
Regulating the stereomicrostructure, circularity and functionality of synthetic PHAs
Ethan C.
Quinn†
 a,
Celine R.
Parker†
a,
Sophie M.
Guillaume
*b and
Eugene Y.-X.
Chen
*a
a,
Celine R.
Parker†
a,
Sophie M.
Guillaume
*b and
Eugene Y.-X.
Chen
*a
aDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA. E-mail: Eugene.Chen@colostate.edu
bInstitut des Sciences Chimiques de Rennes, UMR 6226, Université Rennes, CNRS, Rennes, France. E-mail: sophie.guillaume@univ-rennes.fr
First published on 31st December 2024
Abstract
Biodegradable plastics, especially those that can biodegrade in uncontrolled enviroments, are of importance to help curb the global plastics crisis. Poly(3-hydroxyalkanoate)s (PHAs), which can be either microbially or chemically synthesized, are one of the rare classes of plastics that can biodegrade under both managed and unmanaged conditions. Besides this exceptional upside, PHAs can also be tuned to exhibit thermal, mechanical, and optical properties of commodity polymers including polyolefins, and they can be designed to be chemically recyclable towards a circular PHA economy or functionalized to acquire additional, diverse and/or improved properties. To enable such modularity in chemocatalytic PHAs, the development of stereoselective and controlled molecular catalysts, as well as the design of monomer structures and polymerization processes, is of primary importance. In this context, this Perspective article focuses on three recent advancements, including PHA stereomicrostructural engineering, melt-processability and chemical recyclability, and chemical functionalization.
Introduction
Recently, especially within the past decade, research has focused heavily on searching for practical solutions to addressing the global plastics crisis.1–4 Typical strategies aim to increase mechanical recycling rates, valorize waste plastics, design chemically recyclable polymers, or develop biodegradable plastics.5–13 The development of biodegradable plastics represents an important part of this large effort to combat the plastics problem, especially concerning end-of-life issues and environmental protection. This becomes particularly important for application areas where plastics recovery, recycling, or reuse is highly challenging or nearly impractical.Amongst many biodegradable plastics reported in the literature, the plastics belonging to the large family of poly(3-hydroxyalkanoate)s (PHAs)14–19 are most unique, thanks to their distinct ability to biodegrade in both managed (industrial and home composting) and unmanaged (ocean, freshwater, and soil) environments,20 as well as their largely tunable material properties.21 The biological fermentation route to PHAs is recognized for its ability to take various inputs of biorenewable sources (biomass, fatty acids, plant oils, etc.) and produce stereoperfect (R)-PHAs that typically exhibit high molar mass, high melting points, and strong mechanical properties.21–24 The simplest, most important member in the large PHA family is poly(3-hydroxybutyrate) (P3HB), with the methyl as the side-chain group or exocyclic substituent. A variety of PHA copolymers of P3HB incorporating other short, medium, or long side-chain length moieties can be biologically produced, depending on the growth conditions and feed substrates.15,21,25,26
As these materials are stereoperfect with pure (R) stereo-configurations on the backbone stereogenic centers, they are highly crystalline, especially for short-chain PHAs such as P3HB and poly(3-hydroxyvalerate) (P3HV), imparting these PHAs with high ultimate tensile strength (σB) and melting temperature (Tm) values but also detrimentally making such PHAs extremely brittle, with negligible fracture strain properties or elongation at break (εB) of ∼3%.15 Currently, varying the stereochemistry of PHAs biologically presents a daunting challenge;27 thus, the modulation of material properties of biological PHAs is confined to the copolymer composition, which is typically limited by the functional group tolerance of the bacteria or enzymes.
In contrast to the biological route to PHAs, the chemocatalytic route allows for modulation of the PHA stereochemistry through catalyst-controlled stereoselective polymerization processes, termed stereomicrostructural engineering.14,28–30 Accessing these stereodiverse and functionalized PHAs requires the implementation of advanced molecular catalysts that can regulate the polymerization stereochemistry and tolerate monomer functionality. Herein, we highlight recent advances in the synthesis of stereodiverse and functionalized PHAs utilizing molecular catalysts, as well as thermally robust and chemically recyclable PHAs through monomer design (Fig. 1). We conclude this Perspective by laying out our vision for the future of PHAs, including critical issues that must be addressed to facilitate broader implementation of PHAs in the marketplace.
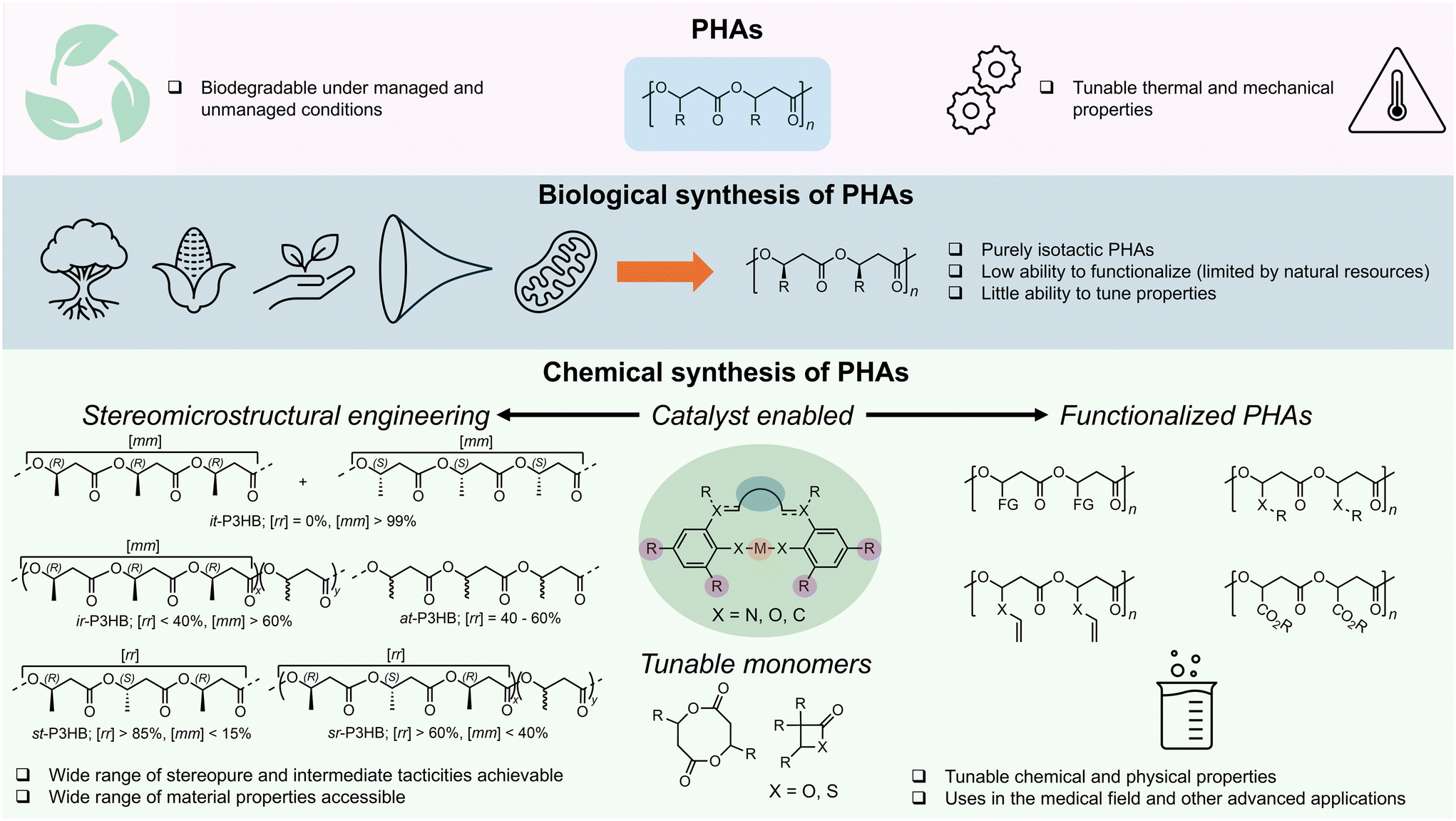 | ||
| Fig. 1 Overview of PHAs, including biological and chemocatalytic routes, stereomicrostructural engineering, and functionalization of PHAs. | ||
Stereomicrostructural engineering of PHAs
For decades, one of the major goals in the chemocatalytic routes to PHAs was to synthesize biomimetic stereoperfect (sp) isotactic (it)-P3HB (characterized as having a probability of meso diads, Pm > 0.99, and a percentage of meso triads, [mm] > 99%) as this P3HB exhibits an it-polypropylene (PP)-like melting temperature (Tm ∼ 175 °C) and desirable tensile strength (σB ∼ 30 MPa). As compared to the biological route to it-P3HB, which has slow kinetics and yields materials with high dispersity (Đ > 2.0) due to its nature of step-growth polymerization (SGP), the chemocatalytic route to P3HB via ring-opening polymerization (ROP) typically has fast kinetics, affords P3HB with low Đ (as low as 1.01) due to its nature of chain-growth polymerization, and is more scalable and cost-effective. In 2018, Chen and Tang reported the first instance of biomimetic sp-it-P3HB through the ROP of the racemic eight-membered dimethyl diolide (rac-8DLMe) with yttrium catalyst 1 (Fig. 2).31 The catalyst system used in this work was also found to perform kinetic resolution on rac-8DLMe. Recently, Coates and coworkers have also developed a bimetallic magnesium catalyst system, which performs kinetic resolution of rac-β-butyrolactone (rac-BBL) with stereoinversion of the polymerized monomer and results in it-P3HB with Pm = 98%, the highest isotacticity achieved in the ROP of rac-BBL to date.32 Notably, this catalyst system does not perform well in polymerizing (S)- or (R)-BBL as it was determined that the opposite enantiomer activates the chiral catalyst to perform the kinetic resolution.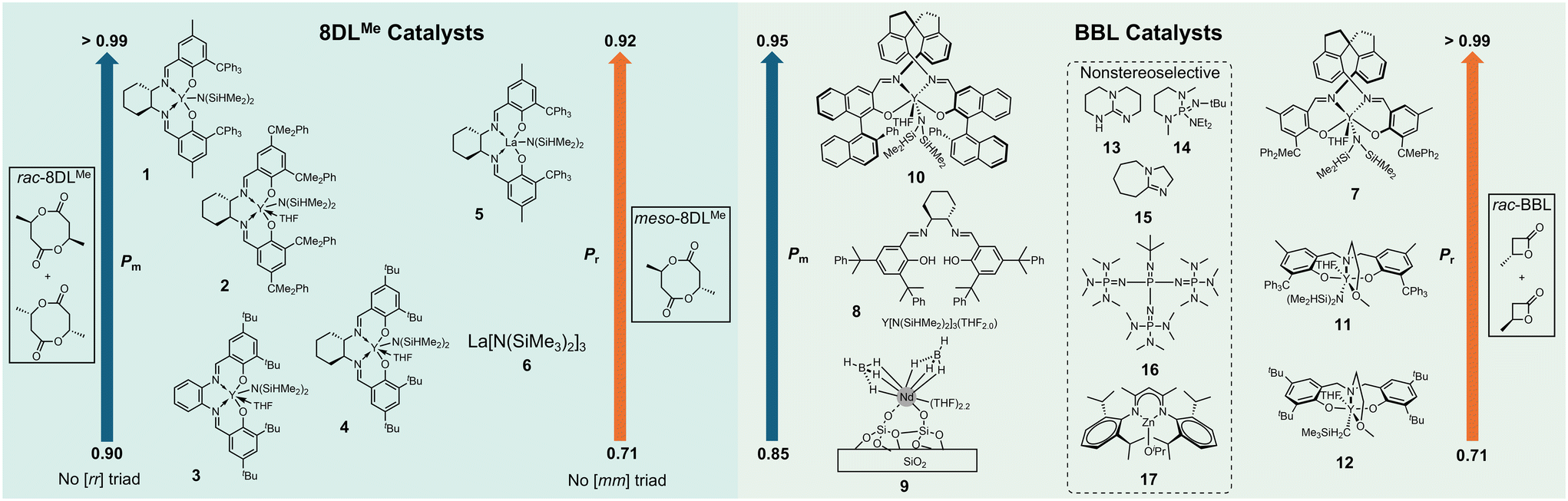 | ||
| Fig. 2 Structures of representative molecular catalysts for ROP engineering of PHA stereomicrostructures. | ||
Work towards the goal of stereoperfection has also been carried out through the polymerization of rac-BBL using 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and 934 as well as towards sp syndiotactic (st) P3HB using 7 (characterized as having a probability of racemic diads, Pr > 0.99, and percentage of racemic triads, [rr] > 99%).35 These works, along with the pioneering report of Carpentier and coworkers, which showed that st-P3HB with Pr = 0.94 can be produced by the ROP of rac-BBL with yttrium catalysts incorporating the tetradentate alkoxy-amino-bis(phenolate) ligand (11, 12),36,37 have advanced our understanding of catalyst structure–polymer stereomicrostructure relationships, more specifically how molecular catalysts use their symmetry as well as steric and electronic effects to control stereomicrostructures of P3HB materials. Even though these contributions are notable, they still do not solve the problem of sp-P3HB's brittleness as both it- and st-P3HB are highly crystalline and extremely brittle. Further catalyst development has been necessary to solve this brittleness issue and enhance the mechanical toughness.
33 and 934 as well as towards sp syndiotactic (st) P3HB using 7 (characterized as having a probability of racemic diads, Pr > 0.99, and percentage of racemic triads, [rr] > 99%).35 These works, along with the pioneering report of Carpentier and coworkers, which showed that st-P3HB with Pr = 0.94 can be produced by the ROP of rac-BBL with yttrium catalysts incorporating the tetradentate alkoxy-amino-bis(phenolate) ligand (11, 12),36,37 have advanced our understanding of catalyst structure–polymer stereomicrostructure relationships, more specifically how molecular catalysts use their symmetry as well as steric and electronic effects to control stereomicrostructures of P3HB materials. Even though these contributions are notable, they still do not solve the problem of sp-P3HB's brittleness as both it- and st-P3HB are highly crystalline and extremely brittle. Further catalyst development has been necessary to solve this brittleness issue and enhance the mechanical toughness.
An abundance of work has also been focused on creating new molecular catalysts, both metallic and organic types, for the ROP of BBL.14 Typically, these catalysts impart some stereocontrol but lead to materials with long or very short segments of stereoperfect regions, which either impart a high degree of crystallinity, making the material brittle, or lead to mostly amorphous softer materials. Recently, Reiger and coworkers developed an in situ generated yttrium catalyst system (8), which yields iso-rich ir-P3HB. Although its isotacticity and Tm are only moderately high (Pm up to 0.89, Tm up to 154 °C), it exhibits good mechanical properties with σB = 21.1 MPa and εB = 392% (Pm = 0.84).33 Robinson and coworkers developed a dual catalysis approach to generate stereoblock it-b-atactic (at) P3HB through irreversible chain transfer ROP of rac-BBL with sequential addition of yttrium and zinc complexes.38 Chen and coworkers previously reported the synthesis of it-b-st P3HB from the diastereoselective polymerization of a mixture of rac-8DLMe and meso-8DLMe.39
To confer polyolefin-like properties to P3HB without employing the copolymerization strategy,40 which does not conform to the mono-material design principle,41 the diolide monomer platform needs to be implemented. In 2023, Chen and coworkers showed that installation of controlled stereodefects into P3HB through the ROP of meso-8DLMe with a commercially available lanthanum catalyst (6) leads to syndio-rich sr-P3HB (Pr = 0.77, no [mm] triads due to the structure of meso-8DLMe), which exhibits a high fracture strain (εB > 400%), high tensile strength (σB = 34 MPa), excellent toughness (UT = 96 MJ m−3), high optical clarity, and a large processing window (difference between Tm and degradation temperature, Td, of 141 °C).28 Furthering this work, high-molar-mass ir- and sr-P3HB synthesized using the diolide platform and complex 2 or 3 resulted in ultra-tough and ductile (εB > 800%) materials with excellent optical clarity.42 Blending these chemocatalytic P3HB materials with biological sp-P3HB gave new P3HB materials with synergistic thermal and mechanical properties. However, when the ir-P3HB obtained from the ROP of rac-BBL was blended with bio-P3HB, enhanced properties were not observed, emphasizing the necessity of synthesizing P3HB from rac-8DLMe for enhancing toughness of bio-P3HB as well as Tm and modulus of ir- and sr-P3HB.42
This stereomicrostructural engineering work has been mainly focused on P3HB, but other PHAs have also been synthesized with diverse stereomicrostructures. For example, P3HV has been synthesized from both 8DLEt and β-valerolactone (BVL) with sr, ir, and intermediate st and it stereomicrostructures.40,43 Increasing the length of the alkyl chain provides access to a wider range of thermal and mechanical properties, such as decreased glass transition temperature (Tg) below −30 °C, enabling packaging applications in cold environments. Again, these stereomicrostructures were imparted by the catalyst used, which has also been shown for PHAs with longer alkyl substituents, aryl substituents, more elaborate substituents, and (alternating) copolymers.14,40,44,45 In an effort to expand the scope of PHAs, stereoselective polymerization of racemic β-thiobutyrolactone (rac-TBL) generated sulfurated PHA poly(3-thiobutyrolactone) (P3TB).46 Increasing the ionic radius (S > O) by utilizing a thiolactone monomer enabled access to topologically diverse, stereocontrolled cyclic st- and it-P3TB using various catalysts (6, 12, etc.).46,47
Tuning the properties of PHAs by regulating their stereomicrostructure is a developed field that is still fast growing. There is still a wealth of knowledge to be gained in this space by further tuning the catalyst and monomer structures and their close covalent- and/or non-covalent interactions during the ROP process. Furthermore, there are application spaces that need to be explored for these stereodiverse PHAs.
Thermally robust and chemically recyclable PHAs
One of the major pitfalls of semi-crystalline it-PHAs is their propensity to degrade (Td5% ∼ 250 °C) near their Tm or in melt (i.e., lack of melt-processability) due to the facile cis-elimination enabled by the presence of reactive α-hydrogens. Four notable advances have been made to enhance the thermal stability and enable chemical recyclability of PHAs.The first was by Liu and coworkers where they designed a four-membered lactone fused with a five-membered ring at the α- and β-carbons of the lactone, effectively eliminating one of the α hydrogens, which enabled chemical recycling through a multi-step process, although the Td values were still similar to a typical PHA (213 °C for the trans-PHA and 268 °C for the cis-PHA; Fig. 3A).48
 | ||
| Fig. 3 Highlighted examples of thermally robust and chemically recyclable PHAs. | ||
Second, through the ROP of α-methylated BBL, Coates and coworkers synthesized an α-methylated PHA that could be chemically recycled through a proposed multi-step process, with noticeably enhanced Td values relative to a typical PHA (269 to 289 °C; Fig. 3B).49 The α-methylated PHA is also found to be intrinsically crystalline, much like the tacticity-independent crystallinity observed in the α,α-dimethylated PHA.50
As a third approach, Chen and coworkers created truly chemically recyclable PHAs from the ROP of α,α-dimethylated BBL, which leveraged the Thorpe–Ingold effect to enable not only depolymerization of the PHA back to the lactone monomer, but also melt-processability with much enhanced Td up to 335 °C and Tm up to 243 °C (Fig. 3C).51 Simple, achiral phosphazene superbase tBu-P4 (16) was used as a catalyst for the ROP of this trimethylated lactone. Polymerization of various stereoisomers of the lactone resulted in semicrystalline at-, ir-, and it-PHAs, demonstrating tacticity-independent crystallinity.50 Lastly, Chen and coworkers extended the α,α-dimethylation strategy to propiolactone, producing α,α-disubstituted propionate PHAs that are not only thermally robust (Td up to 373 °C and Tm up to 266 °C) but also chemically recyclable via two (SGP and ROP) closed-loops (Fig. 3D).52 The α,α-dimethylated PHAs described here solve the critical issues associated with the chemical recycling and thermal resistance of PHAs. These advances should promote the PHA circularity and expand PHA applications to textile fibers and other high-end specialty applications.
Functionalized PHAs
As described above, stereoselective ROP towards aliphatic PHAs has resulted in the production of biocompatible, stereodiverse, high-performance plastics with viable commercial utility, but they are still limited in terms of the breadth of their applications. In particular, the high hydrophobicity of P3HB limits its biomedical applications, making functionalization with hydrophilic groups through post-polymerization modifications or monomer design to introduce hydrophilic pendant groups, the most common approaches to broaden their usage in aqueous environments.Introducing allylic groups onto PHAs that serve as functional handles for further chemical transformations is an effective strategy for producing functional PHAs, which are not directly accessible through bio- or chemo-synthetic routes on account of many catalysts’ functional group intolerances. Allylic PHAs have long been accessible through biological fermentation,53,54 but this work heavily relies upon the synthesis of poly(3-hydroxy-octanoate-co-3-hydroxy-10-undecenoate) (PHOU) with high dispersity values (Đ ∼2.0–3.0) and uncontrolled vinyl incorporation. This was resolved through random copolymerization of rac-BBL with rac-BLallyl using yttrium complex 18a, leading to the corresponding monodisperse st-PHA copolymer (Pr = 0.80–0.84) and up to 80% rac-BLallyl incorporation (Fig. 4).55 Although the homopolymer from rac-BLallyl is amorphous, exhibiting a Tg ∼ −44 °C, the PHA copolymers are semicrystalline with Tm values of 104–109 °C. The vinyl groups in the copolymers can be readily converted to hydroxy and epoxy groups with quantitative yields. The resulting hydroxy- and epoxy-functionalized PHAs showed no change in their stereomicrostructure, but only minor variations in their thermal properties as a result of the chemical modifications, evidencing that the backbone of the polymer remained unaffected by the change in functionality.
 | ||
| Fig. 4 Structures of representative molecular catalysts for functionalized PHAs and PHA post-functionalization. | ||
These types of structural changes are emblematic of the functional utility of an allylic pendant group, and they have since been exploited as an anchoring site for biologically active molecules in drug delivery.56 For example, the dihydroxylated PHA showed increased controlled release kinetics for encapsulated L-leuprolide acetate, which was attributed to the formation of hydrogen bond networks.57 Later it was observed that β-diiminate (BDI) zinc alkoxide catalyst 17 can stereoselectively polymerize rac-BLallyl, resulting in an ir-PHA (Pm = 0.61).58 In seeking to expand the therapeutic applications of PHAs, the allyl-functionalized, ir- and sr-PHAs were next subjected to quantitative hydroboration, which did not alter the backbone length or stereochemistry, but did increase the Tg of the resulting PHAs from ∼−40 °C to −4 °C.58 Boronate substituents are attractive therapeutic targets due to their potential uses in cancer therapy as well as interaction with sugars, which could enable their use for insulin delivery. Most recently an olefin-functionalized PHA copolymer was utilized for accessing reprocessable, recyclable, and biodegradable elastomeric vitrimers through installation of dynamic boronic ester bonds by the thiol–ene “click” chemistry.59
Subjecting a polymer to stoichiometric amounts of chemical reagents to perform post-polymerization modifications could induce cleavage along the backbone and/or other undesired side reactions. Hence, modifications under mild conditions are preferred for their reliability in achieving the desired transformations. A promising example of such a strategy is the mild hydrogenolysis of poly(benzyl β-malolactonate) (PMLABn) to produce hydrophilic poly(malic acid) (PMLA), which can be biometabolized,60 making it attractive for biomedical applications.16,61–63 It was also shown to be possible to polymerize benzyl β-malolactone (MLABn) using organobases 13–15, leading to the corresponding amorphous PHA with Tg ∼ −46 °C.64 This strategy was extended to facilitate the synthesis of random and diblock copolymers from rac-BBL65 or cyclic carbonates,66,67 both of which were utilized to produce defined nanoparticles with low-to-no cytotoxicity with cell types that would make them useful for liver-targeted drug delivery applications.66,68–70 Specifically, PMLA-b-P3HB copolymer nano-objects showed no cytotoxicity in HepaRG and SK-MEL-28 cells at low concentrations (<88 μg mL−1) and mild toxicity at high concentrations (88–320 μg mL−1).66 Similar cell viability was also evidenced using poly(trimethylene carbonate) (PTMC)-b-PMLA68 and polyethylene glycol (PEG)-b-PMLABn copolymers.70 Specific cell-type cytotoxicity is not the only limiting factor in drug delivery, and potential immune responses must be screened in further studies. This open question was addressed for PMLA-b-P3HB and PMLA-b-PTMC, in which favorable uptake by HepaRG cells and reduced macrophage uptake was observed for PMLA-b-P3HB due to the favorable balance of hydrophilic and hydrophobic blocks.69 A series of tetradentate alkoxy-amino-bis(phenolate)-ligated yttrium complexes (11, 18b–e) were next screened for their activity towards MLABn, resulting in the production of a range of semicrystalline st-PMLABn PHAs. The greatest syndioselectivity was achieved using 18d with Cl ortho and para substituents on the ligand backbone, resulting in a PHA with Pr up to >0.95 and Tm up to 117 °C. This syndioselective polymerization strategy was extended to produce semicrystalline st-PMLAall (Pr > 0.95, Tm up to 112 °C; 18c) and st-PMLAMe (Pr up to 0.92, Tm up to 212 °C; 18d),71 demonstrating the functional group tolerance of this series of complexes and their potential utility in producing other functional PHAs (Fig. 4).61
The most direct way to produce hydrophilic PHAs is through polymerization of hydrophilic monomers, which posed a challenge due to functional group intolerance with known catalysts. Lactone monomers of 3-hydroxy-propiolactone (BPLCH2OR) can be stereoselectively polymerized using 18a, 18d, 18f–j, and 18l with an informative trend in stereoselectivity depending on the pendant group of the monomer (Fig. 4). It was found that a given catalyst's ortho-substituent on the ligand backbone dictates the stereoselective polymerization of rac-BPLCH2OR monomers (R = methyl, allyl, benzyl).72–74 Bulky ortho groups such as cumyl (–Me2Ph, 18h, 18i) resulted in sr- to st-PHAs with Pr = 0.81–0.90 across all monomers, while chlorine groups (18d, 18f) yielded ir- to it-PHAs with Pm = 0.90–0.93. Some of these PHAs are semicrystalline; for example, sr-P3HBCH2OAllyl (Pr = 0.81) showed a Tm of 85 °C, and sr-P3HBCH2OMe (Pr = 0.81) displayed a Tm of 116 °C. These improved thermal properties may imply their potential utility in packaging applications and warrant further characterization of their tensile properties, if increased molar masses above their entanglement molecular weights can be reached.
Follow-up studies that expanded the range of stereoregular P3HBCH2OR materials through the use of different pendant groups concluded that the methylene hydrogens on the pendant exocyclic substituent have non-covalent interactions with the ligands of catalysts 18f, 18g, 18i and 18l, which dictate whether tactic functionalized P3HB materials are formed or not (Fig. 4).75–77 This finding was further confirmed in a subsequent report, where a BPL with the pendant group –CH2OCF2CHF2 was polymerized with 18b, 18f, and 18g to produce st-PHA (Pr up to 0.87) and at-PHA using 18l, demonstrating stereoselectivity trends based on the ortho and para substituent identities in the ligand backbone alongside non-covalent interactions with the monomer exocyclic group.78 The continued mechanistic evaluation of the catalyst stereoselectivity represents an important part of ongoing research in this field, aiding in the prediction of material properties of the resulting polymers for specifically targeted applications.
The development of a more diverse range of functionalized PHAs has also been sought to expand their thermal and mechanical properties. Biosynthetic approaches have produced many functionalized PHAs, enlarging their breadth to include PHAs bearing pendant groups such as chlorines,79,80 fluorines,81–83 bromines,84 aromatics,85,86 esters,87 thioesters,88 epoxys,89 phenoxys,90–93 and others.94–96 These routes have relied on copolymerization strategies by which bacteria incorporate unusual functional groups from a given feedstock alongside more traditional feedstocks, creating random PHA copolymers typically with a low amount of functionality.
Fluorinated PHAs were first targeted biosynthetically for their increased hydrophobicity and thermal transitions, but failed to make an impact due to low incorporation of fluorinated feedstocks.82,83 Coates and coworkers sought to produce highly fluorinated PHA homopolymers via a two-step process: carbonylation of fluorinated epoxides to four-membered β-lactone monomers, followed by the ROP using 17 to afford fluorinated PHAs.97 These fluorinated PHAs showed increased Tg over their non-fluorinated counterparts and anticipated higher hydrophobicity.
Aromatic PHAs represent another synthetic target towards high-Tg PHAs imparted by the rigid aromatic groups. The stereoselective ROP of the rac- and meso-dibenzyl eight-membered diolide (8DLBn) using 2 enabled access to it- and st-poly(3-hydroxy-4-phenylbutyrate) (P3H4PhB) as well a series of random and diblock copolymers with other diolide-derived PHAs.44 Stereoperfect it-P3H4PhB (Pm > 99%) and st-P3H4PhB (Pr = 92%) each exhibited a significantly increased Tg up to 43 °C. Rac-8DLBn was copolymerized with meso-8DLBu to provide a soft, elastomeric st-P3HHp polymer, which exhibited a high Td5% of 281 °C, high fracture strain (εB ∼ 191%) and good tensile strength (σB ∼ 22.7 MPa).
Continued innovation in the realm of unusual PHAs derived chemosynthetically with designer monomers can unlock access to a diverse range of novel PHAs with unprecedented material properties.
Future of synthetic PHAs
With the dawn of stereomicrostructural engineering and functionalization of PHAs as well as PHA monomer design, we foresee the future of PHAs as incredibly bright. The amount of versatility that can be imbued in PHAs through these three approaches is vast and will continue to enable PHAs to be used in interdisciplinary applications. We predict the implementation of these PHAs in (multi-layered) packaging applications, textiles, adhesives, agriculture, biomedical fields, and high-end applications, especially in areas where biodegradability and/or biocompatibility are of primary importance.Even with the great success achieved in the last decades, there are still several challenges that need to be overcome for the widespread implementation of PHAs. First, the synthesis of (lactone or diolide) monomers and (metal-based) catalysts needs to be further advanced to be more cost-effective and environmentally benign. Developing full chemical circularity of PHAs to recover their monomers in high selectivity and purity, and combined biocatalytic and chemocatalytic processes, present perhaps the most effective solutions to tackle this challenge. Second, the lack of closed-loop recycling for most of the current PHAs must be overcome to render their chemical circularity towards a circular PHA economy. Regenerating the highly strained lactone or the diolide monomers, essential for the ROP with fast kinetics and full monomer conversion, may be chemically unfeasible for some monomer structures adopting this ideally direct, short circular pathway, but leveraging an additional catalytic step could still close the entire circular loop. Third, to facilitate the industrial adoption of the chemocatalytic PHAs, large-scale product development must be comprehensively investigated via industrial processing and fabrication conditions. The PHA community across academia and industry needs to work together collaboratively on solving these issues, and we are optimistic about them being resolved in the near future so as to promote the applications of sustainable and affordable PHAs.
Author contributions
E. C. Q. and C. R. P.: writing – original draft preparation, writing – review & editing, and investigation; S. M. G. and E. Y.-X. C.: supervision, project administration, funding acquisition, and writing – review & editing.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by the U.S. Department of Energy and CNRS. The work done by E. C. Q., C. R. P., and E. Y.-X. C. was supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Materials and Manufacturing Technologies Office (AMMTO), and Bioenergy Technologies Office (BETO), performed as part of the BOTTLE Consortium, which includes the members from Colorado State University, and funded under contract no. DE-AC36-08GO28308 with the National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy. The work done by S.M.G. was supported by the CNRS and the University of Rennes.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- World Economic Forum, Ellen MacArthur Foundation and McKinsey Company, The New Plastics Economy: Rethinking the Future of Plastics, 2016 Search PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y.-X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS PubMed.
- R. W. Clarke, G. Rosetto, T. Uekert, J. B. Curley, H. Moon, B. C. Knott, J. E. McGeehan and K. M. Knauer, Mater. Adv., 2024, 5, 6690–6701 RSC.
- A. Dhaini, V. Hardouin-Duparc, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Prog. Polym. Sci., 2024, 149, 101781 CrossRef CAS.
- A. H. Westlie, E. C. Quinn, C. R. Parker and E. Y.-X. Chen, Prog. Polym. Sci., 2022, 134, 101608 CrossRef CAS.
- Z. A. Raza, S. Abid and I. M. Banat, Int. Biodeterior. Biodegrad., 2018, 126, 45–56 CrossRef CAS.
- G.-Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- H.-M. Müller and D. Seebach, Angew. Chem., Int. Ed. Engl., 1993, 32, 477–502 CrossRef.
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, e265–e265 CrossRef CAS.
- S. Taguchi, T. Iwata, H. Abe and Y. Doi, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 157–182, DOI:10.1016/B978-0-444-53349-4.00223-5.
- T. Narancic, S. Verstichel, S. R. Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. B. Padamati and K. E. O’Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed.
- K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS.
- R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2005, 6, 1–8 CrossRef CAS.
- H. Park, H. He, X. Yan, X. Liu, N. S. Scrutton and G.-Q. Chen, Biotechnol. Adv., 2024, 71, 108320 CrossRef CAS.
- P. Thamarai, A. S. Vickram, A. Saravanan, V. C. Deivayanai and S. Evangeline, Bioresour. Technol. Rep., 2024, 27, 101957 CrossRef CAS.
- S. A. Acharjee, P. Bharali, B. Gogoi, V. Sorhie, B. Walling and Alemtoshi, Water, Air, Soil Pollut., 2022, 234, 21 CrossRef PubMed.
- F. M. de Souza and R. K. Gupta, ACS Omega, 2024, 9, 8666–8686 CrossRef CAS PubMed.
- B. Laycock, P. Halley, S. Pratt, A. Werker and P. Lant, Prog. Polym. Sci., 2013, 38, 536–583 CrossRef CAS.
- E. C. Quinn, A. H. Westlie, A. Sangroniz, M. R. Caputo, S. Xu, Z. Zhang, M. Urgun-Demirtas, A. J. Müller and E. Y.-X. Chen, J. Am. Chem. Soc., 2023, 145, 5795–5802 CrossRef CAS PubMed.
- H. Li, R. M. Shakaroun, S. M. Guillaume and J.-F. Carpentier, Chem. – Eur. J., 2020, 26, 128–138 CrossRef CAS PubMed.
- G. Adamus, A. Domiński, M. Kowalczuk, P. Kurcok and I. Radecka, Polymers, 2021, 13, 4365 CrossRef CAS.
- X. Tang and E. Y.-X. Chen, Nat. Commun., 2018, 9, 2345 CrossRef.
- M. S. Young, A. M. LaPointe, S. N. MacMillan and G. W. Coates, J. Am. Chem. Soc., 2024, 146, 18032–18040 CrossRef CAS.
- J. Bruckmoser, S. Pongratz, L. Stieglitz and B. Rieger, J. Am. Chem. Soc., 2023, 145, 11494–11498 CrossRef CAS.
- N. Ajellal, G. Durieux, L. Delevoye, G. Tricot, C. Dujardin, C. M. Thomas and R. M. Gauvin, Chem. Commun., 2010, 46, 1032–1034 RSC.
- H.-Y. Huang, W. Xiong, Y.-T. Huang, K. Li, Z. Cai and J.-B. Zhu, Nat. Catal., 2023, 6, 720–728 CrossRef CAS.
- N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS.
- A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS PubMed.
- J. E. Chellali, A. J. Woodside, Z. Yu, S. Neogi, I. Külaots, P. R. Guduru and J. R. Robinson, J. Am. Chem. Soc., 2024, 146, 11562–11569 CAS.
- X. Tang, A. H. Westlie, E. M. Watson and E. Y.-X. Chen, Science, 2019, 366, 754–758 CrossRef CAS.
- X. Tang, A. H. Westlie, L. Caporaso, L. Cavallo, L. Falivene and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2020, 59, 7881–7890 CrossRef CAS.
- E. C. Quinn, K. M. Knauer, G. T. Beckham and E. Y.-X. Chen, One Earth, 2023, 6, 582–586 CrossRef.
- Z. Zhang, E. C. Quinn, J. L. Olmedo-Martínez, M. R. Caputo, K. A. Franklin, A. J. Müller and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202311264 CrossRef CAS.
- A. Pietrangelo, C. R. López-Barrón, M. T. DeRocco, S. Kang, S. J. Mattler and P. J. Wright, Macromolecules, 2023, 56, 5588–5598 CrossRef CAS.
- A. H. Westlie and E. Y.-X. Chen, Macromolecules, 2020, 53, 9906–9915 CrossRef CAS.
- Z. Zhang, C. Shi, M. Scoti, X. Tang and E. Y.-X. Chen, J. Am. Chem. Soc., 2022, 144, 20016–20024 CrossRef CAS PubMed.
- H. Li, J. Ollivier, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2022, 61, e202202386 CrossRef CAS PubMed.
- H. Li, S. M. Guillaume and J.-F. Carpentier, Chem. – Asian J., 2022, 17, e202200641 CrossRef CAS PubMed.
- Y.-T. Li, H.-Y. Yu, W.-B. Li, Y. Liu and X.-B. Lu, Macromolecules, 2021, 54, 4641–4648 CrossRef CAS.
- Z. Zhou, A. M. LaPointe, T. D. Shaffer and G. W. Coates, Nat. Chem., 2023, 15, 856–861 CrossRef CAS PubMed.
- M. Scoti, L. Zhou, E. Y.-X. Chen and C. De Rosa, Macromolecules, 2024, 57, 4357–4373 CrossRef CAS.
- L. Zhou, Z. Zhang, C. Shi, M. Scoti, D. K. Barange, R. R. Gowda and E. Y.-X. Chen, Science, 2023, 380, 64–69 CrossRef CAS PubMed.
- L. Zhou, Z. Zhang, A. Sangroniz, C. Shi, R. R. Gowda, M. Scoti, D. K. Barange, C. Lincoln, G. T. Beckham and E. Y.-X. Chen, J. Am. Chem. Soc., 2024, 146(43), 29895–29904 CrossRef CAS.
- K. Fritzsche, R. W. Lenz and R. C. Fuller, Int. J. Biol. Macromol., 1990, 12, 85–91 CrossRef CAS.
- Y. B. Kim, R. W. Lenz and R. C. Fuller, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1367–1374 CrossRef CAS.
- N. Ajellal, C. M. Thomas and J.-F. Carpentier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3177–3189 CrossRef CAS.
- M. Michalak, P. Kurcok and M. Hakkarainen, Polym. Int., 2017, 66, 617–622 CrossRef CAS.
- N. Ajellal, C. M. Thomas, T. Aubry, Y. Grohens and J.-F. Carpentier, New J. Chem., 2011, 35, 876–880 RSC.
- C. Guillaume, N. Ajellal, J.-F. Carpentier and S. M. Guillaume, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 907–917 CrossRef CAS.
- R. M. Cywar, C. Ling, R. W. Clarke, D. H. Kim, C. M. Kneucker, D. Salvachúa, B. Addison, S. A. Hesse, C. J. Takacs, S. Xu, M. U. Demirtas, S. P. Woodworth, N. A. Rorrer, C. W. Johnson, C. J. Tassone, R. D. Allen, E. Y.-X. Chen and G. T. Beckham, Sci. Adv., 2023, 9, eadi1735 CrossRef CAS.
- T. Kajiyama, T. Taguchi, H. Kobayashi, K. Kataoka and J. Tanaka, Polym. Bull., 2003, 50, 69–75 CrossRef CAS.
- C. G. Jaffredo, Y. Chapurina, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2014, 53, 2687–2691 CrossRef CAS.
- C. G. Jaffredo and S. M. Guillaume, Polym. Chem., 2014, 5, 4168–4194 RSC.
- G. Barouti and S. M. Guillaume, Polym. Chem., 2016, 7, 4603–4608 RSC.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2013, 4, 3837–3850 RSC.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2013, 46, 6765–6776 CrossRef CAS.
- G. Barouti, K. Jarnouen, S. Cammas-Marion, P. Loyer and S. M. Guillaume, Polym. Chem., 2015, 6, 5414–5429 RSC.
- M. Helou, G. Moriceau, Z. W. Huang, S. Cammas-Marion and S. M. Guillaume, Polym. Chem., 2011, 2, 840–850 RSC.
- G. Barouti, A. Khalil, C. Orione, K. Jarnouen, S. Cammas-Marion, P. Loyer and S. M. Guillaume, Chem. – Eur. J., 2016, 22, 2819–2830 CrossRef CAS PubMed.
- E. Vene, G. Barouti, K. Jarnouen, T. Gicquel, C. Rauch, C. Ribault, S. M. Guillaume, S. Cammas-Marion and P. Loyer, Int. J. Pharm., 2016, 513, 438–452 CrossRef CAS PubMed.
- H. Casajus, S. Saba, M. Vlach, E. Vène, C. Ribault, S. Tranchimand, C. Nugier-Chauvin, E. Dubreucq, P. Loyer, S. Cammas-Marion and N. Lepareur, Polymers, 2018, 10, 1244 CrossRef.
- C. G. Jaffredo, Y. Chapurina, E. Kirillov, J.-F. Carpentier and S. M. Guillaume, Chem. – Eur. J., 2016, 22, 7629–7641 CrossRef CAS.
- R. Ligny, M. M. Hänninen, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2017, 56, 10388–10393 CrossRef CAS PubMed.
- R. Ligny, M. M. Hänninen, S. M. Guillaume and J.-F. Carpentier, Chem. Commun., 2018, 54, 8024–8031 RSC.
- R. Ligny, S. M. Guillaume and J.-F. Carpentier, Chem. – Eur. J., 2019, 25, 6412–6424 CrossRef CAS PubMed.
- R. M. Shakaroun, A. Dhaini, R. Ligny, A. Alaaeddine, S. M. Guillaume and J.-F. Carpentier, Polym. Chem., 2023, 14, 720–727 RSC.
- R. M. Shakaroun, H. Li, P. Jéhan, M. Blot, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2021, 12, 4022–4034 RSC.
- A. Dhaini, R. M. Shakaroun, J. Ollivier, A. Alaaeddine, S. M. Guillaume and J.-F. Carpentier, Eur. Polym. J., 2024, 209, 112919 CrossRef CAS.
- A. Dhaini, R. M. Shakaroun, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2024, 15, 999–1014 RSC.
- M. Kamachi, S. Zhang, S. Goodwin and R. W. Lenz, Macromolecules, 2001, 34, 6889–6894 CrossRef CAS.
- Y. Doi and C. Abe, Macromolecules, 1990, 23, 3705–3707 CrossRef CAS.
- K. Hori, K. Soga and Y. Doi, Biotechnol. Lett., 1994, 16, 501–506 CrossRef CAS.
- O. Kim, R. A. Gross, W. J. Hammar and R. A. Newmark, Macromolecules, 1996, 29, 4572–4581 CrossRef CAS.
- Y. Takagi, R. Yasuda, A. Maehara and T. Yamane, Eur. Polym. J., 2004, 40, 1551–1557 CrossRef CAS.
- Y. B. Kim, R. W. Lenz and R. C. Fuller, Macromolecules, 1992, 25, 1852–1857 CrossRef CAS.
- Y. B. Kim, R. W. Lenz and R. C. Fuller, Macromolecules, 1991, 24, 5256–5260 CrossRef CAS.
- J. M. Curley, B. Hazer, R. W. Lenz and R. C. Fuller, Macromolecules, 1996, 29, 1762–1766 CrossRef CAS.
- C. Scholz, R. C. Fuller and R. W. Lenz, Macromol. Chem. Phys., 1994, 195, 1405–1421 CrossRef CAS.
- I. F. Escapa, V. Morales, V. P. Martino, E. Pollet, L. Avérous, J. L. García and M. A. Prieto, Appl. Microbiol. Biotechnol., 2011, 89, 1583–1598 CrossRef CAS.
- M.-M. Bear, M.-A. Leboucher-Durand, V. Langlois, R. W. Lenz, S. Goodwin and P. Guérin, React. Funct. Polym., 1997, 34, 65–77 CrossRef CAS.
- H. Ritter and A. G. von Spee, Macromol. Chem. Phys., 1994, 195, 1665–1672 CrossRef CAS.
- K. Kim, Y. H. Rhee, S.-H. Han, G. S. Heo and J. S. Kim, Macromolecules, 1996, 29, 3432–3435 CrossRef.
- J. J. Song and S. C. Yoon, Appl. Environ. Microbiol., 1996, 62, 536–544 CrossRef CAS PubMed.
- R. A. Gross, O.-Y. Kim, D. R. Rutherford and R. A. Newmark, Polym. Int., 1996, 39, 205–213 CrossRef CAS.
- B. Hazer and A. Steinbüchel, Appl. Microbiol. Biotechnol., 2007, 74, 1–12 CrossRef CAS PubMed.
- D. B. Hazer, E. Kılıçay and B. Hazer, Mater. Sci. Eng., C, 2012, 32, 637–647 CrossRef CAS.
- E. Olivera, M. Arcos, G. Carrasco and J. Luengo, in Plastics from Bacteria, ed. G. Q. Chen, Springer, Berlin, Heidelberg, 2010, vol. 14, pp. 133–186 Search PubMed.
- J. W. Kramer and G. W. Coates, Tetrahedron, 2008, 64, 6973–6978 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |