
Glass-confined carbon dots: transparent afterglow materials with switchable TADF and RTP†
Pengwei
Wang‡
a,
Peixi
Cong‡
b,
Jiachen
Chen
a,
Huaiyuan
Cao
a,
Qi
Yue
a,
Zixiao
Xue
a,
Junji
Zhang
 c,
Long
Zhang
a,
Robert S.
Weatherup
*b,
Jiabin
Cui
*d and
Jin
He
*a
c,
Long
Zhang
a,
Robert S.
Weatherup
*b,
Jiabin
Cui
*d and
Jin
He
*a
aKey Laboratory of Materials for High Power Laser, Shanghai Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: jhe@siom.ac.cn
bDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: robert.weatherup@materials.ox.ac.uk
cSchool of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China
dState Key Laboratory of Radiation Medicine and Protection, School of Radiation Medicine and Protection, Collaborative Innovation Center of Radiological Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China. E-mail: jiabin.cui@suda.edu.cn
First published on 12th March 2025
Abstract
The confined synthesis of carbon dots (CDs) in solid matrixes is a promising avenue for developing new afterglow materials. Benefiting from the advantages of the sol–gel preparation of nanoporous glass, we report transparent glass-confined CDs with tunable afterglow luminescence. Switchable thermally-activated delayed fluorescence (TADF) and room-temperature phosphorescence (RTP) of CDs were achieved by adjusting the sintering temperature and ion doping. Our findings reveal that with an increase in sintering temperature from 500 °C to 600 °C, the energy gap (ΔEST) of CD-nanoporous glass (NG) increased from 0.05 eV to 0.21 eV, while the lifetime increased from 329 ms to 548 ms, which is attributed to the enhanced carbonization degree of the CDs. Pb2+ doping is also shown to achieve switchable TADF and RTP of glass-confined CDs attributed to the alteration of interfacial interactions between the glass and confined CDs. This design concept introduces a new perspective for developing transparent afterglow materials for various unique phosphorescence applications.
 Jiabin Cui | Jiabin Cui received his PhD from the Beijing University of Chemical Technology. He has been working with Prof. Uri Banin as a postdoctoral fellow at the Hebrew University of Jerusalem in Israel since 2017. He joined the School of Radiation Medicine and Protection at Soochow University in 2021. His research focuses on coupled nanocrystal molecules, fluorescent nanoprobes, and new applications in sensors, cancer therapy, and molecular imaging. To date, he has published over 40 papers in journals, such as Nat. Commun., J. Am. Chem. Soc., Angew. Chem., Int. Ed., Adv. Mater., Acc. Chem. Res., Nano Lett., Anal. Chem. and so on. |
Introduction
The emergence of synthetic approaches that provide precise spatial control on the nanoscale inspires materials scientists to develop strategies for creating new material functionalities.1–5 Compared with traditional methods that tune interfacial or surface interactions, leveraging the microenvironment of confined matrix chemistry is a key strategy for optimizing performance.6–8 The interfaces associated with these microenvironments play an essential bridging role, not only in connecting the confined materials with the matrix but also by providing a platform for precise control over the confined materials. Such interface regulation strategies are capable of modifying the chemical composition, physical structure, and optical properties of materials and providing the capacity to design and synthesize materials tailored for various applications.9–14Carbon dots (CDs) stand out among numerous materials due to their low toxicity, environmental friendliness, and tuneable optical behaviours, especially their afterglow properties, which have recently garnered significant attention.15–17 Derived from various luminescent mechanisms, afterglow CDs can be categorized into two main types: those exhibiting room-temperature phosphorescence (RTP) and those distinguished by thermally activated delayed fluorescence (TADF).18–21 Precise control over the energy levels, and in particular, the gap (ΔEST) between the lowest singlet (S1) and triplet (T1) excited states, is crucial for the above-mentioned afterglow behaviour. A multitude of studies have demonstrated that confining CDs within a rigid matrix can effectively generate afterglow luminescence and achieve precise control over this phenomenon.22–25
Glass, with its excellent chemical stability, high optical transmittance, and mechanical strength, plays a key role in quantum communication, information encryption transmission, and precision optical instruments.26–30 Therefore, the deliberate design of the intricate structure of CDs in glass to further expand its optical applications emerges as a promising avenue in photonics. However, few reports exist on the preparation of CDs in glass that exhibit afterglow luminescence or on potential strategies for interface regulation.
Glass synthesis by sol–gel routes (low-temperature chemical pathways) involves the transformation of metal alkoxides through hydrolysis and subsequent condensation within a solvent medium, culminating in the room-temperature assembly of a glassy matrix.31 The formed xerogel is a hybrid material consisting of a glass network and organic compounds. Subsequently, under high-temperature heat treatment, these organic compounds undergo combustion or pyrolysis, thus enabling the direct formation of CDs within the glass. Recently, we have developed a metal carboxylate sol–gel route, leading to a series of novel nanoporous glasses with various chemical compositions, such as AlPO4, AlPO4–SiO2, GaAlPO4, and Al2O3–SiO2.32–35 This opens up the potential for simultaneous in situ formation of CDs uniformly dispersed inside a glass matrix with enhanced interfacial interactions, stabilizing the CDs’ triplet state and enabling afterglow luminescence.
Here, carbon dots confined in nanoporous glass (CDs-NG) with afterglow luminescence were prepared via the metal carbonate sol–gel route assisted by sintering. By tailoring the sintering process, carboxyl groups undergo pyrolysis, thereby generating CDs in situ during the formation of NG. The NG functions not only as a conducive environment for CD formation, suppressing aggregation, but also serves as a superior confining matrix that can stabilize the triplet excited state, thereby facilitating afterglow luminescence. Here, we successfully produced glass-confined CDs with switchable TADF and RTP by adjusting the sintering temperature and employing ion doping. Firstly, we regulated the degree of carbonization of the glass-confined CDs by modulating their calcination process. With the sintering temperature adjusted from 500 °C to 600 °C, the ΔEST of CDs-NG increased from 0.05 eV to 0.21 eV, while the lifetime improved from 329 ms to 548 ms, which is attributed to an enhanced degree of carbonization of the CDs. Furthermore, we achieved switchable TADF and RTP of glass-confined CDs by in situ doping of Pb2+ within the sol–gel process to modulate the interface between the CDs and NG (Fig. 1a). X-ray photoelectron spectroscopy and extended X-ray absorption fine structure measurements reveal that the Pb–O–C bonding at the interface between the CDs and NG not only connects the glass matrix with the CDs but also enables precise regulation of the CDs’ emission characteristics. We introduce the concept of “precise microenvironment chemistry” based on the glass-confined matrix for afterglow CDs, which opens new opportunities for switchable TADF and RTP modulation with diverse applications.
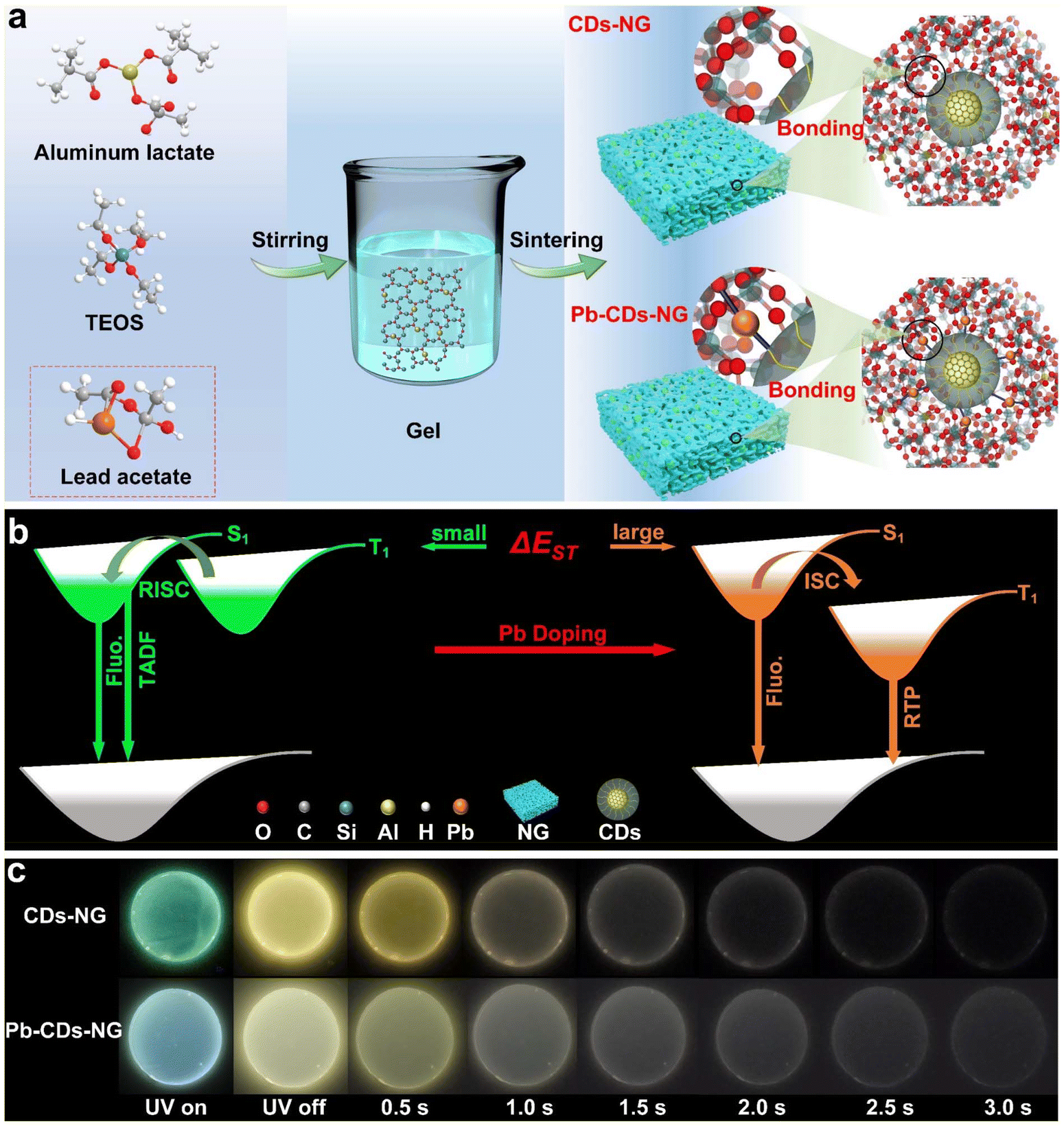 | ||
| Fig. 1 Schematic illustration of the fabrication of CDs-NG and Pb-CDs-NG via the sol–gel route. (a) Schematic illustration of the sol–gel fabrication route for CDs-NG and Pb-CDs-NG. (b) Mechanism diagram of TADF and RTP. (c) Photographs of CDs-NG and Pb-CDs-NG. | ||
Experimental section
Materials
Aluminum lactate (95%), tetraethyl orthosilicate (TEOS) (99%), and lead acetate trihydrate (99.5%) were purchased from Sigma-Aldrich. Ethanol (EtOH) was purchased from Qiangsheng Inc. Jiang Su, China. Sodium hydroxide (97%) was purchased from Aladdin, Shanghai, China.Synthesis of CDs-NG
The carbon dot (CD)-based nanoporous glass (NG) was prepared by the sol–gel method at sintering temperatures of 500 °C, 550 °C, 600 °C or 700 °C. A measured amount of aluminum lactate was first dissolved in an adequate volume of deionized water with continuous stirring, while TEOS, in an equivalent quantity, was diluted in a comparable amount of ethanol using the identical procedure. The two clarified solutions were then mixed with continuous stirring. Following an 8–10 hour stirring period, the mixture was poured into a circular mould and left at ambient temperature for 12 hours. The circular moulds were transferred to an oven at 50 °C, and the oven was slowly heated to 100 °C over three days. Then the xerogel was calcined in a muffle furnace, and the temperature was raised at 1 °C per minute, holding for four hours at each interval of 100 °C to prevent the sample from cracking during the calcination process with a final holding step at the desired sintering temperature. Finally, the CDs-NG was obtained.Synthesis of Pb-CDs-NG
The CDs confined in lead-doped NG (Pb-CDs-NG) were prepared in a similar way, but lead acetate solution was added when the two clarified solutions were mixed. Otherwise, the synthesis process remained unchanged.Characterization
The measurements of surface area and pore size distribution were conducted using a nitrogen adsorption–desorption system (Autosorb iQ, Anton Paar, Austria), operated at a temperature of −196 °C. Prior to analysis, each sample underwent degassing in a vacuum environment at 120 °C for a duration of 6 hours. TEM, HRTEM, and HAADF-STEM micrographs were recorded using a JEOL-2010. The transmittance spectra were recorded using a LAMBDA 750 (PerkinElmer) UV-Vis spectrophotometer. Thermogravimetric (TG) analysis on the powdered, dry-ground samples was performed using a NETZSCH STA-2500 Regulus, with a temperature range starting from ambient to 1000 °C, ramping at a rate of 10 °C per minute. SEM was conducted with an Auriga S40 (Zeiss). XPS measurements were conducted using a K-Alpha spectrometer (Thermo Fisher Scientific), employing non-monochromatic Al Kα X-ray radiation at a constant pass energy of 20.0 eV. The C 1s peak is fitted and assigned based on prior literature. Here, XPSPeak software was employed to perform Gaussian–Lorentzian mixture fitting on the C 1s peak.36,37 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C component was fitted using an asymmetric lineshape due to the metallic bonding of graphitic carbon. We employed a Gaussian–Lorentzian hybrid function, specifically GL(40), for the fitting process. Binding energies for all measurements (±0.2 eV) were energy calibrated by setting the CC feature in the C 1s region to 284.4 eV. Photoluminescence (PL) spectra were recorded using a FLS1000 (Edinburgh Instruments).
C component was fitted using an asymmetric lineshape due to the metallic bonding of graphitic carbon. We employed a Gaussian–Lorentzian hybrid function, specifically GL(40), for the fitting process. Binding energies for all measurements (±0.2 eV) were energy calibrated by setting the CC feature in the C 1s region to 284.4 eV. Photoluminescence (PL) spectra were recorded using a FLS1000 (Edinburgh Instruments).
Results and discussion
In situ formation of glass-confined carbon dots with afterglow luminescence
As shown in Fig. 1a, carbon dots in nanoporous glass (CDs-NG) and those doped with Pb2+ (Pb-CDs-NG) were produced by the sol–gel process, followed by a subsequent sintering step. The first step is the preparation of the gel. The CDs-NG are derived from two precursors, aluminum lactate and tetraethyl orthosilicate (TEOS), while the preparation of Pb-CDs-NG requires the addition of lead acetate as the precursor. Upon the introduction of TEOS into the aluminum lactate aqueous solution, the sol–gel procedure initiates the formation of a silica–alumina framework via hydrolysis and condensation reactions. For Pb-CDs-NG, we need to add a lead acetate solution at this step to form a Pb2+ doped silico-alumina glass network. After the sol–gel process, the resulting gel is subjected to aging and sintering treatments. The even distribution of lactate groups throughout the three-dimensional gel network is critical for the formation of uniform nanopores and the confinement of CDs within the NG during the calcination stage. During the sintering process, CDs-NG are formed, effectively preventing phase separation and stabilizing the triplet excited state, which is crucial for sustained afterglow luminescence. Based on the emission mechanisms, CD-based afterglow luminescence can be categorized as thermally activated delayed fluorescence (TADF) or room-temperature phosphorescence (RTP). As shown in Fig. 1b, RTP materials display persistent emission as excitons move from the lowest triplet state (T1) to the ground state (S0). Conversely, in TADF materials, excitons at T1 absorb thermal energy to shift to the lowest singlet state (S1), and then they return to S0, releasing delayed fluorescence. This primarily hinges on the energy difference (ΔEST) separating the S1 and T1 excited states.18–25 Thus, effective modulation of ΔEST can enable control over the afterglow luminescence behavior. Herein, we have achieved regulation of the afterglow photoluminescence behavior of CDs by doping Pb2+ into NG. As shown in Fig. 1a, we suggest that lead atoms act as a bridge between the glass matrix and the CDs, altering the interfacial interactions between NG and CDs, enabling precise modulation of the CDs, and thereby changing the photoluminescence behavior of the CDs’ afterglow. The luminescence photographs of CDs-NG and CDs-Pb-NG are depicted in Fig. 1c.CDs-NG with high transmittance in the visible light range were prepared via the sol–gel method (Fig. S1 and S2†) at a sintering temperature of 500 °C (CDs-NG-500). Transmission electron microscopy (TEM) observations indicate that monodisperse carbon dots with an average diameter of 5.1 nm, as shown in Fig. S3,† are uniformly dispersed and encapsulated within the NG matrix. High-resolution TEM (HR-TEM) reveals a lattice fringe period of 0.21 nm as shown in Fig. 2a, consistent with a number of prior reports of CDs, although we note that the assignment of this spacing remains under debate.38–42 Then, a series of optical measurements was carried out. As depicted in Fig. 2b, the photoluminescence of CDs-NG-500 under various excitation wavelengths demonstrates a dependence on excitation, with the emission peak shifting from 530 nm to 550 nm as the excitation wavelength grows from 340 nm to 390 nm. This is consistent with the characteristics of triplet-state luminescence. The excitation-dependent fluorescence of triplet-state luminescence is owing to the electronic states of molecules and their transitions. The transition of electrons in molecules between different energy levels, such as singlet states (S) and triplet states (T), affects the luminescence properties. During fluorescence, electrons absorb light energy and move from the ground state (S0) to the excited singlet state (S1) and subsequently return to the ground state through a radiative transition. However, in molecules with TADF properties, the small energy gap between the S1 and T1 states permits triplet excitons to revert to S1via the reverse intersystem crossing (RISC) process, thereby producing delayed fluorescence with longer persistence. In TADF materials, alterations in the excitation wavelength markedly influence the energy gap separating S1 and T1, along with the efficacy of the RISC process, thereby affecting fluorescence. Then, we compared the fluorescence spectrum with the delayed fluorescence spectrum of CDs-NG-500. As illustrated in Fig. 2c, upon excitation at 360 nm, the steady-state fluorescence emission peak is centered at 538 nm, while the delayed fluorescence emission peak is located at 550 nm. As shown in Fig. 2d, the lifetime at 550 nm is determined to be 329 ms, which is comparable to the afterglow lifetimes of CDs in other confined systems. For example, zeolite-confined CDs reported by Yu et al. have a lifetime of ∼350 ms.43–45 In control experiments, we have exfoliated the CDs from the NG, with TEM revealing well-dispersed CDs. HR-TEM again reveals lattice fringes with a spacing of 0.21 nm, as seen in Fig. 2e. As illustrated in Fig. 2f, the emission center of the exfoliated CDs is at 470 nm, exhibiting a significant blue shift compared to the 538 nm of the CDs-NG-500. When irradiated by a UV lamp, the CDs emit a blue-green luminescence, with the sample no longer emitting light once the UV lamp is extinguished (as depicted in the inset of Fig. 2f). In solution, due to the intense intramolecular vibrations and rotations, isolated CDs primarily deactivate through non-radiative pathways. In contrast, we suggest that the rigid structure of the NG with its nanoconfined space can effectively prevent non-radiative relaxation by inhibiting intramolecular motion, thereby playing a crucial role in stabilizing the long-lived triplet state.
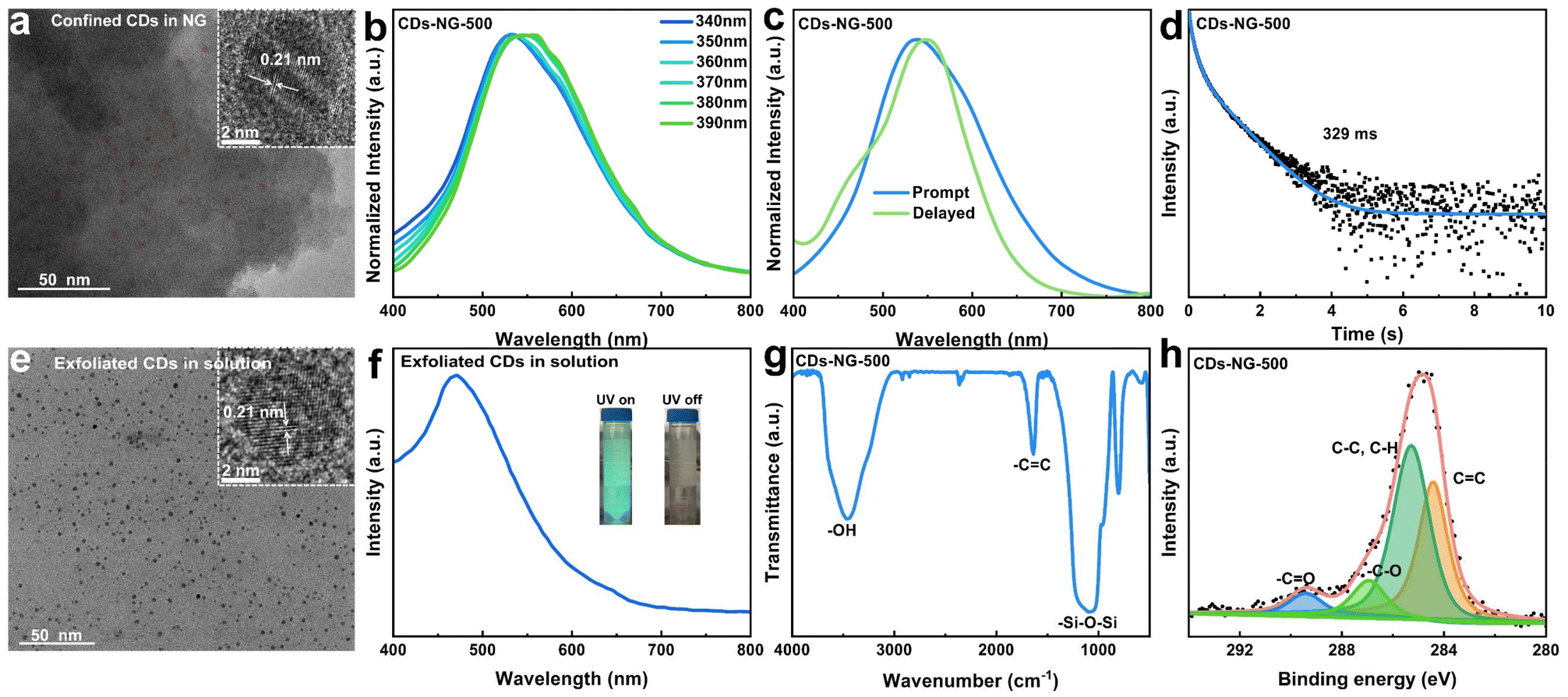 | ||
| Fig. 2 Characterization of glass-confined CDs. (a) TEM and HR-TEM images of the CDs in NG. (b) Excitation-dependent photoluminescence of CDs-NG-500. (c) Normalized steady-state (blue line) and time-delayed (green line) photoluminescence spectra, both excited at 360 nm. (d) Time-resolved decay spectra. (e) TEM and HR-TEM images of the exfoliated CDs in solution from CDs-NG-500. (f) Photoluminescence spectra of the exfoliated CDs in solution under 360 nm excitation from CDs-NG-500 (insets: images captured under 365 nm UV light and in the dark after the UV light is switched off). (g) FT-IR spectra of CDs-NG-500. (h) XPS spectra of C 1s of CDs-NG-500. | ||
To explore the formation process of NG-confined CDs, we conducted thermogravimetric (TG) analysis on the xerogel prepared at 100 °C. As depicted in Fig. S4,† it demonstrates four distinct stages along the thermal gradient: (1) solvent evaporation (50–150 °C). (2) Decomposition of the lactate group (150–400 °C). The in situ formation of NG-confined CDs occurs during the gel–glass transition. (3) Progressive carbonization (400–600 °C). (4) Decomposition of CDs (above 600 °C). To explore the interactions between the CDs and NG, we conducted Fourier transform infrared (FT-IR) and X-ray photoelectron spectroscopy (XPS) analyses on the 500-CDs-NG. The distinctive functional groups are identifiable through FT-IR spectroscopic analysis. As depicted in Fig. 2g, the peak at 3450 cm−1 in absorption is attributed to the O–H stretching vibration, likely arising from the COOH groups in the CDs and the Si-OH groups within the Al2O3–SiO2 glass matrix.44 The peak observed at 1620 cm−1 in absorption is assignable to the CC stretching vibration.46 Additionally, the absorption peak at 1090 cm−1 is the characteristic of Si–O–Si vibrations. Furthermore, XPS was used to identify the chemical species close to the CDs-NG surface. As illustrated in Fig. 2h, the C 1s spectrum shows the existence of CC bonds at 284.4 eV, C–C, C–H bonds at 285.2 eV, –C–O bonds at 286.8 eV, and –CO bonds at 289.4 eV.47,48 This indicates that CDs with a rich array of functional groups are confined within the glass matrix.
The above results indicate enhanced interfacial interactions of confined CDs in NG, which help stabilise the triplet state and afterglow luminescence. Therefore, NG is not only an ideal host for CDs, effectively preventing their aggregation, but also an excellent confining matrix that can stabilize the triplet excited state, enabling afterglow luminescence.
Switchable TADF and RTP by regulating the degree of carbonization
Drawing from diverse luminescent processes, the afterglow emission of CDs is generally divided into two main categories: RTP and TADF. The switchable RTP and TADF of CDs broaden the potential applications of these materials in areas including display technology, bioimaging, and optoelectronics. It is widely acknowledged that effective modulation of ΔEST can regulate the afterglow luminescence behavior of CDs. In this study, we achieved the regulation of ΔEST for CDs-NG by varying the sintering temperature, which results in different degrees of carbonization of the CDs. From the abovementioned Tg testing, it is observed that when the sintering temperature reaches 400 °C, the NG-confined CDs have essentially completed their growth, and their degree of carbonization gradually increases with higher sintering temperatures. Moreover, when the sintering temperature exceeds 600 °C, the NG-confined CDs begin to decompose, which leads to the formation of pure NG. Therefore, as shown in Fig. 3a, by exercising meticulous control over the sintering process, we prepared intact circular samples at sintering temperatures of 500, 550, 600, and 700 °C, respectively. As illustrated in Fig. S5,† intact square glass can also be fabricated employing this method. With the change in sintering temperature, we can observe a significant change in the luminescence of the samples. When the sintering temperature reaches 700 °C, the samples no longer emit light, which indicates the total decomposition of the CDs formed. This observation agrees well with the Tg results. Therefore, the samples with sintering temperatures of 550 and 600 °C were studied in detail and compared to CDs-NG-500. As presented in Fig. 3b and c, the prepared CDs-NG sintered at temperatures of 550 °C (CDs-NG-550) and 600 °C (CDs-NG-600) have high transparency in the visible light range (Fig. S6 and S7†), and both exhibit excitation-dependent fluorescence spectra. Regarding the photoluminescence properties, under 360 nm excitation, the emission center of CDs-NG-550 is approximately 533 nm (Fig. 3b), while that of CDs-NG-600 is around 510 nm (Fig. 3c), and CDs-NG-500 exhibits an emission center at 538 nm (Fig. 2b). Compared to CDs-NG-500, the emission centers of both CDs-NG-550 and CDs-NG-600 exhibit a significant blue shift, which increases with the sintering temperature. This notable blue shift is attributed to the decrease in the size of the CDs (Fig. S3, S8 and S9†). Upon ultraviolet flashlight excitation, the discernible phosphorescent emissions from CDs-NG-550 and CDs-NG-600 are visible to the unaided eye. Upon ultraviolet excitation, CDs-NG-550 emits green fluorescence and subsequently exhibits a yellow afterglow (Fig. 3a). In contrast, CDs-NG-600 displays blue-green fluorescence under ultraviolet lamp excitation, which transitions to a yellow afterglow once the excitation is removed (Fig. 3a). Additionally, we compared their steady-state photoluminescence (PL) and delayed fluorescence (DF) spectra. Exposed to 360 nm radiation, the photoluminescence emission peak for CDs-NG-550 is centered at 533 nm (indicated by the blue line in Fig. 3d), with the phosphorescence emission peak situated at 556 nm (as shown by the green line in Fig. 3d). In the case of CDs-NG-600, the photoluminescence emission peak is positioned at 510 nm (blue line in Fig. 3e), and the phosphorescence emission peak is found at 560 nm (green line in Fig. 3e). The CIE chromaticity coordinates for the CDs-NG-500, CDs-NG-550, and CDs-NG-600 carbon dot samples are (0.36, 0.46), (0.34, 0.46), and (0.28, 0.38), respectively. As depicted in Fig. 3f, with the sintering temperature increasing from 550 °C to 600 °C, the measured phosphorescence lifetime of the CDs was found to increase from 382 ms to 548 ms, while the photoluminescence quantum yield (PLQY) improved from 46.7% to 50.2% (Fig. S11†). By calculation, it can be determined that as the sintering temperature is increased from 500 °C to 600 °C, the ΔEST of CDs-NG rises from 0.05 eV to 0.21 eV, while the lifetime increases from 329 ms to 548 ms (Table 1).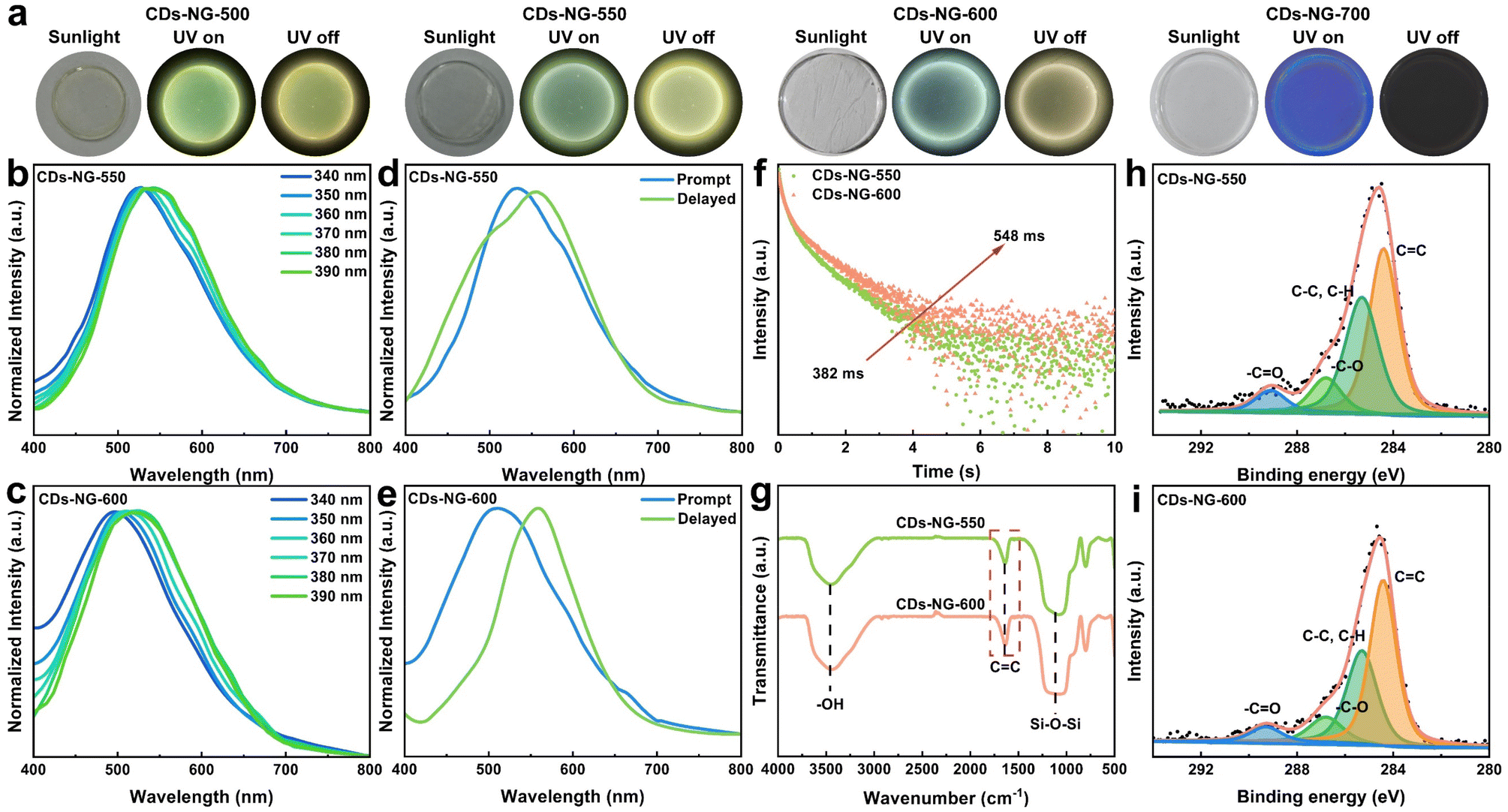 | ||
| Fig. 3 Carbonization modulates TADF and RTP. (a) Images captured in sunlight, under illumination with a 365 nm UV lamp, and in the dark after UV illumination ceases. (b and c) Excitation-dependent photoluminescence of CDs-NG-550 and CDs-NG-600. (d and e) Prompt (blue line) and delayed (green line) spectra of CDs-NG-550 and CDs-NG-600, excited at 360 nm. (f) Time-resolved decay spectra of CDs-NG-550 and CDs-NG-600. (g) FT-IR spectra of CDs-NG-550 and CDs-NG-600. (h and i) XPS spectra of C 1s for CDs-NG-550 and CDs-NG-600. | ||
| Samples | PL (nm) | CIE | QYs (%) | τ (ms) | ΔEST (eV) |
|---|---|---|---|---|---|
| CDs-NG-500 | 538 | (0.36, 0.46) | 46.7 | 329 | 0.05 |
| CDs-NG-550 | 533 | (0.34, 0.46) | 43.7 | 382 | 0.08 |
| CDs-NG-600 | 510 | (0.28, 0.38) | 50.2 | 548 | 0.21 |
To explore the reasons for the variance in luminescence behaviour resulting from the different sintering temperatures, we compared FT-IR and XPS measurements of the CDs-NG-550 and CDs-NG-600 to those of CDs-NG-500. The FT-IR spectra indicate that increasing the temperature from 500 °C to 600 °C leads to a strengthening of the CC bond's stretching vibration peak (circa 1620 cm−1), consistent with a greater degree of carbonization of the CDs (Fig. 3g). Correspondingly, the XPS spectra also suggest that the carbonization degree of the CDs increases with the rise in sintering temperature, based on the greater atomic ratio of CC bonds, compared to other carbon species (Fig. 3h and i). This indicates that the photoluminescence behavior of CDs changes with the sintering temperature, likely due to changes in the degree of carbonization and/or the nature of the CDs-NG interface. The origin of afterglow luminescence is attributable to the effective stabilization of the triplet excited state by the NG matrix, which suppresses intramolecular vibrations and rotations, thereby inhibiting non-radiative decay processes. The increase in the degree of carbonization further enhances the interaction between CDs and NG, leading to changes in the energy gap between the ground state and the triplet state, which in turn affects the emission wavelength and afterglow emission behavior. The enhanced interactions also further suppress intramolecular vibrations and rotations, thereby inhibiting non-radiative decay processes, resulting in an increased lifetime.
To further verify this, we exfoliated the CDs from the CDs-NG-550 and the CDs-NG-600 for comparison. TEM observations confirmed uniformly distributed CDs with average diameters of 4.2 nm and 3.4 nm, respectively. HR-TEM again reveals lattice fringes with a period of 0.21 nm (as depicted in Fig. S8 and S9†). Then we proceeded to perform spectral testing on the CDs. As the sintering temperature increased, under 360 nm excitation, the emission center of the CDs shifted from 450 nm to 390 nm, indicating a pronounced blue shift (Fig. S10†). This significant blue shift is attributed to the changes in the size of the CDs. This further suggests that the sintering temperature has altered the structure of the CDs.
The aforementioned results indicate that by altering the sintering temperature of CDs-NG, we have successfully achieved switchable TADF and RTP afterglow luminescence of CDs, attributable to varying degrees of carbonization within the CDs.
Switchable TADF and RTP by regulating the interface between CDs and nanoporous glass
Doping with heavy metal ions is an effective method for regulating the afterglow luminescence properties of CDs.49 By doping heavy atoms into CDs, the electronic structure and energy level distribution can be altered, thereby affecting the luminescence characteristics, including TADF and RTP. This regulation is primarily based on two principles. Firstly, doping enhances the spin–orbit coupling, which promotes ISC and increases the generation of triplet excitons, crucial for the emission of TADF and RTP. Secondly, doping alters the energy levels and ΔEST values of CDs, enabling switchable TADF and RTP. Through these mechanisms, heavy atom doping provides an effective way to develop efficient TADF or RTP materials with specific luminescence characteristics.50,51 In our work, sol–gel processing offers the advantage of achieving homogeneous mixing at the molecular level, providing a possibility for the in situ doping of heavy metal ions within the glass. A series of Pb2+-doped CDs-NG (Pb-CDs-NG) with varying Pb2+ doping concentrations were prepared by adding lead acetate to the precursor solution, and these samples were also found to exhibit nanoporous characteristics (Fig. S12†). As shown in Fig. 4a, under 360 nm excitation, the emission center of 0.01% Pb-CDs-NG is at 500 nm, a significant blue shift compared to the 538 nm of CDs-NG-500. Moreover, as the doping concentration of Pb2+ increases from 0.01% to 1.00%, the emission center of Pb-CDs-NG further red shifts to 550 nm. This may be due to the strong coupling between Pb2+ and CDs enhancing the stability of the triplet state within the CDs. As the amount of Pb doping increases, the triplet state of the CDs becomes more stable. Then, we tested the delayed fluorescence spectrum of Pb-CDs-NG. Upon excitation at 360 nm, the afterglow emission center shifted from 550 nm to 580 nm with the increase of lead doping concentration from 0.01% to 1.0%. Concurrently, the lifetime decreased from 290 ms to 20 ms. Compared with 500-CDs-NG, the luminescence and lifetime of Pb-CDs-NG have undergone significant changes. Compared with the ΔEST of 0.05 eV for CDs-NG-500, the ΔEST of 0.01% Pb-CDs-NG increased to 0.23 eV, while the PLQY improved from 50.2% to 56.2% (Fig. S13†). Similar effects can also be observed for Pb-CDs-NG sintered at other temperatures (Fig. S14†). This indicates that we have successfully achieved NG-confined CDs with switchable TADF and RTP by Pb2+ doping.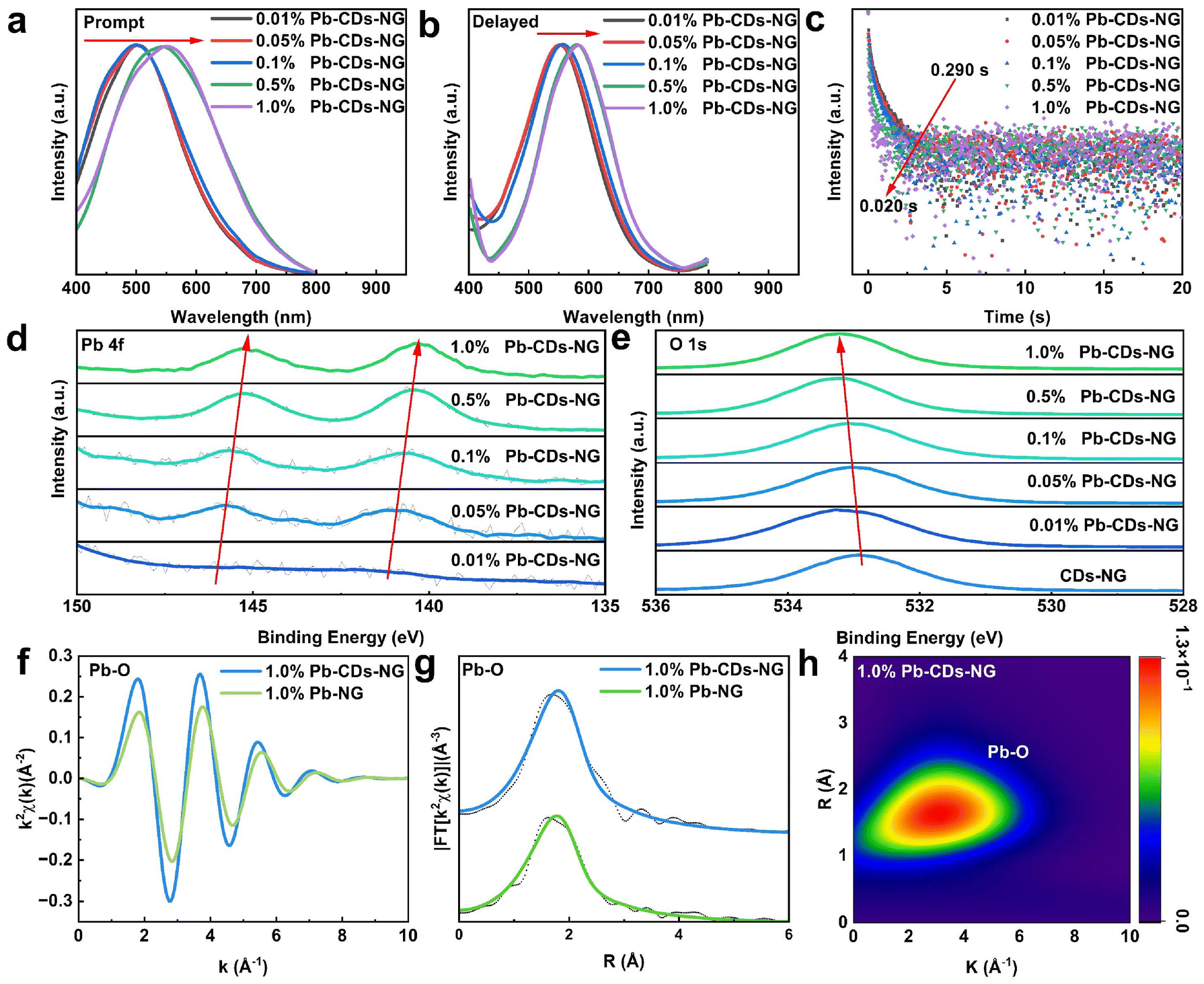 | ||
| Fig. 4 Switchable TADF and RTP by interface regulation. (a) Steady-state photoluminescence spectra of a series of Pb-CDs-NG, excited at 360 nm. (b) Delayed photoluminescence spectra of a series of Pb-CDs-NG, excited at 360 nm. (c) Time-resolved decay spectra of a series of Pb-CDs-NG. (d) XPS spectra of Pb 4f of a series of Pb-CDs-NG. Here, we have smoothed the curve to enhance the clarity of the data presentation. (e) XPS spectra of O 1s of CDs-NG and a series of Pb-CDs-NG. (f) EXAFS in the k-space of 1.0% Pb-CDs-NG and 1.0% Pb-NG. (g) k2-weighted Fourier transform EXAFS moduli of 1.00% Pb-CDs-NG and 1.00% Pb-NG. (h) Counterplot Morlet (5,1) wavelet transform amplitude of 1.0% Pb-CDs-NG. | ||
The mechanism of Pb2+ doping in NG-confined CDs was studied in detail by XPS, ICP and extended X-ray absorption fine structure (EXAFS) spectra. X-ray photoelectron spectroscopy (XPS) of pure Pb2+ doped NG (Pb-NG) (Fig. S15†) clearly reveals the Pb 4f core level peaks, which indicates that Pb2+ is integrated into the Al2O3–SiO2 glass network due to the molecular-level doping uniformity provided by the sol–gel method. Within the entire network of Pb-NG, Pb2+ is uniformly dispersed, hence existing in the form of Pb–O bonds.52Fig. 4d shows the Pb 4f core level for a series of Pb-CDs-NG with different Pb-doping levels. Apparently, as the Pb-doping increases, the binding energy of the Pb 4f peaks correspondingly decreases. Meanwhile, the O 1s core level region of the CDs-NG and Pb-CDs-NG shows a shift to higher binding energies with increased Pb-doping (Fig. 4e). This shift in binding energy may reflect electron density being drawn towards Pb, indicating a new bonding environment between Pb and CDs, which was further investigated by EXAFS. In order to identify the location of Pb2+ within the NG-confined CD system, we have exfoliated CDs from 1.0% Pb-CDs-NG for ICP measurement. 1.0% Pb-CDs-NG was dissolved in 1 M NaOH. Following centrifugation, the CDs were predominantly found in the precipitate. Both the supernatant and the precipitate post-centrifugation were tested by ICP. As shown in Table S1,† the concentration of Pb in the supernatant is 220 times higher than that in the precipitate, indicating that Pb2+ ions are predominantly distributed in the glass network rather than in the CDs. Based on the ICP test, the chemical bonding formed between CDs and Pb2+ revealed by XPS spectra is probably at the confined interface between NG and CDs.
To further validate the interfacial bonding between Pb2+ and CDs, we conducted extended X-ray absorption fine structure (EXAFS) tests on Pb within both Pb-CDs-NG and Pb-NGs. The k2-weighted transmission Pb LIII-edge (k-space) EXAFS spectra of 1.0% Pb-CDs-NG and 1.0% Pb-NG samples are presented in Fig. 4f. Both spectra exhibit negligible intensity in the high k range, indicating that the Pb ions are either highly dispersed or in a highly disordered local environment. This observation is further illustrated in the k2-weighted Fourier transform EXAFS moduli (Fig. 4g), where no significant intensity is detected beyond the first coordination shell. To gain a deeper understanding of the coordination environment around the Pb ions, fitting of the Pb EXAFS spectra was carried out in R space for the first-shell nearest neighbours (NN). Due to oxygen and carbon being close to each other in atomic number (Z), EXAFS is insufficient to resolve their contributions separately. Consequently, a Pb–O single scattering path is utilised to represent their combined contribution. The results (Table S2†) indicate that the Pb first-shell NN bond length in 1.0% Pb-CDs-NG (2.44 ± 2 Å) is significantly longer than that in 1.0% Pb-NG (2.40 ± 2 Å). The alteration of the first-shell coordination of Pb2+ provides direct evidence of Pb bonding to CDs.
Overall, the XPS and EXAFS provide evidence that the doping with heavy metal ions modulates the confined interface between CDs and the NG matrix. The doping of Pb2+ has likely replaced some of the original Si–O–C bonds with Pb–O–C bonds. This modulation of the interfacial interactions between CDs and NG is shown to successfully increase the ΔEST value of NG-confined CDs from 0.05 eV to 0.21 eV, achieving switchable TADF and RTP of NG-confined CDs.
Conclusions
We have successfully demonstrated a facile strategy for the preparation of transparent CD-based afterglow materials with switchable TADF and RTP by a sol–gel derived process that produces CDs-NG. During the gel–glass transition by a sintering process, NG-confined CDs are obtained by the simultaneous formation of NG and CDs, which is a result of the decomposition and carbonization of carboxyl groups. The unique interfacial nano-confined effect not only effectively incorporates CDs into NG but also ensures the stability of the triplet excited state and the afterglow luminescence. Then, we have managed to achieve switchable TADF and RTP of NG-confined CDs by varying the sintering temperature of CDs-NG, attributing this regulation to the differing degrees of carbonization within the NG-confined CDs. The synthesized CDs-NG-600 exhibit superior quantum yields and exceptionally long phosphorescence lifetimes, reaching 50.2% and 548 ms, respectively. Moreover, switchable TADF and RTP of NG-confined CDs have been achieved by Pb2+ doping within the glass, which modulates the interface between CDs and NG. The formation of Pb–O–C interfacial bonding enables the precise regulation of the NG-confined CDs. This discovery of a new class of CD-based transparent afterglow materials with ultralong lifetimes and high efficiency is developed as a proof-of-concept, which we believe will shed new light on both fundamental understanding and future practical applications of afterglow materials.Author contributions
J. H., J. B. C., and R. S. W. oversaw and managed the research. P. W. W., P. X. C., J. C. C, H. Y. C., Q. Y., Z. X. X., J. J. Z., L. Z., J. B. C., R. S. W., and J. H. designed the experiments. P. W. W., J. C. C., P. X. C., Q. Y., and H. Y. C. performed the experiments. P. W. W., P. X. C., R. S. W., J. H., and J. B. C. wrote and revised the manuscript with the help of other authors.Data availability
The findings of this study are underpinned by data accessible in the ESI† provided with this publication.Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this manuscript.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 52272013 and 22104105). J. B. C. also acknowledges the Jiangsu Provincial Department of Science and Technology for the Leading Technology Basic Research Major Project (grant no. BK20232041), the Natural Science Foundation of Jiangsu Province (grant no. BK20210702) and the Leading Talents of Innovation and Entrepreneurship of Gusu (grant no. ZXL2022515). J. H. acknowledges the Royal Society Sino-British Fellowship (IES/R1/241269) and the International Partnership Program of the Chinese Academy of Sciences (111GJHZ2022024FN). R. S. W. and P. X. C. acknowledge funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (EXISTAR, grant agreement no. 950598) and a UKRI Future Leaders Fellowship (MR/V024558/1). The authors acknowledge the B18 Core XAS beamline at Diamond Light Source and Energy Materials BAG (University of Kent) for the provision of synchrotron radiation beamtime (session SP31218-5).References
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y.-H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B.-H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed
.
- J. Liu, N. Wang, Y. Yu, Y. Yan, H. Zhang, J. Li and J. Yu, Sci. Adv., 2017, 3, e1603171 CrossRef PubMed
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed
- O. Bunjes, D. Hedman, A. Rittmeier, L. A. Paul, I. Siewert, F. Ding and M. Wenderoth, Sci. Adv., 2022, 8, eabq7776 CrossRef CAS PubMed
- H. Yang, G. Ji, M. Choi, S. Park, H. An, H.-T. Lee, J. Jeong, Y. D. Park, K. Kim, N. Park, J. Jeong, D.-S. Kim and H.-R. Park, Sci. Adv., 2024, 10, eadm7315 CrossRef CAS PubMed
- M. A. Ahsan, A. R. Puente Santiago, Y. Hong, N. Zhang, M. Cano, E. Rodriguez-Castellon, L. Echegoyen, S. T. Sreenivasan and J. C. Noveron, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed
- J. Cai, A.-A. Liu, X.-H. Shi, H. Fu, W. Zhao, L. Xu, H. Kuang, C. Xu and D.-W. Pang, J. Am. Chem. Soc., 2023, 145, 24375–24385 CrossRef CAS PubMed
- G. Chen, H. Zhu, G. Liu, G. Liu and W. Jin, Angew. Chem., Int. Ed., 2024, e202418649 Search PubMed
- S. Liang, S. He, M. Zhang, Y. Yan, T. Jin, T. Lian and Z. Lin, J. Am. Chem. Soc., 2022, 144, 12901–12914 CrossRef CAS PubMed
- Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Adv. Mater., 2022, 34, 2105958 CrossRef CAS PubMed
- J. Cui, Y. E. Panfil, S. Koley, D. Shamalia, N. Waiskopf, S. Remennik, I. Popov, M. Oded and U. Banin, Nat. Commun., 2019, 10, 5401 CrossRef CAS PubMed
- J. Cui, S. Koley, Y. E. Panfil, A. Levi, Y. Ossia, N. Waiskopf, S. Remennik, M. Oded and U. Banin, J. Am. Chem. Soc., 2021, 143, 19816–19823 CrossRef CAS PubMed
- J. Cui, S. Koley, Y. E. Panfil, A. Levi, N. Waiskopf, S. Remennik, M. Oded and U. Banin, Angew. Chem., 2021, 133, 14588–14593 CrossRef
- P. Wang, Z. Hu, P. Cong, F. Zhou, Q. Yue, Z. Xue, C. Lin, Y. Jiang, J. Du, A. Pan, L. Zhang, J. Cui and J. He, Adv. Opt. Mater., 2024, 12, 2400630 CrossRef CAS
- G.-S. Zheng, C.-L. Shen, C.-Y. Niu, Q. Lou, T.-C. Jiang, P.-F. Li, X.-J. Shi, R.-W. Song, Y. Deng, C.-F. Lv, K.-K. Liu, J.-H. Zang, Z. Cheng, L. Dong and C.-X. Shan, Nat. Commun., 2024, 15, 2365 CrossRef CAS PubMed
- Z. Guo, Y. Bian, L. Zhang, J. Zhang, C. Sun, D. Cui, W. Lv, C. Zheng, W. Huang and R. Chen, Adv. Mater., 2024, 36, 2409361 CrossRef CAS PubMed
- K. Jiang, Y. Wang, C. Lin, L. Zheng, J. Du, Y. Zhuang, R. Xie, Z. Li and H. Lin, Light: Sci. Appl., 2022, 11, 80 CrossRef CAS PubMed
- J. Sun, Z. Sun, Z. Wang, N. Wang, Y. Han, L. Zhang, B. Zhang and X. Zhang, Adv. Opt. Mater., 2024, 12, 2302542 CrossRef CAS
- Q. Lou, N. Chen, J. Zhu, K. Liu, C. Li, Y. Zhu, W. Xu, X. Chen, Z. Song, C. Liang, C. Shan and J. Hu, Adv. Mater., 2023, 35, 2211858 CrossRef CAS PubMed
- K. Wang, L. Qu and C. Yang, Small, 2023, 19, 2206429 CrossRef CAS PubMed
- H. Shi, Y. Wu, J. Xu, H. Shi and Z. An, Small, 2023, 19, 2207104 CrossRef CAS PubMed
- J. Liu, H. Zhang, N. Wang, Y. Yu, Y. Cui, J. Li and J. Yu, ACS Mater. Lett., 2019, 1, 58–63 CrossRef CAS
- Y. Liu, X. Kang, Y. Xu, Y. Li, S. Wang, C. Wang, W. Hu, R. Wang and J. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 22363–22371 CrossRef CAS PubMed
- K. Chen, Y. Jiang, Y. Zhu, Y. Lei, W. Dai, M. Liu, Z. Cai, H. Wu, X. Huang and Y. Dong, J. Mater. Chem. C, 2022, 10, 11607–11613 RSC
- J. Liu, Y. Luo, Z. Ran, F. Wang, M. Sun, Y. Luo, J. Zhuang, X. Zhang, B. Lei, Y. Liu and C. Hu, Chem. Eng. J., 2023, 474, 145597 CrossRef CAS
- J. Ràfols-Ribé, P.-A. Will, C. Hänisch, M. Gonzalez-Silveira, S. Lenk, J. Rodríguez-Viejo and S. Reineke, Sci. Adv., 2018, 4, eaar8332 CrossRef PubMed
- Y. Wei, H. Ebendorff-Heidepriem and J. (Tim) Zhao, Adv. Opt. Mater., 2019, 7, 1900702 CrossRef CAS
- J. Lin, Y. Lu, X. Li, F. Huang, C. Yang, M. Liu, N. Jiang and D. Chen, ACS Energy Lett., 2021, 6, 519–528 CrossRef CAS
- T. Ma, H. Chen, L. Yin, F. Lei, Y. Shi, J. Xie, L. Zhang and L. Fan, J. Rare Earths, 2023, 41, 190–199 CrossRef CAS
- Y. Zhang, L. Fan, X. Wang, J. Xie and Y. Shi, J. Rare Earths, 2023, 41, 1696–1702 CrossRef CAS
- C. J. Brinker and G. W. Scherer, J. Non-Cryst. Solids, 1985, 70, 301–322 CrossRef CAS
- L. Zhang, A. Bögershausen and H. Eckert, J. Am. Ceram. Soc., 2005, 88, 897–902 CrossRef CAS
- C. C. De Araujo, L. Zhang and H. Eckert, J. Mater. Chem., 2006, 16, 1323 RSC
- A. R. Allu, S. Balaji, K. Illath, C. Hareendran, T. G. Ajithkumar, K. Biswas and K. Annapurna, RSC Adv., 2018, 8, 14422–14433 RSC
- J. He, D. Avnir and L. Zhang, Acta Mater., 2019, 174, 418–426 CrossRef CAS
- C. M. E. Phelan, E. Björklund, J. Singh, M. Fraser, P. N. Didwal, G. J. Rees, Z. Ruff, P. Ferrer, D. C. Grinter, C. P. Grey and R. S. Weatherup, Chem. Mater., 2024, 36, 3334–3344 CrossRef CAS PubMed
- E. Björklund, C. Xu, W. M. Dose, C. G. Sole, P. K. Thakur, T.-L. Lee, M. F. L. De Volder, C. P. Grey and R. S. Weatherup, Chem. Mater., 2022, 34, 2034–2048 CrossRef PubMed
- D. A. Sousa, M. N. Berberan-Santos and J. V. Prata, Chem. – Eur. J., 2024, 30, e202302955 CrossRef CAS PubMed
- Q. Zhang, R. Wang, B. Feng, X. Zhong and K. Ostrikov, Nat. Commun., 2021, 12, 6856 CrossRef CAS PubMed
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed
- J. Li, X. Rao, F. Xiang, J. Wei, M. Yuan and Z. Liu, Analyst, 2018, 143, 2083–2089 RSC
- D. W. Boukhvalov, V. Y. Osipov, D. Murzalinov, A. Serikkanov and H. Bi, Carbon, 2024, 225, 119101 CrossRef CAS
- J. Liu, N. Wang, Y. Yu, Y. Yan, H. Zhang, J. Li and J. Yu, Sci. Adv., 2017, 3, e1603171 CrossRef PubMed
- B. Wang, Y. Yu, H. Zhang, Y. Xuan, G. Chen, W. Ma, J. Li and J. Yu, Angew. Chem., 2019, 131, 18614–18619 CrossRef
- B. Wang, Y. Mu, H. Zhang, H. Shi, G. Chen, Y. Yu, Z. Yang, J. Li and J. Yu, ACS Cent. Sci., 2019, 5, 349–356 CrossRef CAS PubMed
- B. Wang, J. Yu, L. Sui, S. Zhu, Z. Tang, B. Yang and S. Lu, Adv. Sci., 2021, 8, 2001453 CrossRef CAS PubMed
- Z. Wang, J. Shen, B. Xu, Q. Jiang, S. Ming, L. Yan, Z. Gao, X. Wang, C. Zhu and X. Meng, Adv. Opt. Mater., 2021, 9, 2100421 CrossRef CAS
- K. Jiang, Y. Wang, C. Cai and H. Lin, Adv. Mater., 2018, 30, 1800783 CrossRef PubMed
- X. Yang, G. I. N. Waterhouse, S. Lu and J. Yu, Chem. Soc. Rev., 2023, 52, 8005–8058 RSC
- H. Shi, Y. Wu, J. Xu, H. Shi and Z. An, Small, 2023, 19, 2207104 CrossRef CAS PubMed
- Y. Sun, X. Zhang, J. Zhuang, H. Zhang, C. Hu, M. Zheng, B. Lei and Y. Liu, Carbon, 2020, 165, 306–316 CrossRef CAS
- C. Zhao, J. Liu, B. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2003619 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures & tables. See DOI: https://doi.org/10.1039/d4nr04835k |
| ‡ These authors have contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |