chains of sulfur atoms connect together organic units that act as crosslinking centres.1 This is because linear inverse vulcanised polymers have a tendency to be less stable to depolymerisation, and more crosslinked polymers have a tendency to be more resistant to depolymerisation.54,60 Special attention should be paid to the lifetime of inverse vulcanised polymers, because it is known that they change with time at different rates based on crosslinking degree, sulfur content, and comonomer identity, and this is particularly prudent to the analysis and application of inverse vulcanised polymers.54,60 The results of analysis techniques may differ depending on how much time has passed since their synthesis, and the properties of inverse vulcanised polymers are known to change as they age, which needs to be assessed before they enter applications such as construction materials.61–64This section will give an overview of how to perform inverse vulcanisation: setting out basic methods, looking at variants, and outlining how certain principles affect the reaction and its products.
4.1. General bulk polymerisation methods
The most common method to perform inverse vulcanisation is bulk polymerisation, with a good general method being as follows (Fig. 4).1,29,65–67 A desired amount of elemental sulfur is heated to its molten state with stirring from a polytetrafluoroethylene (PTFE) stirrer, in a disposable glass reaction vessel. Disposable reaction vessels can be convenient because of the challenge in satisfactorily cleaning glassware of inverse vulcanised polymers, usually requiring aggressive cleaning measures such as concentrated KOH base baths. Besides, if the reaction is to be taken to completion in this glass reaction vessel, then it is likely that the vessel will need to be shattered to retrieve the polymer. | ||
| Fig. 4 A recommended inverse vulcanisation bulk polymerisation set up. | ||
In regards to the temperature used, it is recommended that the minimum temperature should be no lower 135 °C, as below this temperature there is the risk of sulfur struggling to remain in the molten state; and that the maximum temperature used should be no higher than 185 °C, as above this temperature elemental sulfur will become solid, and this can lead to inhomogeneous reactions once the comonomer is introduced.18,22,25
In regards to the method of heating, both oil baths or aluminium heating blocks upon hot plates can be used, however, inverse vulcanisation reactions are particularly sensitive to their conditions, and so if a higher degree of control is required, an aluminium heating block and heating pan may be preferrable, as it allows consistent positioning of the reaction apparatus upon the hotplate, and this ensures consistent stirring between reactions.66,67 Aluminium heating blocks are also better conductors of heat, whereas oil baths are more insulating, with a higher specific heat capacity. This could mean that oil baths are more prone to inducing auto-accelerations in inverse vulcanisation reactions, as it is harder to transfer heat out of the reaction and into the an oil bath in comparison top aluminium heating block. It should be noted that for bulk polymerisation inverse vulcanisation, the stirring has a significant effect upon the reaction, and so careful attention should be paid to the size of the PTFE stirrer bar and the stirring rate.66,67 Whether using an oil bath or an aluminium heating block, good contact with the reaction vessel must be ensured: this allows for good thermal control, minimising the variability in the reaction, but it also allows for effective heat dissipation. Inverse vulcanisation is known to be exothermic and prone to the Trommsdorff–Norrish effect, wherein a reaction will violently auto-accelerate, at best leading to an inconsistent product, and at worst posing a hazard to health.66,67 Good contact with the surrounding medium and therefore good heat dissipation from the reaction vessel into its surroundings, minimises this auto-acceleration risk (Fig. 5), and generally improves reaction consistency.
 | ||
| Fig. 5 An inverse vulcanised polymer that has undergone an auto-acceleration during its synthesis. Bubbles can be seen throughout its structure as auto-accelerations in inverse vulcanisation are associated with increased hydrogen sulfide generation. Note the inhomogeneity in the polymer. | ||
With these initial factors in mind, the elemental sulfur should be left to equilibrate at the desired temperature, in the absence of comonomer. If the comonomer is present, this leaves a poorly defined start point for the reaction, and it is a known principle in controlling polymerisations, that a sharp and well-defined initiation event leads to more consistent polymer products.66–68 Heating the sulfur to the reaction temperature in the presence of the comonomer also promotes self-polymerisation of the comonomer: thiyl radicals do not form while elemental sulfur is solid, so all throughout the heating process up until the reaction reaches the melting point of sulfur at greater than 100 °C, inverse vulcanisation cannot occur, leaving any competing reactions like comonomer self-polymerisation unopposed.18,22,23
Once the sulfur is thermally equilibrated, the desired comonomer can be added. This comonomer needs to have a boiling point sufficiently high that volatilisation at the chosen reaction temperature will not be rapid. Where this is not possible, a later discussed method can be employed. With the comonomer added, the stirring rate again becomes a point of importance: early in the reaction, the comonomer will almost certainly be immiscible with the molten sulfur, and so the stirring strongly influences the intimacy of the mixing of elemental sulfur and comonomer.69–74 Usually, a high stirring rate is preferred to ensure an intimate mix of the two phases.
From this point, it is possible to leave the reaction be, and allow it to reach completion by simply leaving it with heating and stirring for an extended period of time (usually overnight).66,67,75
Comonomer evaporation can be minimised quite effectively by sealing the reaction vessel with a septum, and then affixing a balloon to the septum via a needle for pressure regulation (Fig. 4).66,67,75 This prevents volatilised comonomer from leaving the reaction vessel, thus raising the vapour pressure, and encouraging comonomer to remain in the liquid phase. In the event that an inverse vulcanisation is to be performed under an inert atmosphere, it is not difficult to affix the septum after adding the sulfur to the reaction vessel, then purge the reaction vessel with an inert gas, followed by affixing a nitrogen balloon, thermally equilibrating the molten sulfur at the desired reaction temperature, and then injecting the comonomer (Fig. 4).66,67,75
Observing the reaction after the comonomer has been added, without stirring the reaction mixture will separate into two phases, with the denser sulfur phase sinking to the bottom, whilst the comonomer phase floats to the top (Fig. 6). With stirring, these two phases intersperse, resulting in a light scattering dispersion (Fig. 6).66,67 With continued heating and stirring, the biphasic dispersion will eventually become monophasic and therefore, clear (Fig. 6).66,67 This signifies that reaction mixture is no longer predominantly unreacted sulfur and comonomer, but is instead predominantly inverse vulcanised oligomers.66 These oligomers are of low molecular weight and are not stable to depolymerisation upon cooling. Continued heating will normally result in a darkening of the reaction mixture a ccompanied by an increase in viscosity, corresponding to the oligomers growing in molecular weight and achieving higher degrees of crosslinking if they are able. In many cases, this viscosity can become so great that that the stirrer ceases to rotate, indicating an inverse vulcanised polymer of solid characteristics.66 It is important to note that the reaction may not yet be complete, and further heating, often termed curing, for an extended period of time is required for the polymer to reach a thermodynamic minimum, with the maximum possible consumption of comonomer double bonds.66,67 The exact time needed to reach completion in this way varies with comonomer and reaction/curing conditions, so it is advisable to study the extent of reaction with reaction/curing time. This can be done easily by reacting or curing for different amounts of time and then performing infra-red spectroscopy or DSC on the polymers.66 When there is no change in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch intensity in infra-red spectroscopy, and no change in the Tg from DSC with increasing curing time, the reaction can be taken as complete.
C stretch intensity in infra-red spectroscopy, and no change in the Tg from DSC with increasing curing time, the reaction can be taken as complete.
 | ||
| Fig. 6 An inverse vulcanised polymer at various stages of reaction. | ||
Once a solid inverse vulcanised polymer is obtained, it will usually be insoluble. Thus, often the easiest way to remove it from the reaction vessel is to first cool the reaction vessel with liquid nitrogen, separating the polymer from the reaction vessel walls by thermal shrinkage, and embrittling the polymer, before shattering the reaction vessel so that the polymer can be retrieved.66,67 Mechanical grinding can then powderise the polymer (Fig. 7), convenient for analysis, though it should remembered that low Tg polymers are not amenable to grinding, as the rise in temperature from the mechanical grinding will often result in the polymer exceeding its Tg and becoming a thick viscous paste that is damaging to the grinding apparatus and challenging to clean off.
 | ||
| Fig. 7 Examples of (a) shattered pieces of inverse vulcanised polymer, and (b) powdered inverse vulcanised polymers. Typical colours include red, orange, yellow, and brown. | ||
Shattering a reaction vessel is not always convenient, and is certainly not appropriate in industrial processes, so a common alternative is to carry out the inverse vulcanisation reaction until a prepolymer has formed, and then pour that prepolymer into a preheated silicone mould, which is then placed into an oven for curing.32,76 This is convenient as it can allow melt processing of inverse vulcanised polymers into any number of useful and creative shapes (Fig. 8). However, there is nuance to this prepolymer pouring technique: pour the polymer too early, and an inhomogeneous polymer may result as the denser sulfur sinks toward the bottom of the mould in the absence of stirring; pour too late, and the polymer may be too viscous to effectively pour into and take on the shape of the mould.32,76 To determine when the prepolymer can be poured into a preheated mould, the “dip test” is commonly employed: the tip of a micro-spatula is dipped into the reaction mixture and used to remove an aliquot. This aliquot is observed while it cools, and if it can remain at room temperature without elemental sulfur crystallising within the aliquot, then this is taken as an indication that the prepolymer is ready to be poured (Fig. 9).32,76 There is also the consideration that once poured into a mould, the prepolymer is without stirring, and therefore more susceptible to the Trommsdorff–Norrish effect. However, there is little that can be done to mitigate this beyond keeping the curing temperature as low as possible, using a less reactive comonomer, and keeping the reaction to a small scale. As a side note, one should always consider that when scaling up any bulk polymerisation, including an inverse vulcanisation, the Trommsdorff–Norrish effect becomes more likely, thus typical lab scale inverse vulcanisations are normally performed on the scale of 10 g.32,66,67,75,76
 | ||
| Fig. 8 Inverse vulcanised polymers that have been moulded into various shapes and dimensions, by pouring the pre-polymer into a mould and then curing the pre-polymer until it is shape persistent. Fully reacted inverse vulcanised polymers of low Tg can also be melt processed in this fashion. | ||
 | ||
| Fig. 9 The result of dip tests at different times during an inverse vulcanisation, indicating the appropriate time to pour the reaction mixture into a mould. The mixture can be poured any time after the dip test is clear, provided the reaction mixture is of low enough viscosity to pour. Pouring the reaction mixture into a mould at different times can lead to different results. | ||
Sometimes, inverse vulcanised polymers can contain elemental sulfur entrapped within their structures, and in some cases, it can be desirable to remove this impurity. A relatively simple method to achieve this is Soxhlet extraction, though this requires the polymer to be in powder form in order to be effective.58,75 Toluene can be an effective solvent for Soxhlet extraction, as elemental sulfur does show some solubility in toluene, though this does require that the polymer itself be insoluble in toluene.75,77
Finally, it is recommended that inverse vulcanised polymers be stored in the dark, as it has been shown that exposure to ultraviolet light can degrade the polymers.54,60,67 Storage in cold conditions can slow the aging of inverse vulcanised polymers, and storage in dry conditions can reduce uptake of water into the polymer structure, which also another cause of polymer aging.54,60
4.2. Principal variables in inverse vulcanisation reactions
The chief variables that can be varied in all inverse vulcanisation reactions include: sulfur loading, comonomer identity, and reaction temperature.1,32–37,65 This section will discuss these variables and their effects on the resultant polymer.The sulfur loading, being the mass percentage of sulfur that is input into the reaction, is what gives inverse vulcanised polymers their tuneable properties in many cases.1,45 Generally, the sulfur loading can be varied between 10% and 90% by mass, though this range varies between the different crosslinkers used and how many reactive carbon–carbon double bonds are present. Low loadings of sulfur are often not investigated because the excess sulfur problem incentivises the usage of large percentages of sulfur in inverse vulcanisation, and many of the applications of inverse vulcanised polymers benefit from the polymers having a higher percentage of polymerised sulfur in their structure.4,6,40–44,52,53 Otherwise, extremely low sulfur loadings can lead to non-polymeric products, as there is insufficient sulfur present to react with the comonomer, or polymers that mostly consist of homopolymerized comonomer. At the other end of the range, extremely high loadings of sulfur can lead to unstable polymers that depolymerise with time; an effect called sulfur bloom, where elemental sulfur becomes visually evident in the polymer.54,60 High loadings of sulfur also promote incomplete reactions where elemental sulfur remains unpolymerized and entrapped within the polymer network.54,60
With these extremes in mind, varying the sulfur loading leads to several general trends, such as the effect on the soluble fraction and Tg. This is more intuitively explained from the perspective of the comonomer loading rather than the sulfur loading: as the comonomer percentage is increased, assuming this comonomer has at least two double bonds and acts as a crosslinker, the final polymer product becomes increasingly crosslinked, which lowers its soluble fraction.32,66,67,75,76 The increased crosslinking also raises the Tg, explained by two principles. Firstly, as more crosslinker is added, the crosslink density of the polymers is increased, which makes the chains more immobile. Therefore, they require more input thermal energy to allow them to move more freely.32,66,67,75,77 The second principle is that adding more crosslinker results in shorter crosslinking chains of sulfur, as fewer sulfur atoms are distributed across more reactive carbon–carbon double bonds. It has been shown in the literature, that longer chains of sulfur atoms contain weaker central sulfur–sulfur bonds, which are easier to break.67,79–83 Therefore, with more crosslinker, there are fewer of these weaker bonds, making it harder to break enough sulfur–sulfur bonds that the chains can move over one another. As the comonomer percentage is increased further, the soluble fraction continues to decrease and the Tg continues to increase, however in some cases, there comes a point where past a certain comonomer loading, the trends reverse, and increasing the comonomer loading then begins to increase the soluble fraction and decrease the Tg.66,67,75 This is because there is not enough sulfur present to react with all the double bonds, resulting in increasing linear character to the polymers as the comonomer loading is increased. Note that in the seminal publication, Pyun and coworkers reported that an inverse vulcanised polymer of DIB had high Tg at high DIB loading and low Tg at low DIB loading.1 A recent publication revealed that DIB is a special case and actually forms a mostly linear polymer, so the changes to its Tg and soluble fraction are likely explained by increasing molecular weight with increasing DIB loading.84
The next variable that can be modified in inverse vulcanisation is the reaction temperature, though there are more restraints on how this can be varied. As mentioned before, it is recommended that inverse vulcanisation be performed between 135 °C and 180 °C. At the lower end of this temperature range, inverse vulcanisation reactions are slower and take longer, but this can lead to a thermodynamic product over a kinetic product.32,64,67,75,76 Higher temperatures are associated with faster reactions that can become vulnerable to auto-acceleration, but are necessary for less reactive comonomers to achieve reaction in a reasonable amount of time.32,66,67,75,76 Higher reaction temperatures can also be associated with the evolution of more hydrogen sulfide gas by-product, which is always unfavourable on account of its toxicity and the fact that it reduces the atom economy of the reaction.76,85 In some special cases where the comonomer has two different carbon–carbon double bonds that are reactive, like with DCPD, one bond may have a higher activation energy of reaction than the other, so it is possible to favour the reaction of one double bond over the other by using a lower reaction temperature.76,85
The choice of comonomer is the variable with the broadest scope, and has led to the publication of numerous articles that report the vast array of different properties and applications that inverse vulcanised polymers can possess. Comprehensively covering all permutations of inverse vulcanised polymers with varying comonomer is not practical. What is more useful here, is to outline guidelines for comonomer choice. Trivially, the hydrophilicity of the comonomer has been found to directly influence the hydrophilicity of the resulting polymer, though inverse vulcanised polymers have a tendency to be hydrophobic due to their sulfur content.53 When a comonomer with only one reactive carbon–carbon double bond in used, the resultant polymer is typically linear, less stable to depolymerisation, and soluble.35 A greater proportion of reactive double bonds per unit molecular weight in the comonomer generally results in a polymer that can stabilise more sulfur per gram of polymer.75,86 Comonomers with rigid structures (e.g. DVB and DCPD) have a tendency to produce hard, brittle, glassy polymers with higher Tg's, whereas comonomers with more flexible structures, like vegetable oils, tend to produce more elastic, flexible, rubbery polymers with low Tg's.34,76,85,87 It should be noted that comonomers with electron deficient carbon–carbon double bonds, such as methacrylates like ethylene glycol dimethacrylate (EGDMA), can be unreactive in inverse vulcanisation, and require activation to react effectively.66,86
4.3. Solution phase inverse vulcanisation reactions
Whilst not requiring a solvent is often noted as an advantage of inverse vulcanisation, it can also become a drawback.1 Bulk polymerisations are vulnerable to the Trommsdroff-Norrish effect, a violent auto-acceleration of the reaction rate which becomes an increasingly significant risk at larger reaction scales.88,89 This is relevant to any industrial scale production of inverse vulcanised polymers, where the polymers will need to be produced on the kilograms to tonnes scale to meet market demand, but at these scales, the Trommsdroff-Norrish effect becomes very likely and very dangerous. Performing reactions in solution can mitigate this risk twofold: they dilute the reaction compared to the bulk, thereby lowering reactant concentrations and thus lowering the reaction rate; and they dilute the heat production of the reaction over a larger volume, so it becomes less likely that an internal rise in temperature of the reaction mixture will become sufficiently intense to induce auto-acceleration.75,88,89 It is therefore likely that any scaled up inverse vulcanisation will be done in a solvent, however, such methods can also be useful on the lab scale as well.75 Xylene has been used as a solvent in inverse vulcanisations to slow down excessively rapid reactions as well as provide more homogeneous products than an equivalent bulk polymerisation.75 It is important to remember that solid elemental sulfur is barely soluble in any solvent, but once the sulfur melts, it can become miscible with a range of solvents.77The solubility of sulfur increases as the temperature of the system is increased, where the solvent is either, toluene, xylene, chlorobenzene, or cyclohexane.77 Of these four, toluene and chlorobenzene were the most effective solvent, with the more substituted xylene performing comparably well at lower temperature, but less effectively at higher temperature.77 Meanwhile, cyclohexane was less effective at solvating sulfur compared to the other three, all across the temperature range, suggesting that an aromatic solvent is beneficial, particularly if it is of lower substitution level.77 Ideally, both the sulfur and the comonomer should be soluble in the solvent, which can be limiting to the range of solvents or the choice of comonomer.
A further limitation on solvent choice is a newly discovered chemistry involving amide solvents, the chief example being dimethylformamide (DMF). It has been found that DMF is capable of inducing breakage in the sulfur–sulfur bonds of trisulfides, introducing a new set of dynamic chemistry which could complicate the inverse vulcanisation process further.90 It is advised to avoid solvents like DMF when synthesizing and processing inverse vulcanised polymers, unless the introduction of this dynamic chemistry is explicitly desired.90
Regardless, the synthetic technique follows closely that of the bulk polymerisation, with the exception that the reaction is performed in a round bottom flask equipped with a reflux condenser.75 All other considerations that were present for bulk polymerisation, such as melting and thermally equilibrating the sulfur before comonomer addition, should be kept in mind for solution phase synthesis as well. Often, as a solution phase reaction progresses, the molecular weight and degree of crosslinking of the inverse vulcanised polymer will increase to the point that the polymer can no longer remain in solution, and thus precipitates. This can be advantageous to the separation of the product from the reaction solution, but can be disadvantageous because that precipitated polymer can cure and fuse to form a solid that is hard to remove from the reaction vessel, requiring extensive mechanical effort and embrittlement by liquid nitrogen.75 This process can also trap the stirrer and prevent effective stirring of the reaction solution. Further complicating such a process is that the reaction may form two products: a soluble fraction and an insoluble fraction, which although they can have different properties and applications, also require different treatment and purification.75 The soluble and insoluble fractions can be separated easily by filtration, and rotary evaporation can leave the soluble fraction free of solvent. From there, Soxhlet extraction can purify the insoluble fraction or any residual soluble product. After this, both the soluble and insoluble fractions may benefit from a curing step.75
Given these complications, it may be preferable to stop the reaction before a precipitate begins to form, by removing the reaction mixture from the heat source and allowing it to cool. This can bypass the aforementioned complications, and simplify the processing and handling of the reaction products.
4.4. Reacting comonomers with low boiling points
A limitation of inverse vulcanisation has been that the high temperature of the reaction requires a comonomer that has relatively low volatility at the reaction temperature.32–37 This prohibits the use of low boiling point comonomers, unless the method is adapted to accommodate them. A solution phase polymerisation with a reflux condenser can be sufficient, where the comonomer has a boiling point that is not lower than the reaction temperature, but where the reaction temperature is significantly higher than the boiling point of the desired comonomer, volatilisation of the comonomer can be too rapid for a successful reaction to take place.75–89 In this situation, there are two ways to perform the reaction.The first method was reported by Tonkin et al. and was developed in order to react cyclopentadiene (CPD) as the comonomer, in an inverse vulcanisation.91 DCPD is one of the most well established comonomers in inverse vulcanisation, but when heated, it is known to undergo a retro Diels–Alder reaction, thereby cracking to form CPD, and it was this comonomer that Tonkin et al. wished to exclusively react (Fig. 10).76,85,91 To do this, CPD was heated in a round bottom flask with a reflux condenser to create CPD vapour (Fig. 10). This vapour was allowed into a second round bottom flask by means of a tube connecting the two. There, the CPD vapour came into contact with molten sulfur, where it reacted to form an inverse vulcanised polymer of CPD. To assist with this process, a small pump was connected to the second round bottom flask, with the inlet suspended in the headspace of the flask, and the outlet immersed in the molten sulfur. This pumped CPD vapour directly into the molten sulfur, facilitating reaction. Overall, the reaction was performed at 140 °C, to ensure that any DCPD that might form from the spontaneous Diels–Alder reaction of CPD in the first flask, would be confined to that first flask, where it would be trapped, as DCPD boils at 170 °C.91 This method directly reacted a gaseous comonomer with molten sulfur to successfully form an inverse vulcanised polymer, and it is not hard to imagine this strategy being adapted to any comonomer with a low boiling point. It is worth remembering that the two flasks need not be kept at the same temperature: the temperature of the first flask should be kept to a temperature sufficient to vaporise the comonomer, but not so high that this process is excessively rapid, while the second flask should be kept to a desired reaction temperature.91
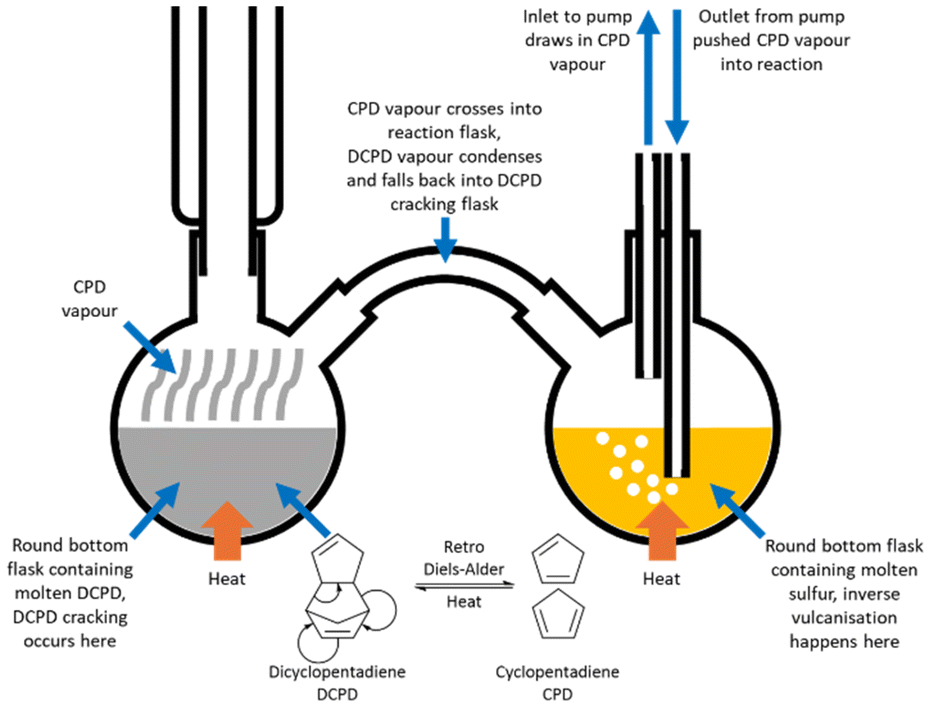 | ||
| Fig. 10 A diagram of the reaction apparatus used to form an inverse vulcanised polymer of cyclopentadiene.91 | ||
The second method that can be used is to employ amine activation.75 It has been shown that amines can initiate inverse vulcanisation by a nucleophilic route; attacking upon the elemental sulfur in some way to generate reactive species from which an inverse vulcanisation reaction can proceed. Crucially, this initiation does not require excessive heat to be made possible, enabling the use of low boiling comonomers. The lowest reported temperature was 70 °C, wherein the elemental sulfur was solid and incapable of generating thiyl radicals, yet the amine activator present was sufficient to drive a reaction.75 This method should be applied to a solution phase inverse vulcanisation, to allow a more homogeneous reaction with solid elemental sulfur, and it should be noted that a relatively large quantity of amine activator may be needed (up to 30% by mass). Tertiary amines are recommended as these activators, with triallylamine (Fig. 11) being a potentially good option, as it doubles as a comonomer itself, being incorporated into the comonomer structure by means of its alkenes.75 To summarise, this amine activation route can be used to access inverse vulcanisation with low boiling point comonomers by using the aforementioned solution phase polymerisation method, with the addition of a tertiary amine into the reaction solution to initiate the elemental sulfur by nucleophilic attack rather than thermally induced homolytic scission of sulfur–sulfur bonds, even when the elemental sulfur is in the solid phase. It is worth noting that in some cases, it may be necessary to perform the initial stage of the reaction at low temperature, to incorporate the low boiling comonomer into oligomers, thereby removing their volatility, and then increase the reaction temperature, in order to drive the reaction to completion.75
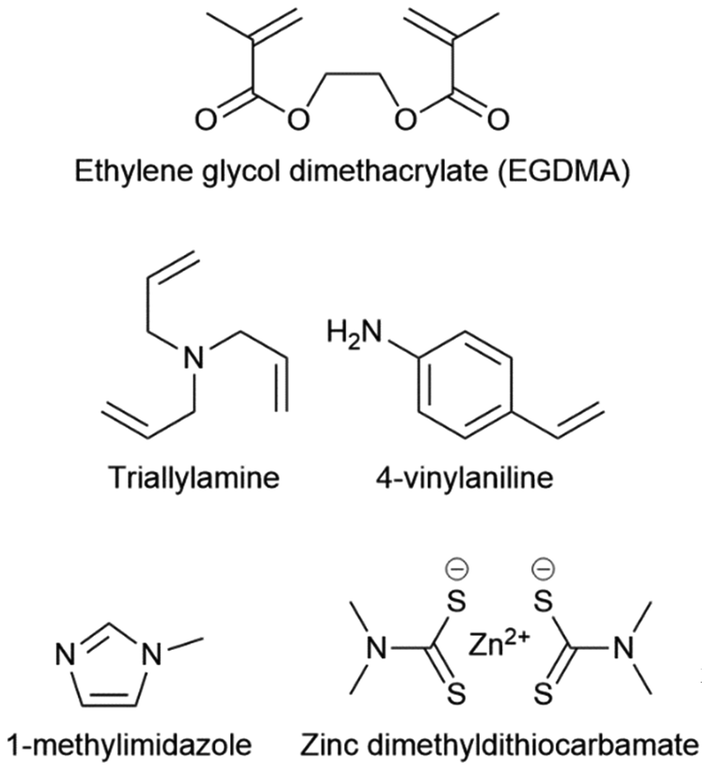 | ||
| Fig. 11 Chemical structures of EGDMA, triallylamine, 4-vinylaniline, 1-methylimidazole, zinc dimethyldithiocarbamate. | ||
4.5. Activation of inverse vulcanisation
Whilst inverse vulcanisation can be remarkable for the production of next generation polymers, it still has drawbacks, such as a relatively high reaction temperature that is energy expensive to maintain, and the production of the toxic by-product hydrogen sulfide.1,66,86 Activation was shown to remedy these drawbacks to a good extent while introducing advantages such as shorter reaction times, increased yields, and increased Tg's.66,86 It also permits lower reaction temperatures, allowing the use of lower boiling point comonomers. Finally, activation allows comonomers that are normally unreactive in inverse vulcanisation, to successfully form inverse vulcanised polymers, with a prime example being EGDMA (Fig. 11).66,86 Electron deficient alkenes are typically unreactive in inverse vulcanisation, perhaps because they do not have sufficient electron density to incite attack from a thiyl radical, however activation introduces new mechanistic routes to the polymerisation which can incorporate such alkenes, thus opening up the highly relevant acrylate and methacrylate families of comonomers.66,86 It is recommended that if a new comonomer is reluctant to polymerise in inverse vulcanisation, activation should be attempted as a simple and easy first solution.There are two main types of activators that can be used in inverse vulcanisation: metal dialkyldithiocarbamates (MDADC's) and amine activators.35,66,86 Because MDADC's are catalysts in conventional vulcanisation, they are often referred to as catalysts in inverse vulcanisation, however, there is no conclusive proof that MDADC's are catalytically regenerated in inverse vulcanisations, so in herein they will be referred to as activators.66,86 Amines and MDADC's can be used in both bulk and solution phase inverse vulcanisations by simply adding them to the elemental sulfur in catalytic quantities (a 1% mass loading is typical). For MDADC's and solid amines it is typical to add them directly to the solid elemental sulfur and then heat to thermal equilibrium, whereas liquid amines are usually mixed with the comonomer and added alongside it.35,66,86 Zinc dimethyldithiocarbamate (Fig. 11) is recommended as an MDADC activator, on account of its good rate enhancements, exceptional cost effectiveness, and its tolerance of many different types of comonomer.66 Typical amine activators include triallylamine, trialkylamines, 4-vinylaniline, and N-methylimidazole (Fig. 11).35,66,75 It should be remembered that excessively rapidly reaction rates in inverse vulcanisation can lead to an auto-acceleration, so activation should be applied with care and consideration: already rapid reactions should not be activated, and the mass loading of the activator should be carefully controlled.35,66,75,86 Activation can also lead to the production of more by-products, like H2S predominantly with amines, and CS2 as a degradation product of MDADC's, so due care and consideration should be given to the impacts of this consequence.54,60
4.6. Comonomer blends
Blending two comonomers in an inverse vulcanisation reaction can be a useful strategy to modify the resultant inverse vulcanised polymer and tune its properties, such as the Tg, molecular weight, solubility, mechanical properties, and colour, all by controlling the ratio of the comonomers.76 An inverse vulcanised polymer of limonene is normally not shape persistent and is of low molecular weight, but was shown to have its properties enhanced when a small amount of DCPD was mixed with limonene and then introduced to molten sulfur. The molecular weight, Tg, and shape persistency were all increased, with the Tg increasing linearly with increasing DCPD content in the blend. This relationship was found to extend to blends of DCPD with other comonomers and multiple weight percentages of sulfur. When DCPD was blended with canola oil to form an inverse vulcanised terpolymer, there was a decrease in compression modulus following a logarithmic trend. Incredibly, inverse vulcanisation of the normally unreactive EGDMA was induced by blending it with DCPD. Finally, terpinolene alone forms a transparent but non-shape persistent polymer when inverse vulcanised alone, rendering it inconvenient for applications, but when DCPD was blended in alongside it, the resultant polymer was shape persistent, but became increasingly opaque with higher proportions of DCPD in the blend.76As such, this blending method can be a convenient way to increase the Tg, molecular weight, shape persistency, and decrease solubility, all primarily through an increase in crosslink density.76 It is simple to apply, requiring only that two comonomers be mixed together without reacting with one another, and then be inverse vulcanised, though this method does come with complications. The most obvious is the increase in complexity of the system, moving from a copolymer to a terpolymer: the ratio of the two comonomers must be considered in relation to the ratio of sulfur, and this creates dramatically more combinations that can be difficult to explore. It can also make characterisation of the resultant polymer notably more complicated. Thus far DCPD has been the flagship comonomer to use with others in terpolymeric blends, but there is no reason that other blends could not also work, and while this creates many possibilities, it also creates a vast number of research avenues that could be very time consuming to explore. There are also practical complications that must be considered. Ideally, the two comonomers should be miscible to minimise the risk of inhomogeneity in the resultant polymer. Inhomogeneity can also arise if one comonomer reacts dramatically faster than the other, leading to its complete consumption before the other.76
4.7. Safety considerations
As with any chemical reaction, safety is paramount when performing inverse vulcanisation. Within inverse vulcanisation, there are two safety considerations that are not present for typical reactions: formation of toxic hydrogen sulfide gas, and auto-acceleration of the reaction which can pose a thermal or pressure explosion hazard, and can liberate hydrogen sulfide gas.35,54,60,62,66,86Reasonable safety measures to address hydrogen sulfide formation can include: using the lowest possible reaction temperature; installing hydrogen sulfide detectors in the premises; where possible, use of comonomers known to produce little hydrogen sulfide; application of activated inverse vulcanisation; out-gassing the reaction through an aqueous hydroxide bath to scrub hydrogen sulfide fumes; and always performing inverse vulcanisations in a high velocity fume hood or better.
Reasonable safety measures to address the auto-acceleration risk include: using the lowest possible reaction temperature; using a solvent to spread heat production over a larger area; always rapidly stirring the reaction; use of an aluminium heating block to dump excess heat into; applying activated inverse vulcanisation only to slow reacting comonomers; where possible, using low reactivity comonomers known to be at low risk of auto-acceleration; performing the reaction at small scale, and scaling up in small increments (auto-acceleration is more likely and more hazardous at large scales); and the use of blast shields and bunds for larger reaction scales.
5. Alternative synthetic routes
Since the seminal publication of inverse vulcanisation, several related polymerisations that give products strongly resembling inverse vulcanised polymers have been noted. These offshoots will be briefly discussed here as they come with their own unique advantages and disadvantages that broaden the chemical toolkit. Note that although these reactions produce polymers very similar to, or even indistinguishable from inverse vulcanised polymers, they are not produced by what is typically deemed to be the inverse vulcanisation route, that being a thermal copolymerisation with elemental sulfur.1 As such, the following polymeric products will be referred to as pseudo inverse vulcanised polymers.5.1. Synthesis by aqueous polysulfide anions
Polymer nanoparticles were synthesized in a route that avoided aggressive heating and the use of thiyl radicals, in a synthesis conducted entirely in water, which is impressive because in a standard inverse vulcanisation both the sulfur and the organic comonomer are hydrophobic.92 Sodium sulfide was added to elemental sulfur in water, which caused the elemental sulfur to react with the sodium sulfide, forming anion capped sulfur chains: polysulfide anions (Fig. 12a). These anions could be of controlled length by simply controlling the ratio of elemental sulfur to sodium sulfide, though whether this control translated to a controlled sulfur rank (the number of sulfur atoms in a chain within the polymeric structure) in the product polymers that were obtained when a divinylic comonomer was added to the solution, is uncertain. Added to the polysulfide anion solution, was divinylsulfone (Fig. 12a), which has electron deficient alkene groups due to the electron withdrawing sulfone group, and though divinylsulfone has not been tested in a classical inverse vulcanisation, it polymerised within minutes as the negatively charged polysulfide anions were able to easily react with the electron deficient alkenes (Fig. 12a). With a steric stabiliser and appropriate surfactant, this synthesis yielded spherical polymer nanoparticles which would disperse in aqueous media. This pseudo inverse vulcanised polymer was made from elemental sulfur and an organic comonomer containing two double bonds (albeit electron deficient alkene bonds rather than electron rich) giving a structure analogous to that of an inverse vulcanised polymer, but despite the similarity, this pseudo inverse vulcanised polymer was synthesized under conditions that would be considered alien to inverse vulcanisation: near room temperature synthesis; aqueous media; and anions over radicals.92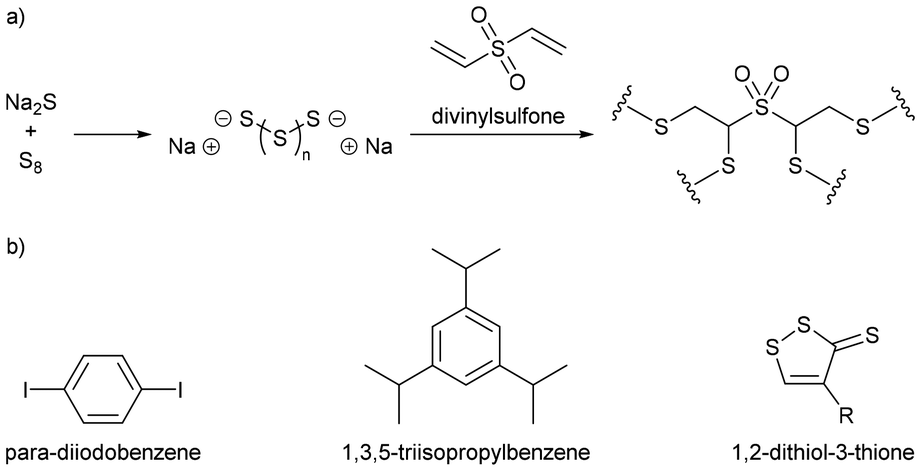 | ||
| Fig. 12 (a) Reaction scheme for forming a pseudo inverse vulcanised polymer by aqueous anionic polymerisation of divinylsulfone and anionic capped sulfur chains, and (b) chemical structures of para-diiodobenzene, 1,3,5-triisopropylbenzene, and a 1,2-dithiol-3-thione ring.92 | ||
5.2. Synthesis from a non-olefinic comonomer
Another reaction that strongly resembled inverse vulcanisation in terms of the products, but started from unusual reagents, was the use of para-diiodobenzene (Fig. 12b), which does not contain any olefinic moieties, yet still reacted with elemental sulfur, and even more surprisingly, formed a crosslinked network, where only a linear polymer would be expected since para-diiodobenzene contains only two functional groups.93 This crosslinking was attributed to the reaction of aromatic hydrogens at the high reaction temperature of 230 °C, with attack upon the hydrogen assisted by the electronic effects of the iodine atoms upon the benzene ring. Active vacuum was required in the synthesis to draw off the elemental iodine by-product, though sometimes this was not sufficient to satisfactorily purify the product polymers of iodine, and Soxhlet extraction was employed. The resultant materials ranged between 33% and 76% by mass sulfur, as determined by combustion microanalysis, which displayed decreasing Tg and decomposition temperature (Td) with increasing sulfur content. These materials could achieve a combination of high extensibility of 300%, and complete strain performance recovery within 2 hours at room temperature. The materials were also scratch-healable under ultraviolet or thermal treatment, and had refractive indices higher than 1.8.93Another example of where a non-olefinic monomer was applied, was the polymerisation of 1,3,5-triisopropylbenzene (Fig. 12b).94 That is, an organic comonomer with only alkyl groups as functionalities. Reactions were performed by normal bulk polymerisation inverse vulcanisation methods, with the rather forcing temperature of 180 °C, and a full 24 hours to achieve a homogenous polymer, with sulfur percentages between 50 and 90 percent by mass. The polymers showed some solubility, suggesting highly branched rather than fully crosslinked polymers, but with the exception of the polymer made from 90% by mass sulfur, all the polymers showed no elemental crystalline sulfur which is evidence towards consumption of sulfur in the reaction. The polymers showed increasing Tg with increasing sulfur content, which is explained by remembering that it is more difficult for the isopropyl groups to react and crosslink than a normal alkene bond in classical inverse, and thus, more sulfur leads to more crosslinking in these reactions, giving a more crosslinked structure that has a higher Tg. A disadvantage to this pseudo inverse vulcanisation is the formation of hydrogen sulfide. In classical inverse vulcanisation, hydrogen sulfide evolution is hypothesized to occur as a by-product of a minor competing pathway, which is hydrogen abstraction by thiyl radicals to give thiols, which can go on to form hydrogen sulfide gas. In this pseudo inverse vulcanisation, hydrogen abstraction would be the main pathway, where instead of attacking a double bond (because none are present) thiyl radicals abstract a hydrogen from 1,3,5-triisopropylbenzene's isopropyl groups, resulting in a carbon radical that can attack on sulfur chains to form an inverse vulcanised polymer network. The evidence of this hydrogen abstraction was obvious in the combustion microanalysis data as the sulfur to carbon and hydrogen to carbon ratios were lower than that of the combined input reagents, indicating that hydrogen sulfide had formed. NMR revealed that all hydrogens of the isopropyl groups could be abstracted: both the tertiary hydrogen which results in a resonance stabilised radical, and the six primary hydrogens which are more abundant so are kinetically easier to abstract but generate a less stable radical. Further evidence of hydrogen abstraction came in the form of thiol signals in the NMR, suggesting polysulfide chains capped with thiols were present in the structure. What is even more interesting is that the NMR detected the presence of 1,2-dithiol-3-thione rings (DT rings) (Fig. 12b), bound to the aryl group at the 4 position, an important reaction by-product that will be discussed in detail later. As a closing remark, evidence of oxidation by means of S–O bond formation was found.94
5.3. Mechanochemical synthesis
A mechanochemical synthesis route was employed to generate pseudo inverse vulcanised polymers by means of ball milling the reagents for the relatively short time of 3 hours.95 This provided several advantages, including: reactions at room temperature; reduced H2S by-product production; avoiding the Trommsdorff–Norrish effect; and a broader monomer range that is not limited by their boiling point. Ten organic comonomers were studied, eight of which were previously reported in inverse vulcanisation, and two never before reported in inverse vulcanisation on account of their prohibitively low boiling points. All ten successfully formed polymers when reacted by mechanochemical synthesis in a ball mill, as indicated by the appearance of a Tg in their DSC thermograms. PXRD revealed that in some cases crystalline elemental sulfur was present in the product polymers, but when Soxhlet extraction was employed to remove the impurity, the exposure to heat caused changes to the polymers by means of thermally induced reaction; undesirable as a purely mechanochemically synthesized polymer was the target. Excitingly, it was demonstrated that these mechanochemically synthesized polymers were effective at removing aqueous mercury from solution, were healable by both application of heat or ultra-violet light, and had superior mechanical properties. Combustion microanalysis revealed decreases in the carbon to hydrogen ratios and the carbon to sulfur ratios. Normally an increase in these ratios is expected, as generation of hydrogen sulfide by-product removes hydrogen and sulfur atoms from the system. Energy dispersive spectroscopy detected an iron content in the polymers (up to 20% by mass indicated by ICP-OES) with traces of chromium, both elements found in the stainless steel container and balls of the ball mill used in the synthesis, and it was hypothesized that chemical binding of the polymer to these elements was the explanation of the raised crosslink density that provided the insolubility, and the decreases in the aforementioned elemental ratios. Evidence of C–O, CO, S–O, and SO bonds was also found.95
5.4. Photochemical synthesis
Jia et al. quoted many of the same limitations of thermally induced inverse vulcanisation as was mentioned in the mechanochemical synthesis publication, but provided a different remedy to these problems, by the discovery of a method for photoinduced inverse vulcanisation using light of either 380 nm or 435 nm (10 W power) at room temperature, which was used to polymerise a variety of previously established crosslinkers as well as new ones like examples of alkynes which have previously received little research attention.95,96 Where photoinduced polymerisation was applied to a crosslinker that had been previously reported to polymerise by thermal means, the products of thermally induced and photochemically induced polymerisation had comparable characterisations by several methods, suggesting the products were of similar nature. PXRD indicated that there was no crystalline elemental sulfur in the product polymers of photoinduced polymerisation, and white lead(II) acetate did not turn into black lead(II) sulfide when left in the presence of an in-progress photoinduced polymerisation, indicating that no hydrogen sulfide gas evolved. Another advantage of the photoinduced reaction route is that thermal polymerisations usually require a curing period, where a polymer is left in a high temperature environment for an extended period of time in order to complete the reaction: this step is not necessary and provides no benefit for a photopolymerised product. The photoinduced reaction route was even capable of reacting gaseous crosslinkers with elemental sulfur. EGDMA, which does not react well in thermally induced inverse vulcanisation, required a photosensitizer or dye to achieve photochemical reaction.96 Several observations about the mechanism were made, which will not be recounted here. Computational chemistry was also employed to further investigate the mechanistic pathway, and aligns well with another publication detailing a computational study of the photoexcitation pathways of elemental sulfur, titled: “ring-to-chain structural relaxation of elemental sulfur upon photoexcitation” which is recommended reading.96,975.5. Electrochemical synthesis
Another alternative synthetic route to inverse vulcanised polymers is an electrochemical synthesis, wherein reducing potentials induce ring opening polymerisation in specially synthesized cyclic trisulfides.98 Inverse vulcanisation by thermal induction suffers problems such as: high temperatures increasing the cost of the reaction; immiscibility between the sulfur and organic reactant phases leading to inhomogeneous products; the serious scale-up hazard of the Trommsdorff–Norrish effect; liberation of toxic H2S gas as a by-product; limited control over sulfur rank; and hard to control C–S stereochemistry. The electrochemical synthesis route circumvents many of these issues. The first stage of the electrochemical synthesis was to synthesize specialised monomers by a literature method of reacting elemental sulfur with an alkene bond in DMF at 120 °C for 16 hours in the presence of a [Ni(NH3)6]Cl2 catalyst (Fig. 13). One disadvantage to the electrochemical synthesis is the fact that a synthetic step is required prior to the polymerisation reaction, which requires prolonged heating at temperatures somewhat lower than inverse vulcanisation, but for a similar timescale, which lessens the advantage of the electrochemical polymerisation itself requiring no heating. This also complicates the synthetic procedure, which is one major advantage of thermal inverse vulcanisation: it merely requires mixing and heating of reagents.1,98 Chronoamperometry of these products at reducing potentials caused a new product to precipitate.98 It was hypothesized that under reducing conditions, the cyclic trisulfides formed polymers by means of electrochemically induced ring opening polymerisation, for which significant evidence was found to support (Fig. 13). It was also found that the polymerisation does not need an inert atmosphere, and that the polymer product becomes increasingly insoluble as the chain grows and the molecular weight increases until it reaches a critical value where the product precipitates, and this limits the molecular weight but also keeps it consistent and protects the polymer product itself from a destructive reduction at the electrode from which it moves away from upon precipitation. The polymerisation kinetics were studied by taking aliquots of the reaction solution and analysing them by gel permeation chromatography (GPC) and 1H NMR. From this study it was found that: the reaction proceeds by pseudo first order kinetics and the molecular weight barely changes over the course of two hours, which was explained by the polymerisation itself being too rapid to see on the minutes timescale, rapidly resulting in fully polymerised products, and that the kinetics instead was probing the rate of formation of polymer chains rather than the rate of growth of the chains. The mechanism of the polymerisation was studied by DFT, with several profound conclusions, including that the reaction has a “self-correcting” mechanism that ensures the ring opening polymerisation results only in a poly-trisulfide: no disulfide or tetrasulfide linkages form from the trisulfide monomer. After the mechanistic study, it was considered that because the formation of S–S bonds should be reversible regardless of whether they are formed via electrochemical polymerisation or thermal polymerisation, if the electrochemically synthesized polymers were heated, then the S–S bonds should reverse in their formation and backbite to yield the cyclic trisulfide precursors, and so the electrochemically synthesized polymer was vacuum distilled with heating to shift the equilibrium towards the monomers, and successfully depolymerised the polymer to its cyclic trisulfide monomer with a yield of 72%, which could then be recycled into another electrochemical synthesis to form the polymer again (Fig. 13). The publication was concluded by comparing the electrochemical route to the thermal route of synthesis, pointing out that it is a good complementary technique to work alongside thermally induced inverse vulcanisation, but one that will not necessarily replace it. While the electrochemical synthesis remedies many issues with the thermal synthesis, it has some drawbacks and caveats in that: it produces only low molecular weight linear polymers, though these can be crosslinked by other means; it requires an initial chemical synthesis to acquire a monomer capable of undergoing the electrochemical synthesis; and that electrochemical polymerisations can sometimes have practical issues with scale up.98 | ||
| Fig. 13 Reaction scheme for forming a pseudo inverse vulcanised polymer by electrochemical synthesis.98 | ||
5.6. Barriers to using elemental selenium
Use of elemental selenium in place of elemental sulfur seems a next logical step in inverse vulcanisation, as selenium could offer higher refractive indices to due to its higher polarizability.46 Selenium would also offer the advantage of NMR analysis as sulfur does not have NMR active isotopes for which spectrometers are easily accessible, and these isotopes are heavily quadrupolar.46 However in the ten years since inverse vulcanisation's discovery, no successful attempts to use elemental selenium in place of elemental sulfur have been reported. This is likely due to the fact that selenium is harder to work with, with an inconveniently high melting temperature of 221 °C (31 °C higher than the temperature where sulfur forms a polymeric solid), with multiple accessible allotropes with raising temperature, each with very different properties, resulting in poorly defined products.46 Selenium also lacks parallel chemistry with sulfur, and so it may be wrong to assume that an analogue of inverse vulcanisation with selenium is possible. Finally, for sulfur inverse vulcanisation, hydrogen sulfide gas can be formed as a by-product, which is toxic (comparable to carbon monoxide) and offensive to the sense of smell, by the description of the odour of rotten eggs, though an added risk is that at hydrogen sulfide levels that are verging on toxicity, humans become nose-blind to the stench.99 Hydrogen sulfide's selenium analogue is gaseous hydrogen selenide, which is vastly more toxic and even more repulsive to olfactory systems.100 As such it may be unacceptably hazardous to directly substitute elemental selenium into an inverse vulcanisation type reaction. The aforementioned publications focused on infra-red optical applications of inverse vulcanised polymers provides to incorporate selenium atoms into an inverse vulcanised polymers.47,486. Characterisation methods
This section will discuss the common characterisation methods that are applied to inverse vulcanised polymers, as well as considering their advantages, drawbacks, and common interpretations. Discussions of how these techniques work will not be given here as they cannot be covered in meaningful detail, and plentiful textbooks exist on the matter. Rather, this section is to orientate the reader on the analyses they may need to perform on their inverse vulcanised polymers.6.1. Differential scanning calorimetry
DSC is a core analysis technique in inverse vulcanisation. Very often, the first action taken upon synthesizing an inverse vulcanised polymer will be to subject it to DSC.32–37,75–89 It is a convenient method with very few requirements of the sample, other than it must be fit into a sample pan, and not liberate excessive quantities of gas within the heating range.101 DSC has two primary uses in the field of inverse vulcanisation: confirmation that a polymeric structure has formed by identifying the Tg; and indicating whether any unreacted crystalline elemental sulfur remains within the polymer by identifying the elemental sulfur's melting peaks or cold crystallisation peaks.32–37,75–89 Beyond this, DSC can be used to compare the degree of crosslinking and molecular weighty qualitatively between inverse vulcanised polymers.32–37,75–89 An excellent resource for a comprehensive understanding of DSC is given in the book: “the handbook of differential scanning calorimetry”.101 An example of a DSC thermogram and its main features is shown in Fig. 14a. Great care must be taken when performing DSC on inverse vulcanised polymers, as if the sample pan ruptures for any reason, then inverse vulcanised polymer residues will be spread over the DSC cell, which can react with and damage the cell at the high temperatures of the DSC analysis (Fig. 14). | ||
| Fig. 14 (a) An annotated diagram of a DSC thermogram of a typical inverse vulcanised polymer, showing all the major features to expect. Note that this is not the thermogram of a real inverse vulcanised polymer.32–37,75–89,101 (b) A clean DSC cell and lid, made of silver metal. (c) A DSC cell and lid where a sample pan has ruptured, leaking inverse vulcanised polymer into the cell, causing the inverse vulcanised polymer to react with the silver metal to form silver sulfide. This requires the cell to be filed down to remove the silver sulfide, which in turn requires full baseline conditioning and cell calibration. | ||
6.2. Thermo-gravimetric analysis
Thermo-Gravimetric Analysis (TGA) is another relatively convenient technique to analyse inverse vulcanised polymers that should ideally be performed before DSC.32–37,75–89 It requires only that the sample be placed onto a pan, preferably in powdered form. TGA will indicate how much mass is lost as a function of temperature when the inverse vulcanised polymer is heated.102 Usually, a two step loss is seen in TGA for inverse vulcanised polymers (Fig. 15), with the first be associated with the loss of sulfur that was not directly bonded to a carbon (the S–S bond is easier to break than the C–S bond).32–37,75–89 By measuring the percentage mass loss of this first step, the mass content of sulfur not directly bonded to sulfur is measured. Some then use this to calculate the average sulfur rank, though it is worth remembering that this is an inferred sulfur rank rather than a directly measured sulfur rank, and it requires several assumptions.32–37,75–89 The second mass loss step is usually attributed to decomposition of the organic component of the inverse vulcanised polymer, leaving behind a final stable mass, known as the char mass, the carbonised remains of the polymer.32–37,75–89,102 It is important to perform TGA before DSC because TGA will identify the temperature where the polymer will begin to emit gas, which is something that should be avoided in DSC. In general, it is advised to set the maximum temperature of a DSC experiment to no higher than thirty degrees Celsius less than the first measured decomposition temperature from TGA.32–37,75–89,102 As a final note, TGA instruments tend to require constant maintenance when they are used to regularly analyse inverse vulcanised polymers, as the sulfurous fumes given off during analysis can be corrosive and detrimental to the instrument.32–37,75–89,102 The TGA exhaust should also be equipped with a sodium hydroxide bubbler to prevent harmful emissions of sulfurous gasses.50,60,62 | ||
| Fig. 15 An annotated diagram of a TGA thermogram of a typical inverse vulcanised polymer, showing all the major features to expect. Note that this is not the thermogram of a real inverse vulcanised polymer.32–37,75–90,102 | ||
6.3. Powder X-Ray diffraction
PXRD is often used as a complementary analysis technique to DSC in the characterisation of inverse vulcanised polymers.32–37,75–89 Inverse vulcanised polymers have, to date, always been observed to take on amorphous structures, resulting in no sharply defined scattering peaks in a PXRD resulting from the polymer itself. Any well-defined scatter peaks are attributed to, and thus used to detect, unreacted crystalline elemental sulfur.32–37,75–89 Note that it has been shown that exposure to X-Rays degrades inverse vulcanised polymers, so care should be taken to minimise the exposure time and intensity in PXRD.756.4. Fourier transform infra-red spectroscopy
Fourier transform infra-red spectroscopy (FTIR) is a mainstay in the field of chemistry and materials research, but it is of limited use in inverse vulcanised polymer analysis.32–37,75–89 Inverse vulcanised polymers may see applications in infra-red optics because of their low infra-red absorbance, so it should come as no surprise that their FTIR spectra show very little.32–37,43–50,75–89 FTIR can be used to identify characteristic peaks for the organic components, indicating whether they have remained intact during the reaction, and it can also be used to detect carbon–carbon double bonds that have not reacted, thus giving an extent of reaction.32–37,75–89 Nevertheless, FTIR is easy and non-destructive to perform, and should be used for the analysis of inverse vulcanised polymers as it does yield some useful information.32–37,75–896.5. Combustion microanalysis
Combustion microanalysis can give estimates of the sulfur content of inverse vulcanised polymers, which is useful information to know, rather than simply inferring the sulfur content from the amount input to the synthesis.32–37,75–89 Another application of combustion microanalysis is to find evidence of hydrogen abstraction and hydrogen sulfide formation in inverse vulcanised polymers. If the ratio of hydrogen to carbon is lower than what is expected, and the ratio of sulfur to carbon is lower than what is expected, then this implies hydrogen and sulfur have been lost as hydrogen sulfide from the polymer.32–37,75–89Unfortunately, combustion microanalysis struggles to accurately determine the sulfur content of inverse vulcanised polymers due to a large extrapolation error in the calibration curve of the instrument (Fig. 16).75,103 Typically, combustion microanalysis instruments combust a sample completely in oxygen, and then perform gas chromatography on the gaseous products. The gas chromatography signal intensities can be related to the elemental content of the sample through the use of a calibration curve, which is constructed by performing combustion microanalysis on samples of known elemental content and relating these elemental contents to the signal intensities provided by the gas chromatography. This requires calibration standards of precisely known elemental contents, and unfortunately in the case of sulfur, there are no suitable and easily obtainable calibration standards with precisely known sulfur contents higher than around one to ten percent.75,103 This results in tremendous extrapolation errors when a calibration curve set up between one and ten percent sulfur content, is used to analyse inverse vulcanised polymers of sulfur contents that are typically between fifty and ninety percent.32–37,75–89 Additionally, it is worth noting that analysis of inverse vulcanised polymers can be unacceptably detrimental to the instrument.75,103
 | ||
| Fig. 16 Illustration of the extrapolation error that occurs in combustion microanalysis when measuring inverse vulcanised polymers.32–37,75–89,103 | ||
6.6. Chromatography
There are several chromatographic methods for the analysis of inverse vulcanised polymers.32–37,50,60,62,75–89 GPC and size exclusion chromatography (SEC) can be used for the qualitative determination of the molecular weight of inverse vulcanised polymers provided they can be solubilised in an appropriate solvent, which is often prohibitive as inverse vulcanised polymers are often insoluble.30–37,50,60,62,75–89 Where GPC and SEC are possible, it is important to remember that the analysis should not be taken as accurate measure of the molecular weight. GPC and SEC rely on the analyte being of similar molecular nature to the standard of known molecular weight.104,105 This standard is often polystyrene with precisely known molecular weight, which leaves the assumption of similarity between the analyte and standard difficult to accept for inverse vulcanised polymers.104,105 Inverse vulcanised polymers are often crosslinked in an unpredictable manner, which already renders them very different to GPC and SEC standards, but inverse vulcanised polymers also contain chains of sulfur which the standards do not.32–37,75–89,104,105 As such, it is best to assume results from GPC and SEC are qualitative.Thin layer chromatography (TLC) and high performance liquid chromatography (HPLC) can be used to extract, detect, and quantify elemental sulfur from inverse vulcanised polymers, regardless of whether that elemental sulfur was crystalline or not.50,60,62 These techniques require that an inverse vulcanised polymer be soaked in an appropriate solvent, such that any elemental sulfur impurities leach out, after which the solvent can be subjected to these chromatographic techniques.50,60,62 This operates on two assumptions: that all of the elemental sulfur is leaching out; and that the solvent is not inducing any degradation or depolymerisation in the polymer.50,60,62
6.7. Less common analyses
NMR can be a powerful analytical technique for discerning the molecular scale structures of inverse vulcanised polymers.106 However, since inverse vulcanised polymers are often insoluble, solution phase NMR is often prohibited.32–37,75–89 Solid state NMR can be used, but it is subject to the availability of such instruments, though where it can be applied it provides useful insights.39 Solution phase NMR can be applied to pre-polymers of inverse vulcanised polymers; aliquots taken when the polymer is not fully reacted and so is still soluble, though the analytical relevance of this is subjective and may not reflect the state of the final product.76 Another way to apply solution phase NMR is to first degrade an inverse vulcanised polymer using lithium aluminium hydride, which destroys the sulfur chains and breaks the polymer down to thiolate capped organic units, which can then be worked up into an NMR sample.84 This technique can be tremendously insightful but work intensive, especially for high throughput synthesis. It also does not work in every case, as some inverse vulcanised polymers can be so crosslinked that they are impervious to lithium aluminium hydride.75Scanning electron microscopy (SEM) can be a useful technique for observing the polymers’ microstructure, where this is desired.57,58 It is commonly applied in the mercury capture application of inverse vulcanised polymers, where particle sizes and porosities have a dramatic effect on performance.57,58 Energy dispersive spectroscopy (EDX), which is a built in functionality to many SEMs, can be a useful complementary technique for the elemental composition analysis of inverse vulcanised polymers.57,58 Nevertheless, because polymer microstructure is not always of interest, it is acceptable not use SEM in every case.57,58
Mass spectrometry can be informative of structural components of inverse vulcanised polymers as well as the sulfur rank, however, to date, there has only been one publication of its use.107 This is indicative of the difficulty of the analysis which is likely why mass spectrometry has not seen wider uptake in the field.107
Raman spectroscopy, where an auto-fluorescent background can be avoided, can yield plentiful information on inverse vulcanised polymers.67 The use of a 1064 nm excitation laser or a Kerr-gated system can suppress or bypass an auto-fluorescent background, allowing clear polymer peaks to be assigned. Unlike FTIR, Raman signals for inverse vulcanised polymers are relatively strong, allowing assignment of peaks for the organic units, the carbon–sulfur bonds, and the sulfur–sulfur bonds. It is possible to quantify the amount of unpolymerized elemental sulfur quickly and easily, regardless of whether it is crystalline or not, and even in very low quantities (approximately 0.5% by mass elemental sulfur content was the detection limit). Raman can be used for reaction tracking, but most importantly, it can quantify the sulfur ranks. Unlike other techniques, 1064 nm Raman spectroscopy can identify the populations of each different sulfur rank, rather than just the average sulfur rank, though this is laborious to do. Qualitative analysis and comparison of sulfur ranks is easy, and this is important to many of the applications of inverse vulcanised polymers. The qualitative analysis can be broadly applied to inverse vulcanised polymers, but the quantitative analysis is still under development and is limited to one polymer system at the current time. Raman analysis can also easily identify the homogeneity of an inverse vulcanised polymer, which is challenging to do by other means. Unfortunately, Ramn instruments that are capable of avoiding auto-fluorescent backgrounds in inverse vulcanised polymers are not widespread.67
7. Post synthetic Modification
Several techniques exist to modify inverse vulcanised polymers towards a desired goal after their initial synthesis. Many of these post-synthetic modifications are driven by the needs of the intended application of the inverse vulcanised polymer, however these techniques could be broadly useful, and will therefore receive some discussion herein.7.1. Friedel–Crafts
Friedel–Crafts chemistry was used to convert a normally unstable polymer, inverse vulcanised styrene, into a crosslinked network.108 A linear polymer of inverse vulcanised styrene was dissolved in chloroform; said solubility owing to the linearity of the polymer. Under nitrogen, aluminium trichloride was added to the chloroform at room temperature, and after 2 hours a suspension had formed. The aluminium trichloride acted as a Lewis acid catalyst, and promoted reaction of the styrene units’ benzene ring with chloroform, thereby alkylating the benzene ring. A further Friedel–Crafts reaction upon the alkylated unit with another styrene unit crosslinks two polymer chains together (Fig. 17). As this crosslinking process continued the polymer eventually became high enough in molecular weight and crosslink density that it was rendered insoluble, resulting in the observed suspension. This polymer was used for the application of water remediation, however this will not be discussed here, as the purpose of reviewing this publication was to point out how the choice of organic comonomer in inverse vulcanisation can open up opportunities to further the modify the product polymer, which in this case yielded a product of radically different properties.108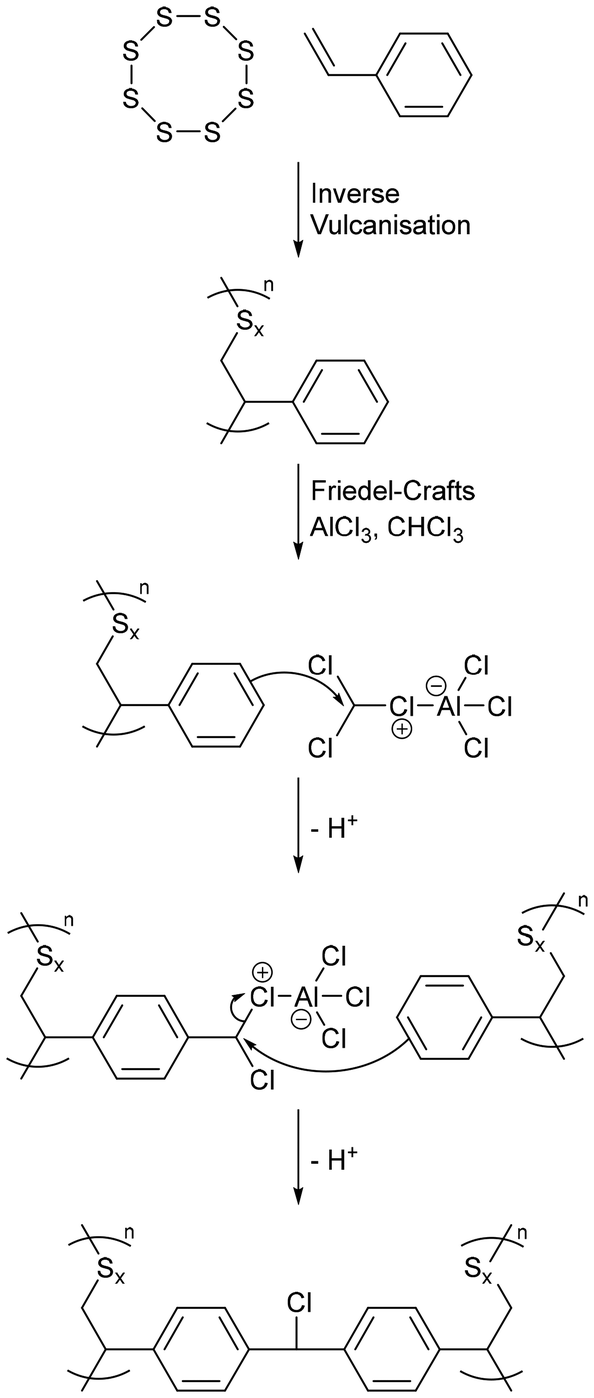 | ||
| Fig. 17 A mechanism for crosslinking a styrene inverse vulcanised linear polymer by Friedel–Crafts alkylation using chloroform and aluminium trichloride.108 | ||
7.2. Super-critical CO2 foaming
The mercury uptake of an inverse vulcanised polymer of DIB was shown to be enhanced by a supercritical CO2 foaming process.57 With appropriate tuning of the conditions, it was found that supercritical CO2 could penetrate well into a DIB inverse vulcanised polymer, and that when the pressure was rapidly released, the CO2 quickly returned to the gas phase and foamed the polymer, introducing pores of sizes in the range of 5 and 100 μm, with most pores being between 10 and 20 μm. This foaming process slightly increased the Tg of the polymers, thought to be the result of the removal of low molecular weight oligomers that acted as plasticisers, by the supercritical CO2 in which they were solubilised and extracted from the polymer matrix. When tested for mercury(II) uptake, it was found that the foamed DIB inverse vulcanised polymer removed approximately 95% of mercury from the solution, whereas the equivalent un-foamed polymer removed only 45%.577.3. Moulding and melt processing
Melt processability in inverse vulcanised polymers is a near ubiquitously useful post synthetic modification. It can be done in two primary ways: producing a low Tg polymer that can be warmed to a liquid like state and reshaped by moulding; or it can be done by pouring a pre-polymer into a pre-heated silicone mould, and then further curing, as discussed earlier in this article.32,76 The latter of these two techniques can form shaped objects from inverse vulcanised polymers that are otherwise too high Tg to be amenable to melt processing. Of note, silicone moulds have been used to create 1 cm3 cubes of inverse vulcanised polymer for anti-bacterial testing, and “dog-bone” samples for mechanical testing, though it is important to remember that the thermal history of a polymer influences its mechanical properties, so mechanical testing can provide different results for two instances of what appears to be the same polymer, because those two instances had different thermal histories.32,53,61,76 A particularly interesting example of moulding an inverse vulcanised polymer is when a sodium chloride porogen was used to introduce porosity with some degree of control.58 Sodium chloride was recrystallised to give controlled crystal sizes, after which, the porogen was heated in an oven. Meanwhile, an inverse vulcanised polymer was synthesized until the reaction mixture became monophasic, at which point it was poured onto the hot sodium chloride porogen. The polymer-porogen mix was returned to the oven, allowing the polymer to penetrate into the porogen and finish reacting to form a salt templated, highly crosslinked network polymer. The sodium chloride was then removed by Soxhlet extraction, and the porous polymer was tested for mercury uptake. It was found that this process was amenable to a variety of different organic crosslinkers although some failed to penetrate into the porogen well due to high viscosity or resulted in polymers with too poor mechanical strength to maintain the templated structure.587.4. Coating
Inverse vulcanised polymers can be coated onto support materials like silica to increase their surface area, ideal for mercury uptake applications.86 The process is simply done by dissolving an inverse vulcanised polymer or a soluble pre-polymer of an inverse vulcanised polymer, then mixing the solution with a desired support, and then subjecting the solution to rotary evaporation to dryness.86 Further drying by vacuum oven can be beneficial, as well as curing, if a pre-polymer was used. Curing can also prevent washing of the polymer off the support, if the composite material is to be exposed to potential solvents.86 An alternative way to coat inverse vulcanised polymers onto supports is by spray coating.109 This entails dissolving an inverse vulcanised polymer and then forcing the solution through a spray nozzle to create an aerosol that is directed onto a surface, leaving the polymer behind once the solvent evaporates.109 This method does however require specialised equipment, making it less common in the laboratory, though spray coating is common in industrial settings.1097.5. Particles by precipitation
Forming particles of materials is a broadly useful technique, and although inverse vulcanised polymers of sufficiently high Tg can be mechanically ground to powder, this technique does not necessarily create controlled particle sizes.66 Where controlled particle sizes are desired, precipitation methods can be employed.110,111In one method, 500 mg of inverse vulcanised polymers of DCPD were dissolved in 10 mL of chloroform. Different volumes of these solutions were dropped into ethanol, forcing precipitation of the polymers as nanoparticles.111 Where too large a volume of polymer solution was added to ethanol, bimodal size distributions were produced, but with lower volumes of polymer solution, more uniform monomodal size distributions were produced. Additionally, smaller nanoparticles were obtained when less polymer solution was used in comparison to the antisolvent. Depending on the formulation, spherical nanoparticles could be obtained, though with out careful consideration, irregular shapes and aggregates can be the result. It was also found that the nanoparticles had higher Tg values than the bulk polymers from which they were made, suggesting that lower molecular weight, lower Tg polymers and oligomers did not precipitate into particles, remained dissolved, and were removed from the population, a conclusion supported by the GPC performed.111
Another publication details two methods to create nanoparticles of inverse vulcanised polymers of geraniol and perillyl alcohol, both of which are soluble in THF and chloroform (Fig. 18).110 Geraniol inverse vulcanised polymers were subjected to the emulsion-solvent evaporation method, by a chloroform in water emulsion stabilised by Tween 80, Brij 80, poly-vinylalcohol or sodium dodecyl sulfate. All of these surfactants were successful at forming stable emulsions, which were left to evaporate and give particles. Brij 80 and sodium dodecyl sulfate gave multimodal particle size distributions and were not studied further, but polyvinyl alcohol and Tween 80 were found to be successful in giving particle sizes of 367.7 nm by dynamic light scattering.110 A nanoprecipitation method was also employed using THF as the solvent for the geraniol inverse vulcanised polymer at a concentration of 5 mg mL−1, and water as the antisolvent in nine times the volume of the THF. Dropping the geraniol solution into the antisolvent with stirring resulted in polymer nanoparticles of diameter 138 nm with monomodal size distributions, as determined by dynamic light scattering. As is the general case, the smallest nanoparticles and the lowest polydispersities were obtained with low polymer concentrations. It was found that inverse vulcanised polymers of perillyl alcohol behaved similarly to those of geraniol in nanoprecipitation, suggesting that the method could have broad applicability.110
 | ||
| Fig. 18 Chemical structures of geraniol and perillyl alcohol. | ||
8. Mechanistic considerations
Despite great research effort, the mechanisms of both conventional vulcanisation and inverse vulcanisation have remained elusive. Conventional vulcanisation has been known for over a century and yet it is only recently that research efforts have begun to prove aspects of its mechanism in any confidence. Inverse vulcanisation has only been known since 2013, and so has not seen such extensive mechanistic studies.1It is however predicted that progress in uncovering the mechanism of inverse vulcanisation will be challenging, due to the difficulty in characterisation of the product polymers and the complicating facet that any sulfur–sulfur bond can undergo scission at any time during the reaction, to break a growing polymer chain in two and generate two new thiyl radicals that are capable of propagation.1,18 Indeed, at the current time there is no mathematical polymerisation model, like the Carother's equation, that has been developed to take into account growing polymer chains that can spontaneously divide and generate new initiator radicals at the same time.112 Nevertheless, what follows is a brief discussion of some of the investigations carried out upon both kinds of vulcanisation, as well as some caveats and potential side-products.
8.1. Conventional vulcanisation
One proposed mechanism for conventional vulcanisation detailed a complex radical mechanism, beginning with homolytic fission of cyclo-sulfur to give a linear diradical, though this mechanism was debated with alternatives proposing ionic mechanisms based on heterolytic cleavage of S8 rings.88 These ionic mechanism theories were substantiated by the fact that vulcanisation reactions were unhindered by the presence of radical acceptors in the reaction, though this could be explained by the fact that any sulfur–sulfur bond is capable of undergoing scission, thus two radical traps would be required for every sulfur–sulfur bond present in order to cause a complete quench of the reaction.113 Attempts to disprove the presence of radicals via a different method: electron paramagnetic resonance, lead to conflicting conclusions, as G. Blokh was not able to detect radicals, but Dondi et al. were, though it may be worth noting that the publication by Dondi et al. is 56 years more modern than Blokh's and was likely performed with technology half a century more advanced.114,115 A more recent study proposes a mechanism that involves both radicals and ions, which may explain the controversy in previous arguments.116 In model reactions, it was found that the lowest temperature that could be used to achieve reaction was 160 °C. This onset temperature was consistent with the temperature found in DSC for homolytic cleavage of cyclo-sulfur. Using FTIR spectroscopy, it was confirmed that the thermal event in the DSC thermogram of sulfur at 159 °C corresponds to homolytic cleavage of cyclo-sulfur, and with a similar method it was possible to identify that the thermal event in the DSC thermogram of sulfur at 230 °C corresponded to heterolytic cleavage of cyclo-sulfur, though this onset temperature shifts to 200 °C in the presence of an activator.116Both temperatures for heterolytic fission of cyclo-sulfur, 230 °C and 200 °C far exceed those of the industrial conditions that are routinely used for the vulcanisation of rubber, so a purely ionic mechanism was therefore ruled out.25 Having confirmed that radicals are involved in the reaction mechanism, it was proposed that vulcanisation proceeds via α-allyl-hydrogen abstraction from a polymer chain by a thiyl radical, as it has been proven that thiyl radicals are capable of this abstraction.114,115 The radicalised polymer can then proceed to attack upon cyclo-sulfur and polymerise. In addition to this, the thiyl radical involved in the initial hydrogen abstraction forms a thiol, which at elevated temperatures (above 160 °C without an activator and above 80 °C with an activator) can cleave to form a thiolate anion. This thiolate anion can also take part in vulcanising the polymer chains and its presence is a possible explanation as to why previous research came to conflicting conclusions in regards to whether the reaction was based on radical or ionic intermediates.25 A possible mechanism, consistent with Lian et al.'s finding is shown in Fig. 19a.
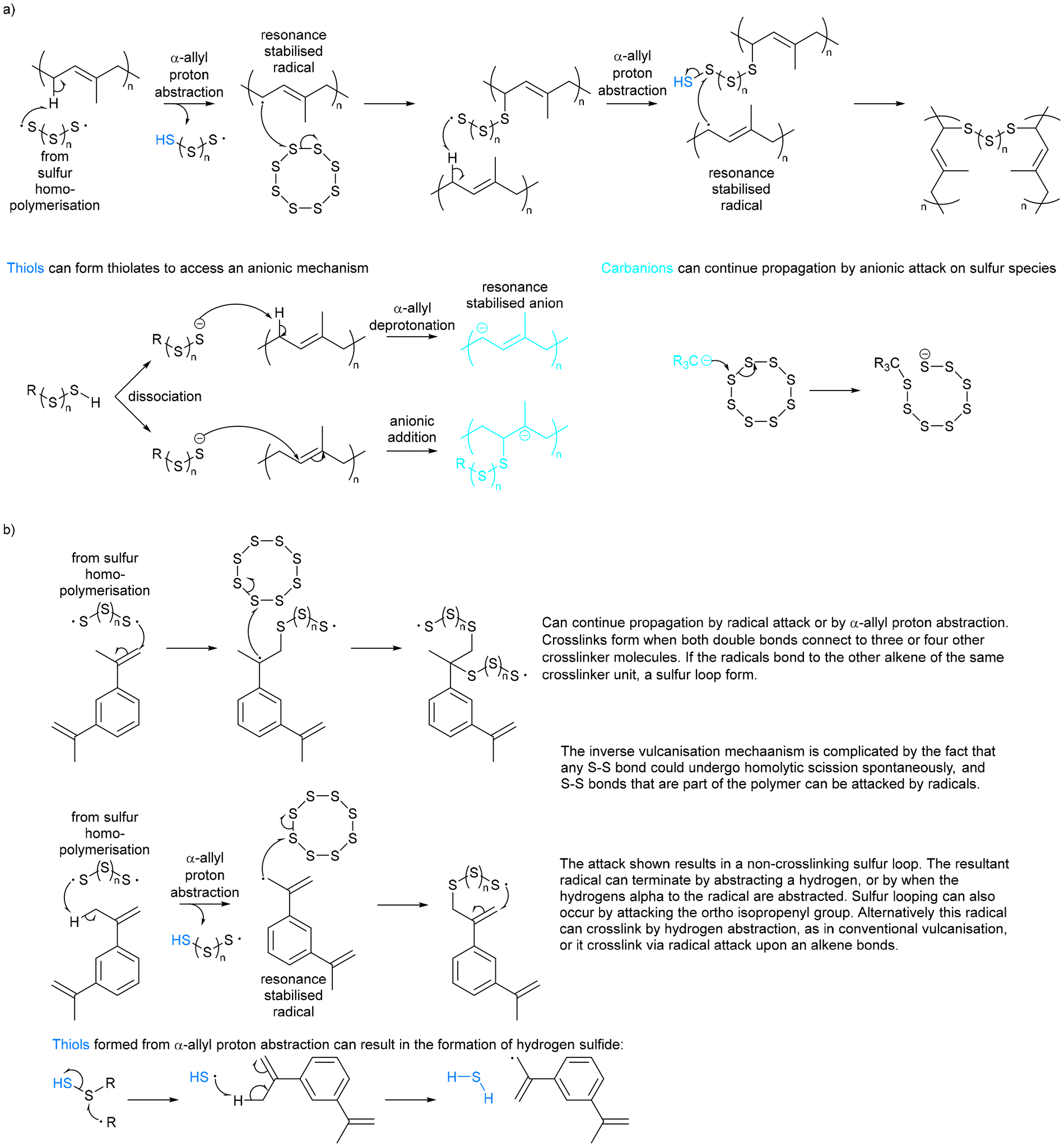 | ||
| Fig. 19 (a) A possible mechanism for the vulcanisation of polyisoprene, and (b) key points of a possible mechanism for the inverse vulcanisation of DIB. | ||
8.2. Inverse vulcanisation
Many theorise that the inverse vulcanisation reaction proceeds via a similar mechanism to conventional vulcanisation. That being said, α-allyl-hydrogen abstraction cannot be the dominant mechanism, because such a hydrogen abstraction leads to hydrogen sulfide as a by-product, and given that because these reactions contain so much sulfur, large amounts of H2S gas would be given off, which is not the case.1 Even further evidence that α-allyl-hydrogen abstraction cannot be the primary mechanism comes in the form of DVB, which is one of the most rapidly polymerising inverse vulcanisation crosslinkers documented, yet it entirely lacks α-allyl-hydrogens.34,65,66,86,117–119 Many current postulates involve the generation of thiyl radicals via homolytic fission of cyclo-sulfur in a manner similar to conventional vulcanisation.59,120 The thiyl radicals then add across the carbon–carbon double bonds of the crosslinker, placing a radical upon the carbon skeleton, which can then continue propagation by attacking on other sulfur rings and polymers.1,120 An alternative to this theory is that the thiyl radicals abstract an α-allyl-proton, thus radicalising the crosslinker molecule.121,122 The latter theory provides an explanation as to why some inverse vulcanisation reactions produce hydrogen sulfide gas, which could form from the thiols formed from hydrogen abstraction. However, not all inverse vulcanisations produce hydrogen sulfide gas, and the production of hydrogen sulfide has been found to vary with temperature, sulfur concentration and crosslinker structure, which points to the conclusion that both radical addition and proton abstraction occur to different extents in different reactions.85 It is also worth noting that if thiols are formed by proton abstraction, then there is the possibility of an anionic mechanism of polymerisation, which utilises thiolates in a similar manner to the mechanism put forward by Lian et al.25 A possible mechanism for inverse vulcanisation is shown in Fig. 19b.25As research progresses, it seems that comparing inverse vulcanisation and conventional vulcanisation mechanistically may not be appropriate, as the two share marked differences. One such difference is that conventional vulcanisation appears not to consume the double bonds involved whereas inverse vulcanisation has been proven beyond reasonable doubt to consume the double bonds of the crosslinker molecules (Fig. 19).1 This points to some measure of fundamental difference between the two reactions that may preclude the idea that the two are mechanistically similar, but unfortunately research projects into the mechanism of inverse vulcanisation carry high risk of being time consuming and ultimately fruitless due to the difficulty in analysing inverse vulcanised polymers. The mechanism of conventional vulcanisation is still debated after one hundred years, and since precise knowledge of the inverse vulcanisation mechanism is not necessary to develop polymers suited to their applications, mechanistic research is sparse.
A recently released publication addresses some of the previous assumptions regarding the mechanism sheds new light on the structure of the original inverse vulcanised polymer of DIB.1,84 The analysis of the inverse vulcanised polymer of DIB was revisited; produced by the original method, from which the previously suggested structure would be crosslinked, though this has been disputed because if the original DIB inverse vulcanised polymers were indeed crosslinked, they should not have shown the solubility that they did.1
Cross polarised magic angle spinning 13C NMR was used to investigate inverse vulcanised polymers of DIB, supported by computationally predicted NMR chemical shifts.84 It was found that the experimental spectra did not completely agree with the predicted chemical shifts: there were only trace amounts of methylene tertiary carbons, which would be expected to common in the original structure, and instead there was twice the intensity of methyl group carbons. Longitudinal relaxation time experiments also supported these conclusions. As such, it was rationalised that crosslinking bis-sulfurated units cannot be the common microstructure in a DIB inverse vulcanised polymers and instead, it was hypothesized that a thiocumyl unit (Fig. 20) was forming as the predominant microstructure; that is, a linear unit, explaining the polymer's solubility. Formation of the usual crosslinking units is not impossible, and did seem to occur, but the overall structure of the polymer seemed closer to that of a branched polymer rather than a fully crosslinked polymer.84
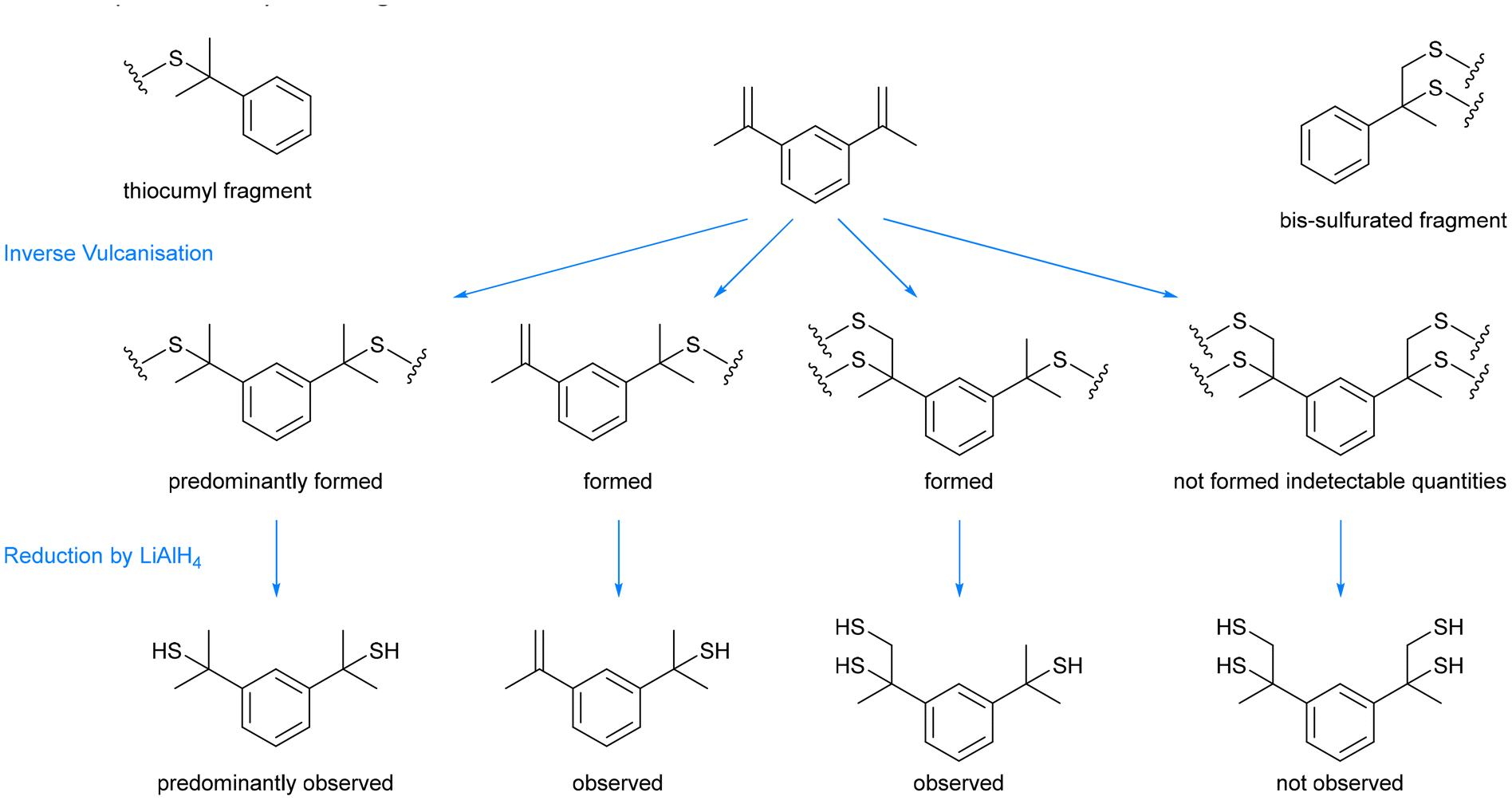 | ||
| Fig. 20 Structures from the study of the mechanism of DIB inverse vulcanised polymer formation.84 | ||
To support these conclusions further, the DIB inverse vulcanised polymers were reduced with LiAlH4 to destroy the sulfur chains and cap the organic units with thiols.84 This yielded a soluble product which was analysed by solution phase NMR, revealing three types of structure in the mixture, which can be seen in Fig. 20. Observed was DIB units with: two thiocumyl thiols, indicative of linear units in the polymer; one thiocumyl thiol and one unreacted double bond, indicative of a terminal units in the polymer; and one thiocumyl thiol with two thiols on the other moiety that would correspond to a bis-sulfurated moiety as expected from the original postulate, indicative of a branching unit in the polymer. What was not detected in a measurable concentration, was reduced units containing the four thiols that would indicate a crosslinking unit. Even still, the branching unit was quite uncommon by comparison to the linear unit, suggesting that DIB inverse vulcanised polymers synthesized in this way are predominantly linear. To help confirm the assignments, chemical syntheses were performed to produce model compounds that mirrored the reductive degradation products of the linear and terminal polymer units.84
Computational chemistry was then employed to assist in finding a mechanism that could lead to the observed products of the thiocumyl fragments.84 The Gibbs free energies of the intermediates and transition states were calculated to find a kinetically viable and thermodynamically feasible pathway. It was found that hydrogen abstraction to yield an allyl radical was the most kinetically demanding pathway, though it was still accessible, and led to a thermodynamically feasible intermediate. More interestingly, it was found that radical attack at the less substituted end of the double bond to yield a primary radical was both less kinetically viable and less thermodynamically feasible than radical attack at the less substituted end of the double bond to yield a tertiary radical, though both pathways are still accessible. This was all surprising, because these reaction pathways would seem to suggest that the originally assumed structure of a bis-sulfurated alkene bond is accessible and expected, but it is not observed experimentally, so some other factor must prevent its formation. Analysis of the tertiary radical revealed that its tendency to undergo elimination depolymerisation is too rapid to allow it to be a viable intermediate in the polymerisation pathway. Thus, formation of the primary radical is the only route towards polymerisation. The calculations showed that termination of the primary radical by hydrogen abstraction is thermodynamically favourable and kinetically easy, provided the hydrogen is abstracted from a H–S bond, though abstraction from an allylic fragment is not kinetically impossible.84
To complete the study, tetrasulfides capped with cumyl groups, and chemically synthesized linear polymers of cumyl tetrasulfides were synthetically prepared in a well-controlled manner.84 The 13C NMR spectra of these well-defined products closely corresponded to those of polymers made from DIB in an inverse vulcanisation method, thereby further confirming the structure. As such, it was concluded that inverse vulcanisation of DIB proceeds by thiyl radical attack upon the alkene bonds of DIB to form primary radicals which terminate by hydrogen abstraction to form a cumyl unit in a linear polymer. These cumyl units are the major component of the DIB inverse vulcanised polymer, and a minor component has branching bis-sulfurated groups.84
Another recent study investigated the structures that formed in inverse vulcanisation.123 Zheng et al. described inverse vulcanisation in terms of three stages: induction, where the reaction mixture is of low viscosity and comprised mainly of oligomers; curing, where alkene groups are further consumed and crosslinking becomes more predominant; and over-cure, where the sample begins to degrade. Note that these stages are determined rheologically, as in conventional vulcanisation. Zheng et al. performed inverse vulcanisations on DVB, DIB, DCPD and soybean oil, and used rheology studies to extract the time periods in which induction, curing, and over-cure occur, however the results are much too extensive to recount here, thus Zheng et al.'s article is recommended further reading.123
Zheng et al. used mono-function alkenyls in inverse vulcanisation and studied the reaction kinetics with NMR.123 DT ring structures were observed (Fig. 21), as well as thiocarbonyls, thiophenes, and carbo(dithioperoxo)thioic acids, indicating the variety of structures that can result from the simple addition of elemental sulfur to a comonomer with heat. Zheng et al. studied the formation of these moieties from different comonomers with different reaction conditions, and ratified their proposals with DFT predicted mechanisms.123
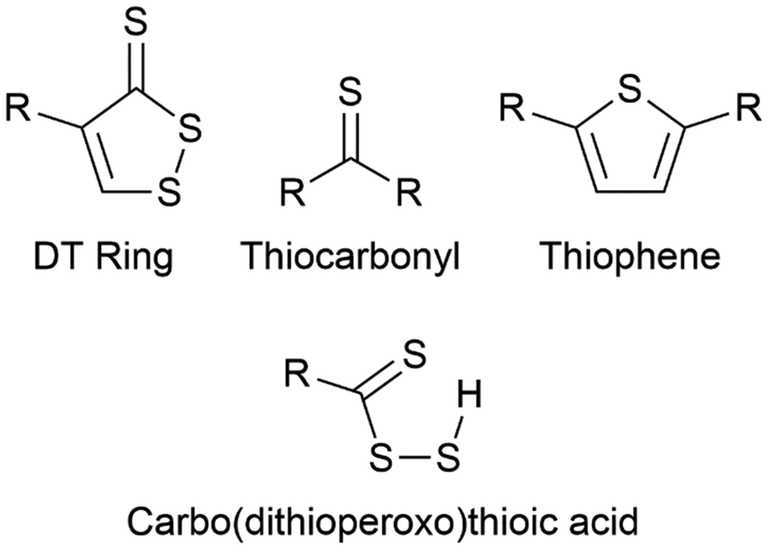 | ||
| Fig. 21 Chemical structures observed by Zheng et al. in different inverse vulcanisations at different times under different conditions.123 | ||
8.3. Thiol–ene and thiol–yne click chemistry
An alternative postulate is that inverse vulcanisation proceeds by a mechanism similar to thiol–ene or thiol–yne click chemistry, though the formation of thiols in inverse vulcanisation has not been proven beyond reasonable doubt.27 Thiol–ene click chemistry is far too vast a field to cover in this article, but numerous reviews exist on the topic.124–126 A basic scheme of thiol–ene click chemistry can be seen in Fig. 22. Typically, thiol–ene reactions occur via a radical process complete with classic initiation, propagation and termination steps, though dependent on the thiol and the alkene it is to be combined with, nucleophilic variants resembling Michael addition are also possible.125 Note that the chain growth propagation by attack of a carbon radical upon an alkene is undesirable, and it is in fact thiol chain transfer that is the desired reaction, to the extent that ideally, chain transfer should occur at a degree of polymerisation of one in thiol–ene click chemistry. That is, one a carbon radical is formed by thiol radical attack upon an alkene, chain transfer should be the next and only reaction, thus terminating what could have otherwise been a polymer chain, to generate a new thiol radical. Generally, for radical thiol–ene click chemistry, bond strain about the alkene bond and increasing electron richness of the alkene bond, promotes reactivity, much in the same way as is observed in inverse vulcanisation: electron poor alkenes like EGDMA do not react well, and the strained norbornene alkene of DCPD reacts at a lower temperature than its cyclopentene alkene.32,76,86 It is also observed that excessive substitution levels about the alkene can also reduce reactivity in thiol–ene click chemistry. | ||
| Fig. 22 Reaction scheme and ba sic mechanism for thiol–ene click chemistry.124–126 | ||
Just as alkynes can be used in inverse vulcanisation, alkynes can be used in thiol–ene click chemistry reactions, though alkynes react cleanly in the latter but not in the former.33,127,128 Thiol–yne click chemistry is much analogous in its mechanism to thiol–ene click chemistry, with the exception that alkynes can react twice sequentially, whereas alkenes react only once with thiols. When thiol–yne click chemistry is capable of forming products such as that shown in Fig. 23, it is not hard to see why a comparison to inverse vulcanisation is so attractive.127 Comparable hyperbranched polymers could also be formed if thiol–ene click chemistry was applied with a thiol and a compound containing two alkene bonds, just like the comonomers of inverse vulcanisation. With that being the case it is attractive to consider they may have similar mechanisms, though it is worth noting that thiol–ene and thiol–yne click chemistry can only form thioether linkages, whereas inverse vulcanisation can form disulfide, trisulfide, and longer linkages.1,67
 | ||
| Fig. 23 Reaction scheme for thiol–yne click chemistry and an illustrative example for how a hyperbranched polymer with resemblance to an inverse vulcanised polymer might form.127,128 | ||
8.4. Organosulfides
Organic sulfide linkages are expected to be a major component of inverse vulcanised polymers’ molecular structure.1,67 As such, a discussion of the general behaviour of organic sulfide compounds is prudent, as much of this behaviour may carry through to inverse vulcanised polymers. Three classes of sulfides will be considered here: thioethers, disulfides, and trisulfides; corresponding to sulfur ranks of one, two, and three, in inverse vulcanised polymers (Fig. 24). Tetrasulfides and pentasulfides do also exist, and are thought to exist in inverse vulcanised polymers, but examples of discrete organic molecular tetrasulfides and longer, that are well characterised in the literature are uncommon (Fig. 24).98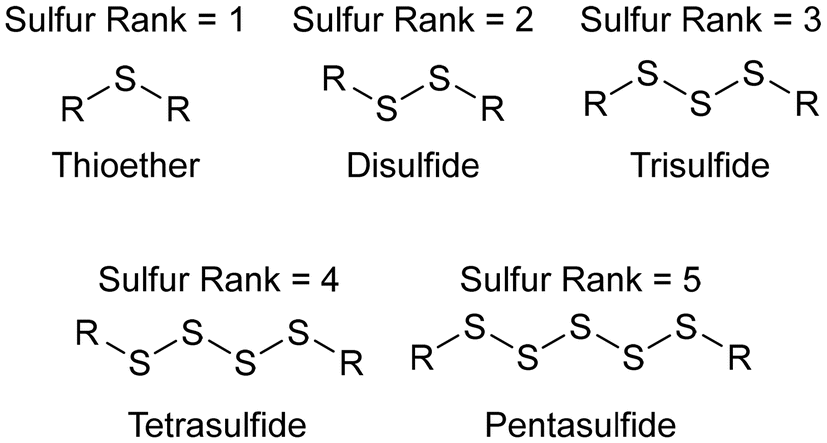 | ||
| Fig. 24 General structures of organosulfides. | ||
Thioethers are the shortest sulfur rank sulfide, and do not contain a sulfur–sulfur bond, thus making them exempt from the dynamic chemistry associated with reversible sulfur–sulfur bond formation that gives inverse vulcanised polymers many of their properties.12 As such they might be considered something of a “dead-end” in inverse vulcanised polymers, due to their irreversibility, but this does not mean they have no important impact upon the structure and behaviour of inverse vulcanised polymers. Because they do not contain a relatively weak sulfur–sulfur bond, they may form the strongest crosslinks in inverse vulcanised polymers, and increase their Tg's. Thioethers can be susceptible to oxidation to sulfones and sulfoxides without particularly forcing conditions.130–133 Many thioethers can oxidise in air, and when heated to inverse vulcanisation temperatures, this reaction may be very feasible.130–133 However, it is important to not that while some sources have reported differences between inverse vulcanisation reactions performed in air and under nitrogen, oxidation in the structures of inverse vulcanised polymers is not widely reported.65,67
Disulfides are most commonly synthesized by oxidation of thiols with relatively gentle oxidisers, so it is not inconceivable that they could form this way in an inverse vulcanisation exposed to air, though this would certainly be a minor process in comparison to the much more dominant scission of longer sulfur chains that is believed to be predominant in inverse vulcanisation.134–136 Disulfides are also readily oxidised in the human body, where the product disulfide bridges connect together two cysteine amino acid units in a protein. Like this, the disulfide bridges can control the shape of proteins by anchoring together two different parts of a protein chain. This is somewhat reminisce of how disulfide bridges form crosslinks in inverse vulcanised polymers, though the contexts are vastly different.137 Disulfides are also prone to oxidation to thiosulfonates and thiosulfinates, though these species can also be formed from thiols under oxidising conditions.129 Disulfides are generally less stable than thioethers on account of the fact that disulfides contain the relatively weak sulfur–sulfur bond. Trisulfides are somewhat more challenging to produce synthetically than disulfides, and as a result, their oxidation has been less explored in the literature, though it is expected that because of their structural similarities, disulfides and trisulfides would share much of the same behaviour in terms of oxidation.138–141 One key difference is that trisulfides have weaker sulfur–sulfur bonds than disulfides, and so are more reactive, as was demonstrated in the literature where it was identified that disulfide linkages in inverse vulcanised polymers have less dynamic chemistry than trisulfides, and that it is the trisulfides that are responsible for the self-healing of inverse vulcanised polymers.78,90
8.5. Dark sulfur, ageing, sulfur bloom, and polymer storage
A relatively recent discovery in the field of inverse vulcanisation is that of dark sulfur: residual sulfur trapped within an inverse vulcanised polymer network but not incorporated as part of that network, which is not crystalline, and so cannot be detected by techniques that rely on crystallinity (i.e. PXRD and DSC).54,60,62,67 This has caused a re-evaluation of whether polymers that were previously assumed to be free of elemental sulfur, do in fact lack this impurity, or whether it was merely undetectable. HPLC and 1064 nm Raman spectroscopy allow for detection and quantification of all residual elemental sulfur within an inverse vulcanised polymer, regardless of whether it is crystalline or not, and so these techniques should be applied where possible and relevant.54,60,62,67Another consideration regarding inverse vulcanised polymers that has largely gone unstudied, is their ageing. It has been shown that some polymers’ properties change with time after their synthesis, particularly their mechanical properties and their Tg; normally the polymers become more brittle and Tg increases with age, as the polymers depolymerise and reduce their average sulfur ranks resulting in shorter, stronger sulfur chains.54,60,62 Factors that accelerate this ageing include being stored in warm environments that are humid. Atmospheric water can penetrate into the polymer network over time, revealing a water peak in DSC that grows as time passes. This water can promote depolymerisation and formation of hydrogen sulfide gas. Different polymers have different tendencies toward these ageing effects: inverse vulcanised polymers that are less heavily crosslinked and contain more sulfur are more prone to ageing by depolymerisation. Some comonomers also give inverse vulcanised polymers that are more prone to depolymerisation, though this is an area warranting more research into structure–property relationships regarding ageing.54,60,62 It has also been shown that inverse vulcanised polymers degrade under UV light, so ideally they should be stored in cold, dry, dark environments, and the ramifications of these factors upon the longevity of inverse vulcanised polymers in practical application settings should be thoroughly explored.54,60,62,67
In relation to the ageing of inverse vulcanised polymers, is the concept of sulfur bloom.54,60,62 When an inverse vulcanised polymer ages, from anything on the minutes timescale due to being unable to stabilise a high sulfur percentage from a high sulfur loading into the reaction, to the months timescale due to storage conditions, depolymerisation leaves elemental sulfur within the polymer.54,60,62 This elemental sulfur, if sufficiently abundant and mobile, can crystallise inside the polymer network, causing sulfur bloom. Sulfur bloom refers to the visual effect that occurs upon this crystallisation: the polymer becomes more opaque; its colour shifts towards yellow; and sulfur crystals may even form on the surface (Fig. 25).54,60,62
 | ||
| Fig. 25 Inverse vulcanised polymers showing visual evidence of the sulfur bloom effect. The polymers on the right show severe sulfur bloom all over the pieces due to being made with excessively high sulfur contents and not being cured. The polymers on the left show moderate sulfur bloom. Note how the sulfur bloom occurs toward the bottom of these polymer cubes, because they were poured into a 1 cm3 mould too early in their reaction, resulting in the more dense sulfur sinking to the bottom of the cube in the absence of stirring. This led to excessively high sulfur concentrations in the bottom of the cube in comparison to the top, which could not be stabilised at the bottom of the cube, resulting in sulfur bloom at the bottom of the cube. | ||
9. Outlook
It may be that inverse vulcanisation has reached a turning point as a field. In its early stages, inverse vulcanisation received a “gold rush” of publications, because of the versatility of the reaction and its capacity to accept a wide variety of comonomers.32–37 This led to numerous publications where research groups would identify a new comonomer, polymerise it, perform analyses, and then publish their findings.32–37 This was valuable in the early stages as it broadened the library of possible comonomers, inherently led to discoveries about the reaction itself, and set a baseline for what properties could be expected from inverse vulcanised polymers. But it may be that inverse vulcanisation should move away from this style of publication, as it is becoming increasingly less valuable: fewer and fewer discoveries can be made through inverse vulcanising every accessible molecule with a carbon–carbon double bond. As a chief example of this issue, there are large numbers of reports detailing the inverse vulcanisation of new kinds of bio-renewable triglyceride oil, all leading to very similar inverse vulcanised polymers, largely because many of these triglyceride oils are in fact distinguished only by containing different ratios of the same or inter-related triglycerides. Instead, it is suggested that the field of inverse vulcanisation should take on a new direction with a two-point approach.The first element of this approach would be to finally derive a comprehensive understanding of what is actually being made when an inverse vulcanised polymer is synthesized. This would require the investigation of new analysis techniques, but also a thorough investigation of how to apply those techniques and what can be gained from them. This task is challenging and likely prone to failures, which is likely why such publications have been less favoured when polymerising new comonomers has a much higher chance of successful research output. But inverse vulcanisation is reaching the stage where such publications are becoming less valuable in terms of what they add to the knowledge pool, and so the increased value of more challenging projects focused upon the analysis of inverse vulcanised polymers become more worthwhile in comparison to their risk of failure. But with successful research into the analysis and understanding of inverse vulcanised polymers, many new avenues become available, especially rational design approaches, which would allow the synthesis of new polymers with superior properties, or optimisation of already existing polymers based on design principles.
The second element to this new direction for inverse vulcanisation would include a more concentrated attempt to control the synthesis and post-processing of inverse vulcanised polymers towards optimisation for specific target applications or scale up. This can reduce batch to batch variability in inverse vulcanised polymers, priming them for industrial uptake, where they can begin making a societal impact, but it also drives them toward being fit for application. Understanding what makes the optimum polymer with the best possible properties for a particular application is crucial for the successful uptake of inverse vulcanised polymers in roles where they benefit society. As part of this, a thorough study of how inverse vulcanised polymers change with time is required. Studies have shown that inverse vulcanised polymers age, and their properties change during this process.54,60,62 It is important to know the lifetime of inverse vulcanised polymers, as this could limit the operational lives of products derived from them. This is especially crucial for the suggested application of using inverse vulcanised polymers as cement substitutes and other building infrastructure applications, as ignorance of the changing properties of inverse vulcanised polymers in these applications could have potentially disastrous implications, such as a solid structure upon construction, that weakens over time toward structural failure or collapse. Additionally, a detailed life cycle analysis could be critical to understanding the cost to performance ratio of inverse vulcanised polymers, and how to make them not only economically viable, but also truly sustainable. Several articles, this one included, make the claim that inverse vulcanisation can be sustainable, but a full investigation of this would add needed proof to the claim. Fig. 26 illustrates a suggestion on where research efforts could be directed in the future, as well as a predicted difficulty of making progress towards certain research goals.
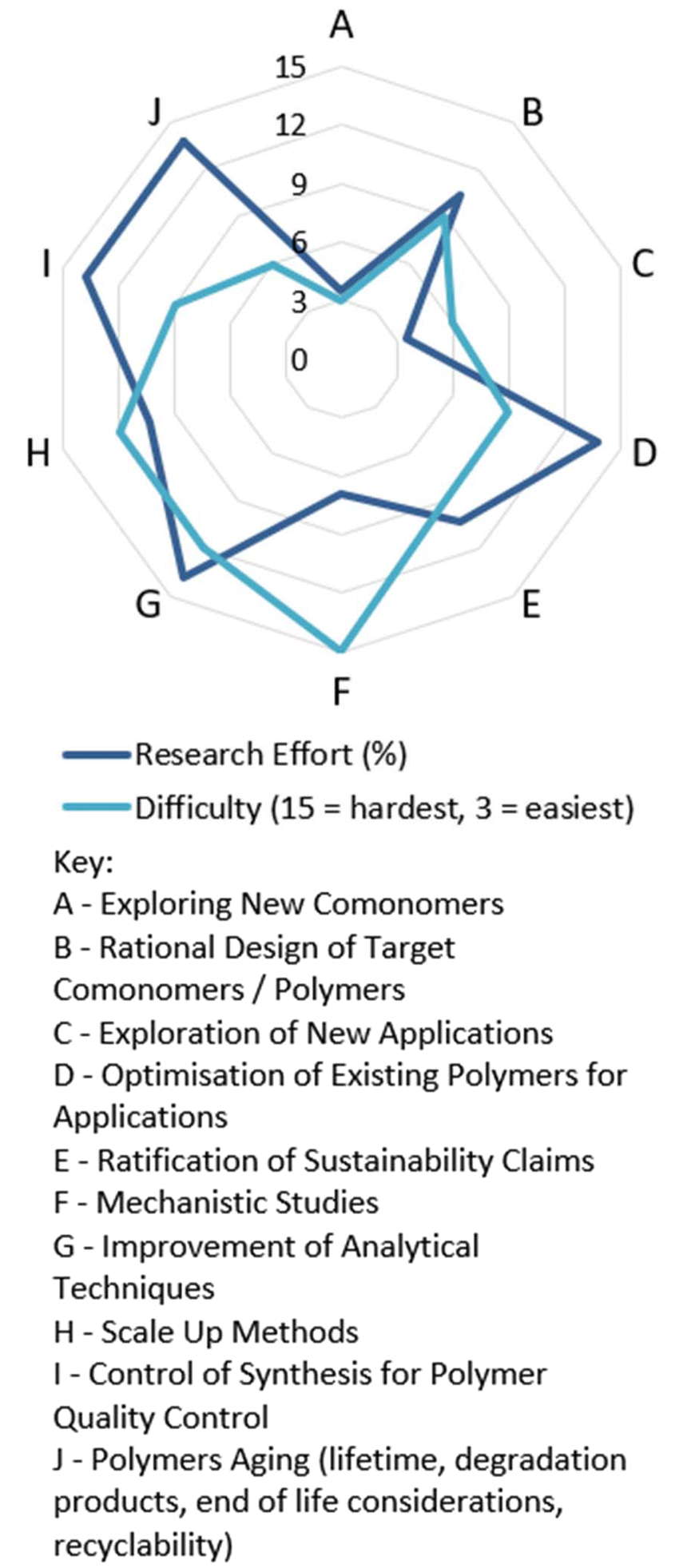 | ||
| Fig. 26 A radar chart proposing how much research effort should be dedicated to different research routes in inverse vulcanisation in the future, as well as possible difficulty levels. | ||
Ultimately, the field of inverse vulcanisation is a small but rapidly growing field, and there is incredible potential for exciting future research and applications. Inverse vulcanisation has had an excellent start as a research field, and with the growing interest, it is hard to imagine that the field's momentum will do anything but grow and diversify. This leads to an outlook of optimism and excitement for all the discoveries yet to be made.
Data availability
The associated article (titled: “Inverse Vulcanisation: a New Starter's Guide to an Emerging Field”) is a review article and so contains no primary research data. All content is cited as per the RSC referencing style, and data associated with those citations can be found from the DOI of those cited articles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to acknowledge Dr Tom Hasell for his useful discussion, proofreading, and feedback on the production of this review article.References
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS.
- J. Y. Li, L. Gao, F. Y. Pan, C. Gong, L. M. Sun, H. Gao, J. Q. Zhang, Y. F. Zhao, G. X. Wang and H. Liu, Nano-Micro Lett., 2023, 16, 12 CrossRef.
- G. R. Li, S. Wang, Y. N. Zhang, M. Li, Z. W. Chen and J. Lu, Adv. Mater., 2018, 30, 1705590 CrossRef PubMed.
- L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2018, 24, 6905–6916 CrossRef CAS PubMed.
- F. L. Zhao, Y. Li and W. Feng, Small Methods, 2018, 2, 1800156 CrossRef.
- F. G. Muller, L. S. Lisboa and J. M. Chalker, Adv. Sustainable Syst., 2023, 7, 2300010 CrossRef.
- T. S. Kleine, R. S. Glass, D. L. Lichtenberger, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259 CrossRef CAS.
- ThioTech website, https://www.thiotech.co.uk, (accessed December 2023).
- Outside the Box Materials website, https://www.otbmaterials.com, (accessed December 2023).
- Uberbinder website, https://uberbinder.com, (accessed December 2023).
- Clean Earth Technology website, https://cleanearth.biz, (accessed December 2023).
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS.
- H. Mutlu, E. B. Ceper, X. H. Li, J. M. Yang, W. Y. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- Y. Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105 RSC.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- R. J. Angelici, Acc. Chem. Res., 1988, 21, 387–394 CrossRef CAS.
- A. L. Kohl and R. B. Nielsen, Sulfur Recovery Processes In Gas Purification, Gulf Publishing, Houston, 5th edn, 1997, pp. 670–730 Search PubMed.
- B. Meyer, Chem. Rev., 1976, 76, 367–388 CrossRef CAS.
- M. J. H. Worthington, C. J. Shearer, L. J. Esdaile, J. A. Campbell, C. T. Gibson, S. K. Legg, Y. Yin, N. A. Lundquist, J. R. Gascooke, I. S. Albuquerque, J. G. Shapter, G. G. Andersson, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Adv. Sustainable Syst., 2018, 2, 1800024 CrossRef.
- J. P. Poirier, Lavoisier: Chemist, Biologist, Economist, University of Pennsylvania Press, Philadelphia, 1998, pp. 107–108 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, 2nd edn, 1997, pp. 654–656 Search PubMed.
- R. Steudel, Elemental Sulfur and Sulfur-Rich Compounds I, Springer-Verlag Berlin, Berlin, 2003, pp. 1–79 Search PubMed.
- I. H. Bell, L. I. Berger, P. E. Bradley, T. J. Bruno, C. E. Carraher, J. Cheng, R. D. Chirico, I. Cibulka, C. J. Cramer, V. Diky, M. Frenkel, J. R. Fuhr, R. N. Goldberg, T. W. Grove, A. H. Harvey, S. R. Heller, N. E. Holden, M. L. Huber, A. Kazakov, D. E. Kelleher, C. A. Koh, E. W. Lemmon, D. R. Lide, F. J. Lovas, Y. Luo, S. N. Lvov, M. Mantina, A. D. McNaught, T. M. Miller, N. Moazzen-Ahmadi, P. J. Mohr, C. D. Muzny, D. B. Newell, I. Ozier, L. I. Podobedova, C. J. Powell, R. Radebaugh, J. Reader, A. J. Remijan, E. D. Sloan, L. E. Snyder, P. D. N. Svoronos, B. N. Taylor, D. G. Truhlar, R. Valero, W. L. Wiese, C. Wohlfarth and D. Zwillinger, Handbook of Chemistry and Physics, CRC Press, Boca Raton, 97th edn, 2017, p. 117 Search PubMed.
- E. Wilberg and A. F. Holleman, Inorganic Chemistry, Academic Press, De Gruyter, 2001 Search PubMed.
- Q. S. Lian, Y. Li, K. Li, J. Cheng and J. Y. Zhang, Macromolecules, 2017, 50, 803–810 CrossRef CAS.
- M. M. Coleman, J. R. Shelton and J. L. Koenig, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13, 154–165 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- S. Penczek, R. Slazak and A. Duda, Nature, 1978, 273, 738–739 CrossRef CAS.
- A. Duda and S. Penczek, Makromol. Chem., 1980, 181, 995–1001 CrossRef CAS.
- L. B. Blight, B. R. Currell, B. J. Nash, T. M. Scott and C. Stillo, Br. Polym. J., 1980, 5–11 CrossRef CAS.
- Y. Ding and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2961–2968 CrossRef CAS.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtarb and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. A. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackay, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718–24722 RSC.
- I. Gomez, D. Mecerreyes, J. A. Blazquez, O. Leonet, H. B. Youcef, C. Li, J. L. Gómez-Cámer, O. Bondarchuk and L. Rodriguez-Martinez, J. Power Sources, 2016, 329, 72–78 CrossRef CAS.
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, J. Mater. Chem. A, 2019, 57, 7–12 CAS.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., 2015, 127, 1–6 CrossRef.
- T. S. Sahu, S. Choi, P. Jaumaux, J. Zhang, C. Wang, D. Zhou and G. Wang, Polyhedron, 2019, 162, 147–154 CrossRef CAS.
- J. L. Wang, J. Yang, C. R. Wan, K. Du, J. Y. Xie and N. X. Xu, Adv. Funct. Mater., 2003, 13, 487–492 CrossRef CAS.
- A. Hoefling, D. T. Nguyen, P. Partovi-Azar, D. Sebastiani, P. Theato, S. W. Song and Y. J. Lee, Chem. Mater., 2018, 30, 2915–2292 CrossRef CAS.
- J. Y. Li, L. Gao, F. Y. Pan, C. Gong, L. M. Sun, H. Gao, J. Q. Zhang, Y. F. Zhao, G. X. Wang and H. Liu, Nano-Micro Lett., 2023, 16, 12 CrossRef.
- G. R. Li, S. Wang, Y. N. Zhang, M. Li, Z. W. Chen and J. Lu, Adv. Mater., 2018, 30, 1705590 CrossRef.
- F. L. Zhao, Y. Li and W. Feng, Small Methods, 2018, 2, 1800156 CrossRef.
- P. W. Atkins and J. De Paula, Atkins’ Physical Chemistry, Oxford University Press, Oxford, 10th edn, 2014 Search PubMed.
- M. E. Mackey, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259 CrossRef PubMed.
- T. S. Kleine, N. A. Nguyen, L. E. Anderson, S. Namnabat, E. A. LaVilla, S. A. Showghi, P. T. Dirlam, C. B. Arrington, M. S. Manchester, J. Schwiegerling, R. S. Glass, K. Char, R. A. Norwood, M. E. Mackay and J. Pyun, ACS Macro Lett., 2016, 5, 1152–1156 CrossRef CAS.
- I. H. Bell, L. I. Berger, P. E. Bradley, T. J. Bruno, C. E. Carraher, J. Cheng, R. D. Chirico, I. Cibulka, C. J. Cramer, V. Diky, M. Frenkel, J. R. Fuhr, R. N. Goldberg, T. W. Grove, A. H. Harvey, S. R. Heller, N. E. Holden, M. L. Huber, A. Kazakov, D. E. Kelleher, C. A. Koh, E. W. Lemmon, D. R. Lide, F. J. Lovas, Y. Luo, S. N. Lvov, M. Mantina, A. D. McNaught, T. M. Miller, N. Moazzen-Ahmadi, P. J. Mohr, C. D. Muzny, D. B. Newell, I. Ozier, L. I. Podobedova, C. J. Powell, R. Radebaugh, J. Reader, A. J. Remijan, E. D. Sloan, L. E. Snyder, P. D. N. Svoronos, B. N. Taylor, D. G. Truhlar, R. Valero, W. L. Wiese, C. Wohlfarth and D. Zwillinger, Handbook of Chemistry and Physics, CRC Press, Boca Raton, 97th edn, 2017, pp. 751–754 Search PubMed.
- D. A. Boyd, C. C. Baker, J. D. Myers, V. Q. Nguyen, G. A. Drake, C. C. McClain, F. H. Kung, S. R. Bowman, W. Kim and J. S. Sanghera, Chem. Commun., 2017, 53, 259–262 RSC.
- L. E. Anderson, T. S. Kleine, Y. Zhang, D. D. Phan, S. Namnabat, E. A. LaVilla, K. M. Konopka, L. R. Diaz, M. S. Manchester, J. Schwiegerling, R. S. Glass, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2017, 6, 500–504 CrossRef CAS.
- T. S. Kleine, T. Lee, K. J. Carothers, M. O. Hamilton, L. E. Anderson, L. R. Diaz, N. P. Lyons, K. R. Coasey, W. O. Parker, L. Borghi, M. E. Mackay, K. Char, R. S. Glass, D. L. Lichtenberger, R. A. Norwood and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 17656–17660 CrossRef CAS PubMed.
- S. J. Tonkin, L. N. Pham, J. R. Gascooke, M. R. Johnston, M. L. Coote, C. T. Gibson and J. M. Chalker, Adv. Opt. Mater., 2023, 11, 2300058 CrossRef CAS.
- K. Saida, Y. Nomoto, H. Okauchi, H. Ogiwara and K. Nishimoto, Sci. Technol. Weld. Joining, 2012, 17, 1–8 CrossRef CAS.
- J. A. Smith, R. Mulhall, S. Goodman, G. Fleming, H. Allison, R. Raval and T. Hasell, ACS Omega, 2020, 5, 5229–5234 CrossRef CAS PubMed.
- R. A. Dop, D. R. Neill and T. Hasell, Biomacromolecules, 2021, 22, 5223–5233 CrossRef CAS PubMed.
- J. J. Dale, S. Petcher and T. Hasell, ACS Appl. Polym. Mater., 2022, 4, 3169–3173 CrossRef CAS.
- M. Ateia, D. E. Helbling and W. R. Dichtel, ACS Mater. Lett., 2020, 2, 1532–1544 CrossRef CAS.
- O. Illa, M. Namutebi, C. Saha, M. Ostovar, C. C. Chen, M. F. Haddow, S. Nocquet-Thibault, M. Lusi, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 11951–11966 CrossRef CAS.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- S. Petcher, D. J. Parker and T. Hasell, Environ. Sci.: Water Res. Technol., 2019, 5, 2142–2149 RSC.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- J. J. Dale, J. Stanley, R. A. Dop, G. Chronowska-Bojczuk, A. J. Fielding, D. R. Neill and T. Hasell, Eur. Polym. J., 2023, 195, 112198 CrossRef CAS.
- V. Hanna, M. Graysmark, H. Willcock and T. Hasell, J. Mater. Chem. A, 2024, 12, 1211–1217 RSC.
- J. J. Dale, V. Hanna and T. Hasell, ACS Appl. Polym. Mater., 2023, 5, 6761–6765 CrossRef CAS.
- V. Hann, P. Yan, S. Petcher and T. Hasell, Polym. Chem., 2022, 13, 3930–3937 RSC.
- M. J. Graham, C. V. Lopez, C. P. Maladeniya, A. G. Tennyson and R. C. Smith, J. Appl. Polym. Sci., 2023, 140, e53684 CrossRef CAS.
- K. Orme, A. H. Fistrovich and C. L. Jenkins, Macromolecules, 2020, 53, 9353–9361 CrossRef CAS.
- L. J. Dodd, Ö. Omar, X. Wu and T. Hasell, ACS Catal., 2021, 11, 4441–4455 CrossRef CAS.
- L. J. Dodd, C. Lima, D. Costa-Milan, A. R. Neale, B. Saunders, B. Zhang, A. Sarua, R. Goodacre, L. J. Hardwick, M. Kuball and T. Hasell, Polym. Chem., 2023, 14, 1369–1386 RSC.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- R. Jalil and J. R. Nixon, J. Microencapsulation, 1990, 7, 25–39 CrossRef CAS PubMed.
- R. Arshady, J. Controlled Release, 1991, 17, 1–22 CrossRef CAS.
- P. Sansdrap and A. J. Moes, Int. J. Pharm., 1993, 98, 157–164 CrossRef CAS.
- W. Chen and D. R. Lu, J. Microencapsulation, 1999, 16, 551–563 CAS.
- F. Gabor, B. Ertl, M. Wirth and R. Mallinger, J. Microencapsulation, 1999, 16, 1–12 CAS.
- W. J. Lin and T. L. Wu, J. Microencapsulation, 1999, 16, 639–646 CAS.
- L. J. Dodd, W. Sandy, R. A. Dop, B. Zhang, A. Lunt, D. R. Neill and T. Hasell, Polym. Chem., 2023, 14, 4064–4078 CAS.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. F. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willcock, J. M. Chalker and T. Hasell, Chem. – Eur. J., 2019, 25, 10433–10440 CAS.
- Y. Ren, H. Shui, C. Peng, H. Liu and Y. Hu, Fluid Phase Equilib., 2011, 312, 31–36 CAS.
- U. Münchberg, A. Anwar, S. Mecklenburg and C. Jacob, Org. Biomol. Chem., 2007, 5, 1505–1518 Search PubMed.
- M. K. Denk, Eur. J. Inorg. Chem., 2009, 1358–1368 CAS.
- M. W. Wong, Top. Curr. Chem., 2003, 231, 1–30 CAS.
- L. Zou, Y. Fu, K. Shen and Q. Guo, J. Mol. Struct.: THEOCHEM, 2007, 807, 87–92 CAS.
- L. Xiao-Hong, G. Xiao-Yang and Z. Xian-Zhou, J. Sulfur Chem., 2011, 32, 419–426 CrossRef.
- Y. Yang, H. Yu, X. Sun and Z. Dang, J. Phys. Org. Chem., 2016, 29, 6–13 CrossRef.
- J. Bao, K. P. Martin, E. Cho, K. Kang, R. S. Glass, V. Coropceanu, J. Bredas, W. Parker, J. T. Njardarson and J. Pyun, J. Am. Chem. Soc., 2023, 145, 12386–12397 CrossRef CAS.
- J. A. Smith, X. F. Wu, N. G. Berry and T. Hasell, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1777–1781 CrossRef CAS.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef.
- A. Nayeem, M. F. Ali and J. H. Shariffuddin, Mater. Today: Proc., 2022, 57, 1095–1100 CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, New York, 1953, pp. 124–129 Search PubMed.
- M. Alger, Polymer Science Dictionary, Elsevier Applied Science, New York, 1989, pp. 28 Search PubMed.
- P. Yan, W. Zhao, S. J. Tonkin, J. M. Chalker, T. L. Schiller and T. Hasell, Chem. Mater., 2022, 34, 1167–1178 CrossRef CAS.
- S. J. Tonkin, L. N. Pham, J. R. Gascooke, M. R. Johnston, M. L. Coote, C. T. Gibson and J. M. Chalker, Adv. Opt. Mater., 2023, 11, 2300058 CrossRef CAS.
- H. Shin, J. Kim, D. Kim, V. H. Nguyen, S. Lee, S. Han, J. Lim and K. Char, J. Mater. Chem. A, 2018, 6, 23542–23549 RSC.
- J. M. Lee, G. Y. Noh, B. G. Kim, Y. Yoo, W. J. Choi, D. Kim, H. G. Yoon and Y. S. Kim, ACS Macro Lett., 2019, 8, 912–916 CrossRef CAS.
- Y. Lai and Y. Liu, Macromol. Rapid Commun., 2023, 44, 2300014 CrossRef CAS.
- P. Yan, W. Zhao, F. McBride, D. Cai, J. Dale, V. Hanna and T. Hasell, Nat. Commun., 2022, 13, 4824 CrossRef CAS.
- J. Jia, J. Liu, Z. Wang, T. Liu, P. Yan, X. Gong, C. Zhao, L. Chen, C. Miao, W. Zhao, S. Cai, X. Wang, A. I. Cooper, X. Wu, T. Hasell and Z. Quan, Nat. Chem., 2022, 14, 1249–1257 CrossRef CAS PubMed.
- E. Cho, S. M. Pratik, J. Pyun, V. Coropceanu and J. Brédas, ACS Mater. Lett., 2022, 4, 2362–2367 CrossRef CAS.
- J. M. M. Pople, T. P. Nicholls, L. N. Pham, W. M. Bloch, L. S. Lisboa, M. V. Perkins, C. T. Gibson, M. L. Coote, Z. Jia and J. M. Chalker, J. Am. Chem. Soc., 2023, 145, 11798–11810 CrossRef CAS PubMed.
- J. Lindenmann, V. Matzi, N. Neuboeck, B. Ratzenhofer-Komenda, A. Maier and F. M. Smolle-Juettner, Diving Hyperb. Med., 2010, 40, 213–217 Search PubMed.
- L. C. Alderman and J. J. Bergin, Arch. Environ. Health, 1986, 41, 354–358 CrossRef CAS.
- J. D. Menczel, R. Andre, W. S. Kohl, V. V. Kronguaz, D. Lőrinczy, M. Reading and J. Grebowicz, Handbook of Differential Scanning Calorimetry, Butterworth-Heinemann, Oxford, 2023 Search PubMed.
- M. K. Singh and A. Singh, in Characterization of Polymers and Fibres, ed. M. K. Singh and A. Singh, Woodhead Publishing, Cambridge, 1st edn, 2022, ch. 10, pp. 223–240 Search PubMed.
- F. Pregl, Quantitative Micro-Analysis of Organic Substances, Springer, Berlin, 1917 Search PubMed.
- S. A. Umoren, M. M. Solomon and V. S. Saji, in Polymeric Materials in Corrosion Inhibition, ed. S. A. Umoren, M. M. Solomon and V. S. Saji, Elsevier, Amsterdam, 1st edn, 2022, ch. 2, pp. 49–81 Search PubMed.
- R. G. Leonard and K. Quigley, in Encyclopedia of Analytical Science, ed P. Worsfold, A. Townshend and C. Poole, Elsevier, Amsterdam, 2nd edn, 2005, ch. 28, pp. 28–37 Search PubMed.
- Y. Onose, Y. Ito, J. Kubawara and T. Kanbara, Polym. Chem., 2022, 13, 5486–5493 RSC.
- J. M. Scheiger, C. Direksilp, P. Falkenstein, A. Welle, M. Koenig, S. Heissler, J. Matysik, P. A. Levkin and P. Theato, Angew. Chem., Int. Ed., 2020, 59, 18639–18645 CrossRef CAS PubMed.
- S. Petcher, B. Zhang and T. Hasell, Chem. Commun., 2021, 57, 5059–5062 RSC.
- R. L. Upton, R. A. Dop, E. Sadler, A. M. Lunt, D. R. Neill, T. Hasell and C. R. Crick, J. Mater. Chem. B, 2022, 10, 4153–4162 RSC.
- R. A. Dop, D. R. Neill and T. Hasell, ACS Appl. Mater. Interfaces, 2023, 15, 20822–20832 CrossRef CAS PubMed.
- B. Zhang, S. Petcher, R. A. Dop, P. Yan, W. Zhao, H. Wang, L. J. Dodd, T. O. McDonald and T. Hasell, J. Mater. Chem. A, 2022, 10, 13704–13710 RSC.
- R. J. Young and P. A. Lovell, Introduction to Polymers, CRC Press, Boca Raton, 2011 Search PubMed.
- J. R. Shelton and E. T. McDonel, Rubber Chem. Technol., 1960, 33, 342–356 CrossRef CAS.
- G. A. Blokh, Dokl. Akad. Nauk SSSR, 1959, 129, 361–364 Search PubMed.
- D. Dondi, A. Buttafava, A. Zeffiro, C. Palamini, A. Lostritto, L. Giannini and A. Faucitano, Eur. Polym. J., 2015, 62, 222–235 CAS.
- Q. S. Lian, Y. Li, T. Yang, K. Li, Y. F. Xu, L. Liu, J. B. Zhao, J. Y. Zhang and J. Cheng, J. Mater. Sci., 2016, 51, 7887–7898 CrossRef CAS.
- S. Diez, A. Hoefling, P. Theato and W. Pauer, Polymers, 2017, 9, 16 CrossRef PubMed.
- M. Arslan, B. Kiskan, E. C. Cengiz, R. Demir-Cakan and Y. Yagci, Eur. Polym. J., 2016, 80, 70–77 CrossRef CAS.
- W. J. Chung, A. G. Simmonds, J. J. Griebel, E. T. Kim, H. S. Suh, I. B. Shim, R. S. Glass, D. A. Loy, P. Theato, Y. E. Sung, K. Char and J. Pyun, Angew. Chem., Int. Ed., 2011, 50, 11409–11412 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2015, 48, 1329–1334 CrossRef CAS.
- V. K. S. Wadi, K. K. Jena, S. Z. Khawaja, K. Yannakopoulou, M. Fardis, G. Mitrikas, M. Karagianni, G. Papavassiliou and S. M. Alhassan, ACS Omega, 2018, 3, 3330–3339 CrossRef.
- B. Zheng, L. Zhong, X. Wang, P. Lin, Z. Yang, T. Bai, H. Shen and H. Zhang, Nat. Commun., 2024, 15, 5507 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- X. Guo, X. Sun, M. Jiang and Y. Zhao, Synthesis, 2022, 1996–2004 CrossRef CAS.
- Z. Cheng, P. Sun, A. Tang, W. Jin and C. Liu, Org. Lett., 2019, 21, 8925–8929 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and M. S. Arabi, J. Org. Chem., 2010, 75, 6208–6213 CrossRef CAS PubMed.
- M. Lutz, M. Wenzler and I. Likthotvorik, Synthesis, 2018, 2231–2234 CAS.
- E. Voutyritsa, I. Triandafillidi and C. G. Kokotos, Synthesis, 2017, 917–924 CAS.
- M. Kirihara, Y. Asai, S. Ogawa, T. Noguchi, A. Hatano and Y. Hirai, Synthesis, 2007, 3286–3289 CrossRef CAS.
- R. Sanz, R. Aguado, M. R. Pedrosa and F. Arnáiz, Synthesis, 2002, 856–858 CrossRef CAS.
- A. Khazaei, M. A. Zolfigol and A. Rostami, Synthesis, 2004, 2959–2961 CrossRef CAS.
- C. S. Sevier and C. A. Kaiser, Nat. Rev. Mol. Cell Biol., 2002, 3, 836–847 CrossRef CAS PubMed.
- S. Xu, Y. Wang, M. N. Radford, A. J. Ferrell and M. Xian, Org. Lett., 2018, 20, 465–468 CrossRef CAS PubMed.
- D. Ali, R. Hunter, C. H. Kaschula, S. De Doncker and S. C. M. Rees-Jones, J. Org. Chem., 2019, 84, 2862–2869 CrossRef CAS.
- S. Lach, M. Sliwka-Kaszynska and D. Witt, Synlett, 2010, 2857–2860 CAS.
- A. Kertmen, S. Lach, J. Rachon and D. Witt, Synthesis, 2009, 1459–1462 CAS.
| This journal is © The Royal Society of Chemistry 2025 |