
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuneable and degradable thermosets possessing dynamic aliphatic disulfide bonds via stereoselective thiol–yne polymerisation†
Daniele
Giannantonio
 ,
Arianna
Brandolese
and
Andrew P.
Dove
*
,
Arianna
Brandolese
and
Andrew P.
Dove
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: a.dove@bham.ac.uk
First published on 20th December 2024
Abstract
The permanent chemical structure that makes thermosets strong and stretchable materials also hinders their reprocessability and leads to their accumulation in the environment. To favour material reprocessability and reuse, polymers have been endowed with dynamic covalent bonds. However, when the dynamic bond is the network-forming bond, a significant trade-off between the robustness of the material and the dynamic behaviour can be encountered. In this study, nucleophilic thiol–yne click polymerisation was used to synthesise different materials possessing tuneable amounts of reversible disulfide bonds, thus achieving diverse thermomechanical properties. Furthermore, by leveraging different catalyst systems, the cis/trans conformer ratio on the backbone could be modulated to provide an additional degree of control over the thermomechanical properties. Finally, the presence of dynamic bonds was used as a handle to enable promotion of degradation and material reprocessing.
Introduction
Thermosets are covalently cross-linked materials that find applications in many industries, such as construction,1 transportation,2 and aerospace,3 largely a result of their outstanding strength, extensibility, and solvent resistance. Their crosslinked nature, which is responsible for many of these useful properties, also causes their intractability and limited reprocessability, contributing to environmental concerns.4 Even when heated at very high temperatures these materials are unable to flow, meaning that they do not melt; hence, the development of reprocessable thermosets constitutes a major goal in polymer science.5Recently, the introduction of dynamic covalent bonds into thermosets established covalent adaptable networks (CANs) as a new class of materials.6 The dynamic bonds within the materials can break and reform on sufficiently short timescales to allow the polymer chains to undergo molecular rearrangement, thus allowing macroscopic flow in a thermoplastic-like fashion. Parameters such as the nature of the dynamic bonds of choice (i.e. associative or dissociative) and the strategy deployed to form the network (step-growth vs. chain-growth polymerisation) influence the final architecture and bond distribution.7,8 Among the different dynamic bonds available, disulfide bonds have received significant attention because of their widespread presence in biological systems and well characterized dynamic behaviour.9,10 With a dissociation energy of around 60 kcal mol−1, lower than that of the aliphatic C–C bond of 80–90 kcal mol−1, disulfide bonds provide a good balance between dynamicity and robustness.11,12
In light of their efficiency, several thiol-based reactions have been used for the synthesis or functionalisation of polymeric materials.13 Among these, the use of thiol–ene Michael addition, radical or nucleophilic, arguably represents the most common approach given its simplicity and functional group tolerance.14–16 Because of the reversibility of the thia-Michael adduct, several CANs built on this reaction have been reported.17–20 Similarly, the use of activated alkynes allows for the nucleophilic thiol addition across the triple bond.21–26 Only recently, Du Prez and co-workers reported the use of nucleophilic thiol–yne addition to activated alkynes to obtain CANs with tuneable relaxation rates depending on the electronic of the alkyne of choice.27 While this work shows that network formation can be achieved through the use of a reversible reaction, only dissociative bonds can be introduced with this approach thus limiting its scope. Moreover, since bifunctional alkynes and thiols are being used, the thiol–yne addition reaction is responsible for the formation of pre-polymers, and only a subsequent thiol–ene addition forms the crosslinked structure.
The use of such a selective reaction represents an advantage because of its efficiency, high yield, and tolerance towards a wide range of functional groups. Additionally, if the presence of the dynamic bonds is independent of the polymerisation method, a greater range of reversible moieties can be introduced.28 Moreover, when a bifunctional alkyne is reacted with a multifunctional thiol (functionalities >2), the prepolymer synthesis step can be bypassed simplifying the network formation. Previous work showed that the ratio of the resulting addition products, the trans and cis isomers, can be modulated by catalyst and/or solvent selection. In fact, cis-enriched products have been synthesised increasing solvent polarity or switching to stronger bases as catalysts.16,29 This is particularly interesting as backbone cis/trans isomerism has been previously used to tailor thermomechanical performances in linear polymers,30–32 hydrogels,33 and 3D printable resin,34 but this concept has not, to date, been applied to CANs. Herein, a bifunctional propiolate possessing an internal dynamic disulfide bond (C2SS) was here successfully introduced into crosslinked materials by reaction with a three-arm thiol (Fig. 1). The thermomechanical properties of the resulting network were found to change according to the ratio of the monomers and cis/trans content. Finally, the presence of dynamic disulfide bonds rendered these materials degradable and reprocessable.
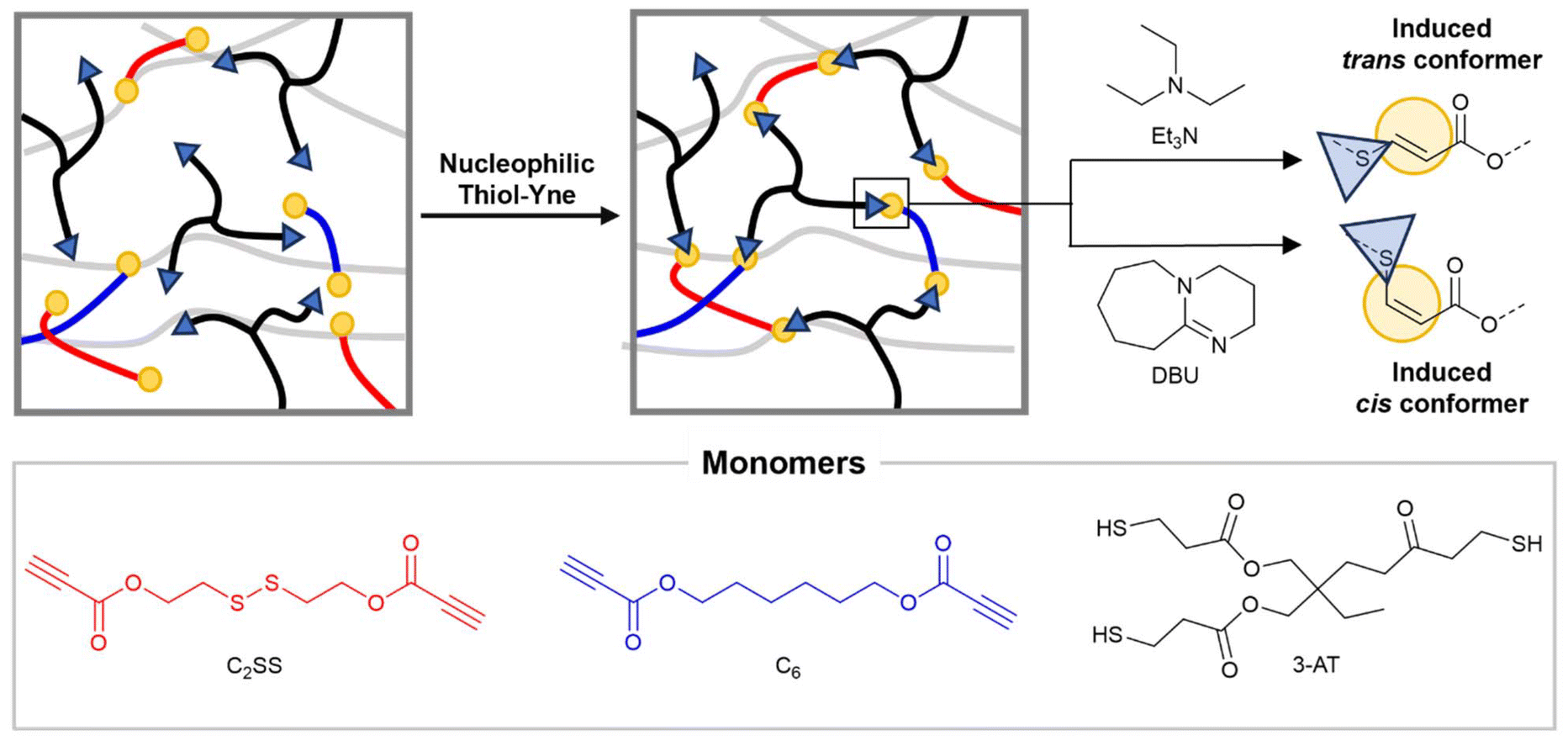 | ||
| Fig. 1 Scheme representing the network formation and the influence of the catalyst on cis/trans isomerism (top) and the monomers used in this work (bottom). | ||
Results and discussion
Network synthesis
To access networks via nucleophilic thiol–yne polymerisation, two monomers with activated terminal alkynes (C2SS and C6) were synthesised via Fisher esterification of commercially available diols with propiolic acid (ESI, Fig. S1–S4†). These monomers were then reacted with a three-arm thiol (trimethylolpropane tris(3-mercaptopropionate) – 3-AT) in different ratios in the presence of an organocatalyst (triethylamine (Et3N) or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)) in chloroform (CHCl3). The mixtures were poured into Teflon beakers; transparent, flexible, and uniform freestanding films were recovered after slow evaporation of the solvent (ESI, Fig. S5†). Residual solvent was removed by heating at 50 °C under vacuum until constant weight was reached. The versatility entailed by the nucleophilic thiol–yne reaction allows the obtainment of diverse networks from a limited pool of monomers by simple tuning of the reaction conditions. To this end, by employing Et3N as the catalyst, trans-rich networks trans-NetC2SS, trans-Net(C2SS-C6), and trans-NetC6 with diverse disulfide content were synthesised by varying the ratio between monomers C2SS and C6 (100% C2SS, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1: C2SS:C6, and 100% C6 respectively) while keeping the stoichiometric ratio between alkyne and thiol groups 1:1. trans-Net(3-AT1.5-C2SS1) was accessed by reacting monomer C2SS with 3-AT in equimolar amounts, to have a thiol:alkyne ratio of 1.5:1. Lastly, cis-rich networks NetC2SS and NetC6 (100% C2SS and 100% C6 respectively) were accessed by employing DBU as the catalyst of the reaction.29,30 Consumption of the alkyne bonds following thiol–yne addition was confirmed by Fourier-transform infrared (FTIR) spectroscopy on the free-standing films, confirming the disappearance of the relative stretching bands (Fig. 2A and B). The formation of networks was further confirmed by performing swelling experiments at room temperature in CHCl3. All networks displayed high gel content (>80%) and a swelling ratio above 250% confirming nucleophilic thiol–yne polymerisation as an excellent tool for the synthesis of crosslinked materials (ESI, Table S1†).
:1: C2SS:C6, and 100% C6 respectively) while keeping the stoichiometric ratio between alkyne and thiol groups 1:1. trans-Net(3-AT1.5-C2SS1) was accessed by reacting monomer C2SS with 3-AT in equimolar amounts, to have a thiol:alkyne ratio of 1.5:1. Lastly, cis-rich networks NetC2SS and NetC6 (100% C2SS and 100% C6 respectively) were accessed by employing DBU as the catalyst of the reaction.29,30 Consumption of the alkyne bonds following thiol–yne addition was confirmed by Fourier-transform infrared (FTIR) spectroscopy on the free-standing films, confirming the disappearance of the relative stretching bands (Fig. 2A and B). The formation of networks was further confirmed by performing swelling experiments at room temperature in CHCl3. All networks displayed high gel content (>80%) and a swelling ratio above 250% confirming nucleophilic thiol–yne polymerisation as an excellent tool for the synthesis of crosslinked materials (ESI, Table S1†).
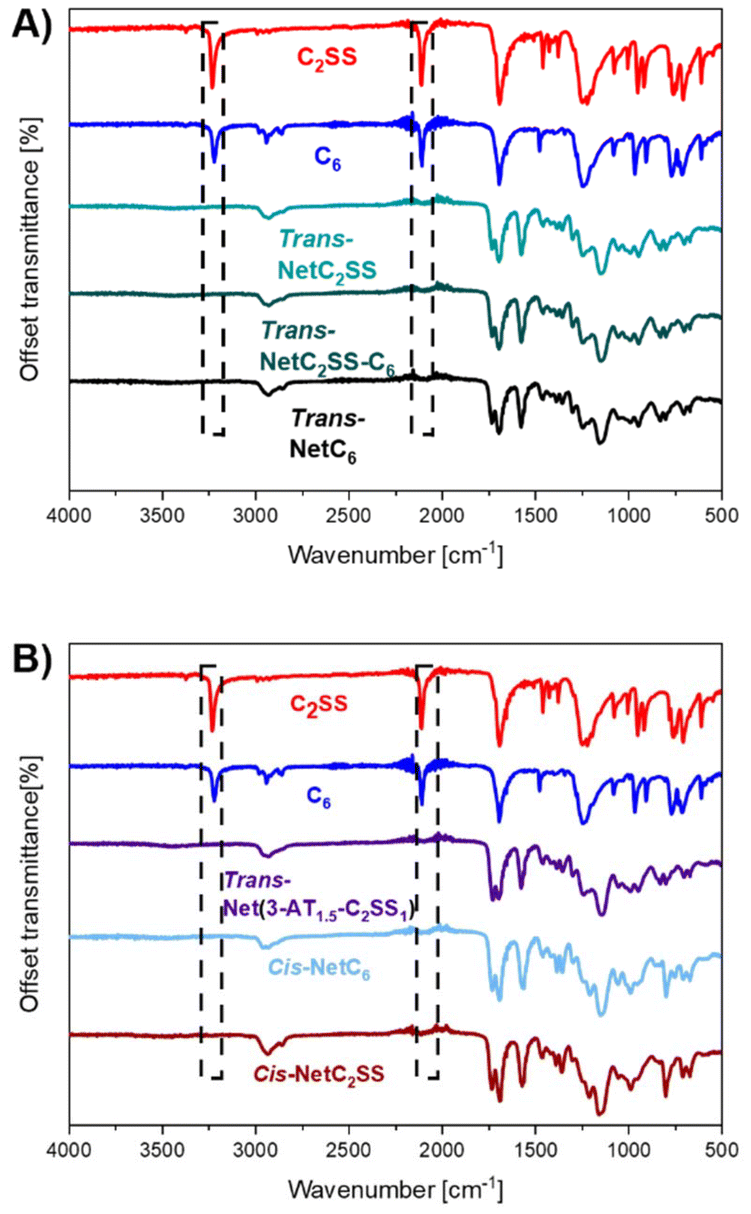 | ||
| Fig. 2 (A) FTIR spectra showing the disappearance of the alkyne stretching bands in the networks trans-NetC2SS, trans-Net(C2SS-C6), trans-NetC6. (B) FTIR spectra showing the disappearance of the alkyne stretching bands in the networks trans-Net(3-AT1.5-C2SS1), cis-NetC2SS, and cis-NetC6. | ||
Mechanical properties
Dynamic mechanical analysis (DMA) was performed to evaluate the temperature dependences of storage modulus (E′) and loss factor (tanδ) of the different networks. The broad and bimodal tanδ curves displayed by the 1:1 thiol:alkyne ratio networks suggest that the networks possess a non-homogeneous structure, however, no drop in modulus was recorded (Fig. 3A) which suggests that all materials showed excellent dimensional stability in the temperature range tested (−80 °C to 130 °C). Notably, trans-NetC2SS did not display any loss in modulus even at temperatures as high as 180 °C (ESI, Fig. S6†). The absence of a significant drop in modulus above Tg, indicative of a gel-to-sol transition and typically found in dissociative CANs, suggests that these materials behave in an associative “vitrimer-like” fashion.35,36 The same behaviour has been observed in many CANs possessing both aliphatic37–39 and aromatic40–43 disulfide bonds. Nonetheless, a significant decrease in stress could be observed for trans-NetC2SS, when stress-relaxation experiments at higher strain (10 μm vs. 1 μm) were performed at 150 °C (Fig. 3B). Full relaxation, determined as the time corresponding to ∼37% of the initial stress value following a Maxwell model,44 was reached in roughly 18 minutes. This behaviour was attributed to the dissociation of the disulfide bonds, as trans-NetC6 modulus remained constant throughout the experiments. The relatively slow dissociation was linked to the aliphatic nature of the disulfide bonds, which display higher stability when compared to aromatic disulfides.11 Even so, this relaxation is consistent with other works displaying aliphatic disulfides. For example, Zhang and colleagues reported variable relaxation rates going from 1.5 s (200 °C) to 90 minutes (60 °C) in a system containing aliphatic disulfide and ester bonds.39 All materials displayed rubbery behaviour above their glass transition temperature (Tg), as the tanδ dropped to 0.
 | ||
| Fig. 3 (A) DMA (−80 to 130 °C, 5 °C min−1, 1 Hz, 1 μm) curves of the networks synthesised via nucleophilic thiol–yne polymerisation. (B) Stress relaxation experiments (150 °C, 10 μm, 20 min) performed on trans-NetC2SS and trans-NetC6 displaying a loss in modulus for trans-NetC2SS exclusively. (C) Exemplary stress vs. strain curves obtained from uniaxial tensile testing (10 mm min−1, 22 °C) of different networks synthesised in this work. | ||
When compared to trans-NetC2SS, trans-Net(3-AT1.5-C2SS1) displayed a monomodal and narrower tanδ curve, suggesting that a 1.5:1 ratio between thiols and alkyne leads to a more homogeneous network. The decreased molecular weight between crosslinking (Mc) (Table 1) and lower swelling ratio (ESI, Fig. S7 and Table S1†) support this hypothesis, however, very few differences regarding Mc were found among the networks, suggesting that these systems are comparable. The limit of the viscoelastic region (LVE) and yield point determined via rheology were found to be similar, as shown by amplitude sweeps performed on the high-trans networks (ESI, Fig. S8†). Frequency sweeps confirmed the reduced influence of different degrees of crosslinks on shear stress as the networks did not display strong differences (ESI, Fig. S9†). It is important to note that the density of the material was assumed to be 1 g mL−1, and the theory of rubber elasticity showed some limitations with highly crosslinked systems.45,46 These data suggest that as different monomer C2SS and C6 ratios were explored, the composition may be responsible for the differences regarding mechanical behaviour, however, when more homogeneous network structures are achieved, these differences can be offset. In fact, trans-Net(3-AT1.5-C2SS1) was able to compete with a fully carbon–carbon based network such as trans-NetC6.
| Name |
T
ga [°C] |
b [MPa] |
M c [g mol−1] |
Ec [MPa] |
σ at break (UTS) [MPa] | ε at break [%] | Strain energy densityd [J m−3] |
T
ge [°C] |
T
d, 5%f [°C] |
|---|---|---|---|---|---|---|---|---|---|
|
a Determined as the peak of the tanδ curve.
b Taken as the value associated with Tg + 50 °C.
c Determined as the slope of the curve at 1% strain.
d Determined as the area below the stress vs. strain curve.
e Determined as the inflexion point of the second heating cycle of the DSC thermograms.
f Determined as the temperature associated with a 5% mass loss.
|
|||||||||
| trans-NetC2SS | 7.25 | 239.7 | 34.5 | 0.73 ± 0.17 | 0.63 ± 0.06 | 94.8 ± 22.2 | 0.34 ± 0.12 | −7.4 | 186.2 |
| trans-NetC6 | 13.2 | 1314.2 | 6.39 | 0.77 ± 0.4 | 1.35 ± 0.39 | 72.5 ± 21.9 | 0.59 ± 0.28 | −7.4 | 337.8 |
| trans-Net(C2SS-C6) | 4.33 | 506.5 | 16.1 | 2.03 ± 0.11 | 2.56 ± 0.15 | 155.4 ± 12.6 | 2.2 ± 0.3 | −10.4 | 300.5 |
| trans-Net(3-AT1.5-C2SS1) | 4.8 | 1186.4 | 6.9 | 3.4 ± 0.7 | 1.3 ± 0.6 | 51.2 ± 25.4 | 0.39 ± 0.31 | −14.4 | 192.2 |
| cis-NetC2SS | 13.25 | 302.4 | 27.7 | 2.5 ± 0.4 | 3.7 ± 1.9 | 235.7 ± 101.7 | 4.9 ± 3.7 | −3.2 | 171.8 |
| cis-NetC6 | 10.5 | 720.3 | 11.6 | 0.93 ± 0.56 | 5.7 ± 2.5 | 471.7 ± 121.9 | 10.7 ± 5.3 | 1.3 | 316.3 |

cis-Rich networks showed different properties when compared to their trans-rich analogous. While cis-NetC2SS displayed a slightly higher rubbery storage modulus ( ) than trans-NetC2SS, cis-NetC6 possessed a lower
) than trans-NetC2SS, cis-NetC6 possessed a lower 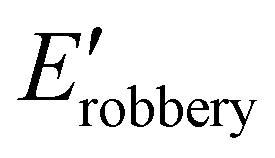 value when compared to trans-NetC6. Taken together, these results suggest that different degrees of cis/trans isomerisation can alter the modulus of thermosets, but this effect is mediated by composition as well (Table 1).
value when compared to trans-NetC6. Taken together, these results suggest that different degrees of cis/trans isomerisation can alter the modulus of thermosets, but this effect is mediated by composition as well (Table 1).
Further probing of the mechanical properties was performed via uniaxial tensile testing on dogbones cut from the cast films (Fig. 3C, Fig. S10–S15† and Table 1). Despite its lower Mc, trans-NetC2SS was found to possess a similar Young's modulus (E) and elongation at break (ε) to trans-NetC6. trans-NetC6 displayed a slightly higher ultimate tensile strength (UTS) (ESI, Fig. S16–S18†), which is likely a result of its higher carbon–carbon bond content.11,12 To our surprise, trans-Net(C2SS-C6) displayed higher UTS and elongation at break than either network formed from a single alkyne monomer, (ESI, Fig. S16–S18†) demonstrating that subtle changes to the chemical composition can significantly alter mechanical performance of the resultant materials. When compared to trans-NetC2SS, trans-Net(3-AT1.5-C2SS1) displayed higher Young's modulus (E = 3.4 ± 0.7 MPa vs. E = 0.73 ± 0.17 MPa, Fig. S16†) and stress at break (σ = 1.3 ± 0.6 MPa vs. σ = 0.63 ± 0.06 MPa, Fig. S17†), but lower elongation (ε = 51.2 ± 25.4% vs. ε = 94.8 ± 22.2%, Fig. S18†). These results are consistent with the higher crosslinking density associated with trans-Net(3-AT1.5-C2SS1), which is also corroborated by its lower swelling ratio (ESI, Table S1†).
Both cis-rich networks showed enhanced UTS than their trans-rich counterparts, confirming that stereochemistry can alter the mechanical properties of thermosets. When compared to trans-NetC2SS, cis-NetC2SS also displayed significantly higher Young's modulus (ESI, Fig. S16†), while cis-NetC6 showed higher elongation at break with respect to trans-NetC6 (ESI, Fig. S18†). Comparing the two, cis-NetC2SS displayed a higher Young's modulus than cis-NetC6 (ESI, Fig. S16†), but no statistically significant differences were observed in stress and strain at break when comparing the results via an unpaired T test. However, it is noticeable from the stress–strain curves that the strain at break of cis-NetC6 is almost double that displayed by cis-NetC2SS, suggesting that further evaluation into the presence of architectural defect could lead to more pronounced differences, underlying once again the trade-off between dynamic behaviour and mechanical performances (ESI, Fig. S17 and S18†). These results suggest that increasing cis-content leads to materials possessing a higher strain energy density (Table 1). This may be explained by mechanically induced cis-to-trans isomerisation, which was probed by Radiom and co-workers.47 The authors described that when cis-rich norbornene polymers were stretched via atomic force microscopy (AFM), an increase in extension was measured because of the isomerisation of the shorter cis linkages into the longer trans conformation, which resulted in relaxation of the force. From a material point of view, this event could lead to energy dissipation, thus allowing the material to withstand greater stress. Notably, trans-Net(C2SS-C6), cis-NetC2SS, and cis-NetC6, displayed ultimate tensile strength and elongation comparable to the likes of styrene butadiene rubber (SBR) and ethylene propylene diene monomer (EPDM), two commonly used elastomers.48
Thermal properties
Thermal stability was investigated via thermogravimetric analysis (TGA) (Fig. 4A and ESI, Fig. S19†). The degradation temperature (Td,5%) was recorded as the temperature associated with a 5% mass loss. The difference in disulfide bond content was found to be the main source of variation among samples, as expected by the lower energy associated with it when compared to the carbon–carbon bond.11,12 Networks trans-NetC6 and cis-NetC6 displayed a Td,5% of 337 °C and 315 °C respectively, which is 151 °C and 145 °C higher than the values recorded for trans-NetC2SS and cis-NetC2SS. trans-Net(3-AT1.5-C2SS1) displayed Td,5% analogous to trans-NetC2SS, which is not surprising given the same chemical composition. trans-Net(C2SS-C6) Td,5% was in-between the ones of trans-NetC2SS and trans-NetC6 at 297 °C. Notably, high trans-content networks displayed better stability than their high cis counterparts, which agrees with the higher stability associated with trans linkages (Table 1). | ||
| Fig. 4 (A) TGA thermogram (25 °C – 600 °C, N2) of different networks synthesised in this work. Td,5% decreases with increasing disulfide bond content and higher cis content. (B) Zoomed in DSC thermograms (−30 °C to 80 °C, N2) of the second heating cycle of networks synthesised in this work. Tg highlighted by vertical dashes. | ||
Differential scanning calorimetry (DSC) revealed all the networks to be amorphous as no melting transition was detected (Fig. 4B and ESI, Fig. S20–S25†). While the higher degree of crosslinking is generally associated with an increase in glass transition temperature (Tg), trans-NetC6 underwent transition at the same temperature as trans-NetC2SS (−7.4 °C). However, when both monomers were used to synthesise trans-Net(C2SS-C6) its Tg was recorded 3 °C lower at −10.4 °C. Tg of trans-Net(3-AT1.5-C2SS1) was determined at −14.7 °C, roughly 7 °C lower than trans-NetC2SS. These observations can be rationalised as a result of unreacted thiols within the network acting as chain-ends, thus increasing the free volume of the network. Both high-cis networks revealed Tgs higher than their high-trans counterparts, confirming that cis/trans isomerism can also be used to modulate the thermal properties of the thermosets (Table 1).
Degradability and reprocessability
The presence of dynamic disulfide bonds was leveraged to degrade the networks in a simple reductive fashion.49trans-NetC2SS and cis-NetC2SS were successfully degraded after treatment with an equimolar amount of DL-dithiothreitol (DTT) with respect to the disulfide bond content in 60 h at room temperature in dimethylformamide (DMF) (Fig. 5A and B). The disulfide bond content was estimated based on the initial C2SS monomer feed (ESI, eqn (S4)†). Control experiments performed by treating trans-NetC6 under the same conditions (DTT in DMF) and trans-NetC2SS without DTT did not show a sign of degradation, confirming that degradation is induced by cleavage of the disulfide bonds (ESI, Fig. S26†). Size-exclusion chromatography (SEC) revealed the formation of low molecular weight peaks only for the samples undergoing degradation (Fig. 5C). The reduced species could be isolated via solvent extraction and proton nuclear magnetic resonance (1H NMR) spectroscopy was used to confirm the different cis/trans ratio of the networks by evaluation of the intensity of peaks in the vinyl region (δ = 5.79 ppm, J = 15.2 Hz, trans conformation; δ = 5.89 ppm, J = 10.1 Hz, cis conformation) (Fig. 5D and ESI, Fig. S27, S28†). The cis content was found to be 26% for trans-NetC2SS and 50% for cis-NetC2SS. However, it is worth noting that temperature can promote cis/trans isomerisation,50 meaning that a more accurate evaluation of the influence of annealing on cis-content is required to understand the impact of cis/trans isomerism on thermosets’ thermomechanical properties. Finally, high-resolution mass spectroscopy (TOF-HRMS-ES+) revealed a molecular ion peak at 789 Da for both samples, which matches the molecular weight of macromonomers (788 g mol−1) resulting from cleavage of the disulfide bond (ESI, Fig. S29 and S30†). | ||
| Fig. 5 (A) Schematic representation of DTT degradation of networks synthesised using monomer C2SS, and formation of the fully reduced species. (B) DTT-mediated reduction of a disulfide bond. (C) SEC (DMF, 5 mM NH4BF4, comparison against PMMA standard) of oligomeric species resulting from networks degradation. (D) Zoomed in section of the vinyl region (between 5.60 and 6.15 ppm) of the 1H NMR spectrum (400 MHz, 300 K, CDCl3) of the products obtained from the degradation of the networks. | ||
Finally, reprocessing of these crosslinked materials was demonstrated by heat compression moulding. trans-NetC2SS fragments could be moulded into a free-standing film by heat compression moulding at 150 °C for 20 minutes at a pressure of 3000 kg. The same was possible for trans-NetC6, albeit the reprocessed films were less structurally stable. This supports the conclusion that the inclusion of disulfide bonds is the primary driving force of the dynamic behaviour of these materials (ESI, Fig. S31†). Neither network displayed the same homogeneity as the virgin analogues, which suggests that the network structure was not perfectly reformed, possibly in light of the thermal degradation of the catalysts or of the network itself.51
Conclusions
The nucleophilic thiol–yne addition was applied in the synthesis of a library of crosslinked materials in which the thermomechanical properties could be controlled over a broad spectrum, while only using a limited pool of monomers. Notably, this method allows for the simple introduction of dynamic disulfide bonds that endowed the materials with degradability without a considerable loss in performance, compared to their fully aliphatic analogues. While the ratio between thiol and propiolate was found to be crucial in obtaining more homogeneous networks, this method nonetheless allowed for the synthesis of robust materials. Finally, the effect of cis/trans isomerism on the thermomechanical properties revealed that increasing cis content can significantly improve the strain energy density of the network. Lastly, the presence of free thiols resulting from the reduction of disulfides allows us to hypothesise the possibility of establishing a closed-loop recycling life cycle for these materials.Author contributions
DG: investigation, analysis, manuscript drafting. AB: revision and supervision. APD: conceptualisation, supervision, revision, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Birmingham for support.References
-
S. Agarwal and R. K. Gupta, in Thermosets, ed. Q. Guo, Elsevier, 2 edn, 2018, pp. 279–302 Search PubMed
.
-
J. Fan and J. Njuguna, in Lightweight Composite Structures in Transport, ed. J. Njuguna, Woodhead Publishing, 2016, pp. 3–34 Search PubMed
-
I. Hamerton and J. Kratz, in Thermosets, ed. Q. Guo, Elsevier, 2 edn, 2018, pp. 303–340 Search PubMed
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed
-
D. Rosato and D. Rosato, in Plastics Engineered Product Design, ed. D. Rosato and D. Rosato, Elsevier Science, Amsterdam, 2003, pp. 381–438 Search PubMed
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed
- N. Darby and T. E. Creighton, Mol. Biotechnol., 1997, 7, 57–77 CrossRef CAS PubMed
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC
- Y.-M. Yang, H.-Z. Yu, X.-H. Sun and Z.-M. Dang, J. Phys. Org. Chem., 2016, 29, 6–13 CrossRef
- A. A. Zavitsas, J. Phys. Chem. A, 2003, 107, 897–898 CrossRef CAS
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC
- J. C. Worch and A. P. Dove, Acc. Chem. Res., 2022, 55, 2355–2369 CrossRef CAS PubMed
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS
- P. Chakma, L. H. Rodrigues Possarle, Z. A. Digby, B. Zhang, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2017, 8, 6534–6543 RSC
- N. Kuhl, R. Geitner, R. K. Bose, S. Bode, B. Dietzek, M. Schmitt, J. Popp, S. J. Garcia, S. van der Zwaag, U. S. Schubert and M. D. Hager, Macromol. Chem. Phys., 2016, 217, 2541–2550 CrossRef CAS
- D. Berne, V. Ladmiral, E. Leclerc and S. Caillol, Polymers, 2022, 14, 4457 CrossRef CAS PubMed
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed
- J. W. Chan, H. Zhou, C. E. Hoyle and A. B. Lowe, Chem. Mater., 2009, 21, 1579–1585 CrossRef CAS
- J. C. Worch, C. J. Stubbs, M. J. Price and A. P. Dove, Chem. Rev., 2021, 121, 6744–6776 CrossRef CAS PubMed
- O. Daglar, S. Luleburgaz, E. Baysak, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Eur. Polym. J., 2020, 137, 109926 CrossRef CAS
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113–4119 CrossRef CAS
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef CAS PubMed
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 22358–22377 CrossRef CAS PubMed
- V. X. Truong and A. P. Dove, Angew. Chem., Int. Ed., 2013, 52, 4132–4136 CrossRef CAS PubMed
- C. A. Bell, J. Yu, I. A. Barker, V. X. Truong, Z. Cao, A. V. Dobrinyin, M. L. Becker and A. P. Dove, Angew. Chem., Int. Ed., 2016, 55, 13076–13080 CrossRef CAS PubMed
- C. J. Stubbs, J. C. Worch, H. Prydderch, M. L. Becker and A. P. Dove, Macromolecules, 2020, 53, 174–181 CrossRef CAS
- J. C. Worch, A. C. Weems, J. Yu, M. C. Arno, T. R. Wilks, R. T. R. Huckstepp, R. K. O'Reilly, M. L. Becker and A. P. Dove, Nat. Commun., 2020, 11, 3250 CrossRef CAS PubMed
- L. J. Macdougall, M. M. Pérez-Madrigal, J. E. Shaw, J. C. Worch, C. Sammon, S. M. Richardson and A. P. Dove, Angew. Chem., Int. Ed., 2021, 60, 25856–25864 CrossRef CAS PubMed
- A. L. Khalfa, M. L. Becker and A. P. Dove, J. Am. Chem. Soc., 2021, 143, 17510–17516 CrossRef CAS PubMed
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. Mcbride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef PubMed
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC
- W.-Q. Yuan, G.-L. Liu, C. Huang, Y.-D. Li and J.-B. Zeng, Macromolecules, 2020, 53, 9847–9858 CrossRef CAS
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed
- B. Lewis, J. M. Dennis and K. R. Shull, ACS Appl. Polym. Mater., 2023, 5, 2583–2595 CrossRef CAS
- L. Zhou, L. Zhou, R. Chen, G. Chang, K. Zhang and M. Chen, ACS Appl. Polym. Mater., 2023, 5, 8442–8449 CrossRef CAS
- B. Lewis, J. M. Dennis, C. Park and K. R. Shull, Macromolecules, 2024, 57, 7112–7122 CrossRef CAS
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Polym. Chem., 2020, 11, 5339–5345 RSC
- C. W. H. Rajawasam, O. J. Dodo, M. A. S. N. Weerasinghe, I. O. Raji, S. V. Wanasinghe, D. Konkolewicz and N. De Alwis Watuthanthrige, Polym. Chem., 2024, 15, 219–247 RSC
- T. R. Long, R. M. Elder, E. D. Bain, K. A. Masser, T. W. Sirk, J. H. Yu, D. B. Knorr and J. L. Lenhart, Soft Matter, 2018, 14, 3344–3360 RSC
- M. Radiom, P. Kong, P. Maroni, M. Schäfer, A. F. M. Kilbinger and M. Borkovec, Phys. Chem. Chem. Phys., 2016, 18, 31202–31210 RSC
- T. M. Nair, M. G. Kumaran and G. Unnikrishnan, J. Appl. Polym. Sci., 2004, 93, 2606–2621 CrossRef CAS
- S. N. Mthembu, A. Sharma, F. Albericio and B. G. Torre, ChemBioChem, 2020, 21, 1947–1954 CrossRef CAS PubMed
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed
- M. A. Alraddadi, V. Chiaradia, C. J. Stubbs, J. C. Worch and A. P. Dove, Polym. Chem., 2021, 12, 5796–5802 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01195c |
| This journal is © The Royal Society of Chemistry 2025 |