
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioderived copolymer alternatives to poly(styrene-co-maleic anhydride) via RAFT-mediated copolymerization†
Lauren Elaine Ball‡
 ,
Michael-Phillip Smith‡ and
Bert Klumperman*
,
Michael-Phillip Smith‡ and
Bert Klumperman*
Department of Chemistry and Polymer Science, Stellenbosch University, Matieland 7602, South Africa. E-mail: bklump@sun.ac.za
First published on 17th January 2025
Abstract
Poly(styrene-co-maleic anhydride) (SMAnh) is a petroleum-based copolymer with desirable properties that afford utility in both industrial and academic fields. The reversible addition–fragmentation chain transfer (RAFT)-mediated polymerization of the bioderived comonomers, indene and itaconic anhydride, was explored using three chain transfer agents with varying activity, and generally well-controlled (Đ < 1.40) polymerizations were observed.
Poly(styrene-co-maleic anhydride) (SMAnh) is synthesized via the copolymerization of the petroleum-derived comonomers, styrene (STY) and maleic anhydride (MAnh) and is readily hydrolysed in alkaline media yielding the amphiphilic copolymer, poly(styrene-co-maleic acid) (SMA). SMA has been employed in a variety of biomedical applications such as drug delivery,1–3 lipid nanodisc formation and membrane protein isolation,4–6 and hydrogel formation.7,8 The copolymerization of STY and MAnh is well-established,9 with ongoing improvements realized through the application of controlled polymerization methodologies e.g. RAFT-mediated polymerization. The synthesis of SMAnh via RAFT-mediated polymerization affords desirable properties such as targeted molecular weights, low Đ and functional chain ends which provide access to various polymer architectures.4–6,10
The increasing need for the development of “greener” synthetic protocols and “green” polymer alternatives can be assuaged through the careful selection of monomers which are bioderived instead of petroleum-derived. The selection of appropriate bioderived monomers, similar in structure to their petroleum-based counterparts, needs to be undertaken with care to retain the targeted chemical composition and physical properties of the copolymer. Ideally suited and poorly explored greener alternatives to STY and MAnh are indene (Ind),11–14 and itaconic anhydride (IAnh),15,16 respectively. These monomers are derived from renewable feedstocks and differ chemically by a single CH2 group from their petroleum-based counterparts. Despite the slight difference in chemical structure, it is hypothesized that well-defined “greener” copolymers with similar properties and biomedical relevance comparable to SMAnh can be synthesized. To the best of our knowledge, there is negligible data available for the RAFT-mediated synthesis of poly(styrene-alt-itaconic anhydride) (SIAnh), poly(indene-alt-itaconic anhydride) (IIAnh), and poly(indene-alt-maleic anhydride) (IMAnh), where these systems have only been investigated via conventional radical polymerizations,17–19 or single-unit monomer insertion (SUMI) reactions.20–23 The investigation presented here provides insight into the unexplored RAFT-mediated copolymerization of these bioderived comonomers, towards the synthesis of well-defined SIAnh, IIAnh, and IMAnh copolymers as SMAnh alternatives. For each SMAnh alternative, systematic replacement of one or both comonomers in the copolymerization reaction was undertaken (summarized in Fig. 1). A representative CTA from each “CTA class” was selected, including a dithiobenzoate (CTA1),5,24–27 trithiocarbonate (CTA3),4,6,28 and universal-type dithiocarbamate (CTA2),29 where the two former classes are commonly used CTAs for SMAnh copolymerizations and the latter CTA has recently been successfully employed for the synthesis of SMAnh and its analogues (Ball et al. – manuscript in preparation). Xanthates were excluded from kinetic experiments in this study, as they do not provide adequate control over the four RAFT-mediated copolymerizations investigated (Table S1†).30
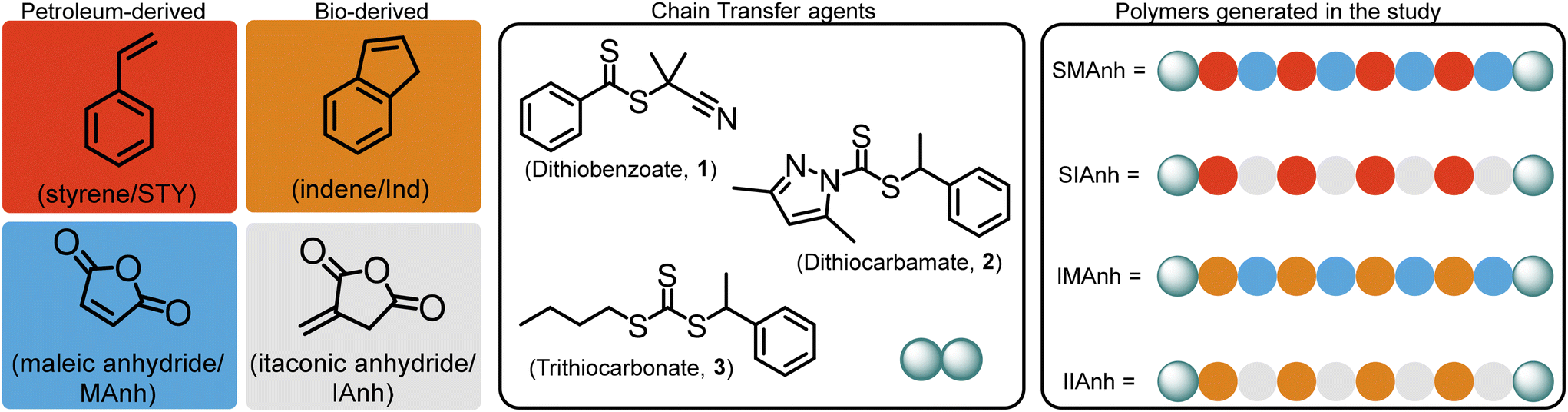 | ||
| Fig. 1 Copolymers synthesized using styrene (STY), indene (Ind), maleic anhydride (MAnh), and itaconic anhydride (IAnh). poly(styrene-alt-maleic anhydride) (SMAnh), poly(styrene-alt-itaconic anhydride) (SIAnh), poly(indene-alt-itaconic anhydride) (IIAnh), poly(indene-alt-maleic anhydride) (IMAnh). | ||
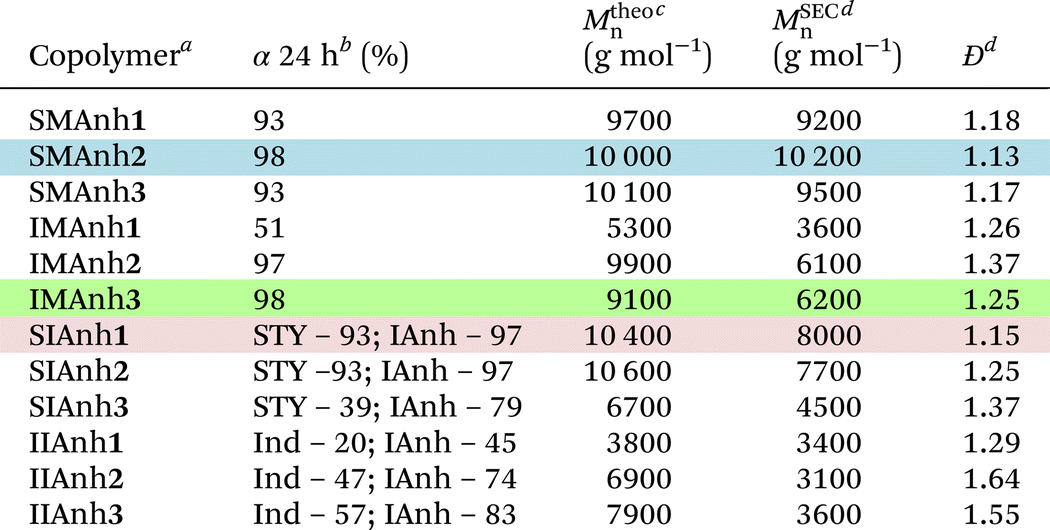
As a basis of comparison, styrene and maleic anhydride were copolymerized to afford SMAnh using two commonly used RAFT agents (CTA1 and 3),4,5 and a universal RAFT agent (CTA2) not typically employed for SMAnh copolymerizations (Table 1). For all SMAnh copolymerizations, Mn increased linearly with increasing monomer conversion (α) and low dispersities were observed for SMAnh1 and SMAnh2 throughout the copolymerization, with a decrease in Đ with increasing α observed for SMAnh3 & 2 (Fig. 2, Fig. S1†). The strong correlation between Mtheon and MSECn would suggest all CTA was converted to macro-CTA. CTA1 appears to retard the RAFT-mediated copolymerization of STY and MAnh significantly, where the kappp of the SMAnh2 and SMAnh3 copolymerization were higher than that observed for SMAnh1 (Table S1†). Furthermore, an initialization period (∼1 h) was observed for SMAnh1, during which only ∼2% comonomer conversion was obtained (Fig. S1†). This behaviour is well-known for dithiobenzoates and has been demonstrated for the RAFT-mediated synthesis of SMAnh previously.24 The reactivity ratios for this comonomer system, and others investigated in this study, were determined (Table 2) and are further discussed in Table S2.† Similar RAFT-mediated copolymerizations (using CTA1, 2 and 3) were investigated, with the replacement of the STY comonomer with Ind, to afford IMAnh. Unsurprisingly, all copolymerizations displayed a strong alternating character,31 where CTA2 and 3 were determined to be most suitable for the synthesis of well-defined IMAnh in good yield. IMAnh1 and IMAnh3 exhibited a generally linear evolution of Mn and decreasing Đ with increasing α, suggesting the copolymerization was well-controlled (Fig. 2, and Fig. S2†). Slower polymerization kinetics were observed for all instances of IMAnh (1–3) copolymerization compared to the respective SMAnh systems (Fig. S2, and Table S1†), potentially due to the increased substitution from a monosubstituted monomer (STY) to a 1,2-disubstituted monomer (Ind), resulting in a more stabilized vinyl bond. A computational analysis of a model IMAnh copolymerization showed that the Gibbs free energy required for a MAnh-based radical to react with Ind is less favourable compared to STY (ΔGSTY, addition = −5.6 kcal mol−1 and ΔGInd, addition = −1.5 kcal mol−1, Fig. S21 & S23†). This is further supported by the higher LUMO energy of Ind (−0.03 Hartree = double bond) compared to STY (−0.05 Hartree = double bond) (Fig. S20†).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AIBN of 1:0.2 (additional experimental data available in the ESI†). The recommended RAFT system for each comonomer pair is indicated by a highlighted row. All copolymers were characterized via SEC, 1H NMR and ATR-FTIR spectroscopy (Fig. S7–13†)
:AIBN of 1:0.2 (additional experimental data available in the ESI†). The recommended RAFT system for each comonomer pair is indicated by a highlighted row. All copolymers were characterized via SEC, 1H NMR and ATR-FTIR spectroscopy (Fig. S7–13†)
| a Copolymer legend provided in Fig. 1, where the number in bold refer to the CTA utilized.b Monomer conversion determined via 1H NMR spectroscopy using 1,3,5-trioxane as internal reference and eqn (S1).†c Calculated using eqn (S2).†d Determined via SEC analysis using THF (5% AcOH) as mobile phase and PS calibration standards. |
|---|
 |
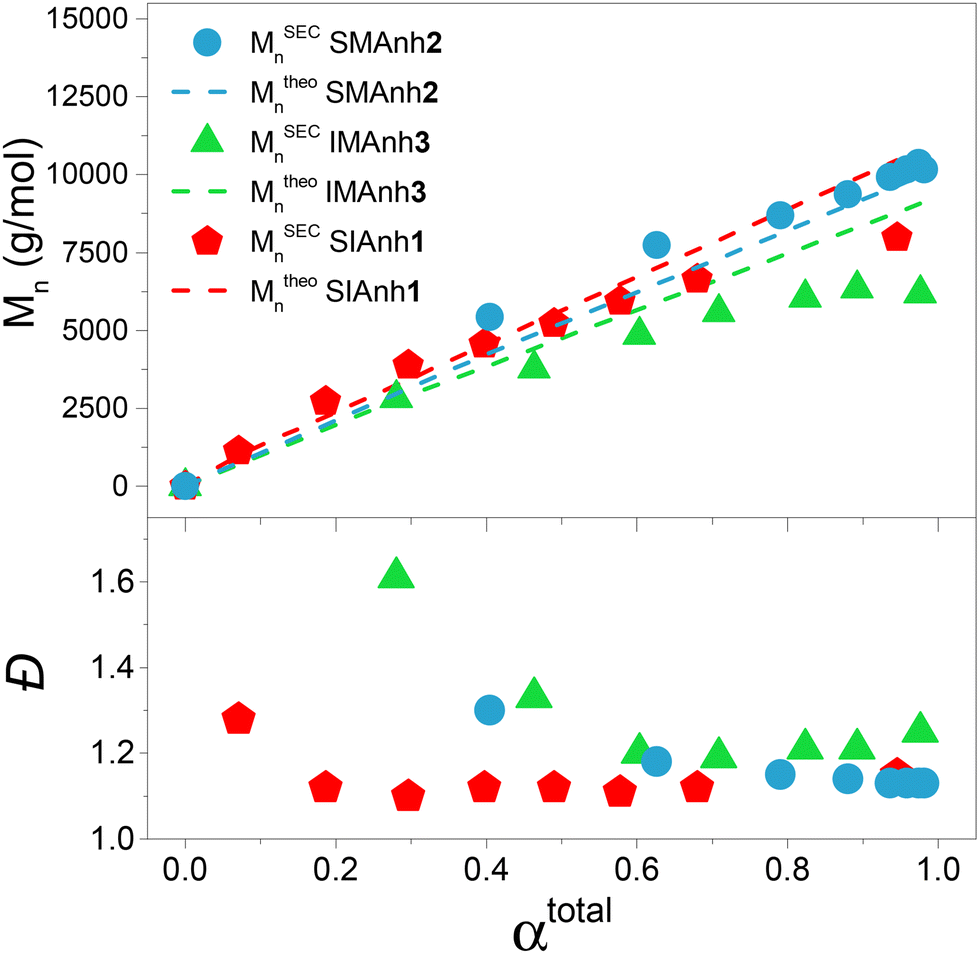 | ||
| Fig. 2 Evolution of MSECn and Đ with increasing monomer conversion for well-controlled SMAnh, SIAnh and IMAnh copolymerizations. | ||
| STY (r1) | Ind (r1) | |
|---|---|---|
| MAnh (r2) | r1 = 0.01; r2 = 0.01 | r1 = 0.01; r2 = 0.01 |
| IAnh (r2) | r1 = 0.12; r2 = 0.01 | r1 = 0.01; r2 = 0.13 |
CTA1 caused significant retardation of the IMAnh copolymerization kinetics (in addition to retardation derived from Ind), with a longer initialization period (∼2 h) observed compared to the SMAnh1 copolymerization (Fig. S2†). As a result, low monomer conversion was obtained (50% within 24 h). Despite this, IMAnh1 copolymers with low Đ and good correlation between Mtheon and MSECn were obtained. For all IMAnh copolymerizations, a deviation of MSECn from Mtheon was observed at higher monomer conversions, accompanied by an increase in Đ (Fig. S2†). 1H NMR spectroscopic analysis of IMAnh copolymerization kinetic samples showed full consumption of CTA1 within 3 h and CTA2/CTA3 within 1 h, which would suggest reasonable Z- and R-group efficiencies. Therefore, the deviation of MSECn at high α is tentatively attributed to inefficient reinitiation of the macro-CTA leaving group, as SEC analysis revealed low molecular weight tailing in the RI eluogram which was also present in the corresponding UV eluogram (320 nm) (Fig. S12†). SMAnh and IMAnh have a strong alternating character as a result of the inability of MAnh to homopolymerize. Contrarily, the homopolymerization of IAnh has been reported in literature.9 SIAnh copolymerizations generally exhibited slower kinetics compared to the corresponding SMAnh copolymerizations, with only slightly higher rates of IAnh consumption compared to STY observed for SIAnh1 and SIAnh2 (Fig. S3†). Inspection of the copolymerization kinetics for SMAnh1 and SIAnh1 shows that similar kappp values are obtained. This result is interesting as it indicates that the substitution pattern of the double bond of IAnh does not impact the rate of polymerization significantly (1,1 vs. 1,2 di-substituted monomers). CTA1 was fully converted to macro-CTA within 1 h (Fig. S5†) without the presence of a prominent initialization period, where MSECn evolved linearly with increasing α and low Đ was maintained throughout the copolymerization (Fig. 2). CTA2 and CTA3 were converted to macro-CTA slowly throughout the copolymerization, where 12% and 7% of CTA2 and CTA3 remained at 24 h, respectively (Fig. S5 and 6†). The RAFT pre-equilibrium for SIAnh3 and SMAnh3 was assessed computationally (Fig. S16 and 17†), where the former exhibited significantly slower consumption of CTA3 than the latter. The addition of IAnh to CTA3 to form the corresponding intermediate radical was energetically unfavourable compared to the addition of MAnh (ΔGMAnh, addition = 13.7 kcal mol−1 vs. ΔGIAnh, addition = 24.7 kcal mol−1). Furthermore, the fragmentation of the MAnh-based intermediate radical favoured the generation of R-group re-initiating radicals (promoting an efficient pre-equilibrium), whereas the fragmentation of IAnh-based intermediate radicals favoured the regeneration of IAnh-based radicals (resulting in an inefficient pre-equilibrium) (Fig. S16 and 17†). SEC analysis of SIAnh2 and SIAnh3 showed low molecular weight tailing in the RI eluograms which had an associated UV signal (320 nm), which in this case likely corresponds to the continual formation of low molecular weight SIAnh as CTA2/3 is slowly converted into macro-CTA throughout the copolymerization (Fig. S10 and 11†). While the linear evolution of MSECn with increasing α was observed for SIAnh2 and SIAnh3, the inefficient conversion of CTA to macro-CTA resulted in poor correlation between MSECn and Mtheon and relatively high Đ throughout the copolymerization (Fig. S3†).
Lastly, attempts were made to copolymerize the fully bioderived comonomer pair (Ind and IAnh) via RAFT-mediated copolymerization. None of the CTAs investigated could provide an adequate level of control over the RAFT-mediated copolymerization of Ind and IAnh (Fig. S4†). Inspection of the kinetic samples via 1H NMR spectroscopy showed that CTA1 was fully converted to macro-CTA within 3 h, while CTA2 and CTA3 were consumed slowly throughout the copolymerization, with a total consumption of 79% and 95%, respectively (Fig. S5 and 6†). The IIAnh3 RAFT pre-equilibrium, and the propagation of Ind- and IAnh-based radicals, were assessed computationally (Fig. S19 & S24,† respectively). Overall, the differences in reactivity of the Ind- and IAnh-based propagating radicals suggest that during the main equilibrium, intermediate radicals predominantly constitute IAnh terminal leaving groups. During the RAFT pre-equilibrium, the fragmentation of the IAnh-based intermediate radical favours the generation of IAnh-based propagating radicals as opposed to R-group re-initiating radicals (critical analysis of the IIAnh RAFT-mediated copolymerization available in the ESI†). Thus, it is plausible that IIAnh is a better polymeric leaving group compared to the 1-phenyl ethyl R-group, resulting in slow consumption of CTA2 and CTA3 throughout the copolymerization. Inspection of the RI eluograms for IIAnh3 shows that a shift towards lower elution volumes with copolymerization time can be observed, but due to the inefficient consumption of the CTA throughout the copolymerization, the Mn remains relatively constant with increasing monomer conversion (Fig. S13†) and consequently, high Đ (>1.5) was observed throughout the copolymerization. The kinetic analyses of all four comonomer systems, consistently suggest that the itaconic anhydride-containing copolymers have slower polymerization kinetics with inefficient conversion of CTA to macro-CTA. While the RAFT-mediated copolymerization of Ind and IAnh yielded promising results, further critical investigation of the system is required to enable the synthesis of a well-defined, and fully bioderived, copolymer. It is possible that an alternative CTA, with an R-group that is more compatible with the IAnh comonomer, would significantly improve the synthesis of the IAnh-based copolymers.
Conclusions
In this work, the ideal CTAs required for the well-controlled RAFT-mediated copolymerization of petroleum-derived and bioderived comonomers to afford SMAnh, SIAnh, and IMAnh were determined (CTA2, 3 and 1, respectively). The fully bioderived copolymer IIAnh could be synthesized successfully, but IIAnh with targeted Mn and low Đ could not be achieved, due to inefficient conversion of CTA to macro-CTA. Nevertheless, this study has effectively expanded the variety of “greener” SMAnh-type copolymers available for utility in biomedical research. An interrogation of solution properties for these bioderived copolymers and how they compare to SMAnh is currently underway within our research group. The results of this interrogation will provide the tools necessary to tune the chemical composition of the bioderived copolymers such that desirable physical properties are realized.Author contributions
Lauren Elaine Ball: data curation, formal analysis, investigation, methodology, visualization, writing. Michael-Phillip Smith: conceptualization, computational investigation, data curation, formal analysis, investigation, methodology, project administration, visualization, writing – first draft. Bert Klumperman: supervision, funding acquisition, resources, reviewing and editing.Data availability
All the data relevant to the current submission has been collated in the ESI.† Raw data, such as NMR, FT-IR and SEC data can be obtained upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge and thank the Wellcome Trust (grant no 223728/Z/21/Z), National Research Foundation (NRF, grant no 46855) and Wilhelm Frank Trust for funding this research. The authors further acknowledge the Centre for High-Performance Computing (NICIS CHPC) for access to the computational software that was utilized in this work.References
- N. Angelova and G. Yordanov, Colloids Surf., A, 2014, 452, 73–81 CrossRef CAS.
- S. Henry, M. El-Slayed, C. Pirie, A. Hoffman and P. Stayton, Biomacromolecules, 2006, 7(8), 2407–2414 CrossRef CAS PubMed.
- R. Richard, M. Schwarz, K. Chan, N. Teigen and M. Boden, J. Biomed. Mater. Res., Part A, 2009, 90(2), 522–532 CrossRef PubMed.
- R. D. Cunningham, A. H. Kopf, B. O. W. Elenbaas, B. B. P. Staal, R. Pfukwa, J. A. Killian and B. Klumperman, Biomacromolecules, 2020, 21, 3287–3300 CrossRef CAS PubMed.
- A. A. A. Smith, H. E. Autzen, T. Laursen, V. Wu, M. Yen, A. Hall, S. D. Hansen, Y. Cheng and T. Xu, Biomacromolecules, 2017, 18, 3706–3713 CrossRef CAS PubMed.
- K. M. Burridge, B. D. Harding, I. D. Sahu, M. M. Kearns, R. B. Stowe, M. T. Dolan, R. E. Edelmann, C. Dabney-Smith, R. C. Page, D. Konkolewicz and G. A. Lorigan, Biomacromolecules, 2020, 21, 1274–1284 CrossRef CAS PubMed.
- J. Hrib, E. Chylikova Krumbholcova, M. Duskova-Smrckova, R. Hobzova, J. Sirc, M. Hruby, J. Michalek, J. Hodan, P. Lesny and R. Smucler, Polymers, 2019, 11(7), 1087 CrossRef CAS PubMed.
- C. Tang, S. Ye and H. Liu, Polymer, 2007, 48, 4482–4491 CrossRef CAS.
- B. Klumperman, Polym. Chem., 2010, 1, 558–562 RSC.
- S. Bag, S. Ghosh, S. Paul, M. E. H. Khan and P. De, Macromol. Rapid Commun., 2021, 42(23), 2100501 CrossRef CAS PubMed.
- E. A. Uslamin, N. Kosinov, G. A. Filonenko, B. Mezari, E. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9(9), 8547–8554 CrossRef CAS.
- B. Zhang, Z. Zhong, J. Zhang and R. Ruan, J. Anal. Appl. Pyrolysis, 2018, 133, 147–153 CrossRef CAS.
- J. Gancedo, L. Faba and S. Ordóñez, ACS Sustainable Chem. Eng., 2022, 10(9), 3057–3065 CrossRef CAS.
- S. Li, Y. Li, J. Wu, Z. Wang, F. Wang, L. Deng and K. Nie, Renewable Energy, 2020, 155, 1042–1050 CrossRef CAS.
- M. Okabe, D. Lies, S. Kanamasa and E. Y. Park, Appl. Microbiol. Biotechnol., 2009, 84, 597–606 CrossRef CAS PubMed.
- T. Willke and K. D. Vorlop, Appl. Microbiol. Biotechnol., 2001, 56, 289–295 CrossRef CAS PubMed.
- J. Drougas and R. Guile, J. Polym. Sci., 1961, 55(6), 161 Search PubMed.
- D. Tohru and M. Yuji, Macromolecules, 1978, 11(1), 270–274 CrossRef.
- L. F. Kim, L. L. Stotskaya, B. A. Krentsel, V. P. Zubov, V. B. Golubev and I. L. Stoyachenko, J. Macromol. Sci., Part A:Pure Appl. Chem., 1978, 12, 1197–1210 CrossRef.
- K. Hakobyan, B. Noble and J. Xu, Chem. Commun., 2024, 60, 7443–7446 RSC.
- K. Hakobyan, B. B. Noble and J. Xu, Polym. Chem., 2023, 14, 4116–4125 RSC.
- Z. Huang, B. B. Noble, N. Corrigan, Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer and J. Xu, J. Am. Chem. Soc., 2018, 140, 13392–13406 CrossRef CAS PubMed.
- R. Liu, L. Zhang, Z. Huang and J. Xu, Polym. Chem., 2020, 11, 4557–4567 RSC.
- E. T. A. van den Dungen, J. Rinquest, N. O. Pretorius, J. M. McKenzie, J. B. McLeary, R. D. Sanderson and B. Klumperman, Aust. J. Chem., 2006, 59, 742–784 CrossRef CAS.
- S. Sirohi, M. Jassal and A. K. Agrawal, Appl. Nanosci., 2018, 8, 1701–1710 CrossRef CAS.
- M. C. Davies, J. V. Dawkins and D. J. Hourston, Polymer, 2005, 46(6), 1739–1753 CrossRef CAS.
- H. De Brouwer, M. A. J. Schellekens, B. Klumperman, M. J. Monteiro and A. L. German, J. Polym. Sci., Part A:Polym. Chem., 2000, 19(38), 3596–3603 CrossRef.
- J. Ma, C. Cheng, G. Sun and K. L. Wooley, Macromolecules, 2008, 41(23), 9080–9089 CrossRef CAS PubMed.
- J. Gardiner, I. Martinez-Botella, J. Tsanaktsidis and G. Moad, Polym. Chem., 2016, 7, 481–492 RSC.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45(13), 5321–5342 CrossRef CAS.
- T. Doiuchi and Y. Minoura, Macromolecules, 1978, 11(1), 270–274 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01227e |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |