
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile route for the chemical functionalisation of polydivinylbenzenes and the application of amphoteric polydivinylbenzene microspheres to the simultaneous solid-phase extraction of acidic and basic drugs from water samples†
Francesc
Borrull
 a,
Peter A. G.
Cormack
*b,
Alan
Corrigan
b,
Calum
Craig
b,
Núria
Fontanals
a,
Rosa Maria
Marcé
*a,
Alberto
Moral
a and
Greg
Smith
b
a,
Peter A. G.
Cormack
*b,
Alan
Corrigan
b,
Calum
Craig
b,
Núria
Fontanals
a,
Rosa Maria
Marcé
*a,
Alberto
Moral
a and
Greg
Smith
b
aUniversitat Rovira i Virgili, Department of Analytical and Organic Chemistry, Sescelades Campus, Marcel lí Domingo s/n, 43007 Tarragona, Spain. E-mail: rosamaria.marce@urv.cat
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, Scotland, UK. E-mail: Peter.Cormack@strath.ac.uk
First published on 28th December 2024
Abstract
Mixed-mode ion-exchange sorbents with amphoteric character are intriguing materials because not only can anions and cations be extracted from liquid samples using one single sorbent rather than two (anion extraction under one set of conditions, cation extraction under a second set of conditions), but it may be feasible to establish extraction conditions where anionic and cationic analytes can be extracted simultaneously. In the present study, an unusual but versatile synthetic route was used to install amphoteric character into polydivinylbenzene microspheres produced through precipitation polymerisation. The key synthetic step used for the chemical functionalisation of the polydivinylbenzenes exploited Diels–Alder cycloaddition chemistry to target the pendent styryl groups that are present in polydivinylbenzenes. With maleic anhydride as a dienophile, Diels–Alder cycloaddition yielded polydivinylbenzenes decorated with anhydride moieties. Whilst such materials are interesting in their own right as reactive resins, ring-opening of the polymer-bound anhydride units with ethylenediamine yielded an amphoteric material with both weak anion-exchange (WAX) and weak cation-exchange (WCX) character. This polymer was evaluated as a pH-tuneable sorbent for the solid-phase extraction (SPE) of acidic and basic pharmaceuticals from water samples. Following optimisation of the analytical method including the SPE, the method was subjected to validation and then applied to the extraction and determination of acidic and basic pharmaceuticals present at low concentrations in river water, effluent wastewater and influent wastewater samples. Simultaneous extraction and determination of acidic and basic compounds was found to be achievable, with method quantification limits down to 1 ng L−1.
1. Introduction
In recent years, the public awareness of contaminants of emerging concern (CECs) has been increasing due to the potential harm to human health and the environment. The CEC classification includes a very diverse range of chemical substances, including per- and polyfluoroalkyl substances, endocrine disruptors, pharmaceuticals and personal care products. There is an urgent need to understand more about their occurrence and their toxicological effects,1,2 which is why several studies have been performed in recent years to determine and monitor emerging contaminants in different regions around the world.1–5 For example, pharmaceuticals reach the environment primarily through wastewater disposal, since wastewater treatment plants cannot effectively remove them from wastewater,6,7 and pharmaceuticals in water present a danger to the environment and human health.8–10 For these reasons, the development of reliable, sensitive methods for pharmaceutical determination in environmental samples is critical.The majority of pharmaceuticals in use around the world have either acidic or basic properties. For instance, many β-blockers, opioids and antidepressants are basic, whereas fluoroquinolones, angiotensin II receptor blockers (“sartan drugs”) and non-steroidal anti-inflammatory drugs tend to be acidic. This acidic/basic character can be exploited in solid-phase extraction (SPE) through using mixed-mode ion-exchange sorbents, which are sorbents that combine reversed-phase interactions and ion-exchange interactions within a single material.11 Indeed, there are many studies reported where acidic and basic pharmaceuticals have been extracted using commercial mixed-mode ion-exchange sorbents.12–14 To determine anions and cations using these commercial sorbents, two different sorbents are normally required – an anion-exchanger to extract acidic pharmaceuticals and a cation-exchanger to extract the basic pharmaceuticals – although anion-exchangers and cation-exchangers have been combined in the same SPE cartridge15 or connected in series16 to streamline extraction processes. In recent times, an intriguing alterative has emerged; amphoteric sorbents offer anion-exchange and cation-exchange properties,17–22 potentially enabling the extraction of acidic and basic compounds using one single sorbent, whether it be silica-based18,20–24 or polymer-based.17,19 Some silica-based sorbents which combine strong anion-exchange and strong cation-exchange character20,23,24 have been evaluated by our group in recent years. In these studies, however, it was observed that acidic analytes were lost in the washing step. In a separate study involving organic polymers, an amphoteric polymeric sorbent combining weak anion-exchange (WAX) and weak cation-exchange (WCX) moieties was developed,25 and in this case it was found that a washing step could be tolerated in the simultaneous determination of acidic and basic analytes. In the latter study,25 the polymers were decorated with sarcosine (N-methylglycine) residues; quaternisation of the tertiary amine groups in sarcosine enabled access into SAX/WCX variants (SAX = strong anion-exchange), whereas functionalisation of the precursor polymers with taurine enabled access into WAX/SCX variants (SCX = strong cation-exchange).17
In the present study, a two-step synthetic methodology is used to install amphoteric character into polydivinylbenzene microspheres produced through precipitation polymerisations, where the key polymer-analogous reaction used is a Diels–Alder (D–A) cycloaddition. A prerequisite for success of the cycloaddition is that the polydivinylbenzenes have accessible pendent styryl groups, and since this is the case for these polymer microspheres as well as most commercial and non-commercial polydivinylbenzenes, including suspension polymerised products, the applicability of the methodology reported here is wide. Indeed, over the years, the pendent styryl groups present in divinylbenzenes have been exploited as functional handles using a range of chemistries, including anti-Markovnikov addition reactions26 and olefin cross metathesis methods.27 Whilst some D–A cycloaddition methods for the functionalisation of suspension polymerised polydivinylbenzene products have been reported in the literature as well,26,28 the methodology has been largely ignored and never applied to the chemical functionalisation of the small, uniform particles that are particularly attractive for separation processes. Use of maleic anhydride as a dienophile in a Diels–Alder cycloaddition with polydivinylbenzene microspheres gives polymer-bound anhydride groups. Ring-opening of the anhydride groups with diamines delivers amphoteric polymer microspheres with WAX and WCX character. To exemplify the amphoteric character and the utility of the polymers in separation processes, we have exploited ethylenediamine-based amphoteric polymer microspheres in SPE followed by liquid chromatography and high resolution mass spectrometry (LC-HRMS) for the simultaneous extraction and determination of acidic and basic pharmaceuticals present in river water, effluent wastewater and influent wastewater samples.
2. Experimental
2.1. Reagents and standards
For the synthesis of the WAX/WCX sorbent, divinylbenzene-80 (DVB-80) (technical grade, 80%), was supplied by Sigma-Aldrich (Saint Louis, MO, USA). Prior to use, it was passed through a column containing aluminium oxide (neutral, activated, from Sigma-Aldrich) to remove the polymerisation inhibitor. The free radical initiator 2,2′-azobisisobutyronitrile (AIBN) (98%) was used as received from Sigma-Aldrich. Acetonitrile (ACN) (99.9%, HPLC grade) was supplied by VWR Chemicals (Portland, OR, USA) and toluene (≥99.3%, lab reagent) and tetrahydrofuran (THF) were supplied by Sigma-Aldrich; they were used as received. For the chemical modification of polydivinylbenzene microspheres, maleic anhydride (MA) (≥98%) and tetracyanoethylene (TCNE) (96%) were used as received from Fluka and Sigma-Aldrich, respectively. For the ring-opening of polymer-bound anhydride units, ethylenediamine (EDA) (≥99%) was purchased from Sigma-Aldrich and used as received.Five basic pharmaceuticals (venlafaxine [VEN], atenolol [ATE], metoprolol [MTO], propranolol [PRO] and trimethoprim [TRI]) and four acidic pharmaceuticals (diclofenac [DIC], bezafibrate [BEZ], valsartan [VAL] and diclofenac [DIC]) and the metabolite of clofibrate (clofibric acid [CLO]) were acquired from Sigma-Aldrich as pure standards (purity >96%). Solutions at 1000 μg L−1 were prepared in methanol (MeOH) and stored in a freezer (−20 °C) in dark bottles. Each week, working solutions of all analytes were prepared in a mixture of ultrapure water and MeOH (90/10, v/v) and stored in a refrigerator (4 °C) in dark bottles.
HPLC and MS grade ACN, HPLC grade MeOH and MS grade water were purchased from Carlo Erba (Val de Reuil, France). Formic acid, HCl, acetic acid and NH4OH were purchased from Sigma-Aldrich. A Millipore purification system (Burlington, MA, USA) was used to obtain ultrapure water.
2.2. Synthesis and characterisation of the WAX/WCX sorbent
The WAX/WCX sorbent was synthesised via a three-step synthetic protocol (Fig. 1): (i) precipitation polymerisation (PP) of DVB-80 to give polyDVB-80 microspheres; (ii) chemical modification of the polyDVB-80 microspheres via a Diels–Alder (D–A) cycloaddition with MA; (iii) ring-opening of polymer-bound anhydride units with EDA.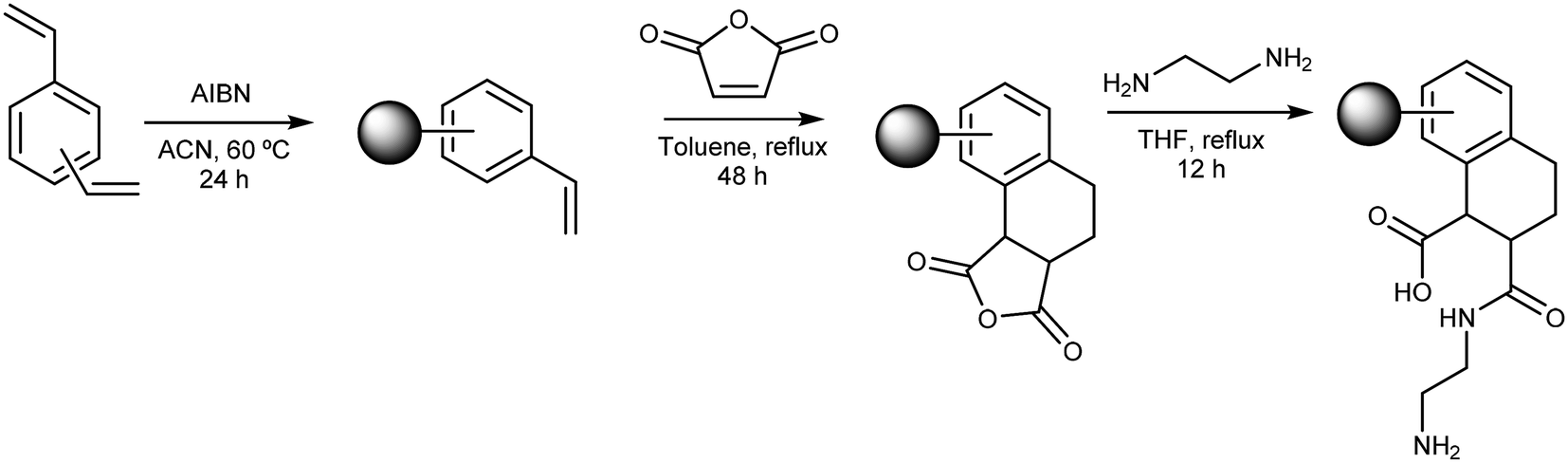 | ||
| Fig. 1 Reaction scheme outlining the synthesis of the WAX/WCX sorbent: Step1. Precipitation polymerisation (PP) of DVB-80 to give polyDVB-80 microspheres; Step 2. Chemical modification of the polyDVB-80 microspheres via a Diels–Alder (D–A) cycloaddition with MA; Step 3. Ring-opening of polymer-bound anhydride units with EDA. | ||
ATR-FTIR was carried out using a Thermo Scientific Nicolet iS5 FTIR spectrometer. Samples were scanned 16 times each, over the range 400–4000 cm−1. Spectra were recorded and analysed using Omnic Series Software from Thermo Scientific. Elemental microanalysis was conducted by the University of Strathclyde Microanalysis Service; C, H, and N contents were determined using a PerkinElmer 2400 Series II analyser. Scanning electron microscopy (SEM) was carried out using a Cambridge Stereoscan 90 Scanning Electron Microscope; prior to imaging, samples were sputter-coated with gold using a Polaron SC500A Sputter Coater. Average particle sizes were determined (n = 100) using ImageJ (v1.54, National Institutes of Health) image processing software. Nitrogen sorption analysis was carried out using a Micromeritics ASAP 2020 Porosimeter.
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) max/cm−1: 3024 (aromatic C–H str.), 2925 (aliphatic C–H str.), 1606 (aromatic C
max/cm−1: 3024 (aromatic C–H str.), 2925 (aliphatic C–H str.), 1606 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C str.), 1525 (aromatic CC str.), 1448 (aliphatic C–H bend), 990 (vinyl C–H def.), 906 (vinyl C–H def.), 844 (1,4-disubstituted aromatic out-of-plane C–H def.), 798 (1,3-disubstituted aromatic out-of-plane C–H def.), 711 (1,3-disubstituted aromatic C–H def.). Elemental microanalysis (found, %): C, 91.4; H, 7.9; N, 0.4. SEM analysis: mean particle diameter = 5.0 ± 0.2 μm, CV = 3.0%. N2 sorption analysis: Langmuir specific surface area = 576 m2 g−1, specific pore volume = 0.23 cm3 g−1, mean pore diameter = 3.1 nm.
max/cm−1: 3034 (aromatic C–H str.), 2935 (aliphatic C–H str.), 1799 (anhydride CO str.), 1608 (aromatic CC str.), 1519 (aromatic CC str.), 1453 (aliphatic C–H bend), 1068 (anhydride OC–O–CO str.), 993 (vinyl C–H def.), 917 (1,2,4-trisubstituted aromatic out-of-plane C–H def.), 802 (1,3-disubstituted aromatic out-of-plane C–H def.), 714 (1,3-disubstituted aromatic C–H def.). Elemental microanalysis (found, %): C, 78.9; H, 6.8; N, 0.3; O (by difference), 14.0; anhydride loading level = 3.3 mmol g−1. SEM analysis: mean particle diameter = 4.2 ± 0.2 μm, CV = 5.6%. N2 sorption analysis: Langmuir specific surface area = 484 m2 g−1, specific pore volume = 0.21 cm3 g−1, mean pore diameter = 3.2 nm.
max/cm−1: 3300 (amine N–H str.), 2927 (aliphatic C–H str.), 2847 (aliphatic C–H str.), 1713 (carboxylic acid CO str.), 1615 (amide I CO str.), 1513 (amide II CN str.), 1350 (amine C–N str.), 835 (1,4- and 1,2,4-trisubstituted aromatic out-of-plane C–H def.), 796 (1,3-disubstituted aromatic out-of-plane C–H def.), 720 (1,3-disubstituted aromatic C–H def.). Elemental microanalysis (found, %): C, 77.4; H, 7.4; N, 4.1; O (by difference), 11.0; WAX/WCX loading level = 2.3 mmol g−1 (from elemental microanalytical data). WAX/WCX loading level (from titration analysis) = 3.5 mmol g−1. SEM analysis: mean particle diameter = 5.1 ± 0.2 μm, CV = 3.4%. N2 sorption analysis: BET specific surface area < 5 m2 g−1.
C str.), 1525 (aromatic CC str.), 1448 (aliphatic C–H bend), 990 (vinyl C–H def.), 906 (vinyl C–H def.), 844 (1,4-disubstituted aromatic out-of-plane C–H def.), 798 (1,3-disubstituted aromatic out-of-plane C–H def.), 711 (1,3-disubstituted aromatic C–H def.). Elemental microanalysis (found, %): C, 91.4; H, 7.9; N, 0.4. SEM analysis: mean particle diameter = 5.0 ± 0.2 μm, CV = 3.0%. N2 sorption analysis: Langmuir specific surface area = 576 m2 g−1, specific pore volume = 0.23 cm3 g−1, mean pore diameter = 3.1 nm.
max/cm−1: 3034 (aromatic C–H str.), 2935 (aliphatic C–H str.), 1799 (anhydride CO str.), 1608 (aromatic CC str.), 1519 (aromatic CC str.), 1453 (aliphatic C–H bend), 1068 (anhydride OC–O–CO str.), 993 (vinyl C–H def.), 917 (1,2,4-trisubstituted aromatic out-of-plane C–H def.), 802 (1,3-disubstituted aromatic out-of-plane C–H def.), 714 (1,3-disubstituted aromatic C–H def.). Elemental microanalysis (found, %): C, 78.9; H, 6.8; N, 0.3; O (by difference), 14.0; anhydride loading level = 3.3 mmol g−1. SEM analysis: mean particle diameter = 4.2 ± 0.2 μm, CV = 5.6%. N2 sorption analysis: Langmuir specific surface area = 484 m2 g−1, specific pore volume = 0.21 cm3 g−1, mean pore diameter = 3.2 nm.
max/cm−1: 3300 (amine N–H str.), 2927 (aliphatic C–H str.), 2847 (aliphatic C–H str.), 1713 (carboxylic acid CO str.), 1615 (amide I CO str.), 1513 (amide II CN str.), 1350 (amine C–N str.), 835 (1,4- and 1,2,4-trisubstituted aromatic out-of-plane C–H def.), 796 (1,3-disubstituted aromatic out-of-plane C–H def.), 720 (1,3-disubstituted aromatic C–H def.). Elemental microanalysis (found, %): C, 77.4; H, 7.4; N, 4.1; O (by difference), 11.0; WAX/WCX loading level = 2.3 mmol g−1 (from elemental microanalytical data). WAX/WCX loading level (from titration analysis) = 3.5 mmol g−1. SEM analysis: mean particle diameter = 5.1 ± 0.2 μm, CV = 3.4%. N2 sorption analysis: BET specific surface area < 5 m2 g−1.
2.3. SPE procedure
150 mg of WAX/WCX sorbent was packed into an empty 6 mL SPE cartridge (Symta, Madrid, Spain) fitted with 10 μm polyethylene frits (Symta) below and above the sorbent bed. A 2 μm stainless steel frit (Sigma-Aldrich) was also placed below the sorbent to prevent sorbent loss.Before the loading of the sample, the sorbent was conditioned with 5 mL of MeOH and 5 mL of ultrapure water adjusted to pH 5. Then, a fixed volume of sample adjusted to pH 5 (100 mL for river water samples, 50 mL for effluent wastewater samples and 25 mL for influent wastewater samples) was loaded at an approximate flow rate of 5 mL min−1. 1 mL of MeOH was used in the cleaning step and the elution step consisted of 5 mL of 5% NH4OH in MeOH. The eluates were evaporated to dryness using a miVac Duo centrifuge evaporator (Genevac, Ipswich, UK) and reconstituted with 1 mL of water/ACN (95/5, v/v). Prior to extraction, river water samples were filtered through a 0.45 μm nylon membrane filter (Scharlab), whereas wastewater samples were filtered through a 1.2 μm glass-fibre membrane filter (Fisherbrand, Loughborough, UK) prior to filtration with a 0.45 μm filter. To establish the optimal loading volume for each sample type, several loading volumes were used for each sample type and then the loading volume that gave acceptable recoveries for each sample type was selected. The loading volumes were higher for cleaner samples (river water, 100 mL) and lower for dirtier samples (influent wastewater, 25 mL).
2.4. Chromatographic conditions
Two liquid chromatographs were used. To optimise the SPE method, an Agilent 1200 (Agilent, Waldbronn, Germany) series equipped with a binary pump, an autosampler, a column oven, an automatic injector and a diode array detector (DAD) was used. To analyse the samples, the method was transferred to LC-HRMS using an Accela 1250 UHPLC, equipped with an Accela Autosampler and a quaternary pump, coupled to an Exactive Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) equipped with a heated electrospray ionisation and a high-energy collision dissociation cell.The chromatographic conditions used were similar to a previous study24 and are included in the ESI.†
3. Results and discussion
3.1. Synthesis of the WAX/WCX sorbent
Precipitation polymerisation (PP) was the polymerisation method of choice for preparing polydivinylbenzene microspheres in the low micron size range to be carried forward to polymer-analogous reactions. This is because PP is a surfactant- and stabiliser-free method of free radical polymerisation that yields high quality polymer microspheres with low mean particle diameters and narrow particle size distributions. Particle diameters, porous morphology and chemical functionality can all be controlled through rational selection of reagents/monomers and the polymerisation conditions (e.g., monomer and initiator concentrations). Typical PP conditions for the synthesis of porous microspheres require the inclusion of a porogen in the solvent mixture. For this reason, toluene was included at 25 vol% in conjunction with ACN. Under these conditions, good quality polyDVB-80 microspheres were obtained, and nitrogen sorption analysis showed that they were porous in the dry state.To maximise the number of pendent styryl groups (i.e., unreacted vinyl groups) which could be exploited in D–A cycloadditions, DVB-80 was selected in preference to DVB-55 since DVB-80 is comprised of 80% divinylbenzene isomers compared to 55% of divinylbenzene isomers for DVB-55. In a study of suspension polymerised DVB resins, Law et al.29 reported that approximately 45% of the DVB-derived vinyl groups remain pendent and unreacted following polymerisation of DVB-80, compared to 32% when using DVB-55. While the microspheres in the present work were synthesised via PP rather than by suspension polymerisation, it can be anticipated that polyDVB-80 microspheres prepared by PP will contain a higher level of pendent styryl groups than a polyDVB-55 prepared using a nominally identical PP method. Pendent vinyl groups give rise to a very diagnostic signal in the FTIR spectrum of the microspheres (at 995 cm−1), and this signal can be used to verify the presence of vinyl groups in the polyDVB-80 microspheres and to monitor the progress of any subsequent reactions that consume pendent vinyl groups.
To install anhydride moieties into the polyDVB-80 microspheres, a D–A cycloaddition with MA was employed. D–A chemistry represents an attractive but under-utilised approach for functional polymer production because polydivinylbenzenes are used in a wide range of applications. Furthermore, there are several dienophiles besides anhydrides that can be exploited as coupling partners in D–A reactions (e.g., styrenes26 and TCNE [see ESI†]) and this versatile approach can be applied to other polymer formats too (see ESI† for a cognate approach using core–shell polydivinylbenzenes). The D–A cycloaddition between polyDVB-80 microspheres and MA was thermally-promoted and conducted in toluene. Toluene is a good wetting/swelling solvent for such materials and allows MA access to the porous network, however it was inevitable that some styryl groups located deep within the pore network would remain inaccessible to the MA. For the subsequent ring-opening of the polymer bound anhydride units, EDA was used to produce the amphoteric WAX/WCX material (for this polymer-analogous reaction, EDA was used in excess to drive the ring-opening reaction to completion and to suppress the likelihood of EDA acting as a crosslinker between neighbouring anhydride groups).
3.2. Characterisation of the WAX/WCX sorbent
FTIR spectroscopy was used to monitor the presence of the pendent vinyl groups in polydivinylbenzenes produced by PP, their subsequent consumption in D–A cycloaddition reactions, and the ring-opening of polymer-bound anhydride groups with EDA. Fig. 2 shows the FTIR spectrum of the WAX/WCX sorbent, whereas Fig. S1† shows the FTIR spectra of polyDVB-80 microspheres, the corresponding D–A adduct with MA and the ring-opened derivative formed upon reaction with EDA. The spectra are consistent with the structures presented in Fig. 1. As can be seen in Fig. S1,† the signal at ∼995 cm−1 that is assigned to pendent vinyl groups in the polydivinylbenzene microspheres diminishes dramatically in intensity as the vinyl groups are consumed by reaction with MA. Simultaneously, new signals appear at 1792 and 1069 cm−1 in the FTIR spectrum of the D–A adduct, and these are assigned to the CO and the OC–O–CO stretches, respectively, of polymer-bound anhydride groups. Following treatment with EDA, the anhydride signals disappear and are replaced by diagnostic signals assignable to the amine (3300 and 1350 cm−1), carboxylic acid (1713 cm−1) and amide (1615 and 1513 cm−1) groups present in the WAX/WCX sorbent (Fig. 2).
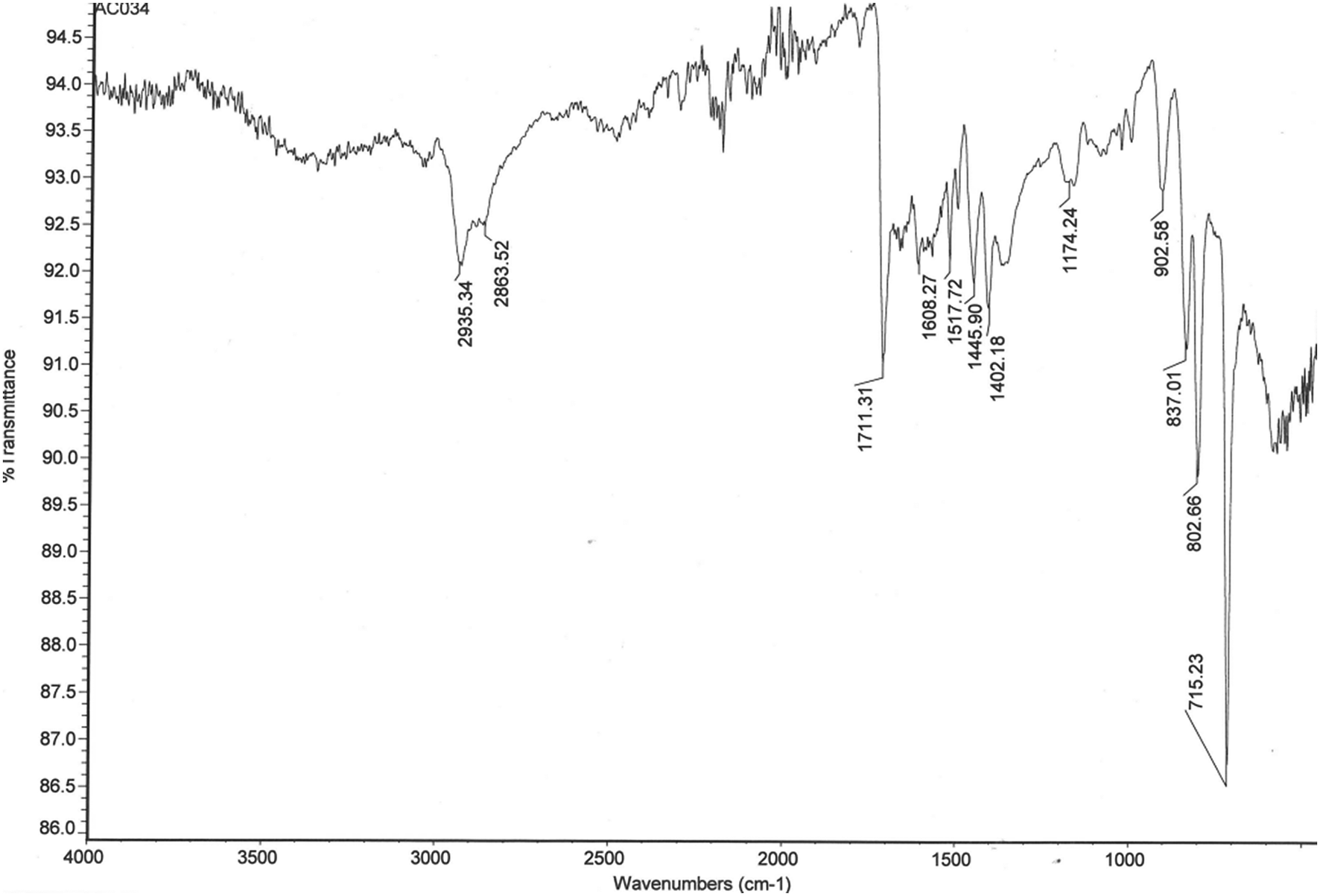 | ||
| Fig. 2 FTIR spectrum of the amphoteric WAX/WCX polymer microspheres. | ||
Nitrogen sorption analysis revealed that the polyDVB-80 microspheres and the D–A adducts were macroreticular, i.e., porous in the dry state. Whilst some drop-off in specific surface area is to be expected as mass is added to the microspheres through polymer-analogous reactions, most dry state porosity was lost as a result of the ring-opening reaction with EDA. This may be due to partial pore-blocking through crosslinking and/or (reversible) collapse of some pores as the amphoteric motifs form and then desolvate upon drying. However, for SPE the WAX/WCX polymer microspheres are exploited in a wet state and not the dry state so were taken forward to SPE nevertheless since access to functional groups in the wet state is the critical consideration.
SEM was used to monitor the particle size and particle size distribution of the polymer microspheres. The high quality of the polymer microspheres was retained throughout the polymer analogous reactions. An SEM micrograph of the final WAX/WCX product is shown in Fig. 3; with a mean particle diameter of 5.1 μm and a size dispersity of 3.4%, such monodisperse microspheres are attractive for use in separation applications since they are small in size and uniform in terms of size distribution.
 | ||
| Fig. 3 SEM micrograph of the amphoteric WAX/WCX polymer microspheres. | ||
Elemental microanalysis was used to measure the carbon, hydrogen and nitrogen contents of the polymers and allow for an estimation of the functional group loading levels (expressed in mmol g−1). Whilst estimating the functional group loading levels from elemental microanalytical data is complicated by the heterogeneity of the polymers, it was found, as expected, that the carbon content falls significantly upon introduction of oxygen-containing anhydride groups by the D–A cycloaddition (from 91.4 to 78.9%) and that the nitrogen-content rises significantly as a result of the ring-opening of anhydrides with nitrogen-containing EDA (from 0.3 to 4.1%). From the elemental microanalytical data, the functional group loading levels were estimated to be 3.3 and 2.3 mmol g−1 for the anhydride-containing polymer and its ring-opened derivative, respectively. However, a more reliable measure of the functional group loading level of the ring-opened derivative (i.e., the WAX/WCX sorbent) was obtained using a titration method; with this method, the ion-exchange capacity of the WAX/WCX polymer microspheres was found to be 3.5 mmol g−1.
3.3. Evaluation of the WAX/WCX polymer microspheres as an SPE sorbent
The extraction of acidic and basic analytes using a sorbent that has both weak anion-exchange character and weak cation-exchange character is a complex task. Both the analytes and sorbent (Fig. 1) have ionisable groups that need to be charged to promote the ion-exchange interactions. In this regard, and considering the pKa values of the analytes (Table S1†) which range from 7.1 to 10.1 for the basic analytes and from 3.2 to 4.5 for the acidic analytes, plus the previous experience gained when working with polymer-based amphoteric sorbents,17,19 it was decided to carry out a pH screening study. 10 mL volumes of ultrapure water with pH adjusted to 3, 5, 6, 7 and 9 were loaded onto the sorbent, 1 mL of MeOH was used as washing step, and 5 mL of 5% NH4OH in MeOH followed by 5 mL of 5% acetic acid in MeOH were used for elution.To evaluate the results, SPE recoveries (%RSPE) were calculated as the ratio between the measured concentration after SPE and the theoretical concentration when working with ultrapure water and analysis by LC-DAD. As expected, pH 3 and pH 9 provided the poorest results. At pH 3, the basic analytes have %RSPE lower than 40% due to the protonation of the carboxylic groups in the sorbent. Meanwhile, at pH 9 the acidic analytes had %RSPE lower than 45% due to the deprotonation of the primary amines in the sorbent. pH 5, 6 and 7 provided the most interesting results, as can be seen in Fig. 4. The best results were obtained at pH 5 for all of the basic compounds (%RSPE higher than 80%) and for most of the acidic compounds (highest %RSPE for DIC, BEZ and FEN). In the case of pH 7, this pH value provided the best results for some of the acidic analytes, such as CLO and VAL (which have the lowest pKa values), and good results for the rest of analytes. BEZ, which has a pKa value close to that of VAL, also had a relatively high %RSPE value at pH 7. pH 6 gave lower recoveries than were obtained at pH 5 or 7, with values ranging from 36 to 71%.
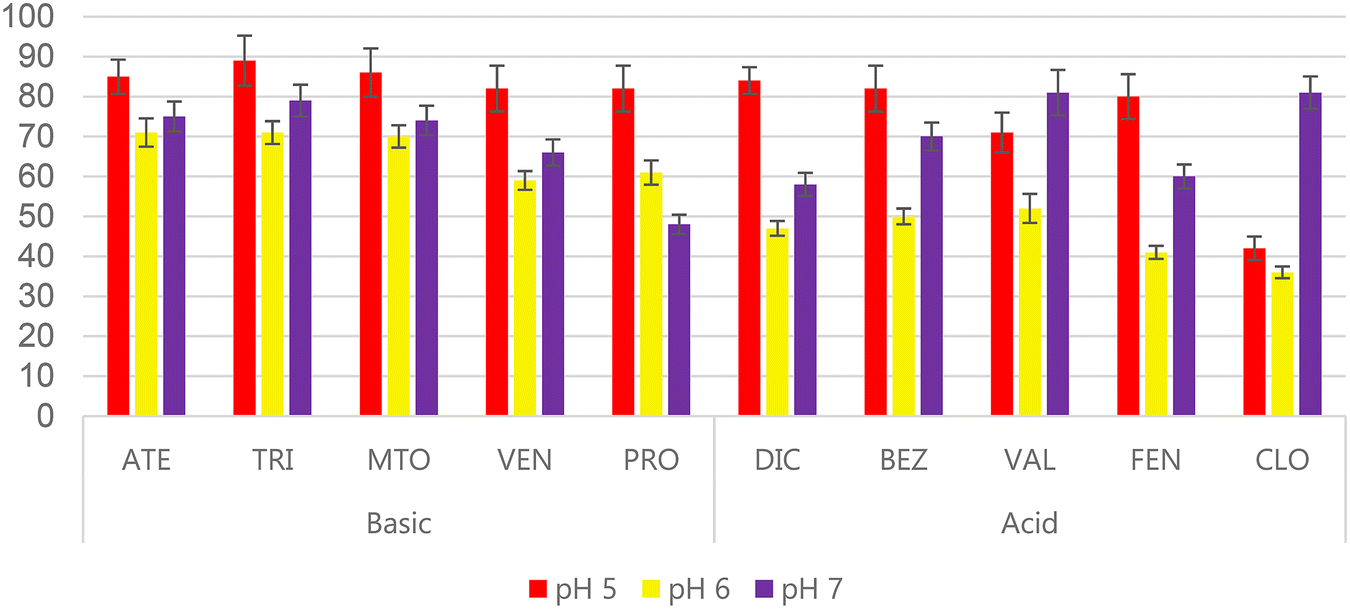 | ||
| Fig. 4 SPE recoveries when 10 mL of ultrapure water at pH 5, 6 and 7 was loaded onto SPE cartridges packed with the WAX/WCX sorbent. | ||
Since the best results for most of the compounds (except for CLO and VAL) were obtained at pH 5, this pH value was selected for the subsequent experiments. In previous studies where WAX/WCX sorbents were used, the pH was slightly acidic; for instance, Jin et al.22 and Nadal et al.19 selected a pH value of 6 when extracting acidic and basic compounds simultaneously. However, in the present study it has been demonstrated that pH 5 yields the best results.
The initial elution step consisted of 5 mL of 5% NH4OH in MeOH followed by 5 mL of 5% acetic acid in MeOH. It was observed during the pH optimisation stage that the acidic and basic analytes were eluted with 5 mL of 5% NH4OH in MeOH, meaning that ammonium hydroxide can disrupt the anion- and cation-exchange interactions. The anion-exchange interactions are disrupted by the neutralisation of protonated amine groups in the sorbent, whereas the cation-exchange interactions are disrupted by the deprotonation of the basic analytes. For this reason, the acidic elution was removed. Other studies where cation-exchange and anion-exchange groups were combined in one sorbent also used solutions of NH4OH in MeOH for the elution step.17,19,21
The volume of MeOH used in the washing step was also evaluated, by using 3 mL of MeOH instead of 1 mL of MeOH. This led to a slight decrease in the %RSPE of the basic compounds; the recovery values ranged from 82 to 89% with 1 mL of MeOH and from 71% to 81% with 3 mL of MeOH. However, the decrease in recovery for the acidic analytes was more evident, ranging from 42% (CLO) to 84% (DIC) with 1 mL of MeOH and from 24% (CLO) to 59% (BEZ) with 3 mL MeOH. For this reason, to avoid high losses of analytes in the washing step it was decided to maintain the washing volumes as 1 mL, similarly to the volumes used in previous studies.17,19
The loading volume was increased from 10 mL to 100 mL to obtain a higher preconcentration factor, without the need for excessively long percolation times. No significant losses were observed when inspecting the %RSPE values, therefore 100 mL was selected as the loading volume for ultrapure water. Due to the complexity of the environmental water samples, the volume of river water samples was kept at 100 mL but was reduced to 50 mL for effluent wastewater samples and to 25 mL for influent wastewater samples to prevent breakthrough.
It is important to compare the performance of a WCX sorbent described previously28 with the performance of the new WAX/WCX polymer microspheres, since both are prepared by polymer-analogous reactions on polymer-bound anhydride motifs. However, only the extraction of cationic analytes can be compared since the WCX sorbent does not contain WAX moieties. In the previous study, ATE, TRI, MTO and PRO were extracted with recoveries ranging from 74 to 106% when 100 mL of ultrapure water was applied to 200 mg of sorbent. In the present study, the recoveries ranged from 82 to 85%, and this may be due to the nearby WAX centres which will repel cations when the WAX centres are protonated. However, this effect is not as marked as it was when sorbents that combine strong anion-exchange and cation-exchange interactions20,24 were evaluated, when the inclusion of a MeOH washing step led to the elution of the acidic analytes. It seems that when a material has anion-exchange and cation-exchange character, to retain both acidic and basic analytes it is helpful if at least one of the two ion-exchange groups is a weak ion-exchanger.
3.4. Validation of the analytical method
After the optimisation of the method using LC-DAD, the method was transferred to LC-HRMS to apply it to the determination of the pharmaceuticals in river water, effluent wastewater and influent wastewater samples. The method was validated using the parameters outlined in the ESI.† The apparent recoveries (%Rapp) were obtained at two concentrations (Table 1 presents the concentrations used for each matrix). As can be observed, good recoveries were obtained for most compounds. Focusing on river samples, %Rapp ranged from 32 to 69%, with most values around 45–50% except for VEN (27 and 29%) and VAL at high concentration (27%). Nadal et al.19 obtained similar recoveries (25–87%) for acidic and basic analytes when percolating 250 mL of river samples through a WAX/WCX polymeric sorbent, while Salas et al.15 obtained higher recoveries (39–109%) when combining two commercial strong ion-exchangers in one SPE cartridge (when loading 100 mL of river water samples). For the effluent wastewater samples, the recoveries ranged from 36 to 78%, which are slightly lower values than reported by Nadal et al.17 when percolating 100 mL of effluent wastewater samples through a SAX/WCX sorbent (24–105%). Lavén et al.16 also obtained slightly higher values when loading 50 mL of effluent wastewater samples onto two strong ion-exchangers connected in series (45–141%). In the case of influent wastewater samples, the recoveries ranged from 34 to 77%, except for TRI (28 and 29%) and MTO (22 and 25%). The results are very similar to the data reported by Salas et al.15 when 50 mL of influent wastewater was loaded (values ranged from 34 to 76%).| River water | Effluent wastewater | Influent wastewater | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| %Rapp 5 μg L−1 | %Rapp 0.5 μg L−1 | %ME 0.5 μg L−1 | R app 10 μg L−1 | R app 1 μg L−1 | %ME 1 μg L−1 | R app 20 μg L−1 | R app 2 μg L−1 | %ME 2 μg L−1 | ||
| Basic | ATE | 61 | 69 | −2 | 54 | 48 | −48 | 35 | 34 | −49 |
| TRI | 43 | 39 | −18 | 36 | 45 | −24 | 29 | 28 | −20 | |
| MTO | 33 | 32 | −9 | 40 | 41 | +10 | 25 | 22 | +57 | |
| VEN | 27 | 29 | +20 | 61 | 52 | −17 | 45 | 34 | +14 | |
| PRO | 46 | 44 | +13 | 56 | 58 | +46 | 35 | 38 | −28 | |
| Acidic | CLO | 54 | 57 | −16 | 78 | 67 | −2 | 77 | 73 | −6 |
| BEZ | 47 | 62 | +3 | 67 | 66 | +11 | 64 | 55 | −38 | |
| VAL | 27 | 37 | −6 | 55 | 46 | +40 | 65 | 72 | +38 | |
| FEN | 47 | 53 | −24 | 76 | 75 | +15 | 40 | 46 | −28 | |
| DIC | 44 | 42 | −31 | 37 | 53 | −17 | 39 | 44 | −34 | |
Coming to the matrix effects (%ME), and considering that ±20% is considered acceptable in terms of matrix effects, the results for river water samples are remarkably good. As can be observed in Table 1, only FEN and DIC had %ME values outside these acceptability limits (%ME values were −24 and −31%, respectively). In the case of effluent wastewater samples, the results were also very good; only ATE, PRO and VAL gave values higher than ±20%. Finally, regarding influent wastewater samples, the use of 1 mL of MeOH as washing step was not sufficient to reduce the matrix effects of the most complex matrix. Only TRI, VEN and CLO had low %ME, ranging from −20 to +14%, however only ATE and MTO presented high %ME values (−49% and +57, respectively). The results with river water and effluent wastewater samples are comparable to the data recorded by Nadal et al.,17 who reported matrix effects ranging from +17 to −30% for river water samples and from +18 to −12% for effluent wastewater samples. Compared with the study by Lavén et al.,16 the results arising from the present study are comparable.
Table S2† shows the MDL and MQL values; these are in the low ng L−1 range and, in some cases, below 1 ng L−1. The exception was FEN, for which the instrumental limits were high due to low ionization yield. In the case of accuracy, the relative recoveries obtained range from 88 to 114%. As for the precision, the %RSD values obtained for repeatability were lower than 12% and for reproducibility between days the values were lower than 18%, which is considered acceptable.
3.5. Analysis of environmental water samples
The five basic and five acidic pharmaceuticals were determined in four samples of river water, effluent wastewater and influent wastewater collected in the province of Tarragona (Spain). The concentrations were determined through external calibration curves and from the apparent recoveries (section 3.4) and following the confirmation parameters.30Table 2 presents the results obtained for the compounds present in the different samples analysed, together with the uncertainty. Starting with river water samples, this was the sample where lowest occurrence was found. Only PRO and MTO were quantified in all samples, whereas TRI was the compound with the highest occurrence in one sample (398 ng L−1) and FEN was not even detected. Similar compounds were found in surface water from China,31 the UK32 and Germany,33 although in the German samples the values of PRO and TRI were lower (<13 and <62 ng L−1, respectively).
| River water | Effluent wastewater | Influent wastewater | ||
|---|---|---|---|---|
| Basic | ATE | <MDL–92 ± 11 | 55 ± 6–3444 ± 376 | 603 ± 7–5424 ± 621 |
| TRI | <MDL–398 ± 48 | 354 ± 35–6132 ± 734 | 322 ± 30–17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 167 ± 2087 167 ± 2087 |
|
| MTO | 4 ± 1–117 ± 15 | 154 ± 18–8324 ± 1176 | 377 ± 44–34464 ± 4270 |
|
| VEN | <MQL–146 ± 17 | 210 ± 24–3654 ± 438 | 196 ± 21–8034 ± 967 | |
| PRO | 15 ± 2–139 ± 16 | 144 ± 16–2367 ± 292 | 98 ± 11–6140 ± 723 | |
| Acidic | CLO | <MDL–45 ± 6 | 34 ± 4–700 ± 86 | 60 ± 15–1257 ± 155 |
| BEZ | <MQL–43 ± 5 | 44 ± 3–1227 ± 146 | 279 ± 34–1532 ± 178 | |
| VAL | <MQL–123 ± 23 | 896 ± 165–5731 ± 1045 | 628 ± 102–21891 ± 2685 |
|
| FEN | <MDL | MQL–557 ± 68 | MQL ± 43–852 ± 122 | |
| DIC | MQL–155 ± 19 | 76 ± 4–789 ± 97 | 108 ± 11–597 ± 77 | |
For the effluent wastewater samples, all of the compounds were quantified, except for FEN. Three of the samples presented significantly lower concentrations (<1000 ng L−1 for all compounds) than the remaining one (for which concentrations were as high as 6132 ng L−1 [TRI] and 8324 ng L−1 [MTO]). When Vergili et al.34 determined TRI, VEN and DIC in effluent wastewater samples from Turkey, DIC was found in significantly higher concentrations, ranging from 300 to 3400 ng L−1, while TRI and VEN were below the MQL (10 ng L−1).
The samples with highest occurrence were the influent wastewater samples. Similarly to effluent wastewater samples, it was possible to quantify all compounds in all samples (except for FEN). One sample presented high concentrations of some compounds, such as TRI (17167 ng L−1), MTO (34464 ng L−1) and VAL (21891 ng L−1). On the other hand, one of the samples presented low concentrations of all compounds, where the compound with highest occurrence in this sample was VAL (628 ng L−1). Van Nuijs et al.35 determined ATE, MTO and VEN in influent wastewater samples from Belgium and similar ATE levels were found, ranging from 41 to 2118 ng L−1, while MTO and VEN were found at even lower concentrations, 252–1190 ng L−1 and 119–480 ng L−1, respectively.
Overall, it can be concluded that this new amphoteric polymer performs very well as an SPE sorbent for the extraction and determination of low concentrations of acidic and basic analytes, even when challenged with the most complex environmental water samples.
4. Conclusions
In this study, polydivinylbenzene-based microspheres with WAX and WCX character have been designed, synthesised and evaluated as an amphoteric SPE sorbent. The D–A cycloaddition chemistry used to functionalise the polydivinylbenzenes with anhydride functional groups represents an unusual strategy for the chemical functionalisation of polymer resins, but it is a facile and versatile method which can, in principle, be applied to a broad range of crosslinked polymers provided that they have pendent diene or dienophile motifs.The amphoteric sorbent was used to determine five acidic and five basic pharmaceuticals in river water, effluent wastewater and influent wastewater samples, and was found to be capable of extracting acidic and basic analytes simultaneously, with good validation parameters. Introduction of a washing step into the SPE protocol reduced the matrix effects. These results highlight the attractiveness of using sorbents that combine weak anion- and cation-exchange interactions for the extraction of acidic and basic pharmaceuticals from complex aqueous samples.
Data availability
The data for this article, including all figures and tables in both the manuscript and ESI,† are available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AC would like to thank the University of Strathclyde and the Engineering and Physical Sciences Research Council (EPSRC) for his Student Excellence Award funded through the Strathclyde Research Studentship Scheme. AM would like to thank Universitat Rovira i Virgili (URV) for his PhD grant (2020PMF-PIPF-33). This research was also part of projects PID2020-114587GB-I00 and PID2023-148939NB-I00 funded by MCIN/AEI/10.13039/501100011033.References
- M. Mofijur, M. M. Hasan, S. F. Ahmed, F. Djavanroodi, I. M. R. Fattah, A. S. Silitonga, M. A. Kalam, J. L. Zhou and T. M. Y. Khan, Advances in identifying and managing emerging contaminants in aquatic ecosystems: Analytical approaches, toxicity assessment, transformation pathways, environmental fate, and remediation strategies, Environ. Pollut., 2024, 341, 122889, DOI:10.1016/j.envpol.2023.122889.
- S. Lee, Y. Choi, D. Kang and J. Jeon, Proposal for priority emerging pollutants in the Nakdong river, Korea: Application of EU watch list mechanisms, Environ. Pollut., 2024, 341, 122838, DOI:10.1016/j.envpol.2023.122838.
- D. Malnes, S. Waara, R. Figuière, L. Ahrens, K. Wiberg, S. J. Köhler and O. Golovko, Hazard screening of contaminants of emerging concern (CECs) in Sweden's three largest lakes and their associated rivers, J. Hazard. Mater., 2023, 453, 131376, DOI:10.1016/j.jhazmat.2023.131376.
- M. Shafi, R. Jan and K. M. Gani, Selection of priority emerging contaminants in surface waters of India, Pakistan, Bangladesh, and Sri Lanka, Chemosphere, 2023, 341, 139976, DOI:10.1016/j.chemosphere.2023.139976.
- S. Zhou, C. D. Paolo, X. Wu, Y. Shao, T. B. Seiler and H. Hollert, Optimization of screening-level risk assessment and priority selection of emerging pollutants – The case of pharmaceuticals in European surface waters, Environ. Int., 2019, 128, 1–10, DOI:10.1016/j.envint.2019.04.034.
- Y. Li, L. Zhang, X. Liu and J. Ding, Ranking and prioritizing pharmaceuticals in the aquatic environment of China, Sci. Total Environ., 2019, 658, 333–342, DOI:10.1016/j.scitotenv.2018.12.048.
- N. Ranjan, P. K. Singh and N. S. Maurya, Pharmaceuticals in water as emerging pollutants for river health: A critical review under Indian conditions, Ecotoxicol. Environ. Saf., 2022, 247, 114220, DOI:10.1016/j.ecoenv.2022.114220.
- E. O. Omotola, A. O. Oluwole, P. O. Oladoye and O. S. Olatunji, Occurrence, detection and ecotoxicity studies of selected pharmaceuticals in aqueous ecosystems- a systematic appraisal, Environ, Toxicol. Pharmacol., 2022, 91, 103831, DOI:10.1016/j.etap.2022.103831.
- L. L. D. De Oliveira, S. C. Antunes, F. Gonçalves, O. Rocha and B. Nunes, Acute and chronic ecotoxicological effects of four pharmaceuticals drugs on cladoceran Daphnia magna, Drug Chem. Toxicol., 2016, 39, 13–21, DOI:10.3109/01480545.2015.1029048.
- C. Vatovec, E. Van Wagoner and C. Evans, Investigating sources of pharmaceutical pollution: Survey of over-the-counter and prescription medication purchasing, use, and disposal practices among university students, J. Environ. Manage., 2017, 198, 348–352, DOI:10.1016/j.jenvman.2017.04.101.
- N. Fontanals, F. Borrull and R. M. Marcé, Overview of mixed-mode ion-exchange materials in the extraction of organic compounds, Anal. Chim. Acta, 2020, 1117, 89–107, DOI:10.1016/j.aca.2020.03.053.
- S. Lardy-Fontan, V. Le Diouron, C. Drouin, B. Lalere, S. Vaslin-Reimann, X. Dauchy and C. Rosin, Validation of a method to monitor the occurrence of 20 relevant pharmaceuticals and personal care products in 167 bottled waters, Sci. Total Environ., 2017, 587–588, 118–127, DOI:10.1016/j.scitotenv.2017.02.074.
- C. Huang, Y. Li, J. Yang, J. Peng, J. Jin, Dhanjai, J. Wang and J. Chen, Preparation of a reversed-phase/anion-exchange mixed-mode spherical sorbent by Pickering emulsion polymerisation for highly selective solid-phase extraction of acidic pharmaceuticals from wastewater, J. Chromatogr. A, 2017, 1521, 1–9, DOI:10.1016/j.chroma.2017.09.021.
- G. Castro, I. Rodríguez, M. Ramil and R. Cela, Selective determination of sartan drugs in environmental water samples by mixed-mode solid-phase extraction and liquid chromatography tandem mass spectrometry, Chemosphere, 2019, 224, 562–571, DOI:10.1016/j.chemosphere.2019.02.137.
- D. Salas, F. Borrull, N. Fontanals and R. M. Marcé, Combining cationic and anionic mixed-mode sorbents in a single cartridge to extract basic and acidic pharmaceuticals simultaneously from environmental waters, Anal. Bioanal. Chem., 2018, 410, 459–469, DOI:10.1007/s00216-017-0736-5.
- M. Lavén, T. Alsberg, Y. Yu, M. Adolfsson-Erici and H. Sun, Serial mixed-mode cation- and anion-exchange solid-phase extraction for separation of basic, neutral and acidic pharmaceuticals in wastewater and analysis by high-performance liquid chromatography-quadrupole time-of-flight mass spectrometry, J. Chromatogr. A, 2009, 1216, 49–62, DOI:10.1016/j.chroma.2008.11.014.
- J. C. Nadal, S. Dargo, F. Borrull, P. A. G. Cormack, N. Fontanals and R. M. Marcé, Hypercrosslinked polymer microspheres decorated with anion- and cation-exchange groups for the simultaneous solid-phase extraction of acidic and basic analytes from environmental waters, J. Chromatogr. A, 2022, 1661, 462715, DOI:10.1016/j.chroma.2021.462715.
- J. C. Nadal, F. Borrull, K. G. Furton, A. Kabir, N. Fontanals and R. M. Marcé, Selective monitoring of acidic and basic compounds in environmental water by capsule phase microextraction using sol-gel mixed-mode sorbents followed by liquid chromatography-mass spectrometry in tandem, J. Chromatogr. A, 2020, 1625, 461295, DOI:10.1016/j.chroma.2020.461295.
- J. C. Nadal, K. L. Anderson, S. Dargo, I. Joas, D. Salas, F. Borrull, P. A. G. Cormack, R. M. Marcé and N. Fontanals, Microporous polymer microspheres with amphoteric character for the solid-phase extraction of acidic and basic analytes, J. Chromatogr. A, 2020, 1626, 461348, DOI:10.1016/j.chroma.2020.461348.
- A. Moral, F. Borrull, K. G. Furton, A. Kabir, N. Fontanals and R. M. Marcé, Development of sol-gel silica-based mixed-mode zwitterionic sorbents for determining drugs in environmental water samples, J. Chromatogr. A, 2022, 1676, 463237, DOI:10.1016/j.chroma.2022.463237.
- T. Wang, Y. Chen, J. Ma, M. Chen, C. Nie, M. Hu, Y. Li, Z. Jia, J. Fang and H. Gao, Ampholine-functionalized hybrid organic-inorganic silica material as sorbent for solid-phase extraction of acidic and basic compounds, J. Chromatogr. A, 2013, 1308, 63–72, DOI:10.1016/j.chroma.2013.08.025.
- S. Jin, Y. Qiao and J. Xing, Ternary mixed-mode silica sorbent of solid-phase extraction for determination of basic, neutral and acidic drugs in human serum, Anal. Bioanal. Chem., 2018, 410, 3731–3742, DOI:10.1007/s00216-018-1037-3.
- A. Moral, P. Clivillé-Cabré, F. Borrull, K. G. Furton, A. Kabir, R. M. Marcé and N. Fontanals, Application of a homemade silica-based mixed-mode ion-exchange sorbent for the determination of drugs in environmental water samples, Adv. Sample Prep., 2023, 7, 100074, DOI:10.1016/j.sampre.2023.100074.
- A. Moral, F. Borrull, K. G. Fourton, A. Kabir, N. Fontanals and R. M. Marcé, Introduction of zwitterionic functional groups in silica sorbent for the simultaneous determination of acidic and basic pharmaceuticals, Anal. Bioanal. Chem., 2025 Search PubMed (submitted).
- J. C. Nadal, K. L. Anderson, S. Dargo, I. Joas, D. Salas, F. Borrull, P. A. G. Cormack, R. M. Marcé and N. Fontanals, Microporous polymer microspheres with amphoteric character for the solid-phase extraction of acidic and basic analytes, J. Chromatogr. A, 2020, 1626, 461348, DOI:10.1016/j.chroma.2020.461348.
- B. R. Stranix, PhD thesis, McGill University, 1997. https://escholarship.mcgill.ca/concern/theses/v405sc01b.
- A. Altuna-Ruiz de Eguino, P. A. G. Cormack and J. Prunet, Olefin cross metathesis as a versatile method for the facile functionalization of porous polymers, Org. Lett., 2020, 22, 2481–2485, DOI:10.1021/acs.orglett.0c00726.
- N. Fontanals, J. Zohar, F. Borrull, S. Ronka and R. M. Marcé, Development of a maleic acid-based material to selectively solid-phase extract basic compounds from environmental samples, J. Chromatogr. A, 2021, 1647, 462165, DOI:10.1016/j.chroma.2021.462165.
- R. V. Law, D. C. Sherrington and C. E. Snape, Quantitative solid state 13C NMR studies of highly cross-linked poly(divinylbenzene) resins, Macromolecules, 1997, 30, 2868–2875, DOI:10.1021/ma9616470.
- EU, Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to, Off. J. Eur. Union, 2021, 180, L180/84–L180/109. https://extwprlegs1.fao.org/docs/pdf/eur203999.pdf.
- Y. Zhang, L. Duan, B. Wang, C. S. Liu, Y. Jia, N. Zhai, L. Blaney and G. Yu, Efficient multiresidue determination method for 168 pharmaceuticals and metabolites: Optimization and application to raw wastewater, wastewater effluent, and surface water in Beijing, China, Environ. Pollut., 2020, 261, 114113, DOI:10.1016/j.envpol.2020.114113.
- M. J. Hilton and K. V. Thomas, Determination of selected human pharmaceutical compounds in effluent and surface water samples by high-performance liquid chromatography – electrospray tandem mass spectrometry, J. Chromatogr. A, 2003, 1015, 129–141, DOI:10.1016/S0021-9673(03)01213-5.
- M. Egli, A. Hartmann, H. R. Wright, K. T. Ng, F. B. Piel and L. P. Barron, Quantitative determination and environmental risk assessment of 102 chemicals of emerging concern in wastewater-impacted rivers using rapid direct-injection liquid chromatography—tandem mass spectrometry, Molecules, 2021, 26, 5431, DOI:10.3390/molecules26185431.
- I. Vergili, Y. Kaya, Z. B. Gönder, A. Boergers and J. Tuerk, Occurence and Prioritization of Pharmaceutical Active Compounds in Domestic/Municipal Wastewater Treatment Plants, Bull. Environ. Contam. Toxicol., 2019, 102, 252–258, DOI:10.1007/s00128-019-02550-z.
- A. L. N. Van Nuijs, I. Tarcomnicu, W. Simons, L. Bervoets, R. Blust, P. G. Jorens, H. Neels and A. Covaci, Optimization and validation of a hydrophilic interaction liquid chromatography-tandem mass spectrometry method for the determination of 13 top-prescribed pharmaceuticals in influent wastewater, Anal. Bioanal. Chem., 2010, 398, 2211–2222, DOI:10.1007/s00216-010-4101-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01312c |
| This journal is © The Royal Society of Chemistry 2025 |