
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal gold-palladium-platinum alloy nanospheres with tunable compositions and defined numbers of atoms†
Sergio
Triviño-Sánchez
 a,
Ren
Xu
a,
Jesús
González-Izquierdo
a,
Luis
Bañares
a,
Israel
Cano
b,
Jorge
Pérez-Juste
c,
Andrés
Guerrero-Martínez
*a and
Guillermo
González-Rubio
*a
a,
Ren
Xu
a,
Jesús
González-Izquierdo
a,
Luis
Bañares
a,
Israel
Cano
b,
Jorge
Pérez-Juste
c,
Andrés
Guerrero-Martínez
*a and
Guillermo
González-Rubio
*a
aDepartamento de Química Física, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: ggrubio@ucm.es; aguerrero@qui.ucm.es
bDepartamento de Química Inorgánica, Universidad Complutense de Madrid, 28040 Madrid, Spain
cDepartamento de Química Física, Universidad de Vigo, 36310 Vigo, Spain
First published on 12th March 2025
Abstract
The combination of different metals into a discrete colloidal nanocrystal (NC) lattice to form solid solutions can result in synergetic and non-additive effects, leading to physicochemical properties distinct from those observed in monometallic NCs. However, these features are influenced by parameters that are challenging to control simultaneously using conventional synthesis methods, including composition, morphology, size, and elemental distribution. In this study, we present a methodology that exploits seed-mediated growth routes and pulsed laser-induced ultrafast heating to synthesize bimetallic and trimetallic colloidal alloy NCs with tailored compositions, well-defined spherical morphologies, and precise control over the number of atoms per NC lattice. Initially, core–shell heterostructures with adjustable compositions and ca. 107 atoms per NC are formed, using Au as the core material and Pd and Pt as the shell metals. In the subsequent stage, ultrafast heating of the heterostructure lattice via nanosecond pulsed laser irradiation facilitates the formation of colloidal AuPd, AuPt and AuPdPt alloy nanospheres. The ability of the proposed synthesis route to produce multimetallic NCs with distinct compositions, consistent morphology, and a fixed number of atoms provides exciting opportunities to investigate how multimetallic NC composition influences catalytic properties. Accordingly, using the catalytic reduction of nitrophenol as a reaction model, we observed a significantly enhanced catalytic performance for AuPdPt NCs compared to AuPd and AuPt NCs.
 Guillermo González-Rubio | Dr González-Rubio studied Chemistry at Complutense University (UCM) of Madrid, earning his PhD in 2017 under Prof. Luis Liz-Marzán and Prof. Andrés Guerrero-Martínez. He completed postdoctoral studies at the University of Konstanz with Prof. Helmut Cölfen, supported by an Alexander von Humboldt Foundation fellowship. Since 2023, he has been an Atracción de Talento Research Fellow at UCM and was recently awarded an ERC Starting Grant to develop advanced methods for synthesizing complex multielemental nanocrystals. His research focuses on designing functional surface ligands and utilizing ultrafast pulsed laser irradiation for synthesizing, stabilizing, and assembling high-quality nanocrystals for optical and catalytic applications. |
Introduction
Metals and their mixtures play a fundamental role in modern societies as critical materials for a wide range of technologies, from energy and communication to transportation.1–4 In nanotechnology, manipulating and combining metals at the nanoscale has been revealed as a fruitful strategy to create materials with functional properties not found in bulk, including strong light–matter interactions or high catalytic activity.5–7 For example, Pt and Ir nanocrystals (NCs) are benchmarking catalysts for green hydrogen production via (electro)catalytic water splitting processes.8–11 Moreover, alloying such noble metals with more abundant transition elements offers the possibility of surpassing the catalytic properties of their monometallic counterparts while reducing their high cost. The high chemical stability of noble metals also facilitates their recovery and reuse, a critical aspect for successfully implementing a circular economy in the quest for sustainable societies.12Despite the potential of this class of nanomaterials, their synthesis represents a significant challenge, especially in the case of multimetallic NCs. An ideal synthesis method should provide simultaneous control over the critical aspects that determine the physicochemical features of metal NCs: shape, size, crystallinity, composition, elemental distribution, and surface chemistry.7,8 Bottom-up wet-chemical methods introduced in the late 1990s and early 2000s have uncovered numerous routes for growing single-component metal NCs.13–15 Among these, colloidal strategies offer unmatched control over the nucleation and growth processes of metal NCs by reducing metal ions and complexes under specific experimental aqueous conditions.16–20 Thereby, the formation of metal NCs with narrow size distributions, defined morphologies and crystal defects, tailored surface chemistry, and distinct self-assembly behaviours (e.g., for their integration in solid-state devices) can be successfully achieved.
However, in multimetallic NCs, the need to control elemental distribution imposes additional requirements that are difficult to meet via colloidal syntheses. The different reduction potentials of metal elements often lead to distinct reduction rates, resulting in elemental segregation.7,21 Indeed, this phenomenon can be advantageous for the synthesis of heterostructured multimetallic NCs such as core–shell or Janus structures, with novel or enhanced catalytic, optical, or magnetic properties.7,22 However, undesired segregation effects can often prevent the growth of targeted colloidal alloy NCs. Moreover, the temperatures reached during wet-chemical synthesis are limited by the solvent boiling point (typically below 350 °C), which can significantly restrict the formation of alloys that require high activation energy for elemental mixing.6,7,23–25
Numerous synthesis strategies have been explored in the last decade for overcoming the kinetic and thermodynamic barriers to alloy NC growth. For example, the growth of colloidal core–shell structures followed by thermal annealing on solid supports facilitates the formation of binary and ternary alloys with random and ordered (intermetallic) atomic distributions.26,27 Although this approach provides fine control over NC size, composition, and elemental distribution, the colloidal nature of the obtained alloy products is often lost. Other strategies involve the direct co-reduction of various metal precursors on solid carbon-based substrates using fast Joule heating.28,29 The thermal shock synthesis route has been demonstrated to be successful for the growth of high-entropy alloy NCs. Unfortunately, governing nucleation and growth events is not straightforward in this case, which may explain the difficulties encountered in tailoring size distributions. Similar challenges are faced by laser synthesis methods, where pulsed laser ablation of alloy targets enables the formation of colloidal alloy NCs with limited size control.30–32 In some cases, colloidal routes have shown great potential to produce alloy NCs with narrow size dispersity and defined compositions in the liquid phase.10,33,34 However, the optimized experimental conditions tend to work for a specific final particle dimensions, making it difficult to tune both composition and size.
Aiming to address current limitations in alloy NC syntheses, we propose in this work a bottom-up wet-chemical route where nanosecond pulsed laser irradiation assists in the production of alloy AuPdPt NCs with tunable compositions while size and shape are tightly controlled. Specifically, we have developed a multistep seed-mediated growth strategy to produce NCs of Au, Pd, and Pt with core–shell and core–shell-shell heterostructures (Au@Pd, Au@Pt and Au@Pd@Pt NCs), a fixed number of atoms per particle, 7 atomic% Au, and varying proportions of Pd and Pt (Fig. 1). Subsequently, nanosecond laser irradiation was used to activate interdiffusion phenomena and reshaping effects, resulting in the formation of smooth AuPd, AuPt, and AuPdPt alloy spheres, as confirmed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray (EDX) spectroscopy. Notably, since the number of atoms per particle is kept approximately constant, we were able to study the impact of alloy composition on the catalytic behaviour of synthesized NCs. The pulsed laser-assisted synthesis route described herein represents an excellent opportunity to fabricate colloidal alloy NCs with on-demand sizes and compositions, which are critical features of alloy nanomaterials directly impacting their potential applications in technology.
 | ||
| Fig. 1 Schematic view of the synthesis of AuPdPt alloy NCs. After the synthesis of the Au core (1), Pd is first deposited to form a cubic Pd shell (2) followed by the reduction of Pt ions, leading to the growth of a dendritic Pt shell (3). The resulting core–shell–shell heterostructures are then rapidly heated with ns-pulsed laser irradiation to induce the formation of alloy NCs. | ||
Experimental
Chemicals
All starting materials were used without further purification: 1-decanol (n-decanol, 98%), hexadecyltrimethylammonium bromide (CTAB, ≥99.9%), hexadecyltrimethylammonium chloride (CTAC, 99.0%), sodium borohydride (NaBH4, 99%), hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O, ≥99.9%), sodium tetrachloropalladate (Na2PdCl4, 98%), silver nitrate (AgNO3, ≥99.0%), potassium hexachloroplatinate(IV) (K2PtClz6, ca. 40% Pt), L-ascorbic acid (≥99%) and 4-nitrophenol, all purchased from Merck. Milli-Q grade water (resistivity 18.2 MΩ cm at 25 °C) was used in all experiments.Synthesis of 30 nm Au NCs
A 10 mL aqueous solution of CTAC (25 mM) was prepared for the synthesis of single-crystal Au nanospheres. To this solution, 100 μL of a 50 mM HAuCl4 solution (0.5 mM) was added under magnetic stirring. The Au3+ ions were allowed to form a complex with CTAC by heating the mixture at 30 °C for 5 minutes (min). Subsequently, 40 μL of a 100 mM ascorbic acid solution was added, changing the colour from deep yellow to pale yellow. Small volumes (2 or 1 μL) of the 100 mM ascorbic acid solution were then added to reduce ca. 99% Au3+ ions to Au+, as monitored via the decrease in absorbance at 320 nm (corresponding to Au3+-CTAC complex absorbance band maxima) remained below 0.05 (1 cm path length). This procedure ensures the presence of a small concentration of Au3+ ions (0.5 mM HAuCl4vs. 0.495 mM ascorbic acid) to avoid undesired homogeneous nucleation phenomena. Then, 25 μL of a 10 mM AgNO3 solution (0.025 mM) was added, followed by the injection of 9.5 × 4.8 nm Au nanorods (final Au0 concentration of 0.006 mM; 9.5 × 4.8 nm Au nanorods were synthesized according to a modified protocol described by González-Rubio et al.35) as seeds to trigger the formation of single-crystal Au nanospheres. The mixture was shaken by hand and heated to 80 °C in a water bath while stirring to ensure homogeneous heating. After the growth mixture reached the targeted temperature (ca. 10 min), the magnet was removed, and the solution was left at 80 °C in the oven overnight. The obtained deep red colloid was centrifuged in 2 mL tubes at 9000 rpm for 60 min, and the precipitate was redispersed with 5 mM CTAC solution. This process was repeated twice, and the precipitated Au nanospheres were redispersed in 5 mM CTAC solution to obtain a final [Au0] of 8 mM (as determined from the Abs400 nm: 1.92, optical path of 0.1 cm).Synthesis of Au@Pd NCs
The seeded growth method, with some modifications, was used to prepare core–shell Au@Pd NCs.36 Specifically, 8 mL of a 5 mM Na2PdCl4 (0.4 mM) solution and 1 mL of a 100 mM ascorbic acid (1 mM) solution were added to 100 mL of a freshly prepared CTAB solution (50 mM). Then, a given volume of the prepared 8 mM 30 nm single-crystal Au nanospheres solution was added to obtain Au@Pd with different compositions: 375 μL for Au7@Pd93, 550 μL for Au10@Pd90, and 1.05 mL for Au18@Pd82 (see Table S1† for more details on the experimental feeding ratios). The growth mixture was then heated on an aluminium block or in a water bath at 65 °C under stirring at 300–500 rpm. After 30 min, the stirring was stopped, and the growth solution was left undisturbed on an aluminium block or in an oven at 65 °C for at least 12 hours. The resulting Au@Pd NCs were sedimented at 6600–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 RFC for 15 min and redispersed in 5 mM CTAC solution. This procedure was repeated twice, and the NCs were finally redispersed in a 2 mM CTAC solution.
600 RFC for 15 min and redispersed in 5 mM CTAC solution. This procedure was repeated twice, and the NCs were finally redispersed in a 2 mM CTAC solution.
Synthesis of Au@Pt and Au@Pd@Pt NCs
A procedure similar to that described for Pd shell growth was used for Pt shell growth.36 Specifically, 8 mL of a 5 mM K2PtCl6 (0.4 mM) solution and 5 mL of a 100 mM ascorbic acid (5 mM) solution were added to 100 mL of a freshly prepared CTAB solution (50 mM). Then, a given volume of the Au nanospheres ([Au0] = 8 mM; 376 μL), Au18@Pd82 NCs ([Pd0] = 8 mM; 2.75 mL) or Au10@Pd90 NCs ([Pd0] = 8 mM; 10.5 mL) was added to grow Au7@Pt93 NCs, Au7@Pd33@Pt60 NCs or Au7@Pd63@Pt30 NCs, respectively (see Table S1† for more details on the experimental feeding ratios). The growth mixture was then heated on an aluminium block at 65 °C under stirring at 300–500 rpm. After 30 min, the stirring was stopped, and the growth solution was left undisturbed on an aluminium block or in an oven at 65 °C for at least 12 hours. The resulting Au@Pt NCs were sedimented at 6600–10600 RFC for 15 min and redispersed in 2 mM CTAC solution. This procedure was repeated twice, and the NCs were finally redispersed with 5 mM CTAC solution. To determine the concentration of Pd0, UV-vis-NIR spectra of the as-synthesized Au10@Pd90 and Au18@Pd82 NCs were recorded, and the absorbance at 500 nm (or other wavelength above 300 nm, where ascorbic acid absorption band does not interfere with the plasmon bands) was determined. Since the reduction of Pd2+ was found to be quantitative (as supported by the EDX analysis of the Au:Pd ratios of the final Au@Pd NCs, corresponding to the feeding ratios; see Results and discussion section), the measured absorbance at 500 nm corresponds to 0.4 mM of Pd (i.e., the concentration of Pd2+ in the growth solution). This relationship was then exploited to adjust the concentration of Pd0 to 8 mM in the Au18@Pd82 and Au10@Pd90 NCs solutions after the purification stage via centrifugation.
Irradiation experiments
Au@Pt, Au@Pd and Au@Pd@Pt NCs with different compositions were irradiated using 5 ns laser pulses at 532 nm, generated by an amplified Nd:YAG laser system. Non-focused beam irradiation regimes were applied. The selected irradiation fluence was 64 J m−2 with a beam diameter of ca. 1 cm and irradiation times of 30 min. The NCs were first redispersed in a 5 mM CTAC solution. The absorbance was adjusted to a range between 0.4 and 0.6 at a wavelength of 500 nm. Subsequently, 30 mL of the sample was irradiated in an Erlenmeyer flask by directing the laser from above under constant stirring at 300 rpm using a magnetic stir bar, and at room temperature.Catalytic reduction of 4-nitrophenol to 4-aminophenol
In a 1 cm quartz cuvette, 1.34 mL of Milli-Q water and 13 μL of a 9.61 mM aqueous 4-nitrophenol solution (54.32 μM) were added. Then, 750 μL of a freshly prepared 9.1 mM NaOH solution (2.967 mM) containing either 1.74 mg (20 mM), 3.48 mg (40 mM), 5.22 mg (60 mM), 6.96 mg (80 mM) or 8.7 mg (100 mM) of NaBH4 was added, followed by the addition of 150 μL of a 5 mM CTAC solution and 50 μL of a 1.34 mM AuPt, AuPd or AuPdPt NCs solution (0.0292 mM). The reduction process was monitored via UV-vis spectroscopy. The consumption of 4-nitrophenol was identified by tracking the absorbance changes over time at 400 nm, where the maximum variations occur. The apparent rate constants were calculated from the absorbance vs. time plots using the first-order rate law equation:37| At = Af + ΔA·e−kt | (1) |
An exact concentration of metal atoms, 0.0292 mM, was used for all experiments. The concentration of metal atoms was calculated by determining the mass of NCs per mL of solution using an ultra-precision mass balance. After determining the weight of a 2 mL Eppendorf tube, 1 mL of the selected NC dispersion was centrifuged and redispersed twice in water to remove any remaining CTAC. Subsequently, the sample was redispersed in ethanol and centrifuged again to facilitate the removal of CTAC and water. The sample was then dried to evaporate the ethanol, and the Eppendorf tube was finally weighed to determine the mass of NCs. This process was repeated three times to ensure reliable results.
Results and discussion
The envisaged methodology for alloy NC synthesis seeks simultaneous control over several critical aspects: composition, elemental distribution, size and morphology. Among advanced synthesis methods, colloidal seed-mediated growth routes are particularly suitable for controlling the size or number of atoms per NC.20 This is achieved through spatiotemporal separation of the early stages of NC formation, i.e., homogeneous nucleation and the growth event. When different metals are consecutively deposited onto the initial seeds, such a bottom-up wet-chemical strategy also facilitates the growth of multimetallic heterostructures with targeted compositions. For these reasons, we have adopted a seed-mediated approach to synthesize Au@Pd@Pt NCs, which constitutes one of the two cornerstones of the proposed methodology (Fig. 1). The other involves the use of nanosecond pulsed laser irradiation to ensure a homogeneous atomic distribution of Au, Pd and Pt within the NC lattice, facilitating the formation of colloidal alloy AuPdPt NCs.Additionally, laser excitation enables the removal of facets and roughness, favouring the formation of NCs with smooth spherical morphology.38 It is important to note that metals such as Au, Pd and Pt crystallize in a highly symmetric fcc lattice and, accordingly, they tend to grow into highly faceted shapes typically enclosed by low-Miller index facets such as cubes ({100}), triangles ({111}), or cuboctahedra ({100} and {111}).20,25,39 Not surprisingly, most protocols for the synthesis of noble metal smooth nanospheres are limited to dimensions typically below 10 nm or involve etching processes to remove facets, corners and vertices.40–42
Morphology control in multimetallic NCs, including the growth of colloidal alloy nanospheres, can be even more challenging.7,22,43 The main reasons for this can be related to the interactions between different metal constituents (i.e., having distinct bond energies, crystal structures or atomic radii) and metal-surface ligands (i.e., molecules and capping agents used to control colloidal stability and surface energies), leading to morphology evolution strongly dependent on the metal NC composition.7,25,43–45 Moreover, due to the coexistence of different reduced and oxidized metal species during the colloidal growth process, galvanic replacement reactions can also play an active role in the evolution of multimetallic NC morphology.7,46,47 For these reasons, pulsed laser irradiation of multielemental NCs represents an exciting approach to overcome the limitations of bottom-up wet-chemical methods in the synthesis of alloy NCs with homogeneous elemental distributions and smooth spherical morphology.
Hence, we combined the capacity of seed-mediated routes to control NC quality, composition and number of atoms per lattice with the ability of pulsed laser irradiation to induce the alloying process.48,49 A number of atoms per NCs of ca. 107 was selected, and the material of choice for the core was Au owing to its high reduction potential: (i) ensuring high chemical stability, and (ii) preventing galvanic replacement reactions during the growth of Pd and Pt shells (metals with lower reduction potentials), which could eventually complicate composition control.
Moreover, Au offers the possibility of finely controlling the nucleation and growth of Au NCs with narrow size distributions between 1 and 200 nm, i.e., a size tunability not offered by other noble metals.35,40,50 Thus, we hypothesized that using single-crystal Au NCs as the core could eventually allow us to synthesize alloy NCs of a wide range of sizes. This is a significant advantage compared to most alloy NCs reported in the literature, which typically display sizes between 2 and 20 nm due to challenges in simultaneously controlling nucleation, growth and co-reduction processes. To maintain the fraction of Au atoms close to 7 atomic%, we specifically synthesized Au NCs with pseudo-spherical morphology (as confirmed by the analysis of transmission electron microscopy (TEM) images, Fig. 2A and Fig. S1†) containing (7.5 ± 0.7) × 105 atoms (29 ± 2 nm, Table 1). The synthesis process was carried out in water using CTAC as a colloidal stabilizer and ascorbic acid as a reductant. The growth was seeded with single-crystal Au nanorods of 4.8 × 9.5 nm, which facilitated the formation of high yield single-crystal Au NCs.35 Control over the core crystal habit is crucial to ensure homogeneous Pd shell growth, as the presence of twin-plane defects can promote distinct shape evolutions and potentially lead to a broad distribution of compositions (i.e., different amounts of Pd deposited on the Au core depending on the crystal habit due to different growth kinetics of twinned NCs compared to single-crystal NCs).20,51,52
 | ||
| Fig. 2 Synthesis of Au@Pt, Au@Pd and Au@Pd@Pt NCs. (A) Low-magnification TEM image of Au nanospheres used as seeds during the coating process. (B–E) TEM images of Au@Pd@Pt NCs with different compositions of metals: Au7@Pd93 NCs (B), Au7@Pt93 NCs (C), Au7@Pd63@Pt30 NCs (D), and Au7@Pd33@Pt60 NCs (E). Scale bars: 100 nm. | ||
| NCs | Number of metal atoms | ||
|---|---|---|---|
| Au | Pd | Pt | |
| Au7@Pd93 NCs | 7.5 × 105 | 9.9 × 106 | |
| Au10@Pd90 NCs | 6.7 × 106 | ||
| Au18@Pd82 NCs | 3.5 × 106 | ||
| Au7@Pd63@Pt30 NCs | 6.7 × 106 | 3.2 × 106 | |
| Au7@Pd33@Pt60 NCs | 3.5 × 106 | 6.4 × 106 | |
| Au7@Pt93 NCs | 9.9 × 106 | ||
In the next stage, Pd2+ was deposited on the synthesized 30 nm single-crystal Au NCs at 65 °C using CTAB as the colloidal stabilizer (and shape-directing agent) and ascorbic acid to reduce Pd2+ on the Au core surface (seeds for the heterogeneous nucleation of the Pd shell). Three distinct Au:Pd atomic ratios were explored: 7:93 (Au7@Pd93 NCs), 10:90 (Au10@Pd90 NCs) and 18:82 (Au18@Pd82 NCs) by experimentally tuning the concentration of Au cores in the growth solution (i.e., at a constant [Pd2+]; see Experimental section, Fig. 2B and Fig. S2–4†). These ratios correspond to Pd shells containing ca. 9.95 × 106, 6.75 × 106 and 3.5 × 106 Pd atoms, respectively (Table 1). Analysis of TEM images of the three different Au@Pd NCs revealed the growth of NCs with cubic morphology in yields above 98%. In the presence of bromide ions and using single-crystal seeds, the Pd shell growth follows a conformal mode, where {100} facets are preferentially stabilized, forming Pd shells enclosed by {100} facets.53,54 The presence of Au@Pd NCs with different morphologies and larger dimensions could be attributed to a minor fraction of Au cores having lattice defects. Notably, the use of single-crystal Au NCs as core allowed us to prepare Au@Pd NCs with narrow size distributions, successfully facilitating control over compositions. The observed increase of the edge length from 48 ± 3 nm in Au18@Pd82 NCs to 60 ± 3 nm in Au10@Pd90 NCs and 73 ± 2 nm in Au7@Pd93 NCs nm shows that the Au:Pd atomic ratio was effectively varied (Fig. 2B and Fig. S2–4†). Quantitative elemental composition analysis via EDX spectroscopy revealed experimental Au:Pd ratios very close to the feeding ratios, indicating quantitative deposition of the Pd precursors and reinforcing the suitability of the investigated synthesis route to control NC compositions (Tables S1 and S2†).
In the next stage, we investigated the growth of Pt shells on the synthesized Au, Au18@Pd82 and Au10@Pd90 NCs. Three samples were prepared with distinct Au:Pt atomic ratios: 7:93 (Au7@Pt93 NCs, Fig. 2C), 7:30 (Au7@Pd63@Pt30 NCs, Fig. 2D) and 7:60 (Au7@Pd33@Pt60 NCs, Fig. 2E). The growth process was carried out following a similar strategy to that used for the Pd shell, using CTAB and ascorbic acid as colloidal stabilizer and reductant, respectively (see Experimental section and Fig. S5–7†). TEM images of the products revealed Pt shells with dendritic morphology, suggesting a growth mechanism that deviates from the conformal growth mode observed in the case of Pd (Fig. 2).7,43 This shape evolution of Pt shell is typically observed in aqueous-based growth synthesis of Pt NCs.36,55,56 Notably, the resulting NCs displayed narrow size distributions ranging from 95 ± 3 nm (Au7@Pt93 NCs) and 78 ± 4 nm (Au7@Pd33@Pt60 NCs) to 73 ± 5 nm (Au7@Pd63@Pt30 NCs).
Moreover, compositions were found to be close to the theoretical values, supporting the suitability of the designed methodology to grow multimetallic heterostructures with defined elemental contents (Tables S1 and S2†). As mentioned above, the final step in the synthesis of alloy NCs proposed herein relies upon the excitation of the prepared bimetallic and trimetallic heterostructures with nanosecond pulsed laser irradiation to promote the combination of the different metals at atomic level (i.e., by increasing the mobility of the metal atoms and enhancing interdiffusion).
This approach offers several advantages, including the ability to preserve the colloidal stability of excited NCs and achieve temperatures well above the melting point of most metals (>1000–3000 K), which are critical to favour desired alloying processes in colloidal NCs.38,57–59 The fast heating (between 103 and 1014 K s−1 depending on the pulse width) and cooling (up to 1013 K s−1 depending on the environment) dynamics help prevent aggregation and represent conditions that are challenging to achieve with standard colloidal heating methods (0.1–10 K s−1).20,33,60 In previous studies, we demonstrated the formation of alloyed AuAg and hollow AuAgPd NCs through excitation with 800 nm femtosecond laser pulses.48,49 However, the noble metal heterostructures obtained in this work exhibit broad plasmon bands in the UV-vis wavelength region (Fig. S8†). Therefore, the alloying process was investigated using 532 nm pulsed laser irradiation from a 5 ns-pulsed 10 Hz Nd:YAG laser.
In this scenario, pulse fluence was found to be the most critical parameter for the success of the alloying process. For instance, excessively high fluences (i.e., 96 J m−2) resulted in undesired NC fragmentation, an effect well reported in Au NCs excited with ns-laser pulses (Fig. S9–13†).38,59 The optimal pulse fluence and irradiation time were determined to be 64 J m−2 and 30 min, respectively. The dimensions of the irradiated NCs ranged between 70 and 75 nm: 75 ± 8 nm (Au7Pt93 NCs), 70 ± 4 nm (Au7Pd33Pt60 NCs), 73 ± 4 nm, (Au7Pd63Pt30 NCs) and 75 ± 5 nm (Au7Pd93 NCs) (Fig. 3 and 4). Moreover, the size dispersity was practically maintained after the reshaping process, with only the Au7@Pd93 NCs showing a slight increase in size dispersity. It is important to note that the colloidal stability of the excited NCs was preserved due to the presence of an excess of CTAC molecules in solution (5 mM). This excess allowed for the replacement of surfactant molecules that stabilize the NCs and may have degraded during the irradiation process. As a result, it was not observed an undesirable loss of colloidal stability and z-potential measurements showed no difference in the charge of the NCs before (40 mV) and after (41 mV) irradiation with nanosecond laser pulses.
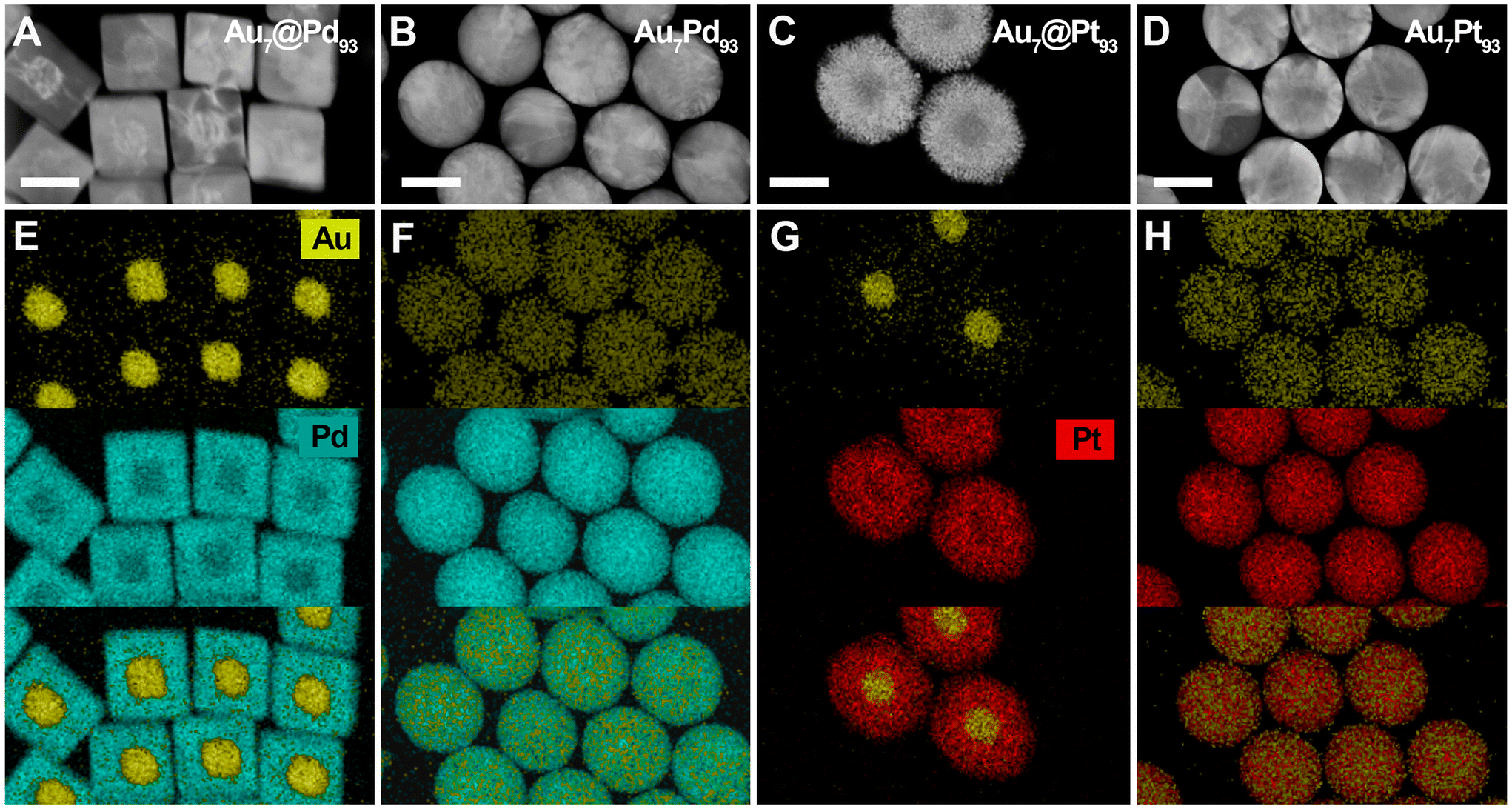 | ||
| Fig. 3 Synthesis of bimetallic alloy NCs via excitation of bimetallic core–shell heterostructures using 30 min of excitation with a 532 nm, 5 ns-pulsed laser irradiation from a Nd:YAG laser at a fluence of 64 J m−2. HAADF-STEM images (A–D) and EDX maps (E–H) of Au7@Pd93 (A and E) and Au7@Pt93 (C and G) heterostructures, along with the resulting Au7Pd93 (B and F) and Au7Pt93 (D and H) alloy NCs obtained after laser irradiation. Scale bar: 50 nm. | ||
 | ||
| Fig. 4 Synthesis of trimetallic alloy NCs via excitation of trimetallic core–shell–shell heterostructures using 30 min of excitation with a 532 nm, 5 ns-pulsed laser irradiation from a Nd:YAG laser at a fluence of 64 J m−2. HAADF-STEM images (A–D) and EDX maps (E–H) of Au7@Pd33@Pt60 NCs (A and E) and Au7@Pd63@Pt30 NCs (C and G) heterostructures, along with the resulting Au7Pd33Pt60 NCs (B and F) and Au7Pd63Pt30 (D and H) alloy NCs obtained after laser irradiation. Scale bar: 50 nm. | ||
To determine whether the mixing of the constituents occurred alongside the NC laser-mediated reshaping, we employed scanning transmission electron microscopy STEM-EDX imaging (Fig. 3 and 4) and line scans (Fig. S14†). The heterostructured nature of the as-synthesized Au, Pd and Pt-based NCs was effectively confirmed, and at the applied pulse fluences, the alloying process was successfully achieved for all compositions. This is particularly significant for the Au–Pt system, as Au is highly insoluble in Pt at room temperature (already at temperatures below 400 °C, in the composition range from ca. 15 to 100 weight% of Pt).61 Likely, the formation and stabilization of such a metastable AuPt alloy could be ascribed to: (i) the capacity of ns-pulsed laser irradiation to heat NC lattice to high temperatures where Au and Pt are miscible (>1073–1173 K), and (ii) the fast quenching of the metastable phase due to the ultrafast cooling (at rates up to 1012–1013 K s−1) of the colloidal NC lattice after the laser excitation.38,59 However, other effects associated to the nanoscale dimensions of the studied AuPt NCs should not be discarded. These results validate the advantages of the proposed synthesis strategy for preparing alloy NCs with precisely tailored compositions along with control over morphology. More importantly, the synthesized alloy NCs exhibit nearly identical number of atoms, which implies similar amount of surface atoms. Alloy NC with distinct compositions and exact number of surface atoms will always present different volumes and surface areas due to the distinct densities of their component (i.e., Pd, Au and Pt in this work).
In this scenario, this methodology offers an exciting opportunity to disentangle the role of composition from those of shape and number of atoms per NC lattice on the catalytic behaviour of multimetallic NCs. In this regard, we investigated the catalytic properties of the synthesized AuPd, AuPt, and AuPdPt alloy NCs to gain preliminary insights into this concept. Specifically, we selected the reduction of 4-nitrophenol using sodium borohydride, a reaction commonly used to determine the catalytic activity of relevant nanocatalysts.62 This reaction serves as a model because it can proceed at ambient temperature and pressure in aqueous solution, and it does not proceed in the absence of a catalyst.62,63
This reaction evolution can be quantitatively monitored using UV-vis spectroscopy, as nitrophenolate ions (the reaction is carried out at basic pH, see Experimental section) and the primary product, 4-aminophenol, have distinct absorption bands at 400 nm and 300 nm, respectively. During the reduction of nitrophenolate ions, the intensity of the 400 nm band gradually decreases while the 4-aminophenol band at 300 nm concurrently increases.64 In the present case, the time-dependent evolution of the 400 nm band was strongly influenced by the alloy NC composition. Au7Pd63Pt30 and Au7Pd33Pt60 NCs showed higher catalytic activity than Au7Pd93 and Au7Pt93 NCs (Fig. 5A). Notably, the concentration of metal atoms was 2.92 × 10−3 mM in all experiments, which corresponds to NC masses of 5.70 × 10−3 mg ml−1 (Au7Pt93 NCs), 4.87 × 10−3 mg ml−1 (Au7Pd33Pt60 NCs), 4.07 × 10−3 mg ml−1 (Au7Pd63Pt30 NCs), and 3.29 × 10−3 mg ml−1 (Au7Pd93 NCs). It must be noted that the observed catalyst-dependent induction period has been attributed to the time interval required for the concentration of dissolved oxygen (which outcompetes 4-nitrophenol for borohydride) to fall below a critical threshold.65
 | ||
| Fig. 5 Investigation of the catalytic activity of synthesized alloy NCs in the reduction of 4-NP with sodium borohydride. (A) Evolution of the absorption maxima of 4-NP (at 400 nm) with time during the reduction reaction catalysed by Au7Pd93 (black), Au7Pt93 (yellow), Au7Pd33Pt60 (cyan), and Au7Pd63Pt30 (red) NCs. Reaction conditions: [4-NP] = 0.05 mM, [metal atoms] = 0.029 mM and [NaBH4] = 0.08 M, T = 25 °C. (B) Time evolution of the absorbance at 400 nm during the reduction of 4-NP catalysed by Au7Pd63Pt30 NCs and linearized data for first-order analysis corresponding to (A). (C) Comparison of the different knor obtained for the investigated alloy NCs. | ||
It is worth noting that the concentrations of 4-nitrophenol and sodium borohydride were fixed at 54.32 μM and 80 mM, respectively. The large excess of sodium borohydride with respect to 4-nitrophenol was necessary for the reaction to advance in a pseudo-first-order regime (Fig. S15–18†):37,66
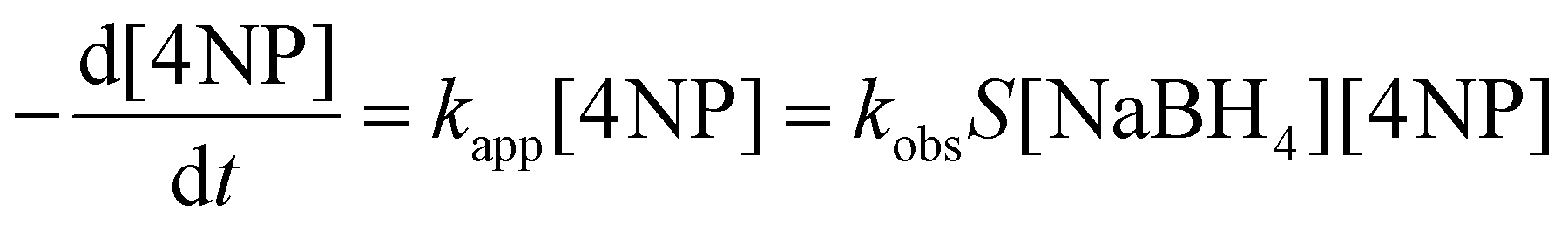 | (2) |
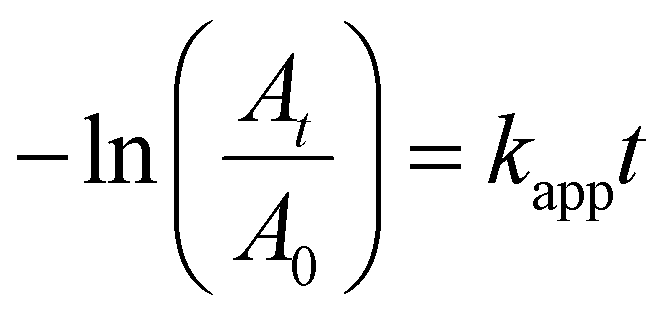 | (3) |
| Catalyst | k nor (mol−1 s−1 M−1) | Size (nm) | Morphology | Ref. |
|---|---|---|---|---|
| Au7Pt93 NCs | 3.70 × 105 | 75 | Sphere | This work |
| Au7Pd93 NCs | 9.00 × 105 | 75 | Sphere | This work |
| Au7Pd33Pt60 NCs | 1.30 × 107 | 70 | Sphere | This work |
| Au7Pd63Pt30 NCs | 1.10 × 107 | 73 | Sphere | This work |
| Pt NCs | 4.95 × 104 | 5 | Sphere | 70 |
| Pd NCs | 7.70 × 104 | 58 | Spherical Aggregates | 70 |
| Au NCs | 2.35 × 104 | 4.6 | Sphere | 70 |
| Pt NCs | 4.55 × 104 | 21 | Nanoflowers | 66 |
| Au@Pd4 NCs | 2.89 × 103 | 36 | Sphere | 71 |
| Au12@Pd88 NCs | 5.29 × 105 | 77 | Nanoflower | 72 |
| Pd NCs | 5.67 × 103 | 60 | Cubic | 73 |
| Pd NCs | 9.47 × 105 | 2 | Sphere | 74 |
| Au NCs | 4.80 × 105 | 2.6 | Sphere | 74 |
| Au NCs | 4.35 × 106 | 5.6 | Sphere | 75 |
| Au NCs | 2.55 × 107 | 13.4 | Sphere | 75 |
In this context, the normalized rate constant (knor) was determined to gain insights into our results compared with previous reports:
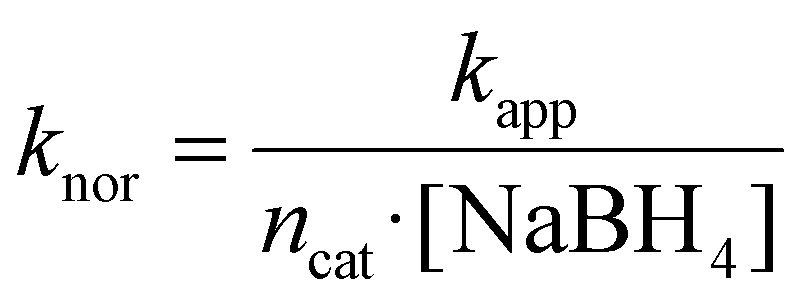 | (4) |
It is worth noting that the number of moles of catalyst, rather than their masses, is provided here, as we aimed to compare the catalytic performance of NCs containing multiple elements. The distinct atomic masses of Au (196.97 g mol−1), Pt (195.08 g mol−1) and Pd (106.42 g mol−1) imply that when knor is expressed in mass units, it is systematically higher for Pd-enriched catalysts as compared to those with higher Pt content. This is also in agreement with the fact that the concentration of catalyst atoms was kept constant across all the performed catalytic experiments. Moreover, utilization of moles of catalyst enables a more direct evaluation of atomic efficiency of distinct nanocatalysts, regardless, for example, of their dimensions, morphology, surface area and composition (Table 2).
The normalized rate constant values obtained for the synthesized bimetallic and trimetallic alloy NCs were approximately (3.7 ± 0.5) × 105 mol−1 s−1 M−1 for Au7Pt93 NCs, (9 ± 2) × 105 mol−1 s−1 M−1 for Au7Pd93 NCs, (1.3 ± 0.1) × 107 mol−1 s−1 M−1 for Au7Pd33Pt60 NCs, and (1.1 ± 0.2) × 107 mol−1 s−1 M−1 for Au7Pd63Pt30 NCs (Fig. 5C). These values are among the highest reported to date in literature for the reduction of 4-nitrophenol catalysed by metal NCs with NaBH4 as reducing agent, especially for the case of trimetallic NCs, that is, Au7Pd33Pt60 and Au7Pd63Pt30 NCs, which highlight the potential of alloying different metals at the nanoscale to produce a new generation of advanced catalysts. This fact is particularly important when we note that the dimensions of the synthesized trimetallic AuPdPt NCs are significantly larger than those of typical nanocatalysts. This results in a smaller surface area available for the catalytic process, which could potentially decrease atomic efficiency (Table 2). Finally, we investigated the reusability of the Au7Pd33Pt60 NCs, the composition displaying the highest activity. Importantly, the obtained results indicate that only a slight loss of activity occurs after eight cycles, suggesting a relatively high stability of the trimetallic alloy nanocatalyst (Fig S24†).
The presence of three different elements homogeneously distributed within the NC lattice can significantly modify the electronic structure of the NC (i.e., d-band center position) and, consequently, the catalytic behaviour. Various effects can be enhanced as the complexity of NC increases, including ligand effects (arising from composition modification by introducing other transition metals), geometric (due to introduction of lattice strain by metals with distinct atomic radii), and/or ensemble (synergistic effect resulting from the presence of more thanone reactive site) effects.3,67–69 Nevertheless, further investigations are needed to fully understand the high catalytic activity exhibited by alloy AuPdPt NCs in the reduction of 4-nitrophenol using sodium borohydride.
Conclusions
In summary, we have developed a methodology to address a major synthesis challenge in nanoscience: the fabrication of alloy multielemental NCs with smooth morphology, tunable compositions, and a fixed number of atoms per NCs. The proposed strategy combines colloidal growth methods and ns-pulsed laser irradiation. Wet-chemical bottom-up approaches offer fine control over composition and the number of metal atoms per particle, while pulsed laser irradiation facilitates the alloying process and the formation of smooth spherical NCs.The process begins with the synthesis of 30 nm single-crystal Au nanospheres, which serve as seeds for the growth of core–shell Au@Pd NCs with distinct Au:Pd ratios: Au7@Pd93, Au7@Pd63 and Au7@Pd33 NCs. In the next stage, different amounts of Pt atoms were deposited on the Au7@Pd63 and Au7@Pd33 NCs, as well as on the 30 nm Au nanospheres, resulting in the formation of Au7@Pd63@Pt30, Au7@Pd33@Pt60, Au7@Pt93 and Au7@Pd93 NCs. This process led to the synthesis of four types of heterostructure NCs with distinct compositions but an equal number of metal atoms per NC lattice, as confirmed by TEM and EDX analysis. Subsequently, the interdiffusion and mixing of the different metals within the distinct NCs was triggered by irradiation with 532 nm, 8 ns Nd:YAG laser pulses at a fluence of 64 J m−2, inducing the formation of alloy Au7Pd93, Au7Pt93 Au7Pd63Pt30 and Au7Pd33Pt60 NCs, as confirmed by EDX elemental analysis. The excitation with ns-pulsed laser irradiation also modified the NC morphology, enabling the formation of alloy NCs with smooth, spherical shapes. Therefore, the major difference between the obtained alloy NCs lies solely in their composition, which facilitates reliable studies on the composition-catalytic activity relationship of colloidal NCs. A preliminary exploration of this idea revealed that the catalytic reduction of 4-nitrophenol to 4-aminophenol with sodium borohydride was significantly enhanced by trimetallic Au7Pd33Pt60 and Au7Pd63Pt30 NCs as compared to bimetallic Au7Pt93 and Au7Pd93 NCs.
Thereby, we have productively demonstrated a wet-chemistry approach to fabricate multimetallic spherical NCs with unprecedented control over both the composition and number of atoms per particle. The combination of seed-mediated growth routes with ns-pulsed laser irradiation may be applicable for synthesizing more complex alloys, for example high entropy alloys, as far as core-multishell metal NCs can be synthesized successfully.
Author contributions
G. G.-R. and A. G.-M. devised the project, the main conceptual ideas and proof outline. S. T.-S. carried out the synthesis, laser irradiation, characterization and catalysis experiments. J. G.-I. and L. B. participated in the laser irradiation experiments. R. X. contributed to the characterization experiments. J. P.-J. and I. C. helped with the design and analysis of catalysis experiments. G. G.-R., A. G.-M. and S. T.-S. wrote the manuscript in consultation with J. P.-J. and I. C.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the ERC Starting Grant Time4Nano (101164205), the Spanish Ministry of Science, Innovation and Universities (grants PID2021-123228NB-I00, PID2021-122839NB-I00 and PID2022-138724NB-I00) and the International Research Network-IRN Nanoalloys. G.G-R. acknowledges the Atracción de Talento Fellowship from the Madrid Regional Government (Grant 2022-T1/IND-23908) and. The facilities provided by the Centro de Microscopia Electrónica and Center for Ultrafast Lasers (CLUR) at the Complutense University of Madrid are gratefully acknowledged.References
- L. Galdun, V. Vega, Z. Vargová, E. D. Barriga-Castro, C. Luna, R. Varga and V. M. Prida, ACS Appl. Nano Mater., 2018, 1, 7066–7074 Search PubMed.
- J. V. Calazans Neto, C. A. S. Celles, C. S. A. F. de Andrade, C. R. M. Afonso, B. E. Nagay and V. A. R. Barão, ACS Biomater. Sci. Eng., 2024, 10, 6029–6060 Search PubMed.
- Y. Nakaya and S. Furukawa, Chem. Rev., 2023, 123, 5859–5947 CrossRef PubMed.
- E. A. Starke and J. T. Staley, Prog. Aeronaut. Sci., 1996, 32, 131–172 CrossRef.
- W. Albrecht, J. E. S. van der Hoeven, T.-S. Deng, P. E. de Jongh and A. van Blaaderen, Nanoscale, 2017, 9, 2845–2851 RSC.
- T. Chen and V. O. Rodionov, ACS Catal., 2016, 6, 4025–4033 CrossRef.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef PubMed.
- Y. Shi, Z. Lyu, M. Zhao, R. Chen, Q. N. Nguyen and Y. Xia, Chem. Rev., 2021, 121, 649–735 CrossRef PubMed.
- H. Kim, T. Y. Yoo, M. S. Bootharaju, J. H. Kim, D. Y. Chung and T. Hyeon, Adv. Sci., 2022, 9, 2104054 CrossRef CAS.
- H. Guo, Z. Fang, H. Li, D. Fernandez, G. Henkelman, S. M. Humphrey and G. Yu, ACS Nano, 2019, 13, 13225–13234 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- A. Zupanc, J. Install, M. Jereb and T. Repo, Angew. Chem., Int. Ed., 2023, 62, e202214453 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 Search PubMed.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 Search PubMed.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 Search PubMed.
- J. De Roo, Chem. Mater., 2023, 35, 3781–3792 CrossRef CAS.
- Y. Xia, K. D. Gilroy, H.-C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- M. Ha, J.-H. Kim, M. You, Q. Li, C. Fan and J.-M. Nam, Chem. Rev., 2019, 119, 12208–12278 CrossRef CAS.
- J. Guo, S. Jiao, X. Ya, H. Zheng, R. Wang, J. Yu, H. Wang, Z. Zhang, W. Liu, C. He and X. Fu, Chem. – Eur. J., 2022, 28, e202202221 CrossRef CAS PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS.
- Y. Kang, J. B. Pyo, X. Ye, T. R. Gordon and C. B. Murray, ACS Nano, 2012, 6, 5642–5647 CrossRef CAS PubMed.
- J. T. L. Gamler, H. M. Ashberry, S. E. Skrabalak and K. M. Koczkur, Adv. Mater., 2018, 30, 1801563 CrossRef PubMed.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed.
- Y. Liao, Y. Li, R. Zhao, J. Zhang, L. Zhao, L. Ji, Z. Zhang, X. Liu, G. Qin and X. Zhang, Natl. Sci. Rev., 2022, 9, nwac041 CrossRef CAS PubMed.
- V. Amendola, S. Scaramuzza, F. Carraro and E. Cattaruzza, J. Colloid Interface Sci., 2017, 489, 18–27 CrossRef CAS.
- F. Waag, Y. Li, A. R. Ziefuß, E. Bertin, M. Kamp, V. Duppel, G. Marzun, L. Kienle, S. Barcikowski and B. Gökce, RSC Adv., 2019, 9, 18547–18558 RSC.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS.
- W. Chen, S. Luo, M. Sun, X. Wu, Y. Zhou, Y. Liao, M. Tang, X. Fan, B. Huang and Z. Quan, Adv. Mater., 2022, 34, 2206276 CrossRef CAS PubMed.
- D. Wu, K. Kusada, Y. Nanba, M. Koyama, T. Yamamoto, T. Toriyama, S. Matsumura, O. Seo, I. Gueye, J. Kim, L. S. Rosantha Kumara, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 3365–3369 CrossRef PubMed.
- G. González-Rubio, P. Llombart, J. Zhou, H. Geiss, O. Peña-Rodríguez, H. Gai, B. Ni, R. Rosenberg and H. Cölfen, Chem. Mater., 2024, 49, 1982–1997 CrossRef.
- F.-R. Fan, D.-Y. Liu, Y.-F. Wu, S. Duan, Z.-X. Xie, Z.-Y. Jiang and Z.-Q. Tian, J. Am. Chem. Soc., 2008, 130, 6949–6951 CrossRef PubMed.
- N. E. Larm, N. Bhawawet, J. A. Thon and G. A. Baker, New J. Chem., 2019, 43, 17932–17936 RSC.
- G. González-Rubio, A. Guerrero-Martínez and L. M. Liz-Marzán, Acc. Chem. Res., 2016, 49, 678–686 CrossRef PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC, 89 edn., 2008 Search PubMed.
- C. Hanske, G. González-Rubio, C. Hamon, P. Formentín, E. Modin, A. Chuvilin, A. Guerrero-Martínez, L. F. Marsal and L. M. Liz-Marzán, J. Phys. Chem. C, 2017, 121, 10899–10906 CrossRef.
- X. Lin, S. Lin, Y. Liu, M. Gao, H. Zhao, B. Liu, W. Hasi and L. Wang, Langmuir, 2018, 34, 6077–6084 CrossRef.
- M. He, L. Protesescu, R. Caputo, F. Krumeich and M. V. Kovalenko, Chem. Mater., 2015, 27, 635–647 CrossRef.
- K. D. Gilroy, X. Yang, S. Xie, M. Zhao, D. Qin and Y. Xia, Adv. Mater., 2018, 30, 1706312 CrossRef.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef PubMed.
- N. Ortiz, R. G. Weiner and S. E. Skrabalak, ACS Nano, 2014, 8, 12461–12467 CrossRef PubMed.
- E. González, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377–1380 CrossRef PubMed.
- H. Cheng, C. Wang, D. Qin and Y. Xia, Acc. Chem. Res., 2023, 56, 900–909 CrossRef CAS PubMed.
- G. González-Rubio, P. Díaz-Núñez, W. Albrecht, V. Manzaneda-González, L. Bañares, A. Rivera, L. M. Liz-Marzán, O. Peña-Rodríguez, S. Bals and A. Guerrero-Martínez, Adv. Opt. Mater., 2021, 9, 2002134 CrossRef.
- V. Manzaneda-González, K. Jenkinson, O. Peña-Rodríguez, O. Borrell-Grueiro, S. Triviño-Sanchez, L. Bañares, E. Junquera, A. Espinosa, G. Gonzalez-Rubio, S. Bals and A. Guerrero-Martínez, Chem. Mater., 2023, 35, 9603–9612 Search PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- Y. Wang, H.-C. Peng, J. Liu, C. Z. Huang and Y. Xia, Nano Lett., 2015, 15, 1445–1450 CrossRef CAS PubMed.
- Y. Xiong, H. Cai, B. J. Wiley, J. Wang, M. J. Kim and Y. Xia, J. Am. Chem. Soc., 2007, 129, 3665–3675 CrossRef CAS PubMed.
- S. F. Tan, G. Bisht, U. Anand, M. Bosman, X. E. Yong and U. Mirsaidov, J. Am. Chem. Soc., 2018, 140, 11680–11685 CrossRef CAS PubMed.
- M. Jin, H. Liu, H. Zhang, Z. Xie, J. Liu and Y. Xia, Nano Res., 2011, 4, 83–91 CrossRef.
- N. C. Bigall, T. Härtling, M. Klose, P. Simon, L. M. Eng and A. Eychmüller, Nano Lett., 2008, 8, 4588–4592 CrossRef PubMed.
- M. Grzelczak, J. Pérez-Juste, F. J. García de Abajo and L. M. Liz-Marzán, J. Phys. Chem. C, 2007, 111, 6183–6188 CrossRef.
- D. Werner and S. Hashimoto, J. Phys. Chem. C, 2011, 115, 5063–5072 CrossRef.
- G. González-Rubio, P. Díaz-Núñez, A. Rivera, A. Prada, G. Tardajos, J. González-Izquierdo, L. Bañares, P. Llombart, L. G. Macdowell, M. A. Palafox, L. M. Liz-Marzán, O. Peña-Rodríguez and A. Guerrero-Martínez, Science, 2017, 358, 640–644 CrossRef PubMed.
- A. Pyatenko, H. Wang, N. Koshizaki and T. Tsuji, Laser Photonics Rev., 2013, 7, 596–604 CrossRef.
- S. García, L. Zhang, G. W. Piburn, G. Henkelman and S. M. Humphrey, ACS Nano, 2014, 8, 11512–11521 CrossRef PubMed.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1985, 6, 46–56 CrossRef.
- J. Strachan, C. Barnett, A. F. Masters and T. Maschmeyer, ACS Catal., 2020, 10, 5516–5521 CrossRef.
- N. Pradhan, A. Pal and T. Pal, Langmuir, 2001, 17, 1800–1802 CrossRef.
- S. Gu, S. Wunder, Y. Lu, M. Ballauff, R. Fenger, K. Rademann, B. Jaquet and A. Zaccone, J. Phys. Chem. C, 2014, 118, 18618–18625 CrossRef.
- E. Menumerov, R. A. Hughes and S. Neretina, Nano Lett., 2016, 16, 7791–7797 CrossRef CAS PubMed.
- S. Mourdikoudis, T. Altantzis, L. M. Liz-Marzán, S. Bals, I. Pastoriza-Santos and J. Pérez-Juste, CrystEngComm, 2016, 18, 3422–3427 RSC.
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 1–13 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed.
- J. W. M. Crawley, I. E. Gow, N. Lawes, I. Kowalec, L. Kabalan, C. R. A. Catlow, A. J. Logsdail, S. H. Taylor, N. F. Dummer and G. J. Hutchings, Chem. Rev., 2022, 122, 6795–6849 CrossRef CAS PubMed.
- R. Grzeschik, D. Schäfer, T. Holtum, S. Küpper, A. Hoffmann and S. Schlücker, J. Phys. Chem. C, 2020, 124, 2939–2944 CrossRef CAS.
- L. Srisombat, J. Nonkumwong, K. Suwannarat, B. Kuntalue and S. Ananta, Colloids Surf., A, 2017, 512, 17–25 CrossRef CAS.
- A. G. M. da Silva, T. S. Rodrigues, L. S. K. Taguchi, H. V. Fajardo, R. Balzer, L. F. D. Probst and P. H. C. Camargo, J. Mater. Sci., 2016, 51, 603–614 CrossRef CAS.
- S. Swain, B. M. B. V. Kandathil, P. Bhol, A. K. Samal and S. A. Patil, Langmuir, 2020, 36, 5208–5218 CrossRef CAS PubMed.
- F. Liu and X. Liu, Colloids Surf., A, 2024, 685, 133225 CrossRef CAS.
- R. D. Neal, R. A. Hughes, P. Sapkota, S. Ptasinska and S. Neretina, ACS Catal., 2020, 10, 10040–10050 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04898a |
| This journal is © The Royal Society of Chemistry 2025 |