
TBD-catalyzed anionic ring-opening polymerization of hexamethylcyclotrisiloxane: a new route for the controlled synthesis of PDMS†
Alice
Corfa
,
Sylvain
Caillol
 ,
Julien
Pinaud
* and
Vincent
Ladmiral
*
,
Julien
Pinaud
* and
Vincent
Ladmiral
*
ICGM, University of Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: Julien.pinaud@umonpellier.fr; vincent.ladmiral@enscm.fr
First published on 5th December 2024
Abstract
The controlled anionic ring-opening polymerization of hexamethylcyclotrisiloxane (D3) was optimized for a commercial and easy-to-use organic catalyst, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), employing various types of initiators. Alcohols and silanols were compared for the synthesis of monofunctional and telechelic PDMS. Polymerizations were monitored by 1H NMR spectroscopy, MALDI-TOF mass spectroscopy and SEC to compare the effects of the initiator structure and catalyst concentration. TBD was shown to control the AROP of D3 when using silanols as the initiator, thus affording well-defined PDMS structures. When alcohols were used as initiators, the polymerization proceeded with a lower level of control due to slower initiation. These differences in initiation rates likely originated from the differences in pKa between silanols and alcohols.
Introduction
Polydimethylsiloxanes (PDMS) are high-performance industrial polymers ubiquitous in daily life, from cosmetics to construction materials.1 They have unique properties (mechanical, resistance to ageing and hydrophobicity) that make them irreplaceable in many applications. They also exhibit specific properties such as flame retardancy, chemical inertness, exceptional flow properties and very low Tg due to their siloxane backbone.1 Depending on the application, different topologies and molar masses are targeted. Industrially, PDMS are provided as oligomeric cyclic siloxanes for use as lubricants, as network for coating resins, or as crosslinked material for sealing elastomers or molds.1,2 The properties and behavior of the final material are directly related to the topology, molar mass, and chain-ends structure. Consequently, finding synthetic pathways leading to PDMS with controlled molar masses and structures is of high interest. The most common synthetic route to PDMS is the anionic ring-opening polymerization (AROP) of hexamethylcyclotrisiloxane (D3) or octamethylcyclotetrasiloxane (D4). The polymerizations of D3 and D4 proceed very differently due to the higher ring tension in D3 than in D4.3 D3 is less thermodynamically stable and rapidly opens to produce linear polysiloxanes via kinetically controlled AROP. However, the ROP of cyclosiloxanes is an equilibrated polymerization, and depolymerization (back-biting, Scheme 2) is driven by the formation of stable macrocycles. The polymerization of D3 should, thus, be stopped at the highest D3 conversion value. Under kinetic control, the polymerization is driven by the fast polymerization rate that allows the polymerization to reach high conversions before any thermodynamic equilibrium takes place. Unlike the aforementioned AROP, equilibrium polymerization of D4 is carried out under a thermodynamical condition due to the low ring tension of D4.4 In this case, propagation and back-biting are in competition, and the polymerization reaches a thermodynamic equilibrium driven by the temperature, the monomer concentration and the nature of the catalyst/initiator used.5,6 Usually, the AROP of D4 is performed in bulk at relatively high temperature, whereas D3 can be polymerized readily at room temperature in solution.6 The formation of cyclic compounds occurs via the back-biting reaction, and cyclic compounds Dx (x = 4, 5, or 6, mostly) are formed.7–9The AROP of cyclosiloxanes can be performed using different types of initiators or catalysts, but the method mainly adopted by industry so far is the use of an alkali hydroxide such as KOH, where HO− initiates the polymerization and K+ catalyzes the propagation. However, the AROP of cyclosiloxanes suffers from a back-biting reaction that limits polymer-chain length and results in the formation of undesired macrocycles that must be separated from the desired PDMS product. The counter cation has proven to have a strong effect on the back-biting and polymerization rates. The rate of polymerization increases strongly in the series Cs+ < Rb+ < K+ < Na+ < Li+ < N(Et)4+, with almost no macrocycles formed with the ammonium counter cation.6 This phenomenon hints at the advantages of organic compounds for the synthesis of PDMS.
In the early 1990s, phosphazene bases were found to be very effective for the organocatalytic ROP of cyclosiloxanes such as D4.10–12 In 2022, Shi et al. focused on the use of the highly basic (pKb = 33.3) trisphosphazene catalysts (3a in Fig. 1) for the polymerization of D3 initiated by benzyl alcohol,13 and were able to produce PDMS ranging from 3 to 16 kg mol−1. N-heterocyclic carbenes (NHC) also exhibit high catalytic activity for the synthesis of PDMS.14 In 2006, NHC (4a and 4b in Fig. 1) were reported for the conversion of D4 into PDMS of high molar mass (Mn = 100–200 kg mol−1), albeit with relatively high dispersity (∼1.6).14 However, in 2023, NHC were shown to promote the ROP of D4 without producing cyclic oligomers.4 Phosphazenes and NHC are very efficient catalysts, providing very high polymerization rate of D3 and D4 due to their high catalytic activity and high pKb. However, this high catalytic activity also leads to stability issues of the catalyst that can easily react with moisture or CO2.15
 | ||
| Fig. 1 Organocatalysts used for the AROP of D3. | ||
Guanidines have also proven to be very efficient catalysts for the synthesis of PDMS from D3.16–18 In 2018, Fuchise et al. published an important work on the scope of organocatalysts able to polymerize cyclosiloxane monomers, and showed that initiation by water could be used to produce telechelic PDMS via ROP even with non-distilled solvents.16 In the case of lactones, guanidine-based catalysts are known to have two activation modes: chain-end activation of the hydroxyl group or monomer activation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the ester.19 In the guanidine-catalyzed mechanism, initiation and propagation steps occur via chain-end activation with a proton shuffle as shown in Scheme 1. In 2020, Fuchise et al. optimized the conditions of the AROP of D3 catalyzed by 1,3-trimethylene-2-n-propylguanidine (TMnPG) or 1,3-trimethylene-2-ethylguanidine (TMEG), and initiated by water and silanols.17 Moreover, they demonstrated that organocatalysis with guanidines is a suitable method for the polymerization of many cyclosiloxanes, allowing the preparation of telechelic or monofunctional polymers.
O of the ester.19 In the guanidine-catalyzed mechanism, initiation and propagation steps occur via chain-end activation with a proton shuffle as shown in Scheme 1. In 2020, Fuchise et al. optimized the conditions of the AROP of D3 catalyzed by 1,3-trimethylene-2-n-propylguanidine (TMnPG) or 1,3-trimethylene-2-ethylguanidine (TMEG), and initiated by water and silanols.17 Moreover, they demonstrated that organocatalysis with guanidines is a suitable method for the polymerization of many cyclosiloxanes, allowing the preparation of telechelic or monofunctional polymers.
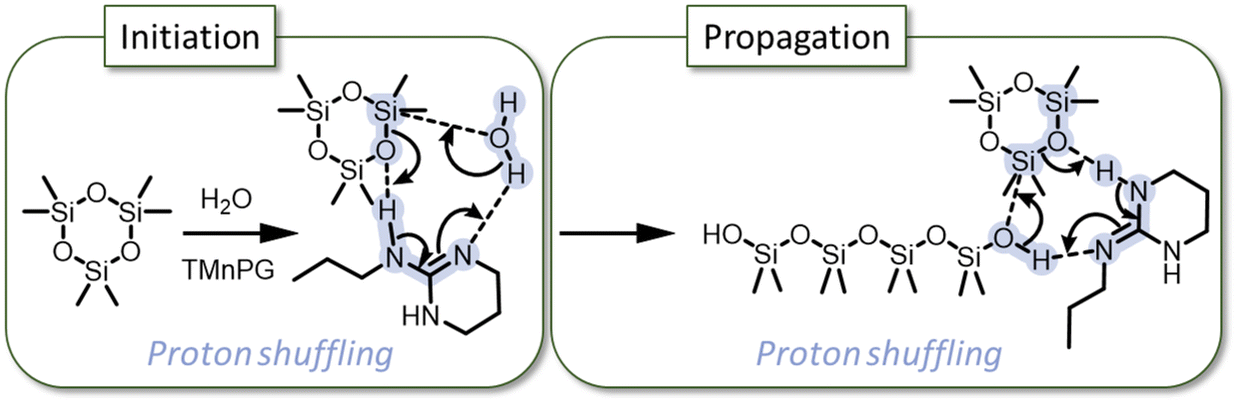 | ||
| Scheme 1 “Proton shuffling” mechanism of initiation and propagation of D3via TMnPG-catalyzed AROP.16 | ||
These previous reports open the way to the preparation of various types of polysiloxanes, which is a key issue for the production of polymers endowed with superior properties. In addition, organocatalysis appears to be a valuable alternative to the usual AROP because it can proceed at ambient temperature, with relatively short reaction times and easy handling of reactants. However, it should be noted that this last feature is not achieved if using NHC or phosphazene bases because of their strong basicity and reactivity. The guanidine-based organic catalysts recently studied for the ROP of D3 or D4 are not commercially available and require rigorous purification steps. Thus, it would be more convenient to turn towards less reactive and commercially available organic catalysts.
The present work, thus, aims at studying and optimizing the AROP of D3 catalyzed by a stable and readily commercially available catalyst: 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). TBD has proven to be a very efficient catalyst for many types of reactions, among which the ROP of lactones and cyclic carbonates.20 As a guanidine-based catalyst, TBD can undergo monomer activation and chain-end activation for lactones. However, in the case of cyclosiloxanes, TBD is expected not to be nucleophilic enough to perform a nucleophilic attack upon the silicon atom. As a commercial and stable catalyst, it appears as a potentially viable alternative for the AROP of cyclosiloxanes. The present work reports kinetic studies of the ROP of D3 using TBD as a catalyst with different types of initiators, as shown in Scheme 3. It highlights the influence of the pKa and structure of the initiator on the kinetics of the AROP of D3. Finally, it demonstrates the potential of TBD as a catalyst for the synthesis of PDMS with molar masses up to 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and low dispersity.
000 g mol−1 and low dispersity.
Results and discussion
The organocatalyzed AROP of D3 proceeds via three steps: initiation, propagation and termination. In parallel, side reactions such as condensation, redistribution or back-biting (shown in Scheme 2) can take place depending on the reaction temperature as well as the structure and concentration of the catalyst. Initiation is usually achieved by hydroxyl group-containing molecules such as water, alcohol, or silanol when activated by basic catalysts. A controlled AROP is characterized by low dispersity, first-order kinetics behavior, and linear evolution of molar mass with monomer conversion. These are the consequence of a fast initiation compared with propagation and the absence of any chain termination, implying that all the chains start and grow at the same pace.21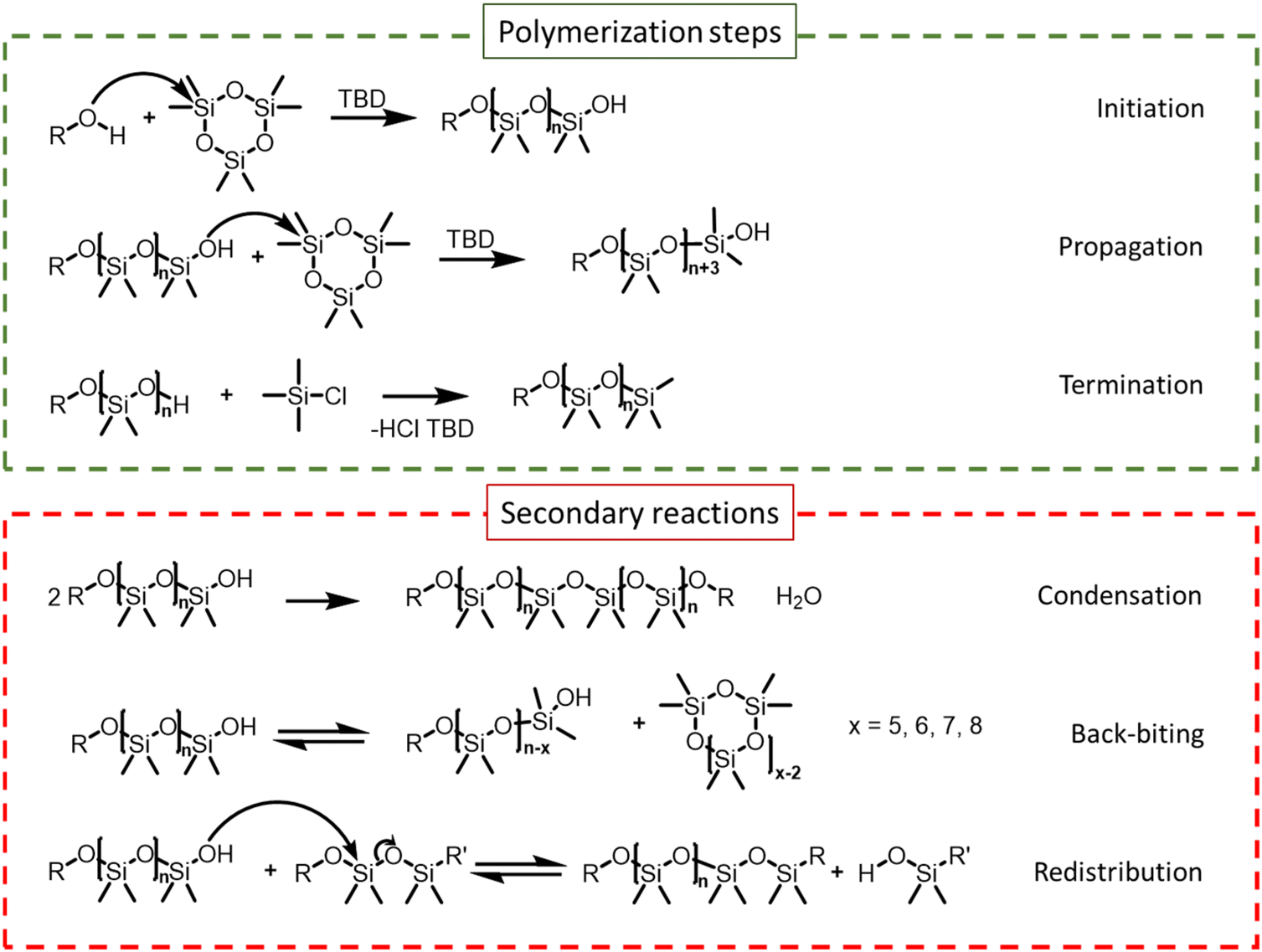 | ||
| Scheme 2 Polymerization steps and side reactions occurring during ring-opening polymerization (ROP) of hexamethylcyclosiloxane using different types of initiators and TBD as a catalyst.6,16 | ||
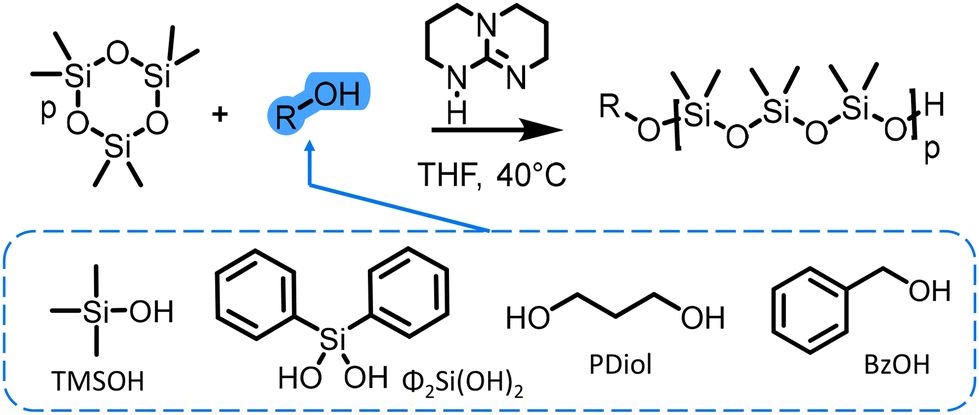 | ||
| Scheme 3 Ring-opening polymerization (ROP) of hexamethylcyclotrisiloxane (D3) using different types of initiators and TBD as a catalyst. | ||
In our study, silanols and alcohols initiators were compared to investigate the impact of their structure on the initiation step. We also examined the efficiency of TBD as an organocatalyst for the AROP of cyclosiloxanes. Both difunctional (diphenylsilanediol and propanediol) and monofunctional (trimethylsilanol and benzyl alcohol) initiators (Scheme 1) were employed to determine the best TBD-catalyzed AROP conditions combining an easy setup and high control (Đ < 1.15) of the polymerization.
As mentioned previously, PDMS can be synthesized with various topologies depending on the target application. For many industrial uses, PDMS with high molar masses are needed to achieve the desired mechanical properties. In consequence, we attempted to optimize the experimental conditions to produce PDMS with molar masses up to 50000 g mol−1.
Characteristics of polymerization
In a first set of experiments (entries 1–8, Table 1), TBD was used as an organocatalyst for the AROP of D3 in THF at 40 °C using the conditions depicted in Table 1. Four initiators were tested: benzyl alcohol (BnOH), trimethylsilanol (TMSOH), diphenylsilane diol (Φ2Si(OH)2) and propane diol (PDiol). The crude products were precipitated in methanol and dried under high vacuum to give transparent oils. PDMS were obtained with molar masses and conversions ranging between 5000 and 12000 g mol−1 (1.1 < Đ < 1.3) and between 95 and 99% respectively, as shown in Table 1. Telechelic PDMS were obtained in similar polymerization times as those reported by Fuchise et al. (i.e., 117 min for [D3]0/[H2O]0/[TBD]0 = 50/1/0.50). The resulting PDMS were characterized by MALDI-ToF mass spectrometry (Fig. 5) and 1H NMR spectroscopy (Fig. 2).
 | ||
| Fig. 2 1H NMR spectra in CDCl3 of precipitated PDMS initiated by (a) diphenylsilanediol, (b) trimethylsilanol, (c) benzyl alcohol, and (d) propane diol. | ||
| Entry | Initiator (I) | [D3]0/[I]0/[TBD]0 | [D3]0/[OH]0/[TBD]0a | Conv.b (%) | Time (min) | M n (kg mol−1) | Đ | k app (min−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CALCc | NMRd | SECe | ||||||||
a [D3]0 = 1.8 mol L−1.
b Calculated from 1H NMR normalizing D3 and PDMS signals to 100.
c
 .
d Calculated from the 1H NMR signals of PDMS and of the initiator according to the following equations: for polymers initiated by Φ2Si(OH)2, BnOH and PDiol: .
d Calculated from the 1H NMR signals of PDMS and of the initiator according to the following equations: for polymers initiated by Φ2Si(OH)2, BnOH and PDiol:  (NΦ2Si(OH)2 = 10 and NBnOH = NPDiol = 2); and for polymers initiated by TMSOH: (NΦ2Si(OH)2 = 10 and NBnOH = NPDiol = 2); and for polymers initiated by TMSOH: 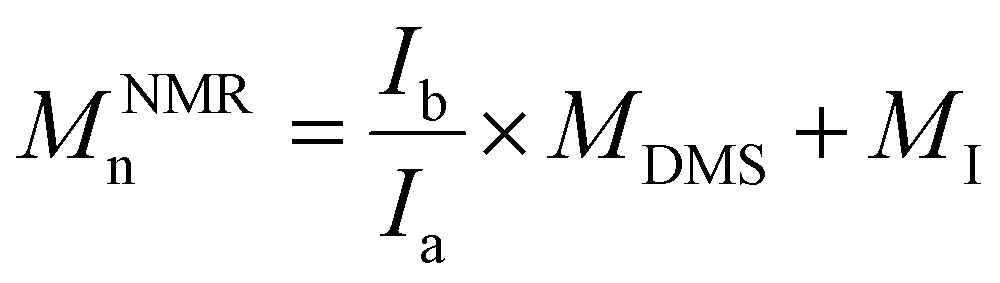 (MDMS = 74.02 g mol−1, MΦ2Si(OH)2 = 216 g mol−1, MBnOH = 108 g mol−1, MTMSOH = 90 g mol−1, MPDiol = 76 g mol−1).
e Determined by SEC in THF (1 ml min−1) using PMMA calibration. (MDMS = 74.02 g mol−1, MΦ2Si(OH)2 = 216 g mol−1, MBnOH = 108 g mol−1, MTMSOH = 90 g mol−1, MPDiol = 76 g mol−1).
e Determined by SEC in THF (1 ml min−1) using PMMA calibration.
|
||||||||||
| 1 | TMSOH | 50/2/0.2 | 25/1/0.1 | 97 | 130 | 5.5 | 5.6 | 5.7 | 1.16 | 0.024 |
| 2 | TMSOH | 50/2/1 | 25/1/0.5 | 94 | 25 | 5.3 | 5.5 | 6.6 | 1.20 | 0.108 |
| 3 | BnOH | 50/2/0.2 | 25/1/0.1 | 95 | 130 | 5.4 | 6.4 | 5.6 | 1.34 | — |
| 4 | BnOH | 50/2/1 | 25/1/0.5 | 96 | 33 | 5.4 | 7.3 | 6.9 | 1.30 | — |
| 5 | Φ2Si(OH)2 | 50/1/0.2 | 25/1/0.1 | 96 | 90 | 10.9 | 10.5 | 9.7 | 1.11 | 0.033 |
| 6 | Φ2Si(OH)2 | 50/1/1 | 25/1/0.5 | 95 | 18 | 10.5 | 12.2 | 11.0 | 1.18 | 0.159 |
| 7 | PDiol | 50/1/0.2 | 25/1/0.1 | 99 | 90 | 11.0 | 11.1 | 9.9 | 1.27 | 0.044 |
| 8 | PDiol | 50/1/1 | 25/1/0.5 | 95 | 22 | 10.5 | 11.3 | 12.0 | 1.30 | 0.124 |
In all 1H NMR spectra of PDMS samples, dimethylsiloxane units in the PDMS chain exhibited a signal at 0.10 ppm and terminal dimethylsiloxane units were observed at 1.69 ppm. Additionally, for PDMS initiated by BnOH, a benzylic chain-end was observed with an aromatic resonance at 7.32 ppm and a methylene signal at 4.70 ppm (Fig. 2c) whereas, for PDMS initiated by TMSOH, the TMS chain-end was observed at 0.07 ppm (Fig. 2b). For PDMS initiated by Φ2Si(OH)2, the diphenyl group exhibited a signal at 7.50 ppm (Fig. 2a) and for those initiated by PDiol, the propylene group showed two signals at 3.80 ppm and 1.78 ppm (Fig. 2d).
Moreover, for each experiment, aliquots were withdrawn from the reaction media at specific times in the course of the polymerization, quenched with acetic acid and analyzed by 1H NMR spectroscopy to determine monomer conversion and by steric exclusion chromatography (SEC) in THF to determine the average molar mass (Mn) and dispersity (Đ) of the produced PDMS.
Conversion was calculated from 1H NMR spectra (Fig. S3†) using the integration of the signals of D3 at 0.20 ppm and of PDMS at 0.10 ppm. The signal of D4 (∼0.15 ppm) resulting from backbiting side reaction (Scheme 1) was not observed for any polymerization at any conversion.
For each polymerization performed with monofunctional and telechelic initiators, pseudo first-order kinetic plots were constructed (Fig. 3 and 6). For both silanol and silane diol initiators (entries 1, 2 and 5, 6, Table 1), the pseudo first-order kinetics plots were linear, indicating good control of the polymerization and allowing the determination of kapp, the apparent rate of polymerization. kapp decreased with an increasing [I]0/[TBD]0 ratio (Table 1, entries 1, 2 and 5, 6). For PDiol, kapp could be determined (0.0444 and 0.1245 min−1) and was comparable with the value determined for the silane diol-initiated AROP (0.0332 and 0.1595 min−1). However, the BnOH-initiated AROP did not lead to a linear first-order kinetic plot.
 | ||
| Fig. 3 Kinetic plot of the AROP of D3 for (a) entry 1, (b) entry 2, (c) entry 3 and (d) entry 4, Table 1. | ||
The evolution of the average number molar masses and dispersity with conversion were plotted (Fig. 4 and 7) to better understand the differences in kinetics between each initiator. PDMS produced by the silanol-initiated AROP (entries 1, 2 and 5, 6, Table 1) displayed low dispersities (Đ < 1.2), thus confirming the control of the polymerization. In contrast, PDMS produced by the alcohols-initiated AROP (BnOH and Pdiol) (entries 3, 4 and 7, 8, Table 1) exhibited higher dispersity (Đ > 1.27).
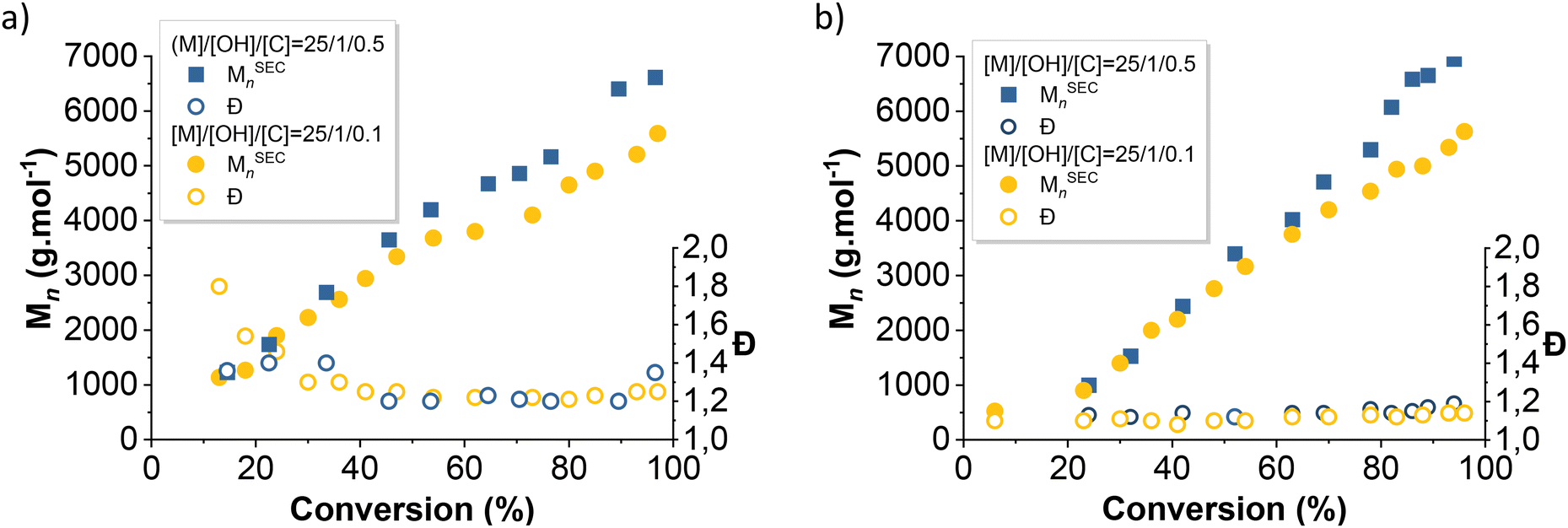 | ||
| Fig. 4 Evolution of number average molecular weight and dispersity with conversion of D3 initiated with (a) BnOH and (b) TMSOH. | ||
In order to better understand the difference between the initiators, a parametric study was undertaken. First, the effect of the difference in pKa between alcohols and silanols was examined. TMSOH and BnOH were compared to highlight the difference between an alcohol- and a silanol-initiated AROP. Second, the effect of monofunctional and difunctional initiators was studied.
Influence of pKa
Interestingly, the kinetics of the AROP conducted using alcohols as initiators did not follow perfect first-order kinetics. Indeed, when benzyl alcohol was used as an initiator (entries 3 and 4, Table 1), kapp increased during the polymerization, indicating that the concentration of the propagating species increased over the course of the polymerization. Nevertheless, at conversion >70%, the kapp values of the BnOH-initiated polymerization were close to those of the TMSOH-initiated AROP (Table 1), suggesting that a similar concentration of active species was reached in both cases. This observation can be explained by the slower initiation of BnOH compared with silanol groups caused by the higher pKa of BnOH (pKa, BnOH = 15, pKa, SiOH = 12). This was confirmed by the evolution of the 1H NMR spectra of the reaction mixtures for the AROP of D3 initiated by BnOH (Fig. S3a†) and TMSOH (Fig. S3b†) at various reaction times, which showed a slow decrease of the signal at 4.64 ppm assigned to CH2–OH in Fig. S3a,† as opposed to the very fast disappearance of the silanol signal at 0.11 ppm (completely absent after only 2 min) in Fig. S3b.†Additionally, the conversion of BnOH reached plateaus of 75% and 90% for [BnOH]0/[TBD]0 = 1/0.1 = 10 and 1/0.5 = 2, respectively. In both cases the plateau was reached at ∼60% conversion of D3. These observations indicated that initiation by BnOH occurred throughout the polymerization, whereas it was almost instantaneous in the case of TMSOH. Consequently, in the case of initiation using BnOH, active centers were generated all along the polymerization, leading to the increase of kapp with time. It is nonetheless important to note that, as in the case of AROP initiated with TMSOH, increasing the TBD concentration leads to faster initiation with BnOH.
As expected, this slower initiation rates directly translated into PDMS with higher dispersity, although this effect was particularly pronounced in the early stages of polymerization (Đ = 1.8 after 10 min vs. Đ = 1.2 after 40 min of reaction) (Fig. 4a). Moreover, for a similar conversion, the Mn values of the BnOH-initiated PDMS were higher than those of the silanol-initiated PDMS (Fig. 4).
Decreasing the [BnOH]0/[TBD]0 ratio from 10 to 2 resulted in a faster initiation and, thus, in the production of PDMS with lower dispersity in the early stages of polymerization (Fig. 4a). However, decreasing this ratio also promoted condensation reactions at lower monomer conversion (∼80%). This could not be observed unambiguously on the AROP initiated by monofunctional initiators, but was clear on the experiments carried out with difunctional ones (vide infra).
To sum-up, because of the similar nature and pKa to the propagating chain ends, silanols were more efficient initiators than the primary aliphatic alcohol for the AROP of D3 using TBD as a catalyst. Nevertheless, the variety of alcohol initiators offers the opportunity to design PDMS with specific functionalities. As such, when employing a slower initiator, such as aliphatic alcohols, higher catalyst/initiator ratios should be employed, although high monomer conversion should not be reached to minimize the extent of side reactions, such as condensation and potentially chain scrambling.
The MALDI-ToF spectra of PDMS initiated by TMSOH or BnOH allowed more precise determination of the structure of the polymers (Fig. 5). The spectrum of the TMSOH-initiated PDMS (Fig. 5, bottom) exhibited a main peak at m/z = 3966 g mol−1, which corresponded to ∼52 DMS units, and translated into an average degree of polymerization of 17. The BnOH-initiated PDMS spectrum (Fig. 5, top) showed a main population at m/z = 3239 g mol−1 (∼42 DMS units, average degree of polymerization of 14). The spectrum exhibited two secondary distributions corresponding to telechelic PDMS with silanol end-groups (distribution 2 in Fig. 5), and to α,ω-dibenzyl-PDMS (distribution 3). The small population 3 was ascribed to telechelic BnOH-capped PDMS likely formed by termination of the growing chain with excess BnOH (Scheme 4) or by a condensation reaction (Scheme 2).
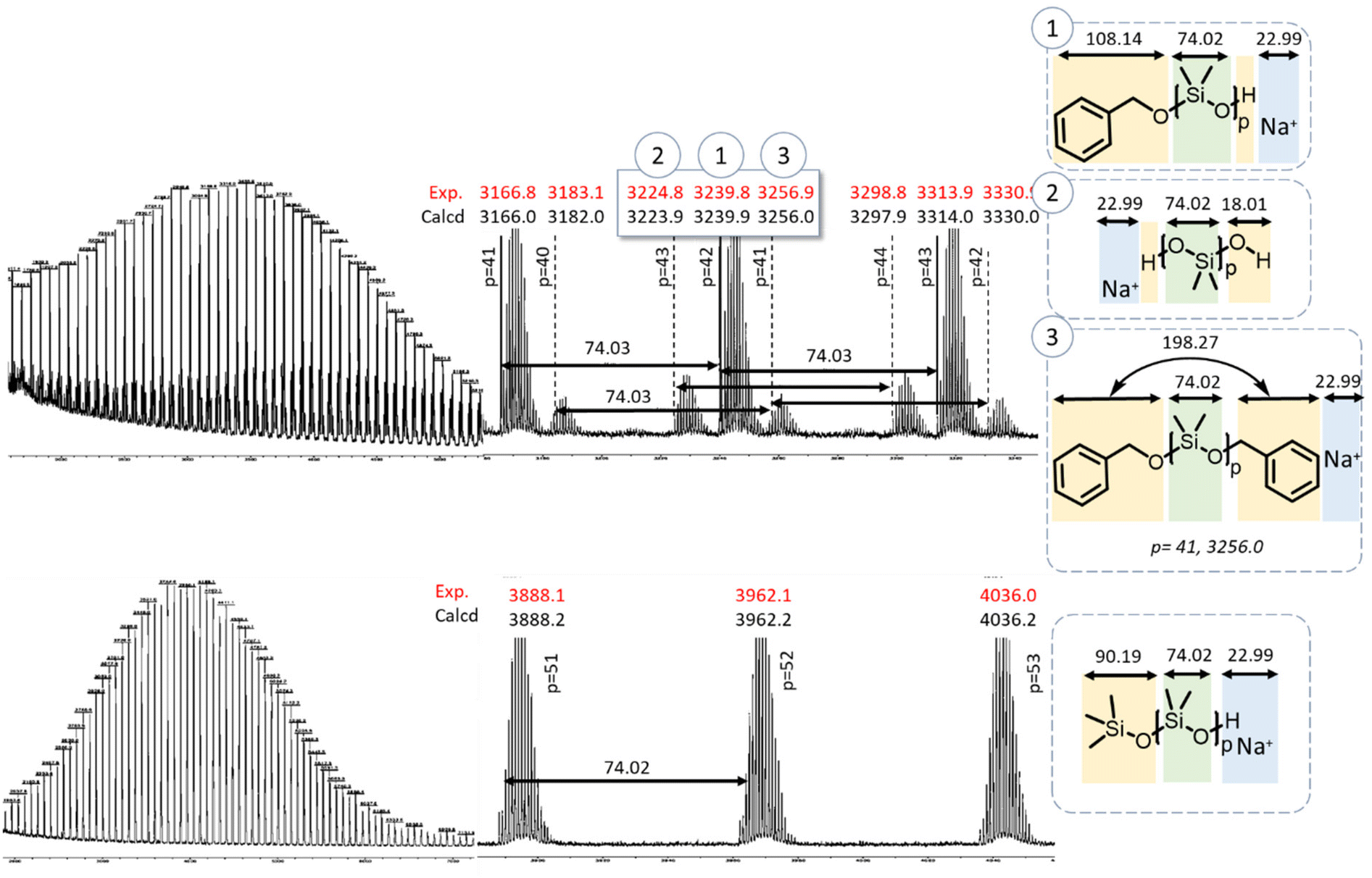 | ||
| Fig. 5 MALDI-TOF mass spectra of precipitated PDMS initiated by benzyl alcohol (top) and trimethyl silanol (bottom). | ||
 | ||
| Scheme 4 Mechanism of BnOH initiated AROP of D3 catalyzed by TBD. | ||
The MALDI-ToF spectrum shown in Fig. 5 corresponds to a PDMS sample taken at the early stages of polymerization (low conversions), when condensation reactions are minimal (vide infra). The hypothesis of termination by excess BnOH, thus, seemed more probable. Indeed, the slow initiation led to significant residual quantity of BnOH (33% free BnOH after 50 min and 50% conversion of D3) available for such a chain-termination reaction. In Fig. S5 and S7† (MALDI-ToF of Φ2Si(OH)2 and PDiol-initiated PDMS, respectively), water initiation could be detected as a second population (No. 2) corresponding to chains with m/z = n × MD3 + MH2O + MNa. The third population detected in the MALDI-ToF spectrum of the PDiol-initiated PDMS corresponded to the chains terminated by excess Pdiol via the mechanism proposed above for the BnOH-initiated PDMS.
Influence of functionality: telechelic vs. monofunctional
Telechelic initiators (1,3-propane diol, Pdiol and diphenyl silane diol, Φ2Si(OH)2) were tested in identical conditions ([D3]0/[OH]0/[TBD]0 = 25/1/0.1 or 0.5) to be compared with their monofunctional analogs (BnOH and TMSOH). It is worth mentioning that, during an AROP with a monofunctional initiator, the presence of adventitious water produces a second polymer population of twice the molar mass of the expected polymer population. Moreover, condensation occurs more prominently towards the end of an AROP (when the monomer concentration is low), also generating a secondary population with twice the molar mass of the desired polymer. However, using a telechelic initiator will effectively mask the effect of initiation by adventitious water. The two different telechelic PDMS populations will effectively be very difficult to distinguish from each other. In consequence, a difunctional initiator will permit the study of only the effect of condensation reaction.As seen above, the initiation using alcohols was slower than that using silanols. The use of a silane diol as an initiator for the TBD-catalyzed AROP of D3 led to rigorously linear pseudo first-order kinetics (Fig. 6). However, unlike the AROP initiated with BnOH, initiation using PDiol did not lead to a frank double slope in the first-order kinetic plot (Fig. 6). 1H NMR titration of difunctional initiators was not possible due to overlapping signals with the reaction solvent (THF). However, Fig. 7b shows that the evolution of dispersity with monomer conversion behaved as in the case of the BnOH-initiated AROP, with high dispersity at low conversion, decreasing as the polymerization progressed (Fig. 7b). The effect of the quantity of catalyst was also very similar (faster initiation and lower dispersity at higher catalyst loading).
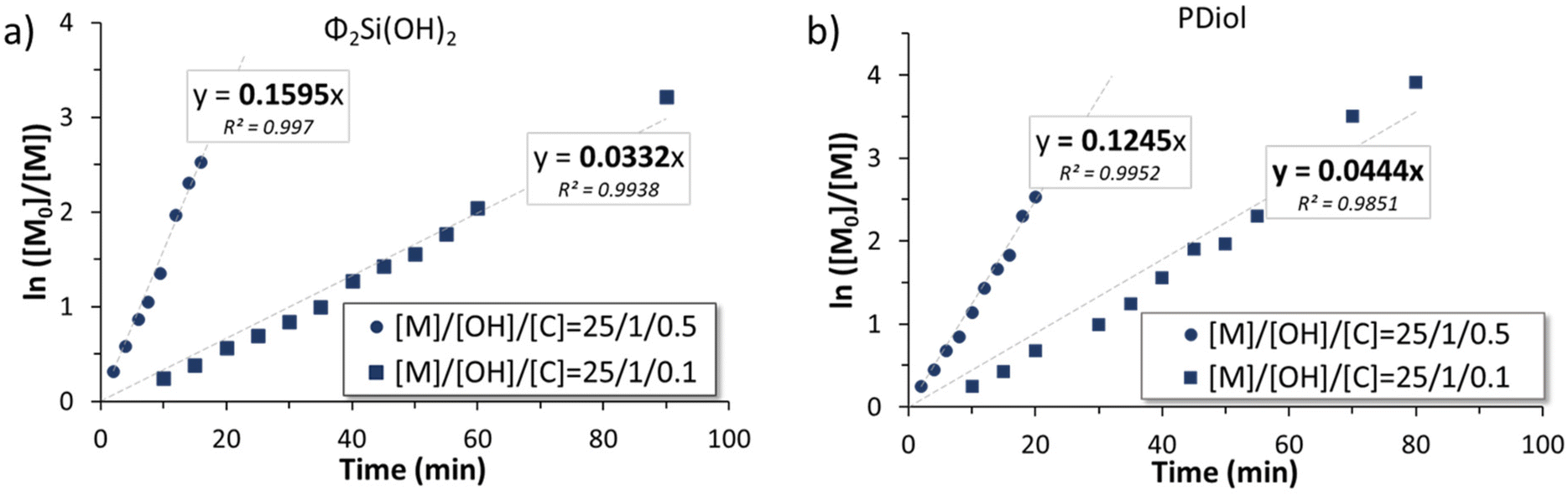 | ||
| Fig. 6 First-order kinetics plot of the ROP initiated by (a) diphenylsilanediol and (b) propanediol. | ||
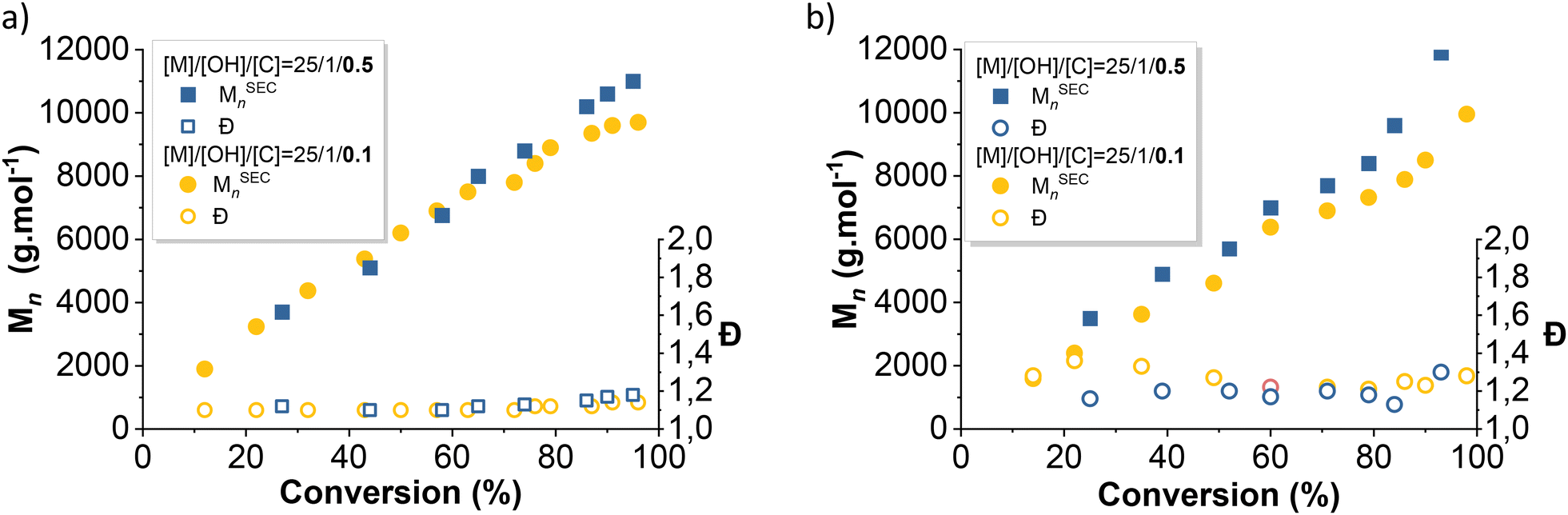 | ||
| Fig. 7 Evolution of the number average molar mass and dispersity with D3 conversion of PDMS prepared by a TBD-catalyzed AROP of D3 initiated with (a) Φ2Si(OH)2 and (b) Pdiol. | ||
Adventitious water is an issue in an AROP initiated with alcohols or silanols because it generates a secondary population of polymer chains. Water is very hard to remove completely from the components of the polymerization (particularly from TBD that favors hydrogen bonds with H2O). Shi et al. showed that in the case of monofunctional initiation, a secondary population is always observed due to initiation by water.22 In the case of monofunctional initiators, a more drastic drying procedure for TBD than for telechelic initiator was implemented to obtain monomodal SEC chromatograms. Even with rigorous distillation and drying steps, adventitious water remained, and led to the production of a secondary population of chains, albeit in small proportion, but that induced a noticeable increase of dispersity in the case of monofunctional initiators (entries 1 and 2). Moreover, the kinetics of the polymerization was influenced by the quantity of residual water because of its ability to initiate polymerization. It decreased the real [M]/[OH] ratio, and increased the kapp of the polymerization (see the kapp values for entries 1, 2, 5 and 6 in Table 1).
As described previously, increasing the catalyst-to-initiator ratio induces faster initiation, lowers initial dispersity, but also promotes condensation reactions and also likely other secondary reactions such as redistribution or back-biting. Indeed, using telechelic initiators enabled highlighting of the condensation occurring towards the end of the polymerization at higher catalyst loading, as shown in Fig. S4 and S6.†Fig. 8 clearly shows this secondary population, which appeared at 92% conversion for the AROP carried out at [I]0/[TBD]0 = 0.5, whereas it was significantly lower at 95% conversion for the polymerization carried out at [I]0/[TBD]0 = 0.1. Chain scrambling (redistribution) and back biting were also likely promoted by an increase in catalyst loading, but these effects were not investigated.
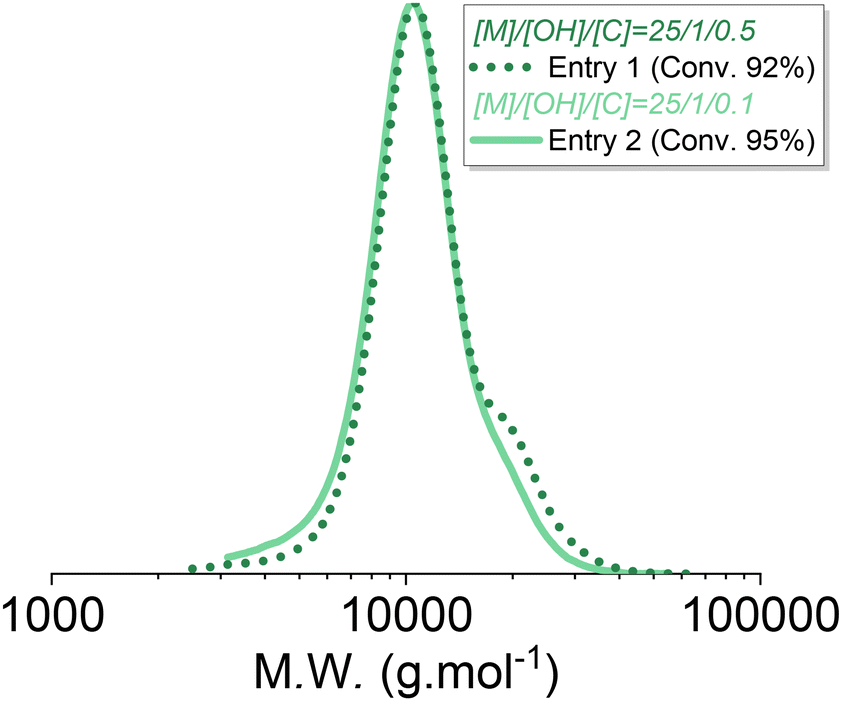 | ||
| Fig. 8 SEC traces of the PDMS corresponding to entry 1 (straight line) and 2 (dashed line) of Table 1. | ||
One of the aims of the present study was to evaluate the capacity of TBD to yield PDMS of high molar masses. Thus, the silane diol was chosen as the initiator in order to use the best experimental conditions for controlled AROP of D3 with TBD as a catalyst.
[D3]0/[I]0 ratios ranging from 100/1 to 500/1 were tested (Table 2). The polymerizations were carried out using a fixed monomer concentration (1.8 mol L−1). The catalyst concentration was also kept constant for each polymerization (0.008 mol L−1) and corresponded to the one used for entry 5 in Table 2. As a consequence, the [TBD]0/[I]0 ratio increased for polymerizations targeting PDMS with higher degree of polymerizations.
| Entry | [D3]0/[I]0/[TBD]0a | Conv.b (%) | Time (min) |
M
Calcnc (kg mol−1) |
M
SECnd (kg mol−1) |
Đ |
|---|---|---|---|---|---|---|
a [D3]0 = 1.8 M.
b Calculated from 1H NMR normalizing D3 and PDMS signals to 100.
c
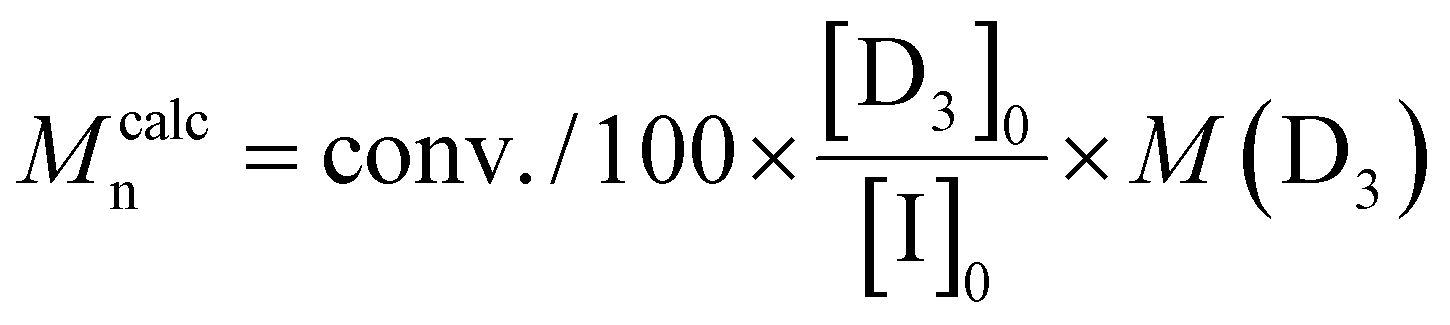 .
d Determined by SEC in THF (1 ml min−1) using a PMMA standard calibration. .
d Determined by SEC in THF (1 ml min−1) using a PMMA standard calibration.
|
||||||
| 1 | 100/1/0.4 | 94 | 60 | 21.0 | 16 | 1.07 |
| 2 | 200/1/0.8 | 95 | 80 | 42.4 | 22 | 1.19 |
| 3 | 350/1/1.4 | 93 | 100 | 72.5 | 27 | 1.26 |
| 4 | 500/1/2 | 95 | 120 | 105.7 | 33 | 1.26 |
| 5 | 350/1/0.7 | 96 | 120 | 74.8 | 35 | 1.17 |
PDMS of high molar mass ranging from 16 to 35 kg mol−1 were synthesized under these conditions. Nevertheless, the increasing [TBD]0/[I]0 ratio led to a noticeable increase in dispersity for PDMS of high molar mass. Indeed, condensation was observed on the GPC traces of these PDMS (Fig. 9). However, decreasing the proportion of catalyst-to-initiator in this case afforded PDMS with relatively high molar masses (35 kg mol−1) and low dispersity (1.17). Fig. 9b displays the GPC traces of PDMS from entries 3 and 5 of Table 2, and shows that less condensation occurred during the polymerization of Entry 5, in Table 2 with [I]0/[TBD]0 = 1/0.7, despite reaching a higher conversion. Indeed, Đ decreased with the catalyst concentration, which was in good agreement with the conclusion of the kinetics study.
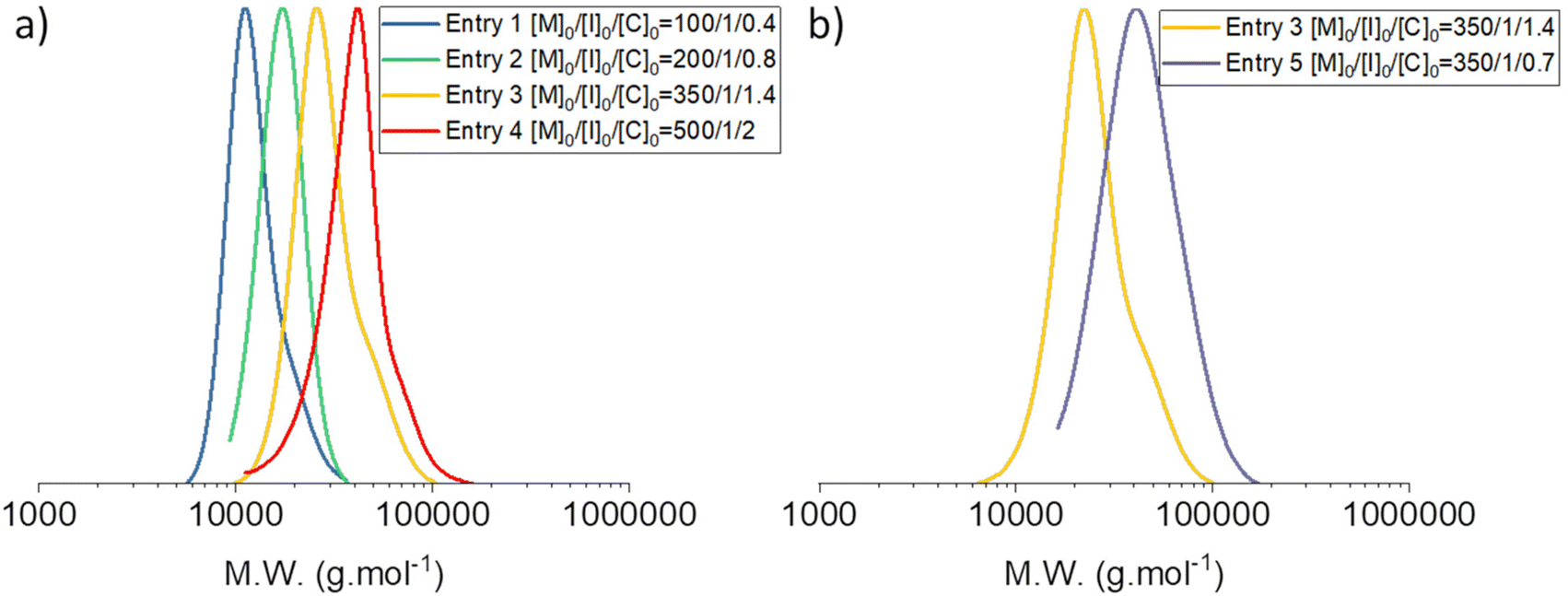 | ||
| Fig. 9 Size-exclusion chromatograms of the PDMS prepared by the AROP of D3 and shown in Table 2. | ||
Polymerization of D4
As mentioned in the Introduction section, D4 can be another monomer of interest for the obtention of PDMS via thermodynamical control of the polymerization.A series of D4 polymerizations were performed to ascertain if polysiloxanes could be prepared under TBD-catalyzed AROP conditions. D4 is a larger and more stable cyclosiloxane than D3, so the AROP of this monomer under the conditions used for the polymerization of D3 may be possible albeit in a longer time, at higher temperature or at higher catalyst concentration. The polymerizations of D4 were attempted in THF at 40 °C (except for entry 4, for which T = 60 °C) using Φ2Si(OH)2 as an initiator and increasing [TBD]0/[I]0 from 1 to 10 in entry 2 while keeping [D4]0 = 1.8 mol L−1 (Table 3). The monomer conversion was followed by 1H NMR spectroscopy by quenching samples with a solution of acetic acid in THF. The polymers were characterized by 1H NMR spectroscopy and size-exclusion chromatography.
| Entry | [D4]0/[I]0/[TBD]0a | T (°C) | Conv.b (%) | Time (h) | D5c (mol%) |
M
Calcnd (kg mol−1) |
M
SEC
n
(kg mol−1) |
Đ |
|---|---|---|---|---|---|---|---|---|
a [D4]0 = 1.8 M.
b Calculated from 1H NMR normalizing D4 and PDMS signals to 100.
c Calculated from 1H NMR.
d
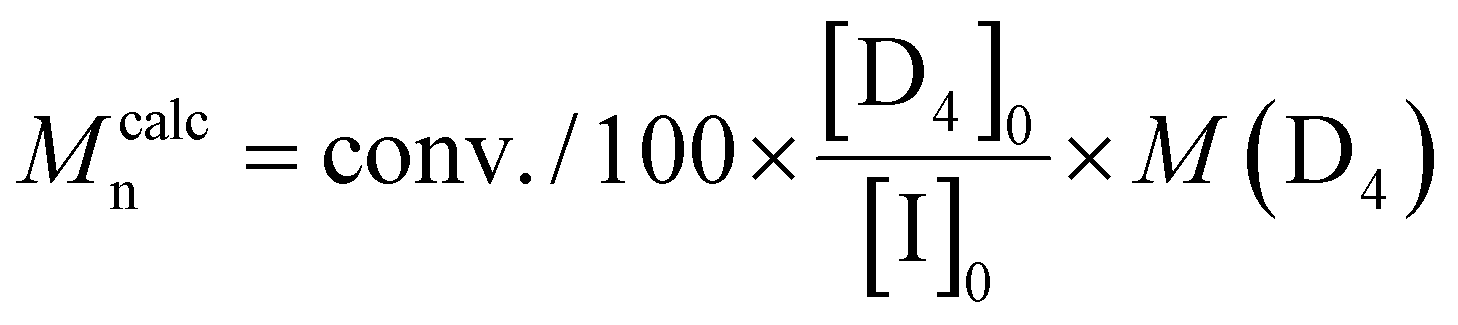 .
e Determined by SEC in THF (1 mL min−1) using a calibration PMMA standard. .
e Determined by SEC in THF (1 mL min−1) using a calibration PMMA standard.
|
||||||||
| 1 | 50/1/1 | 40 | 75 | 18 | 10 | 11.3 | 10.1 | 1.41 |
| 2 | 50/1/10 | 40 | 86 | 4 | 18 | 13.0 | 16.0 | 1.60 |
| 3 | 50/1/5 | 40 | 84 | 6 | 15 | 12.6 | 15.6 | 1.56 |
| 4 | 50/1/5 | 60 | 88 | 4 | 16 | 13.2 | 13.1 | 1.58 |
These polymerizations of D4 produced PDMS with molar masses ranging from 10100 to 15600 g mol−1. For [TBD]0/[I]0 = 1 (entry 1, Table 3), the conversion reached 75% after 18 h and the resulting polymer had a molar mass of 24000 g mol−1 and a relatively high dispersity (1.41). However, in the course of this polymerization, a non-negligible quantity of D5 was formed (10 mol% of the final mixture of polymer and macrocycles) (Fig. S8†). When the catalyst concentration was increased to 5 eq. and then 10 eq. to accelerate the polymerization, the reaction time was decreased from 18 to 6 and 4 h, respectively, while the conversion increased from 75% to about 85%. However, both the dispersity and production of D5 increased, reaching 1.6 and 18 mol%, respectively (for [TBD]0/[I]0 = 10). This production of D5 may explain the important decrease in the PDMS molar masses when high catalyst contents were used.
Experimental part
Materials
Hexamethyltricyclosiloxane (D3) (97%), diphenylsilanediol and trimethylsilanol (TMSOH) were purchased from ABCR. D3 was dried over CaH2 at 90 °C for 48 h, and sublimated under reduced pressure prior to use. 1,3-Propane diol, benzyl alcohol and THF (non-stabilized) were purchased from MilliporeSigma. 1,3 propane diol and benzyl alcohol were dried over CaH2 for 48 h, distilled under reduced pressure and stored over 4A molecular sieves. THF was dried over Na/benzophenone and distilled prior to use under reduced pressure at ambient temperature. TMSOH was distilled under reduced pressure at 40 °C and stored over 4A molecular sieves. TBD was purchased from TCI. TBD and diphenylsilanediol were dried under vacuum (7.5 × 10−2 mbar) at 60 °C for 2 h prior to the preparation of solution in dry THF.Size-exclusion chromatography
SEC was recorded using a triple-detection GPC from Agilent Technologies with its corresponding software, dedicated to multidetector GPC calculation. The system used two PL1113-6300 ResiPore 300 × 7.5 mm columns with THF as the eluent with a flow rate of 1 mL min−1. The detector was a 390-LC PL0390-0601 refractive index detector. The entire SEC-HPLC system was thermostated at 35 °C. Polymethylmethacrylate (PMMA) standards were used for calibration between 540 and 2210000 g mol−1. Samples were prepared by diluting 0.1 mL (1.8 M) aliquots from the polymerization in 1 mL of THF.
NMR
NMR samples were prepared with CDCl3 as a solvent and the analyses were performed using a Bruker Avance I 400 MHz spectrometer at 25 °C. The 1H NMR spectra were recorded at 8 kHz for spectral width, 3 kHz for transmitter frequency offset, 4 s for acquisition time and four scans were performed.MALDI-ToF
For MALDI-ToF analysis, a Bruker RapifleX Instrument with a RapifleX computer were used. Structures of the obtained PDMS were analyzed by positive-ion MALDI-TOF mass spectroscopy using dithranol as a matrix and sodium iodide as a cationization agent.Polymerizations
A typical PDMS synthesis was performed. Each piece of glassware was dried thoroughly using a heat gun under reduced pressure. D3 (1 g, 4.5 mmol, 50 eq.) was directly sublimated in a Schlenk flask equipped with a magnetic stirrer bar. Next, 1.8 mL of a solution of diphenylsilanediol in THF (0.05 M) was added. Experimental [M]0/[I]0 was determined before the catalyst was added by 1H NMR. TBD was solubilized in THF at 60 °C to obtain a perfectly clear solution. Then, 0.1 mL of a solution of TBD in THF (0.14 M) was added to start the polymerization at 40 °C. Aliquots were sampled out of the reaction medium at different reaction times, and the polymerization was quenched using an acetic acid/THF solution.Conclusions
This study demonstrated that TBD, an easy-to-handle and commercially available catalyst, could catalyze the AROP of D3 in a controlled manner depending on the initiator employed. Two types of initiators (alcohols and silanols) were compared under similar polymerization conditions. The pKa of the initiator was shown to strongly influence the course of the polymerization. Initiators with a higher pKa than those of the propagating silanol-derived species led to slow and incomplete initiation. Consequently, alcohol-initiated polymerizations were not perfectly controlled. This effect can be mitigated (at least in part) by increasing the catalyst concentration. However, increasing the catalyst concentration also promoted undesired condensation reactions, especially at high monomer conversion. Indeed, higher catalyst loading also likely promoted chain redistribution and back-biting reactions, but these effects were not investigated. In addition, both monofunctional and telechelic initiators were assessed. The pKa seemed to be the key for a fast initiation, so the catalyst concentration allowed an increase in the initiation rate. In the same manner, the pKb of the catalyst should be another key to fast initiation. However, a higher pKb of the catalyst is often correlated with lower stability. The use of monofunctional initiators (BnOH and TMSOH) makes the need to eliminate any incidental water critical. Indeed, difunctional initiators produce telechelic PDMS that mask the water-initiated chains. The use of difunctional initiators also makes it easier to observe condensation reactions if they occur. Remarkably, TBD could also polymerize D4 although these polymerizations led to PDMS with relatively broad distributions of molar mass (1.54 < Đ < 1.6) and significant formation of D5. In conclusion, TBD was found to be an efficient, commercially available and affordable organocatalyst for the polymerization of D3. Its evaluation as a catalyst for other cyclosiloxanes, and for the synthesis of silicones copolymers, is underway in our research team.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the French National Research Agency ANR (FLAMS project: ANR-20-CE06-0024). The authors thank Professor E. Richaud, Associate Professor S. Roland, and Dr I Iliopoulos at the PIMM laboratory for very useful discussions.References
- C. Julian and C. Marek, in Silicon-Containing Polymers, Springer, Netherlands, Dordrecht, 1st edn, 2000. DOI:10.1007/978-94-011-3939-7.
- J. E. Mark, Silicones and Silicone-Modified Materials, ACS Symposium Series, 2000, vol. 729. DOI:10.1021/bk-2000-0729.ch001.
- Z. Zhang, Y. Zhang, Q. Wang and Z. Xie, Kinetics of the Anionic Ring-Opening Polymerization of Octamethylcyclotetrasiloxane Initiated by Potassium Isopropoxide, J. Appl. Polym. Sci., 2006, 102(4), 3510–3516, DOI:10.1002/app.24665.
- L. Shi, A. Boulègue-Mondière, D. Blanc, A. Baceiredo, V. Branchadell and T. Kato, Ring-Opening Polymerization of Cyclic Oligosiloxanes without Producing Cyclic Oligomers, Science, 2023, 381(6661), 1011–1014, DOI:10.1126/science.adi1342.
- S. C. Greer, Physical Chemistry of Equilibrium Polymerization, J. Phys. Chem. B, 1998, 102(28), 5413–5422, DOI:10.1021/jp981592z.
- T. C. Kendrick, B. M. Parbhoo and J. W. White, Comprehensive Polymer Science and Supplements, Elsevier LtdPergamon, 1996, vol. 4. DOI:10.1016/B978-0-08-096701-1.00140-3.
- J. Chojnowski, Kinetically Controlled Siloxane Ring-Opening Polymerization, J. Inorg. Organomet. Polym., 1991, 1(3) DOI:10.1007/BF00702495.
- J. E. McGrath, J. S. Riffle, A. K. Banthia, I. Yilgor and G. L. Wilkes, Initiation of Polymerization, ed. F. E. Bailey Jr., E.J. Vandenberg, A. Blumstein, M. J. Bowden and J. C. Arthur, ACS, Washington, DC, 1983, Vol. 212. DOI:10.1021/bk-1983-0212.ch013.
- W. T. Grubb and R. C. Osthoff, Kinetics of Polymerization of a Cyclic Dimethylsiloxane, J. Am. Chem. Soc., 1995, 77(6), 1405–1411, DOI:10.1021/ja01611a003.
- P. C. Hupfield and R. G. Taylor, Ring-Opening Polymerization of Siloxanes Using Base Catalysts, J. Inorg. Organomet. Polym., 1999, 9(1), 17–33 CrossRef CAS.
- B. Epwein, M. Marlin and A. Moller, Use of Polyiminophosphazene Bases for Ring-Opening Polymerization, Macrornol. Symp., 1996, 107(1), 331–340, DOI:10.1002/masy.19961070131.
- A. Molenberg and M. Moller, A Fast Catalyst System for the Ring-Opening Polymerization of Cyclosiloxanes, Macromol. Rapid Commun., 1995, 16, 449–453 CrossRef CAS.
- J. Shi, Z. Liu, N. Zhao, S. Liu and Z. Li, Controlled Ring-Opening Polymerization of Hexamethylcyclotrisiloxane Catalyzed by Trisphosphazene Organobase to Well-Defined Poly(Dimethylsiloxane)s, Macromolecules, 2022, 55(7), 2844–2853, DOI:10.1021/acs.macromol.1c02654.
- M. Rodriguez, S. Marrot, T. Kato, S. Stérin, E. Fleury and A. Baceiredo, Catalytic Activity of N-Heterocyclic Carbenes in Ring Opening Polymerization of Cyclic Siloxane, J. Organomet. Chem., 2007, 692(4), 705–708, DOI:10.1016/j.jorganchem.2006.10.006.
- D. Margetic, Superbases for Organic Synthesis: Guanidines, Amidines and Phosphazenes and Related Organocatalysts, ed. T. Ishikawa, Wiley, Chiba, 2009. DOI:10.1002/9780470740859.ch2.
- K. Fuchise, M. Igarashi, K. Sato and S. Shimada, Organocatalytic Controlled/Living Ring-Opening Polymerization of Cyclotrisiloxanes Initiated by Water with Strong Organic Base Catalysts, Chem. Sci., 2018, 9(11), 2879–2891, 10.1039/c7sc04234e.
- K. Fuchise, T. Kobayashi, K. Sato and M. Igarashi, Organocatalytic Ring-Opening Polymerization of Cyclotrisiloxanes Using Silanols as Initiators for the Precise Synthesis of Asymmetric Linear Polysiloxanes, Polym. Chem., 2020, 11(48), 7625–7636, 10.1039/d0py01251c.
- K. Fuchise, K. Sato and M. Igarashi, Organocatalytic Controlled/Living Ring-Opening Polymerization of 1,3,5-Triphenyl-1,3,5-Tri-: P-Tolylcyclotrisiloxane for the Precise Synthesis of Fusible, Soluble, Functionalized, and Solid Poly[Phenyl(p-Tolyl)Siloxane]s, Polym. Chem., 2021, 12(36), 5178–5190, 10.1039/d1py00652e.
- M. T. Martello, A. Burns and M. Hillmyer, Bulk Ring-Opening Transesterification Polymerization of the Renewable δ-Decalactone Using an Organocatalyst, ACS Macro Lett., 2012, 1(1), 131–135, DOI:10.1021/mz200006s.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Organocatalytic Ring-Opening Polymerization, Chem. Rev., 2007, 107(12), 5813–5840, DOI:10.1021/cr068415b.
- A. P. Dove, Organic Catalysis for Ring-Opening Polymerization, ACS Macro Lett., 2012, 1(12), 1409–1412, DOI:10.1021/mz3005956.
- J. Shi, Z. Liu, N. Zhao, S. Liu and Z. Li, Controlled Ring-Opening Polymerization of Hexamethylcyclotrisiloxane Catalyzed by Trisphosphazene Organobase to Well-Defined Poly(Dimethylsiloxane)s, Macromolecules, 2022, 55(7), 2844–2853, DOI:10.1021/acs.macromol.1c02654.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01254b |
| This journal is © The Royal Society of Chemistry 2025 |