
Photoinduced Ullmann-type cross-coupling reactions: mechanistic insights and emerging challenges
Ahmed Th.
Abdulghaffar†
 ad,
Haolong
Zhang†
a,
Qiankun
Zhang†
a,
Qian
Tong†
a,
Ruirui
Tian†
a,
Hao
Xu
*ab,
Jiawei
Yang
*bc and
Yuanqing
Xu
*a
ad,
Haolong
Zhang†
a,
Qiankun
Zhang†
a,
Qian
Tong†
a,
Ruirui
Tian†
a,
Hao
Xu
*ab,
Jiawei
Yang
*bc and
Yuanqing
Xu
*a
aInstitute of Functional Organic Molecular Engineering, College of Chemistry and Molecular Sciences, Henan University, Kaifeng 475004, China. E-mail: xuhao@henu.edu.cn; xuyuanqing@henu.edu.cn
bKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: yangjiawei@mail.tsinghua.edu.cn
cThe Future Laboratory, Tsinghua University, Beijing 100084, China
dChemistry Department, Faculty of Science, Fayoum University, Fayoum, 63514, Egypt
First published on 20th November 2024
Abstract
Photoinduced Ullmann-type cross-coupling reactions have emerged as a significant advancement in organic synthesis, providing an efficient means to form C–C and C–heteroatom bonds under milder, light-driven conditions. Utilizing copper catalysis, these reactions present considerable benefits over traditional thermal methods by improving reaction efficiency and promoting more sustainable processes. This review evaluates recent mechanistic developments, focusing on the nonchain single-electron transfer (SET) mechanism, which is central to the success of these transformations. The discussion includes an up-to-date overview of both homogeneous and heterogeneous catalytic systems, addressing their practical applications and inherent limitations. In addition, this review identifies key challenges, such as catalyst stability, scalability, and the difficulty of activating less reactive substrates like aryl chlorides. To address these limitations, we propose future research directions aimed at overcoming these obstacles.
 Ahmed Th. Abdulghaffar | Ahmed Th. Abdulghaffar was born in Beni Suef, Egypt, in 1994. He received his Bachelor's degree in Chemistry in 2015 from the Faculty of Science at Fayoum University. Since 2016, he has worked as a faculty member at Fayoum University. In 2020, he completed his Master's degree in Organic Chemistry under the supervision of Professor Mamdouh Taha. He is currently undertaking his Ph.D. in photocatalytic organic chemistry under the supervision of Professor Xu Hao at the College of Chemistry and Molecular Sciences, Henan University. His research interests are focused on the development of photoinduced cross-coupling reactions and the investigation of their underlying mechanisms. |
 Haolong Zhang | Zhang Haolong was born in Zhengzhou, Henan, China, in 2000. He obtained his Bachelor's degree in Chemistry from Zhengzhou University of Light Industry in 2022. In the same year, he began his Master's studies in Photocatalytic Organic Chemistry at the College of Chemistry and Molecular Sciences, Henan University, under the supervision of Professor Xu Yuanqing. His research interest primarily focuses on the development of photoinduced transition-metal-catalyzed organic syntheses. |
 Qiankun Zhang | Zhang Qiankun was born in Henan, China, in 2001. He graduated with a Bachelor of Science degree in Applied Chemistry from Xuchang University in 2023. He is currently pursuing a Master's degree in Photocatalytic Materials and Chemical Engineering at the School of Chemistry and Molecular Sciences, Henan University, under the supervision of Professor Xu Hao. His research focuses on photocatalytic cross-couplings for the formation of P(O)–N bonds. |
 Hao Xu | Xu Hao was born in Henan, China, in 1985. He received his Bachelor of Science degree in Applied Chemistry from Tianjin University in 2006 and completed his Ph.D. in Organic Chemistry at Tsinghua University in 2012 under the supervision of Professor Fu Hua. Since 2012, he has been engaged in independent research and became a Lecturer in Organic Chemistry at Henan University. In 2023, he was promoted to Professor at Henan University. His research interests are primarily focused on photoinduced organic transformations and the development of synthetic processes for fine chemicals. |
 Jiawei Yang | Jiawei Yang was born in Hebei, China, in 1985. He obtained his Bachelor's degree from the School of Chemical & Environmental Engineering at China University of Mining & Technology-Beijing in 2005. He earned his Ph.D. in Physical Chemistry from Tsinghua University in 2019 under the supervision of Professor Bo-Qing Xu. Since 2019, Dr Yang has been working as a postdoctoral researcher at The Future Laboratory, Tsinghua University, and became a research assistant professor in 2023. His research interests include heterogeneous catalysis and the synthesis of inorganic and organic–inorganic hybrid materials for gas sensors. |
 Yuanqing Xu | Xu Yuanqing was born in Xinyang, Henan, China, in 1976. He received his Bachelor of Science degree from the Beijing Institute of Technology in 1997 and his Ph.D. in Materials Science from Soochow University in 2006 under the supervision of Professor Lu Jianmei. He completed a one-year postdoctoral fellowship at Laurentian University and became a professor of organic chemistry at Henan University in 2015. His research focuses on the preparation of photofunctional materials and their application in photoinduced organic transformations. |
1 Introduction
In recent decades, transition metal-catalyzed cross-coupling reactions have revolutionized organic synthesis by enabling efficient and selective bond formation. Among these, the Ullmann coupling reaction stands out for its versatility as a copper-catalyzed method for forming C–C and C–heteroatom bonds such as C–N, C–O, and C–S. First described by Fritz Ullmann in 1901, this reaction involves the self-coupling of aryl bromides using stoichiometric amounts of copper powder, producing biphenyl compounds (Scheme 1a).1 This foundational discovery laid the groundwork for Ullmann-type reactions, which have since been expanded by Ullmann,2,3 Goldberg,4 and Hurtley,5 to include the formation of various C–N, C–O, and C–C bonds (Scheme 1b–e).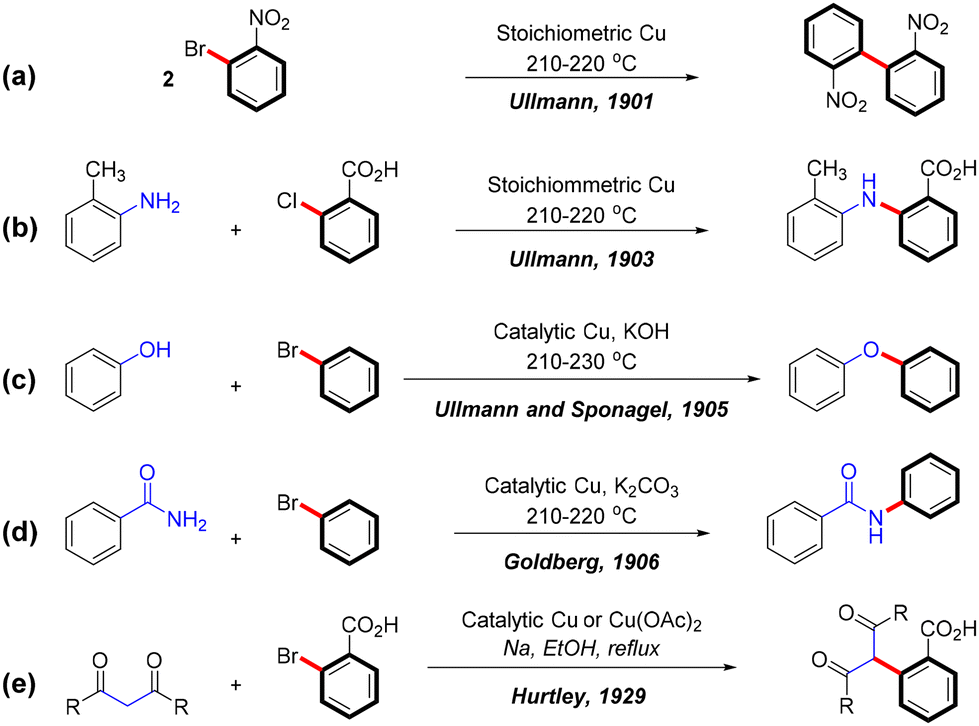 | ||
| Scheme 1 Ullmann-type coupling reactions. | ||
Copper catalysts are highly valued in green chemistry for their low toxicity, affordability, abundance, and redox activity,6 as well as their compatibility with aqueous conditions.7 These attributes have established Ullmann cross-coupling reactions as a cornerstone in the synthesis of essential compounds in pharmaceuticals,8,9 natural products,9 agrochemicals,10–12 and functional materials.13 However, the high electrophilicity of copper, while advantageous in certain reactions, often necessitates elevated temperatures or specialized ligands in C–C and C–heteroatom cross-couplings due to significant energy barriers. This limitation is particularly evident in the ineffectiveness of copper catalysts with aryl chlorides, where the high bond dissociation energy of the carbon–chlorine bond presents a significant challenge. In contrast, palladium catalysts are more effective in facilitating cross-coupling reactions with aryl chlorides, as demonstrated by the Buchwald–Hartwig reaction,14,15 where aryl chlorides react smoothly with amines. Despite their effectiveness, palladium catalysts pose sustainability challenges, including high costs, toxicity, and difficulties in recycling use.16 Consequently, conventional Ullmann couplings often require rigorous conditions, such as high temperatures, strong bases, and stoichiometric amounts of copper catalysts, resulting in a limited substrate scope and low reaction yields.1–3,17 These harsh conditions restrict the broader application of these reactions and highlight the need for more sustainable alternatives.
Recent advancements in photoinduced copper-catalyzed methodologies offer promising alternatives to conventional Ullmann coupling reactions, directly addressing the limitations of traditional methods.18 These innovative processes harness light as a sustainable energy source to form C–C and C–heteroatom (C–X) bonds under milder conditions, enhancing efficiency while advancing greener synthetic techniques.19 The introduction of photoredox catalysis has further revolutionized organic synthesis by enabling single-electron transfer (SET) reactions,20–22 which not only improves bond formation efficiency but also significantly reduces the environmental and economic impacts associated with traditional thermal methods. Additionally, photoinduced techniques that bypass the demanding oxidative addition step in copper-catalyzed reactions simplify the bond formation process via the SET mechanism, broadening the scope of cross-coupling reactions and increasing yields.23
This review provides an in-depth overview of the latest advancements in photoinduced copper-catalyzed Ullmann-type C–C and C–X cross-coupling reactions. By summarizing key studies and examining both the achievements and limitations, we aim to emphasize the potential of these methods to advance sustainable chemistry and contribute to the ongoing development of more efficient and environmentally friendly synthetic processes.
2 Mechanism evolution in photoinduced Ullmann cross-couplings
The evolution of Ullmann cross-coupling reaction has been marked by significant advancements in reaction conditions that increasingly align with green chemistry principles. The first generation of this reaction utilized a stoichiometric amount of copper as a homogeneous catalyst at elevated temperatures.12 The most widely accepted mechanism under thermal conditions involved the oxidative addition of aryl halide to the CuI complex, resulting in the formation of a CuIII intermediate,24–27 followed by reductive elimination to form the coupled product and regeneration of CuI catalyst. The creation of photoinduced Ullmann mechanism is based on the oxidative addition/reductive elimination (OA/RE) pathway. In this mechanism, the copper salt catalyst interacts with a nucleophile in the presence of a base to generate the copper-nucleophile complex B. Upon exposure to light, this complex transitions into the excited state complex C. Following this, an aryl halide undergoes oxidative addition, resulting in the formation of the CuIII intermediate D. The subsequent reductive elimination yields the desired coupling product while concurrently regenerating the CuI catalyst (Scheme 2I).28 During the oxidative addition step, this high-valent CuIII complex is inherently unstable compared to its CuI and CuII counterparts. As a result, the CuIII intermediate, being in a higher energy state, is prone to reduction or decomposition. This instability can limit the effectiveness of the OA/RE mechanism under certain conditions. In the absence of stabilizing factors such as appropriate ligands or solvents, the CuIII intermediate may decompose or undergo side reactions before reductive elimination can occur, making the process less efficient.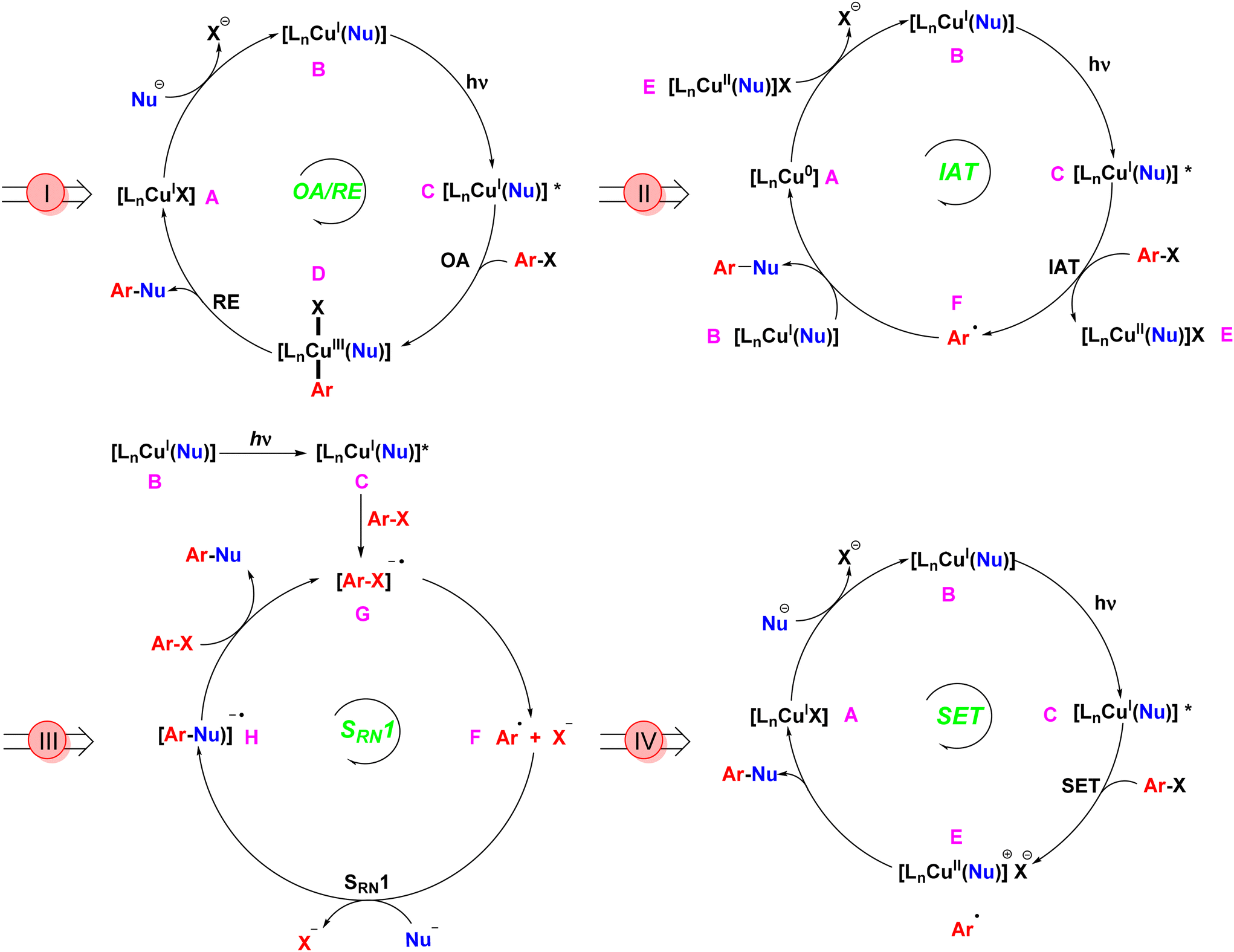 | ||
| Scheme 2 Plausible mechanisms for photoinduced Ullmann cross-couplings. | ||
Bunnett and Wamser later introduced an alternative iodine atom transfer (IAT) mechanism.29 This mechanism demonstrated the radical abstraction of an iodine atom from aryl iodides during the decomposition of bis(p-chloro-benzoyl) peroxide. This breakthrough paved the way for further innovations in Ullmann reactions, but it couldn't be applied to aryl bromides. In 2010, Jones conducted a computational investigation of Ullmann-type reaction mechanisms, including the IAT mechanism.30 In this mechanism (Scheme 2II), a copper nucleophile complex B is excited by light to form an excited state C. This triggers the transfer of a halide atom from the aryl halide, producing a copper(II) nucleophile halide E and an aryl radical F. Unlike the OA/RE mechanism, which operates through a CuIII intermediate, the IAT mechanism involves a more stable CuII intermediate. The cross-coupled product forms when the copper nucleophile complex B couples with the aryl radical F, regenerating the copper catalyst Avia electron transfer from the CuII intermediate E. Because CuII reacts with aryl radicals faster than CuI,31 the CuI/CuII redox cycle efficiently facilitates iodine transfer and radical formation, making this pathway energetically feasible under photoinduced conditions. Moreover, the CuII intermediate typically exists in a lower energy state than CuIII,32 resulting in milder reaction conditions and greater substrate tolerance.
Kornblum33 and Russell34 introduced an electron transfer radical mechanism for aliphatic nucleophilic substitution via a radical anion intermediate. Bunnett later extended this concept to aromatic substitution, which is termed as a radical chain mechanism of nucleophilic substitution (SRN1).35 This mechanism, not involving transition metal catalysis, provided vital insights that facilitated future advancements. Arai et al. later introduced a copper catalyst (CuBr) and demonstrated the formation of the radical anion intermediate through EPR experiments.36 The Cu-catalyzed SRN1 Ullmann coupling mechanism, as described by Kochi,37 involves initiation, propagation, and termination steps. The process begins with photo-stimulated electron transfer from the CuI complex to the aryl halide (Ar–X), generating a radical anion intermediate, which then forms an aryl radical. The aryl radical interacts with a nucleophile to form a new radical anion, which ultimately leads to the coupling product and regeneration of the Cu catalyst.38 The SRN1 mechanism, depicted in Scheme 2III, involves a copper(I) nucleophile complex B that, upon light irradiation, reaches an excited state C. This complex transfers an electron to an aryl halide, forming an aryl halide radical anion G, which subsequently generates an aryl radical F. The aryl radical couples with a nucleophile to produce the aryl nucleophile radical anion H. Another aryl halide is then introduced to yield the final coupled product and another aryl halide radical anion G.28 A distinguishing feature of the SRN1 mechanism is its chain propagation process, whereby electron transfer and radical formation can be repeated, significantly increasing the overall reaction efficiency. Once the aryl radical is formed, it reacts with another substrate molecule, continuing the electron transfer cycle. This chain propagation enables the reaction to proceed with minimal catalyst loading, which is a major advantage of the SRN1 mechanism compared to others. Furthermore, the chain process reduces the need for continuous energy input, lowering the overall energy consumption. The propagation feature also allows the reaction to proceed at lower catalyst concentrations, making the process more economical and environmentally friendly. This mechanism also broadens the substrate scope, enabling reactions with less reactive or sterically hindered aryl halides. Additionally, the absence of high-energy intermediates like CuIII ensures that the reaction can occur under milder conditions, reducing the overall energy barrier and enhancing sustainability.
In 2012, G. C. Fu expanded the scope of photoinduced Ullmann cross-couplings through a single-electron transfer (SET) mechanism (Scheme 2IV).18 In this process, a copper-nucleophile complex B is formed in the presence of a base. Upon exposure to light, the complex transitions to an excited state C, allowing SET with the aryl halide and generating a copper(II) complex and an aryl radical E. The aryl radical then couples with the nucleophile, forming the desired product and regenerating the copper catalyst in its initial CuI state A, thereby continuing the catalytic cycle. This mechanism showcases the advantages of photocatalysis, including lower energy requirements and improved efficiency compared to traditional thermal methods. Furthermore, the CuII intermediate in the SET mechanism is relatively stable, much like in the IAT pathway, but with a key difference: the SET mechanism can bypass the need for oxidative addition entirely. The energy profile of SET mechanisms is characterized by lower activation energies, as they avoid high-energy CuIII intermediates, making SET particularly favourable for green chemistry applications, especially in the formation of C–N and C–O bonds under mild conditions. Additionally, SET offers high reaction efficiency, even with sterically hindered or less reactive substrates.
In conjunction with the aforementioned reaction mechanisms, understanding the reactivity order of aryl halides is crucial for optimizing catalytic processes. This reactivity is determined by carbon-halide bond energies, following the order: C–F < C–Cl < C–Br < C–I. While iodobenzenes exhibit the highest reactivity due to their lower bond dissociation energy, the importance of employing aryl chlorides as substrates in the Ullmann reaction is increasingly recognized. Aryl chlorides are more cost-effective and widely available compared to aryl iodides, but their higher bond dissociation energy presents challenges under copper-catalyzed conditions. Therefore, developing catalytic systems that can efficiently utilize aryl chlorides is of great significance. The use of heterogeneous catalysts offers a promising approach to achieving this goal. By providing stronger activation capabilities and greater stability, heterogeneous catalysts may enable the effective use of aryl chlorides in copper-catalyzed reactions, all while adhering to the principles of green chemistry.39,40
3 Photoinduced Cu-catalyzed Ullmann-type reactions
Photoinduced Cu-catalyzed Ullmann-type reactions have emerged as a powerful tool in organic synthesis, offering a sustainable and efficient alternative to conventional thermal methods. By leveraging light energy, these reactions facilitate the formation of a wide array of carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds under milder conditions. This section delves into the various types of photoinduced Ullmann reactions, exploring their applications in the synthesis of C–C, C–N, C–O, and C–S bonds. Each type of reaction demonstrates the versatility and effectiveness of photoinduced copper catalysis in expanding the scope of cross-coupling chemistry.3.1 Photoinduced C–C cross-couplings
Copper-catalyzed Ullmann-type C–C bond cross-couplings via C-arylation of CH-acids with aryl iodides, first discovered nearly a century ago, have undergone substantial development and are now extensively employed in the synthesis of natural products and pharmaceutical compounds.41 This approach has proven particularly effective in the synthesis of commercially significant drugs or their intermediates, including the nonsteroidal anti-inflammatory Flurbiprofen42 and the intermediate of the anti-inflammatory biomolecular SEGRA (Scheme 3).43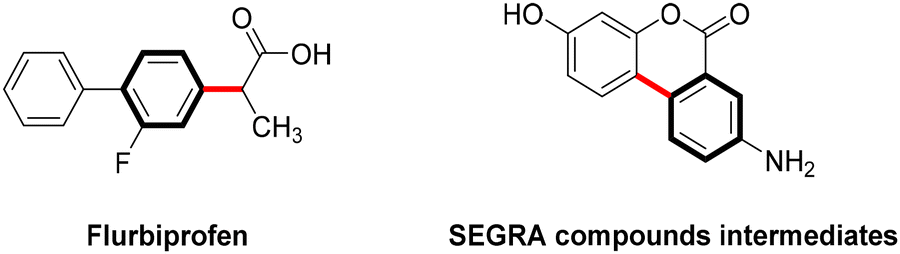 | ||
| Scheme 3 Synthesis of real drug molecules via Ullmann C-arylation. | ||
Nevertheless, during the single-electron transfer process in Ullmann C–C coupling reactions, carbon nucleophiles often encounter difficulties in losing electrons under mild conditions due to the high energy state of the carbon radical, which leads to a significant energy barrier.44 Consequently, these photoreactions are challenging to execute and are currently infrequently reported.
 | ||
| Scheme 4 Photoinduced CuI-catalyzed cross-coupling of aryl iodides with azoles. | ||
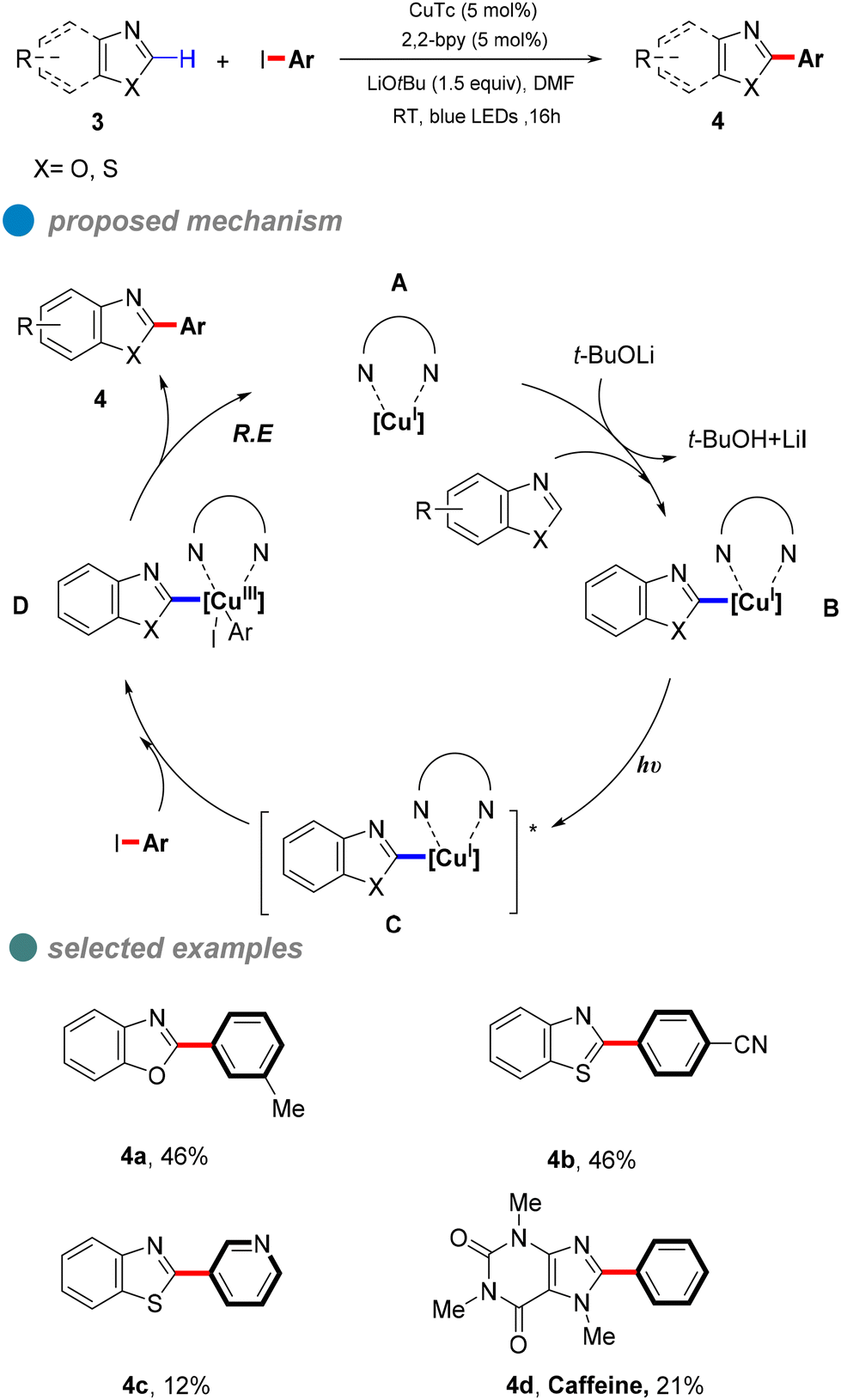
In 2020, Ma et al. proposed a visible-light-induced copper-catalyzed C–C cross-coupling of aryl halides with benzoxazoles (Scheme 5).46 Compared to traditional CuI salt catalysis, this reaction proceeds at room temperature without heating, utilizing blue light to minimize energy input. While the method demonstrates broad substrate compatibility, its overall yield is limited by the high electron-loss barrier of carbon nucleophiles. UV-visible absorption and fluorescence quenching experiments provided mechanistic insights, suggesting that a ligand forms complex A with copper(I) thiophene-2-carboxylate (CuTc). In the presence of a base and benzoxazole, this complex generates LnCu(I)-benzoxazole B, which, upon excitation, forms an excited-state species [LnCu(I)-benzoxazole]* C. The excited complex undergoes a double electron transfer process, oxidizing CuI to CuIII. Subsequent oxidative addition with the aryl halide yields intermediate D, followed by reductive elimination to afford the desired product 4.
 | ||
| Scheme 5 Photoinduced CuTc-catalyzed C–C cross-coupling with azoles. | ||
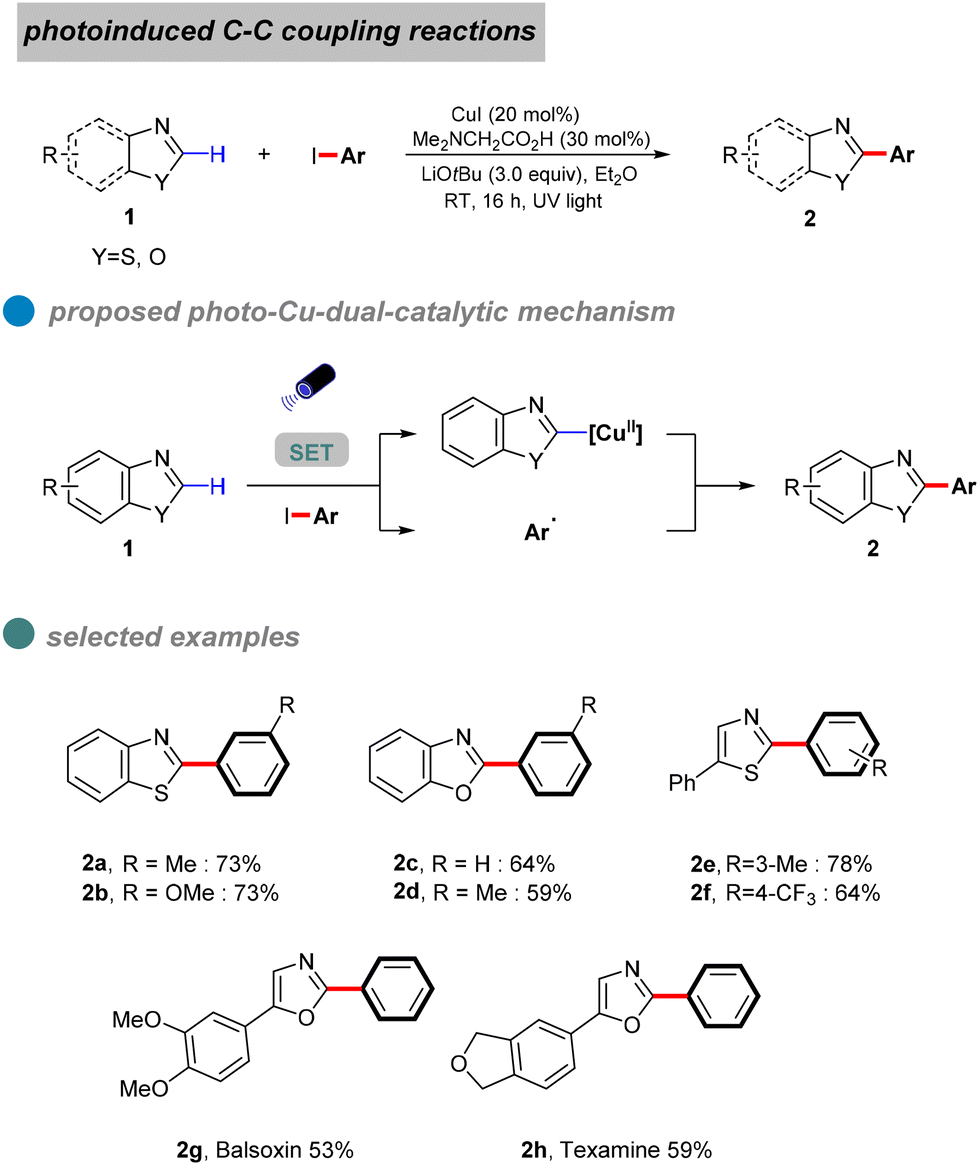
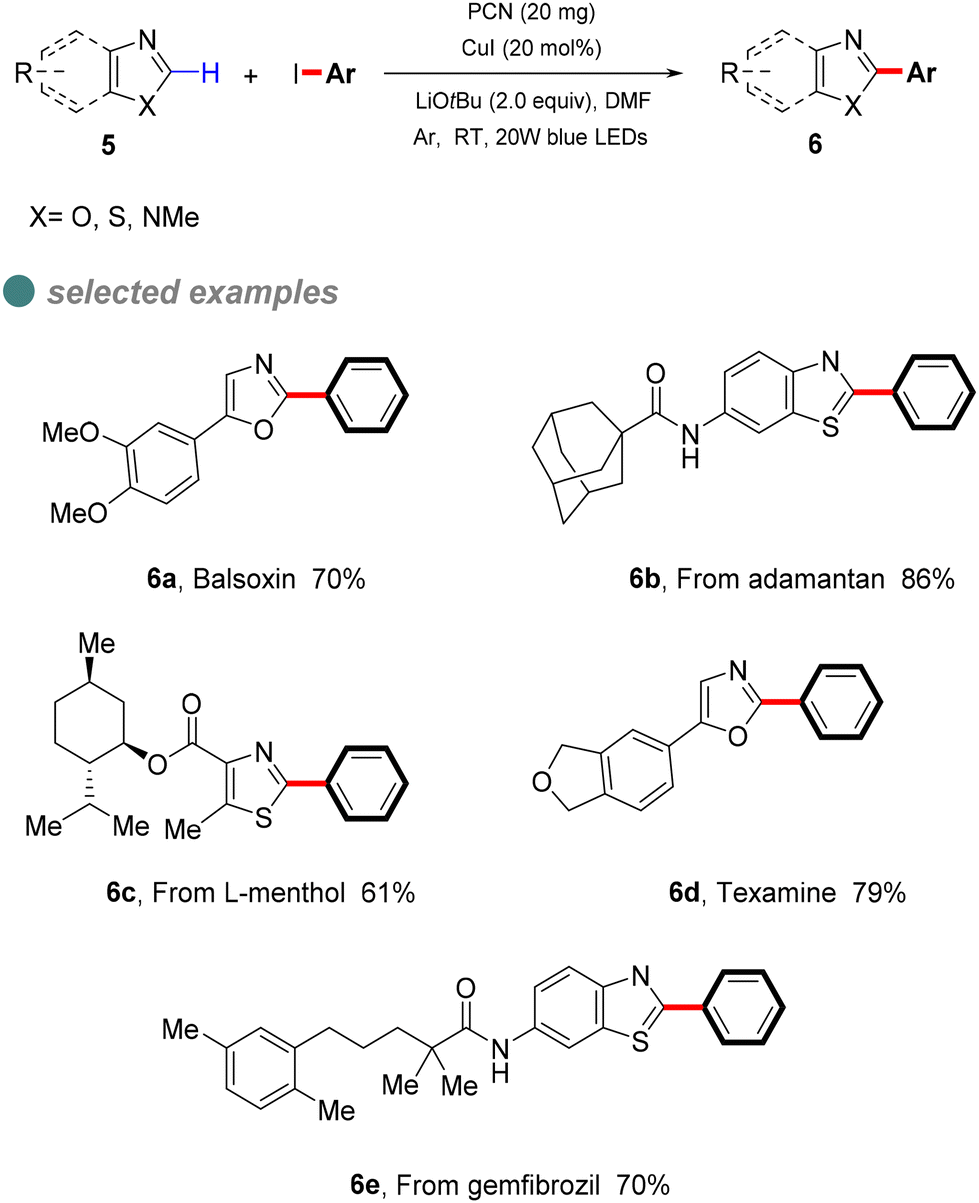
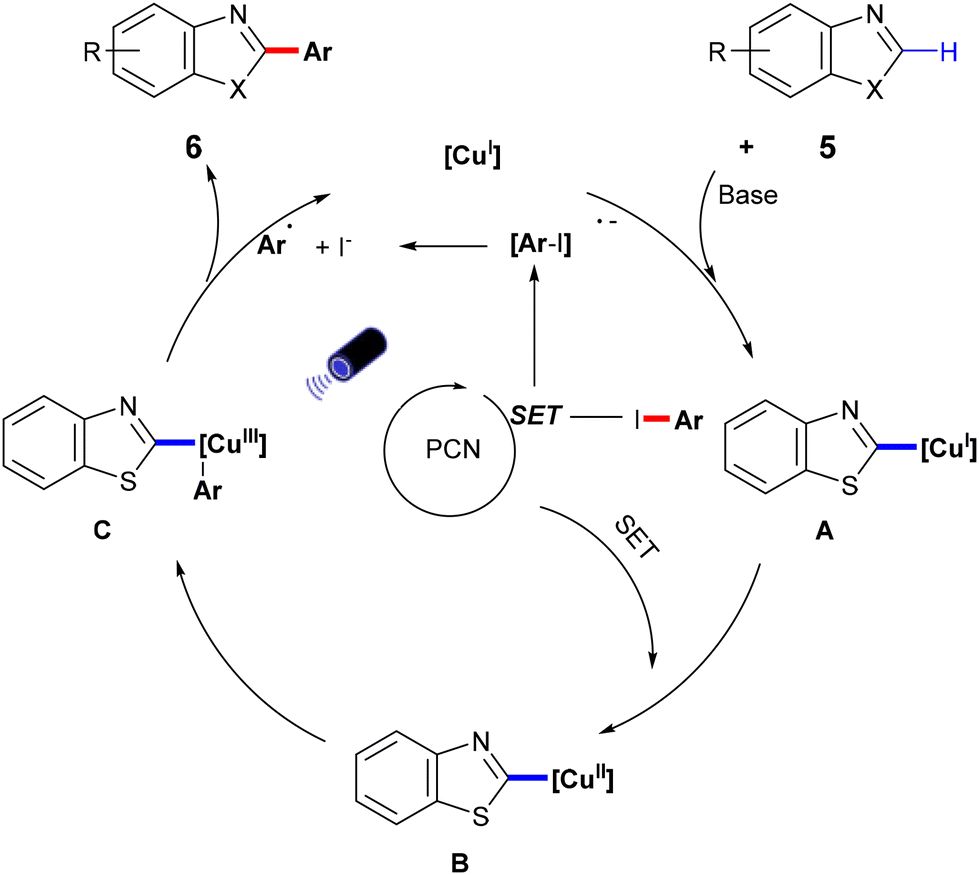
In 2022, Zhang et al. developed a semi-heterogeneous catalytic system utilizing polymeric carbon nitride (PCN) for photoinitiated copper-catalyzed C–C bond cross-coupling reactions (Scheme 6).49 This CuI/PCN dual catalytic system not only initiates reactions under mild conditions but also exhibits superior photocatalytic activity compared to traditional transition metal complexes. The heterogeneous catalyst PCN displays remarkable photochemical stability, generating and rapidly separating charges under illumination, which provides a surface redox center. Subsequently, aryl iodides undergo single-electron transfer to form corresponding anionic radicals, which are then disproportionate to produce aryl radicals. The abundant active sites on the PCN surface accelerate the single-electron transfer process, particularly the conversion of the CuI complex A to the CuII complex B. The CuII complex B then captures the phenyl radical to form the unstable CuIII complex C, which is eliminated by reduction to obtain the final product 6 (Scheme 7).
 | ||
| Scheme 6 Photo-Cu-dual-catalytic cross-coupling with azoles. | ||
 | ||
| Scheme 7 The proposed photo-Cu-dual-catalytic mechanism. | ||
In this section, the coupling of aromatic azoles and non-aromatic oxazolines, acting as CH-acids, with aryl iodides to form C–C bonds via a photoinduced single-electron transfer (SET) mechanism is discussed. In Ullmann C–C coupling reactions, carbon nucleophiles often encounter difficulties in losing electrons under mild conditions due to significant energy barriers. However, the interaction between copper catalysts and the heteroatoms in azoles enhances the efficiency of the SET process, facilitating electron transfer and promoting effective C–C bond formation.
3.2 Photoinduced C–N cross-couplings
Nitrogen-containing compounds are vital in pharmaceuticals, agriculture, and functional materials.50,51 For example, compounds such as carbamazepine, nefiracetam, and propanil (Scheme 8) have been synthesized via Ullmann N-arylation under thermal conditions,52,53 underscoring the demand for efficient and direct synthetic methods. Recently, photoinduced copper-catalyzed Ullmann-type C–N couplings have emerged as promising alternatives, enabling the synthesis of target compounds under mild and environmentally benign conditions and offering significant advantages over traditional thermal methods. This section provides an overview of key advancements in photoinduced copper-catalyzed Ullmann C–N coupling reactions, emphasizing both homogeneous and heterogeneous catalysts.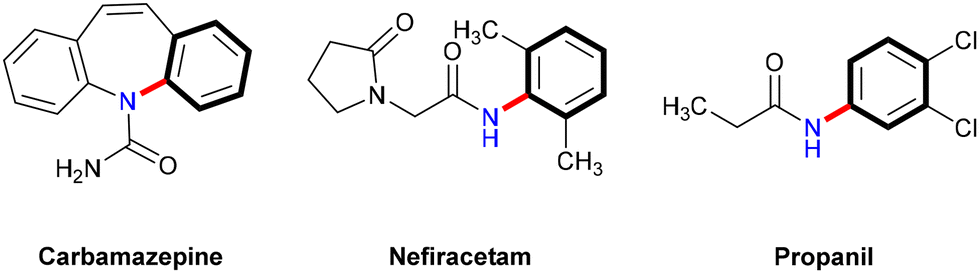 | ||
| Scheme 8 Pharmaceutical application of Ullmann N-arylation. | ||
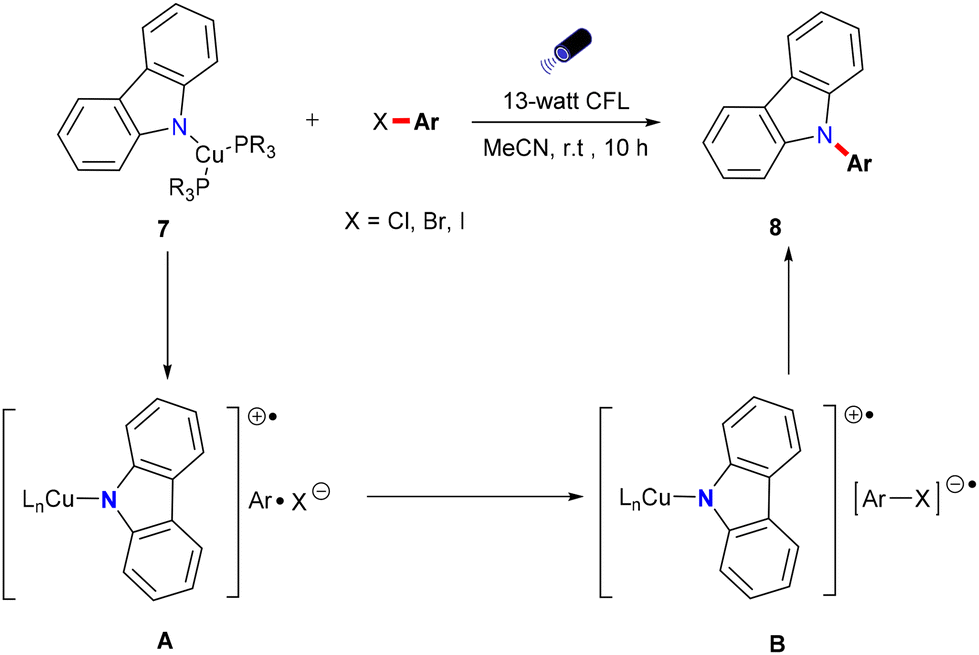 | ||
| Scheme 9 Photoinduced C–N cross-coupling with carbazolide. | ||
The core innovation of this work lies in the proposal of a novel photoinduced single-electron transfer mechanism. Unlike the copper-catalyzed SRN1 mechanism proposed by Kochi31 and Arai,36 where single-electron transfer exists as part of a chain reaction, the SET mechanism proposed by the Fu group is a non-chain, photoinduced single-electron transfer process. Upon irradiation, the copper-carbazolide complex forms a C(sp2)–N bond by absorbing light, which excites an electron that undergoes SET to the aryl halide, creating a radical anion. This radical anion dissociates into an aryl radical and a halide anion, with the aryl radical subsequently reacting with the copper complex to yield the C–N bond. Controlled experiments confirm that C(sp2)–N bond formation occurs via the SET process, highlighting the emergence of this non-chain, photoinduced SET mechanism in photocatalyzed Ullmann C–N coupling. However, this approach has a limited substrate scope, primarily involving carbazole salts with strong basicity.
In 2013, Fu and Peters expanded their methodology for photoinduced Ullmann C–N couplings at room temperature, successfully applying it to a variety of nucleophiles 9, including indoles, benzimidazoles, imidazoles, and carbazoles (Scheme 10), thereby broadening the scope of copper-catalyzed reactions for various nitrogen heterocycles in synthetic applications.54 The reaction employed specific light wavelengths (254 nm) to facilitate the process. This wavelength selection is due to the high bond energy of the C–I bond in aryl iodides and the limited interaction of organic molecules with visible light wavelengths. Notably, this reaction does not require ligands and can directly utilize carbazole instead of carbazole salts, resulting in high yields. Additionally, the study demonstrated the method's effectiveness in accommodating hindered, deactivated, and heterocyclic aryl iodides, as well as an aryl bromide and an activated aryl chloride.
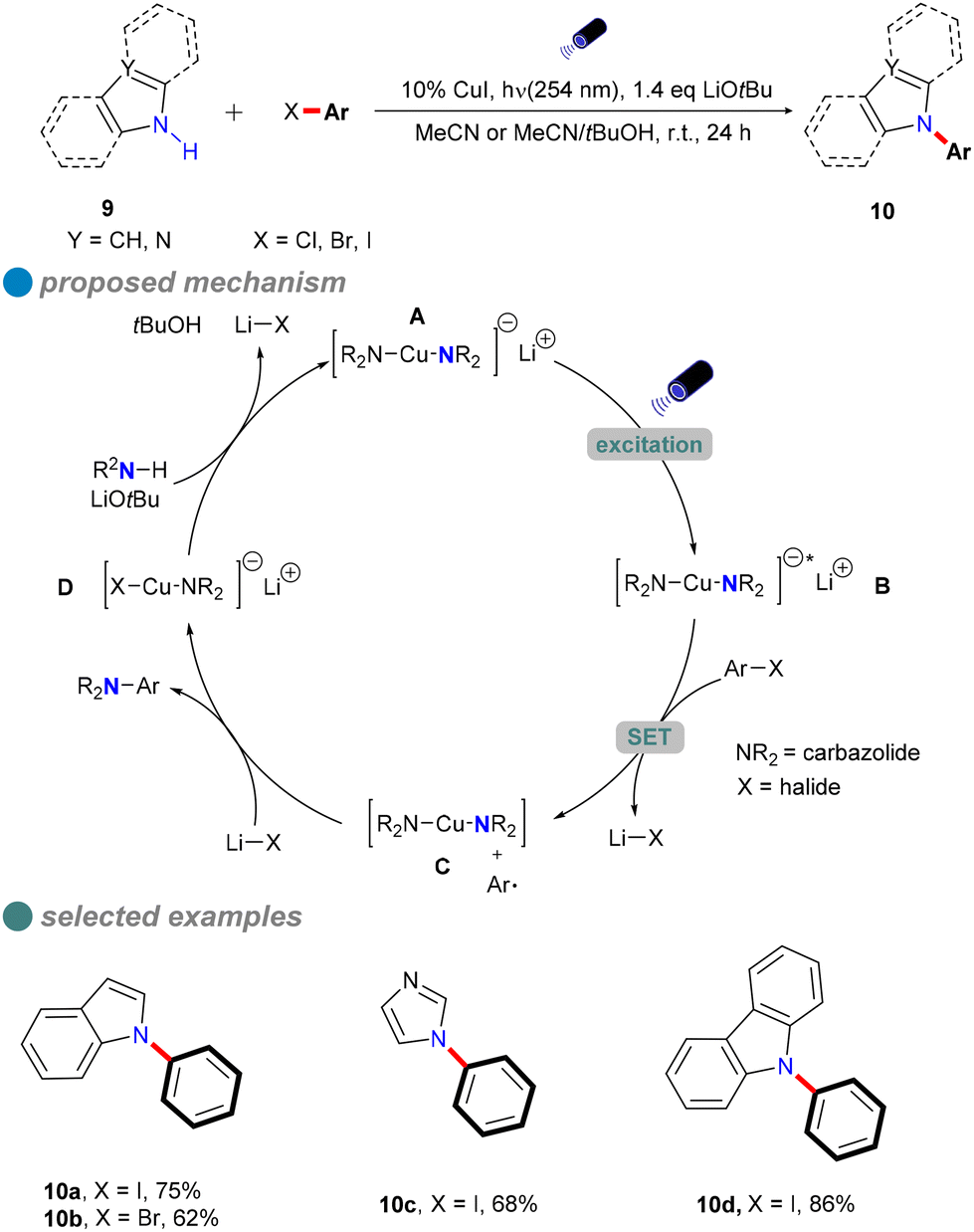 | ||
| Scheme 10 Photoinduced C–N cross-coupling with aromatic N-nucleophiles. | ||
The author postulated that these reactions may proceed through the initial photoexcitation of a copper-carbazolide complex A. After photoexcitation, the excited-state copper-carbazolide complex B undergoes a single-electron transfer with an aryl halide, generating an aryl radical and a copper-carbazolide complex C. Subsequently, the copper-carbazolide complex C undergoes ligand exchange with Li–X and then reacts with the aryl radical to form the C–N coupling product and a halide-copper complex D, completing the catalytic cycle.
In 2015, Kobayashi and colleagues developed a visible-light-induced Ullmann-type C–N coupling reaction using carbazole derivatives 11 and aryl iodides (Scheme 11).55 To overcome the limitations of using UV light in the N-arylation of carbazoles, they employed Ir(ppy)3 as a photosensitizer alongside a CuI catalyst. The iridium-based photosensitizer was crucial for energy transfer, allowing the reaction to utilize visible light instead of UV light. As shown in Scheme 11, Ir(ppy)3 undergoes metal-to-ligand charge transfer (MLCT) to generate the excited state complex Ir(ppy)3* upon visible light irradiation. The excited Ir(ppy)3* photosensitizer is then quenched by the copper(I) amide complex B, resulting in the formation of excited carbazole copper C and ground-state Ir(ppy)3. Notably, Ir(ppy)3 does not replace CuI as it cannot directly participate in inner-sphere bond-forming processes. However, this method was only tested for the coupling of iodobenzene with carbazoles, and its reliance on costly iridium-based photocatalysts limited its scalability and cost-effectiveness.
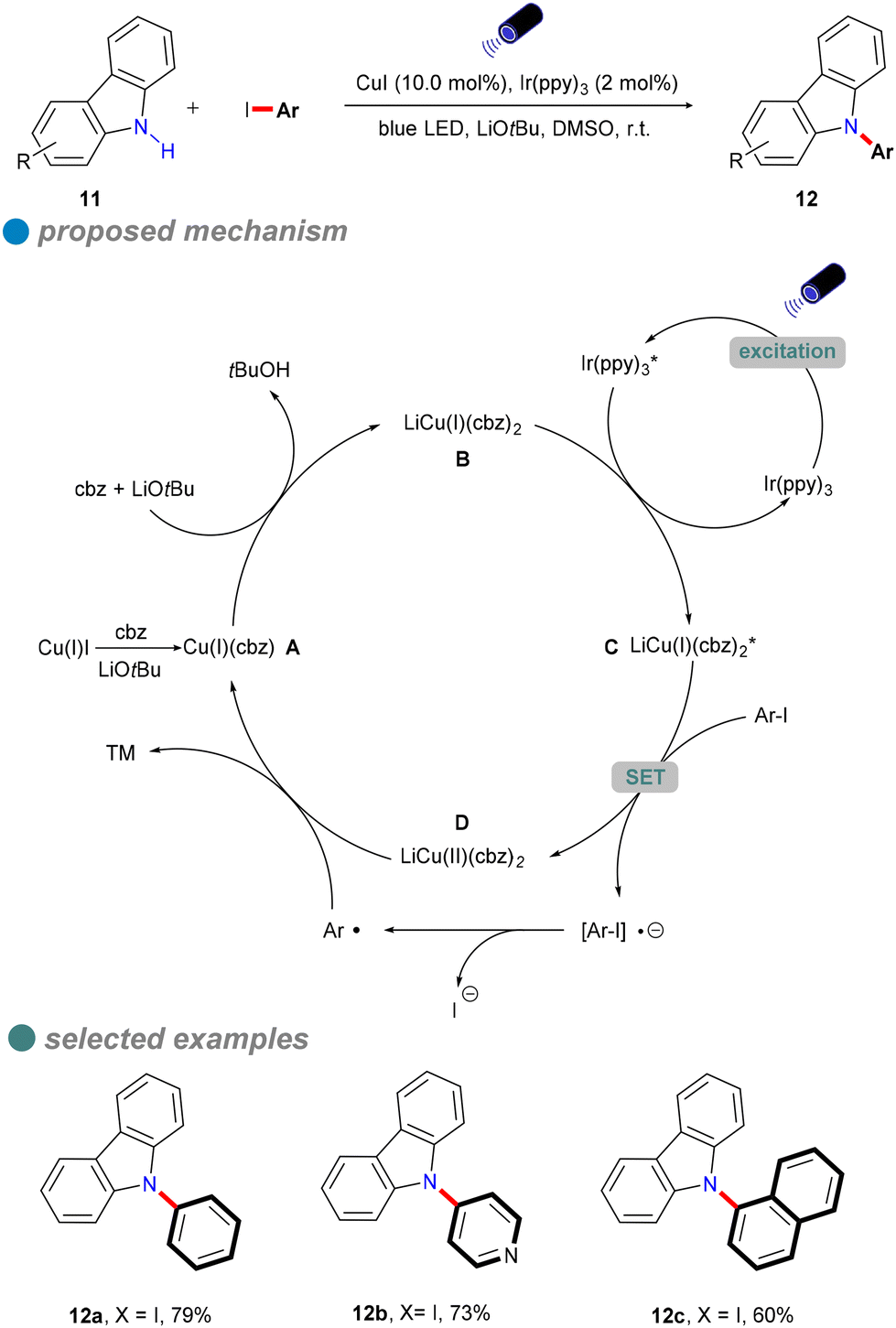 | ||
| Scheme 11 Visible-light-induced C–N cross-coupling with carbazoles. | ||
In 2023, Nagula Shankaraiah's research group introduced a sustainable methodology for the construction of unsymmetrical diarylamines 14 or 15 through a photoredox-mediated dual catalytic system that incorporates copper and ruthenium (Scheme 12).56 This innovative approach entails a sequential two-step reaction. The initial step involves a photoinduced Cu-catalyzed N-arylation of ammonia 13 with aryl halides (Ullmann-type reaction), facilitated by a ruthenium complex photocatalyst, which serves as a cost-effective alternative to iridium photocatalysts. The second step comprises a Chan–Lam C–N cross-coupling reaction employing aryl boronic acids, as depicted in Scheme 12. Additionally, this methodology has been successfully applied in the synthesis of imatinib drugs.
 | ||
| Scheme 12 Visible-light-induced sequential N-arylation for unsymmetrical diarylamines and imatinib drug. | ||
The authors performed several controlled experiments that underscored the importance of photoinduced conditions, the copper catalyst, and the presence of an oxygen atmosphere. Based on these experiments, the proposed mechanism is as follows: an electron is transferred from the Ru photocatalyst to the Cu(II) catalyst, generating Cu(I). This is followed by the oxidative addition of the aryl halide, resulting in the formation of the Cu(III) complex A. Subsequently, a ligand substitution with ammonia leads to the elimination of hydrogen halide in the presence of DIPEA, yielding the intermediate C. Thereafter, the introduction of aryl boronic acid results in the formation of intermediate D, ultimately leading to the target product along with Cu(I), which is oxidized to Cu(II) in the presence of O2.
In 2018, Vandana Bhalla's research group synthesized ultra-fine Cu2O–Fe2O3 hybrid nanoparticles using supramolecular assemblies of hexaphenylbenzene (HPB) derivatives as nanoreactors and stabilizers (Scheme 13).57 The group reacted HPB derivative 18 with 4-(2-aminoethyl) morpholine 19 in a mixed solution of dichloromethane and ethanol to obtain HPB derivative 20. Then, gradually add aqueous solutions of FeCl3 and CuCl2 to the supramolecular assembly solution of HPB derivative 20. Using the supramolecular assemblies of HPB derivative 20 as nanoreactors and stabilizers, ultrafine Cu2O–Fe2O3 nanoparticles are formed. These nanoparticles served as efficient heterogeneous photocatalysts for C–N formation between aryl iodides and a range of nucleophiles 16, including primary and secondary amines, under visible light irradiation. The reaction exhibited minimal sensitivity to water, utilizing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/EtOH solvent mixture. The catalyst's narrow band gap enabled effective absorption of visible light, thereby eliminating the need for costly iridium-based photosensitizers.55 This innovation effectively addressed limitations of previous methods, such as the requirement for an inert atmosphere, additional reagents, strong alkaline conditions, and organic solvents. The catalyst was straightforward to prepare and demonstrated significant biological relevance. Furthermore, a ‘dip strip’ created by coating a filter paper with the supramolecular ensemble functioned as a reusable hetero-catalytic system for C–N bond formation reactions, effective for up to three cycles. Although the catalyst exhibited high photo-utilization efficiency and reusability, its effectiveness was notably reduced with bromobenzenes and chlorobenzenes.
:1 H2O/EtOH solvent mixture. The catalyst's narrow band gap enabled effective absorption of visible light, thereby eliminating the need for costly iridium-based photosensitizers.55 This innovation effectively addressed limitations of previous methods, such as the requirement for an inert atmosphere, additional reagents, strong alkaline conditions, and organic solvents. The catalyst was straightforward to prepare and demonstrated significant biological relevance. Furthermore, a ‘dip strip’ created by coating a filter paper with the supramolecular ensemble functioned as a reusable hetero-catalytic system for C–N bond formation reactions, effective for up to three cycles. Although the catalyst exhibited high photo-utilization efficiency and reusability, its effectiveness was notably reduced with bromobenzenes and chlorobenzenes.
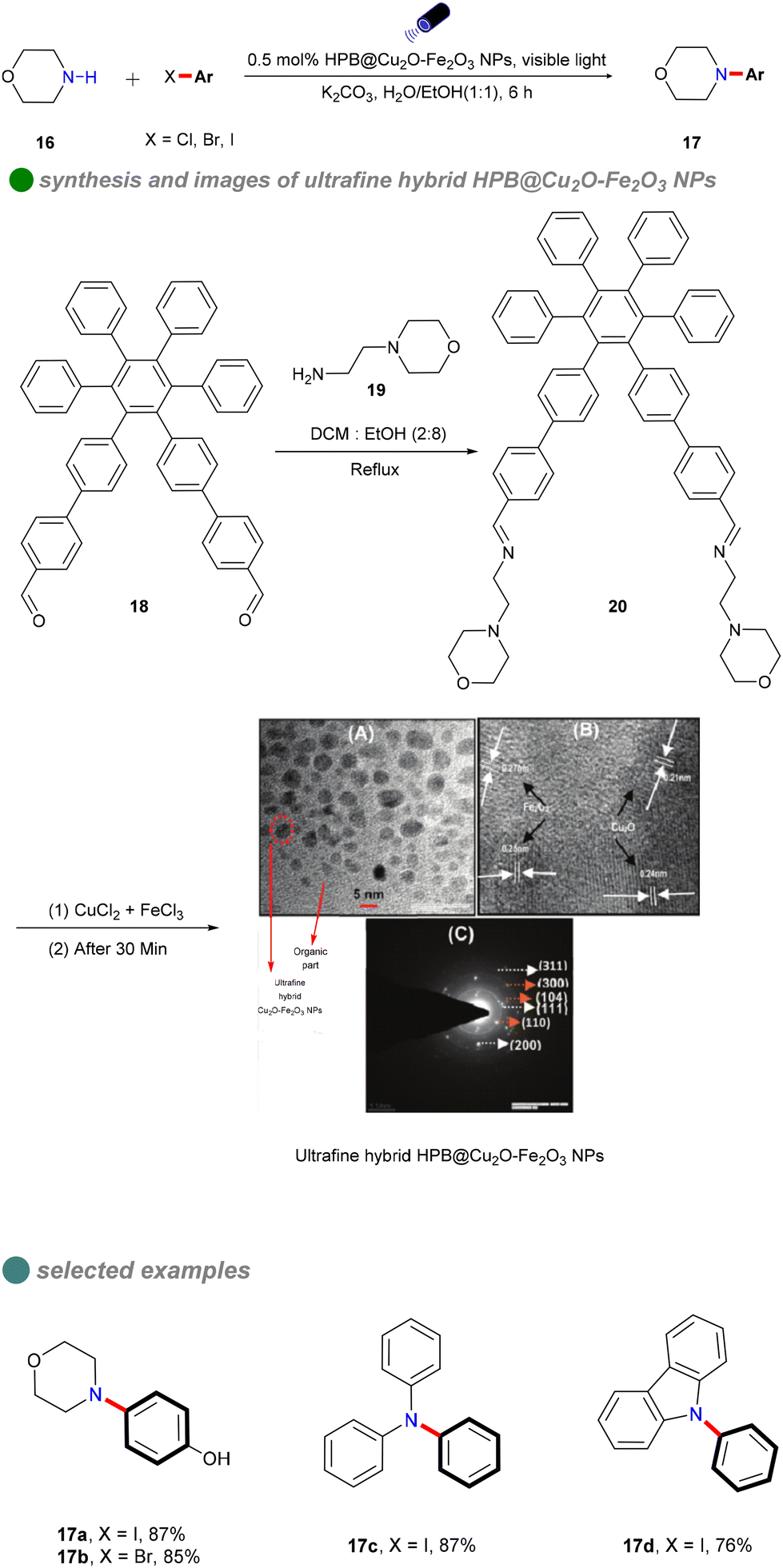 | ||
| Scheme 13 Visible-light-induced N-arylation catalyzed by HPB@Cu2O-Fe2O3 NPs, including (A) TEM image (B) SAED, and (C) HR-TEM of HPB@Cu2O-Fe2O3 NPs. | ||
In 2018, Li's group introduced a novel method for synthesizing composition-tunable (111)-faceted Cu/Cu2O nanoparticles, which exhibit significant photocatalytic activity in ligand-free and solvent-free C–N Ullmann coupling reactions (Scheme 14).58 By varying the amount of glucose, the researchers were able to control the evolution of nanoparticle shapes, including octahedra, tetrahexahedra, and stars. This method addresses the high temperatures and energy demands typically associated with conventional Ullmann reactions, while also promoting environmentally sustainable chemical synthesis. The nanoparticles’ low toxicity, appropriate band gap, and potential for reusability further enhance their appeal. Cu/Cu2O nanoparticles efficiently catalyzed reactions with both aliphatic and cyclic amines 21, achieving high yields without the need for ligands or solvents. However, the substrate scope for aryl halides was limited to o-nitroiodobenzene and o-nitrobromobenzene. The reaction mechanism follows a single-electron transfer (SET) process, involving an electron transfer between the aryl halides and amines.
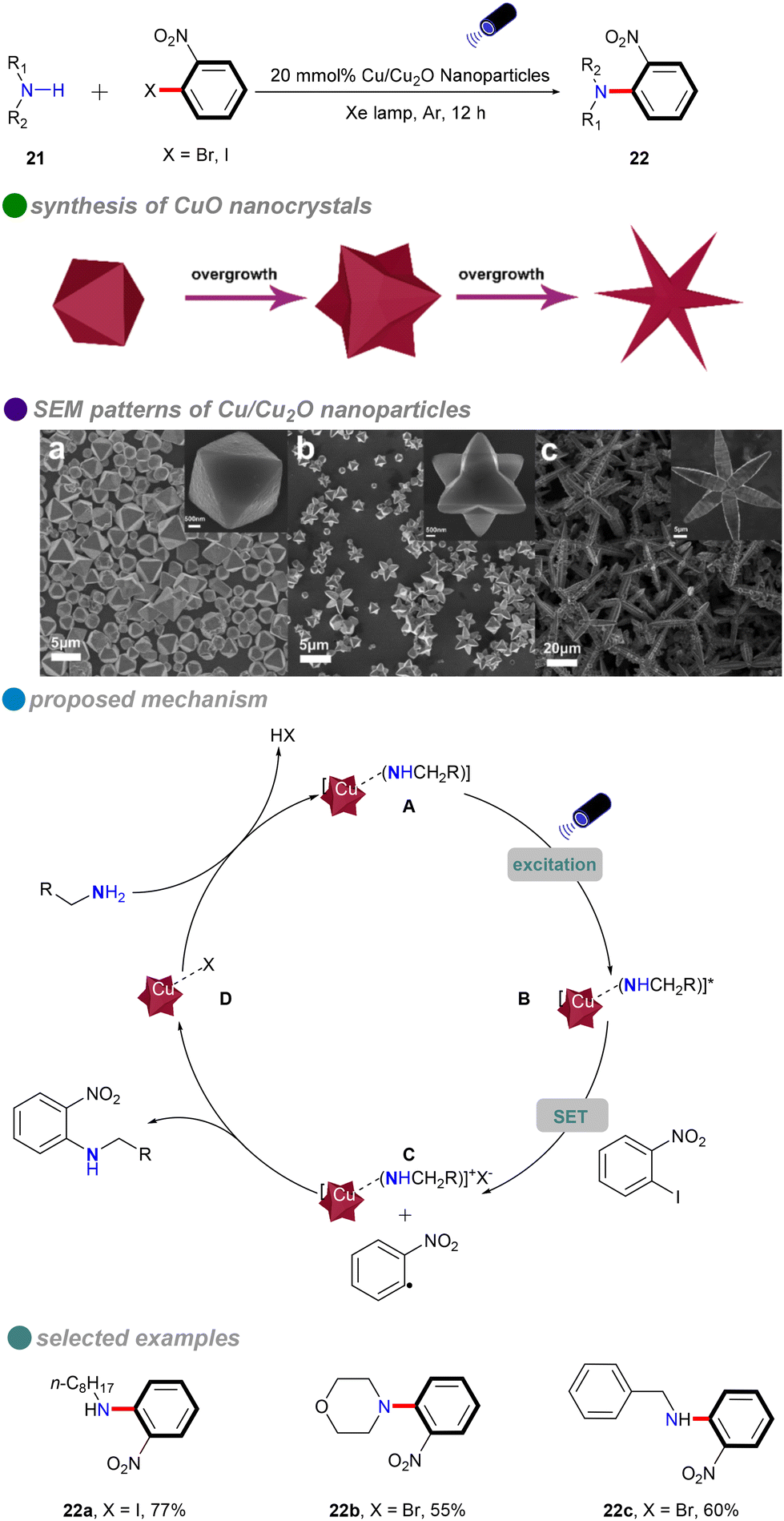 | ||
| Scheme 14 Visible-light-induced N-arylation catalyzed by Cu/Cu2O NPs, with SEM patterns of various Cu/Cu2O NPs shapes: (a) octahedron, (b) tetrahexahedron, (c) star-shaped. | ||
In 2021, Chen and colleagues synthesized a porous polymeric ligand (PPL) and complexed it with copper to form a novel, highly porous heterogeneous polymeric catalyst, Cu@PPL (Scheme 15).59 This catalyst, under visible light irradiation and heating at 80 °C, was employed for C–N cross-coupling of aryl chlorides with ammonia/amines. The reaction achieved remarkable yields of up to 98%, even with chlorobenzenes substituted with electron-donating groups. The catalyst was characterized using various techniques, including Fourier-transform infrared spectroscopy (FT-IR), ultraviolet-visible spectroscopy (UV-Vis), thermogravimetric analysis (TGA), Brunauer–Emmett–Teller (BET) surface area analysis, nonlocal-density-functional-theory (NLDFT) pore size distribution analysis, high-resolution transmission electron microscopy (HR-TEM), and scanning transmission electron microscopy (STEM). These analyses confirmed the porous structure of the catalyst, which facilitated efficient substrate adsorption and enhanced its catalytic performance. Moreover, this method enabled the modular synthesis of unsymmetrically substituted aromatic diamines from conjugated dihalides and nucleophiles through tandem reactions, offering significant potential for drug discovery and materials science. However, the study did not investigate the mechanism underlying the cleavage of the high-energy C–Cl bonds. It is plausible that synergistic photothermal catalysis played a role, although this aspect remains unexplored.
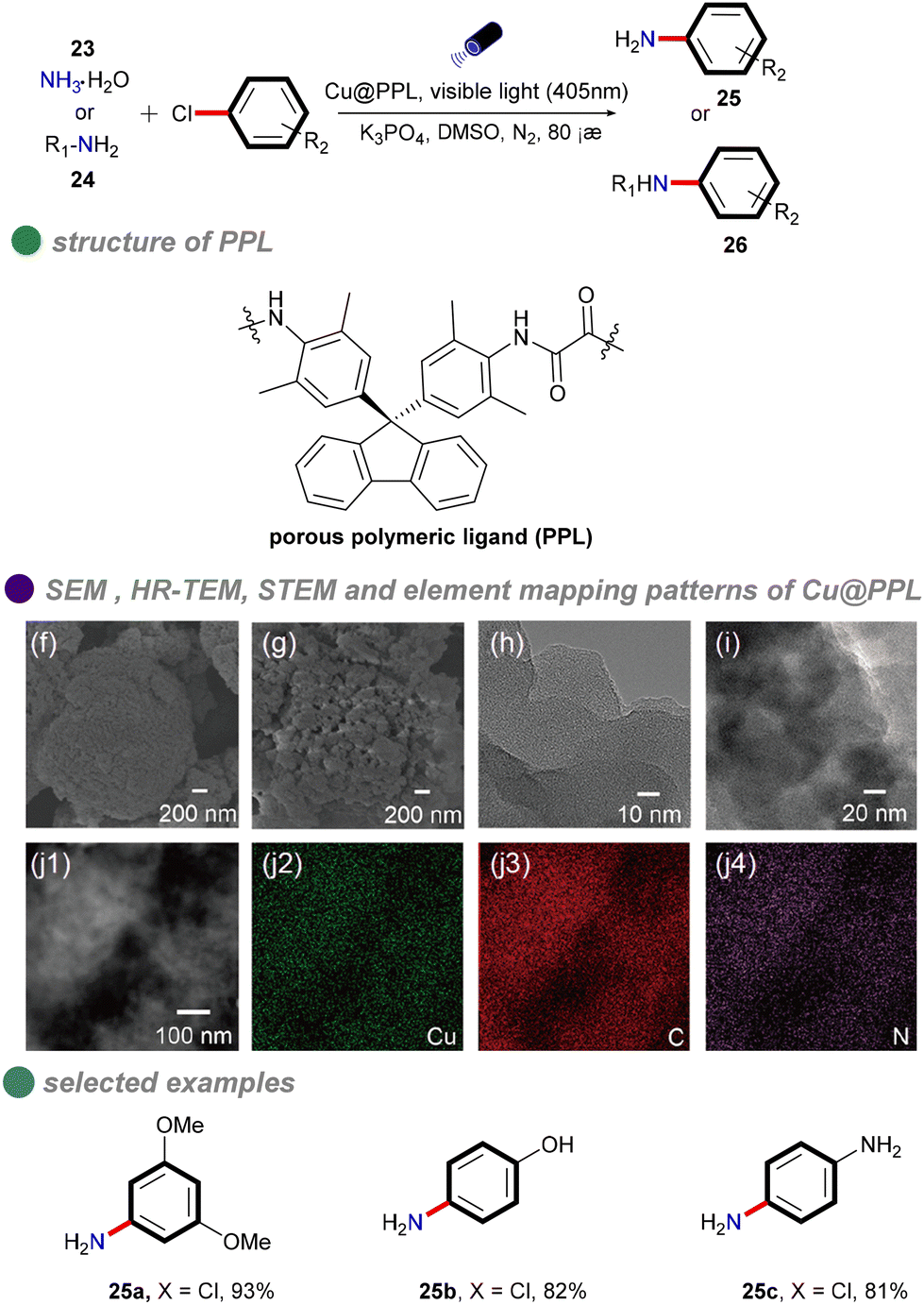 | ||
| Scheme 15 Visible-light-induced N-arylation catalyzed by Cu@PPL. SEM images of PPL and Cu@PPL are shown in (f, g), while (h, i) present HR-TEM images of PPL and Cu@PPL, respectively. Additionally, (j1) features an SEM image of Cu@PPL, and (j2–j4) provide element mapping images based on (j1) (color online). | ||
In 2022, Jing and colleagues developed novel single-molecule metallaphotocatalysis (SMPs) from cuprous clusters designed to catalyze aryl halides through microenvironment modulation (Scheme 16).60 The aromatic microenvironment in cuprous cluster C1 facilitated effective electron transfer via π–π interactions between SMPs and aryl halides. This innovative design enabled the efficient cross-coupling of aryl halides with carbazole 27 under visible light, without the need for a photosensitizer. The ability of cuprous cluster C1 to absorb visible light, combined with the proximity of metal centers in SMPs, enhanced electron transfer during aryl halide activation. The substrate scope included aryl iodides, aryl bromides, and an activated aryl chloride.
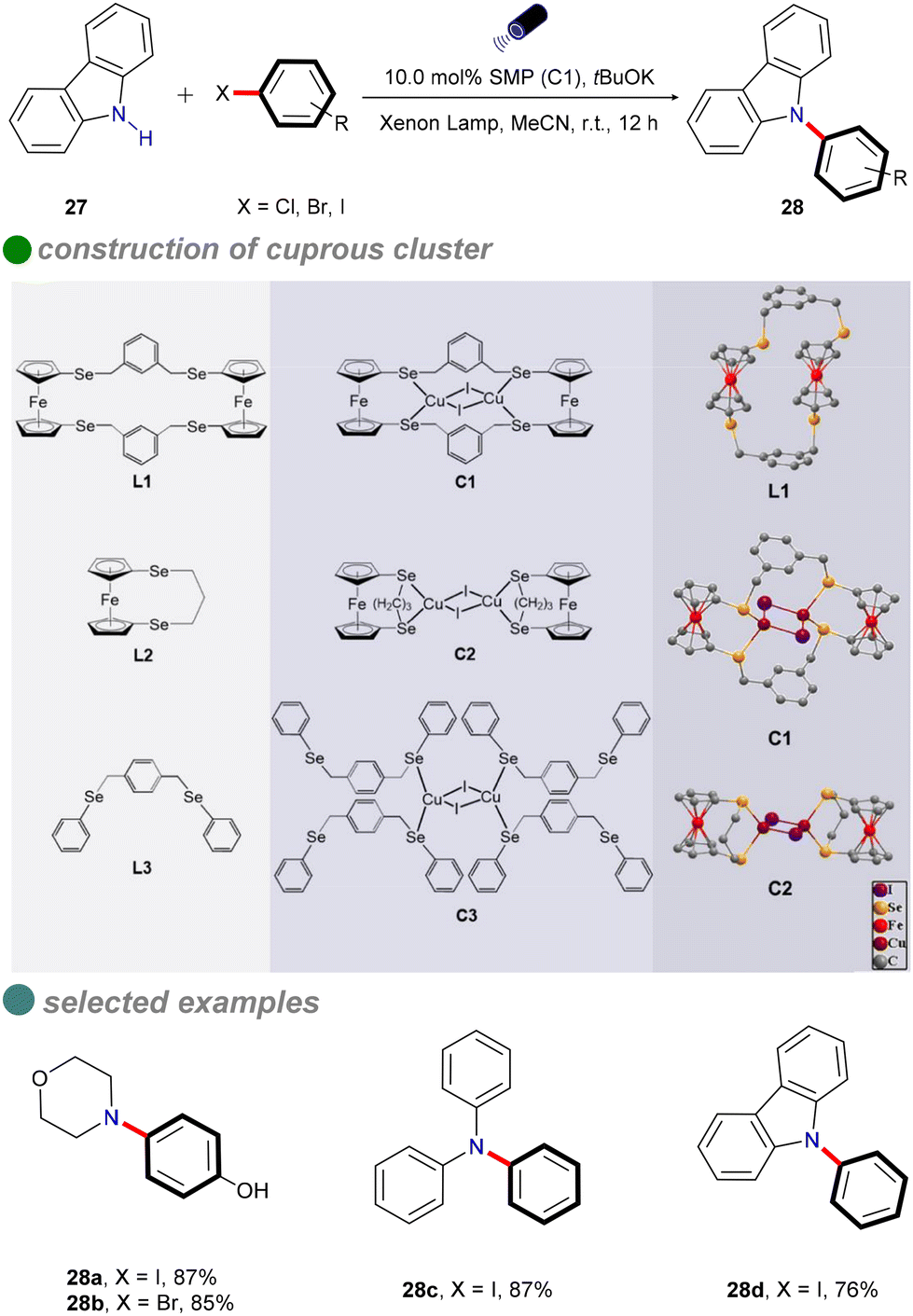 | ||
| Scheme 16 Visible-light-induced N-arylation catalyzed by cuprous cluster. | ||
In situ electron paramagnetic resonance (EPR) spectroscopy and density functional theory (DFT) calculations revealed that electron transfer occurs through π–π interactions between SMPs and aryl halides. Based on the photophysical and electrochemical properties of the aryl halides, a SET pathway was proposed. Aryl iodides (Ph–I) and bromides (Ph–Br), possessing more favorable redox potentials, are likely to undergo a thermodynamically favorable single-electron transfer (SET) mechanism (Scheme 17). Upon irradiation, the excited SMP forms a π–π complex with aryl halides, facilitating the SET process and generating aryl radicals.
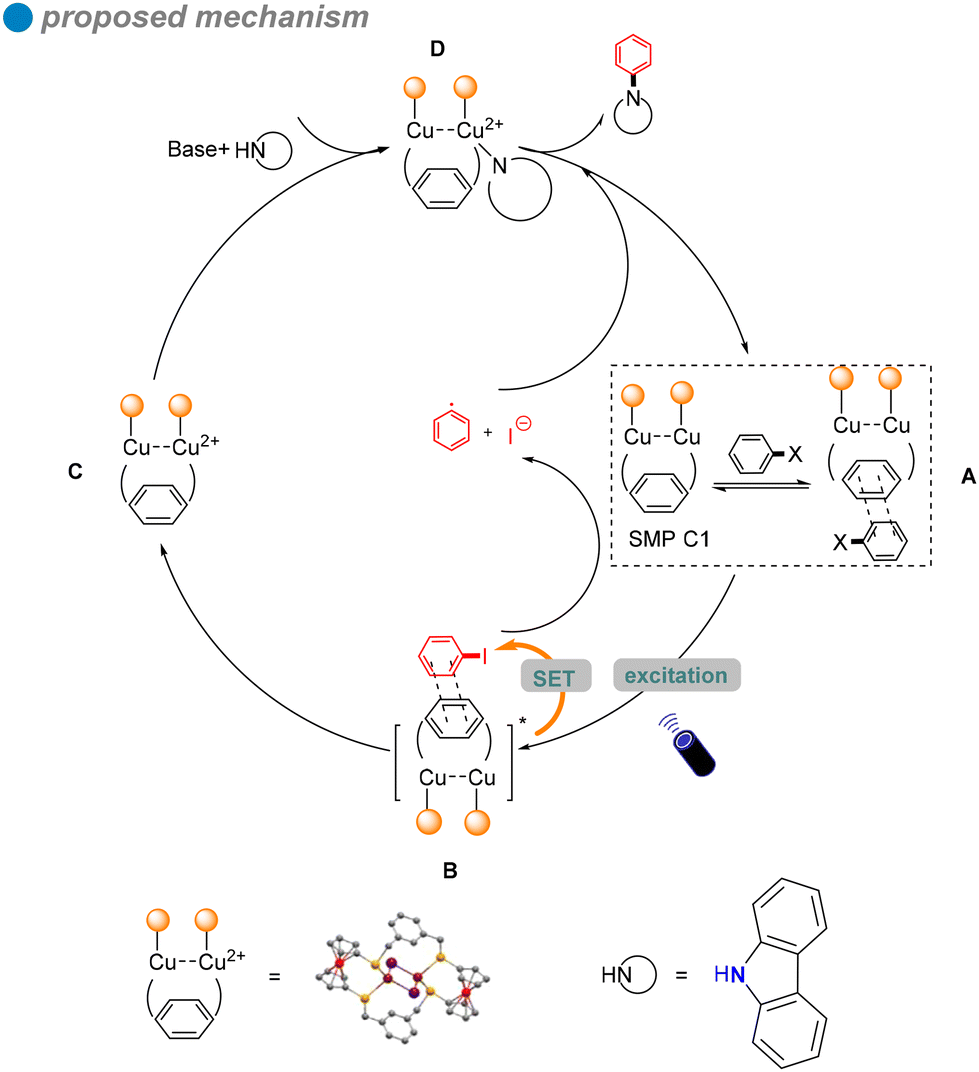 | ||
| Scheme 17 Proposed mechanism for cuprous cluster photocatalysis. | ||
The development of a robust copper catalyst capable of activating aryl chlorides in N–heteroatom cross-coupling reactions under mild conditions is of significant interest. In 2022, Rueping's research group introduced ultrasmall, atomically precise nanoclusters, specifically [Cu61(StBu)26S6Cl6H14] (Cu61NC), as a heterogeneous photocatalyst for the N-arylation of carbazole derivatives 29 using both activated and inactivated aryl chlorides (Scheme 18).61 Under visible light irradiation at room temperature, Cu61NC effectively facilitates the formation of C(sp2)–N bonds between aryl chlorides and various N-heterocycles, such as carbazole, indole, azaindole, and imidazole. The catalyst's efficiency is attributed to its large absorption cross-section and long excited-state lifetime, enabling high yields and a broad substrate scope with low-energy blue LEDs. For instance, p-methoxy chlorobenzene, containing an electron-donating group, achieves a yield of 54%.
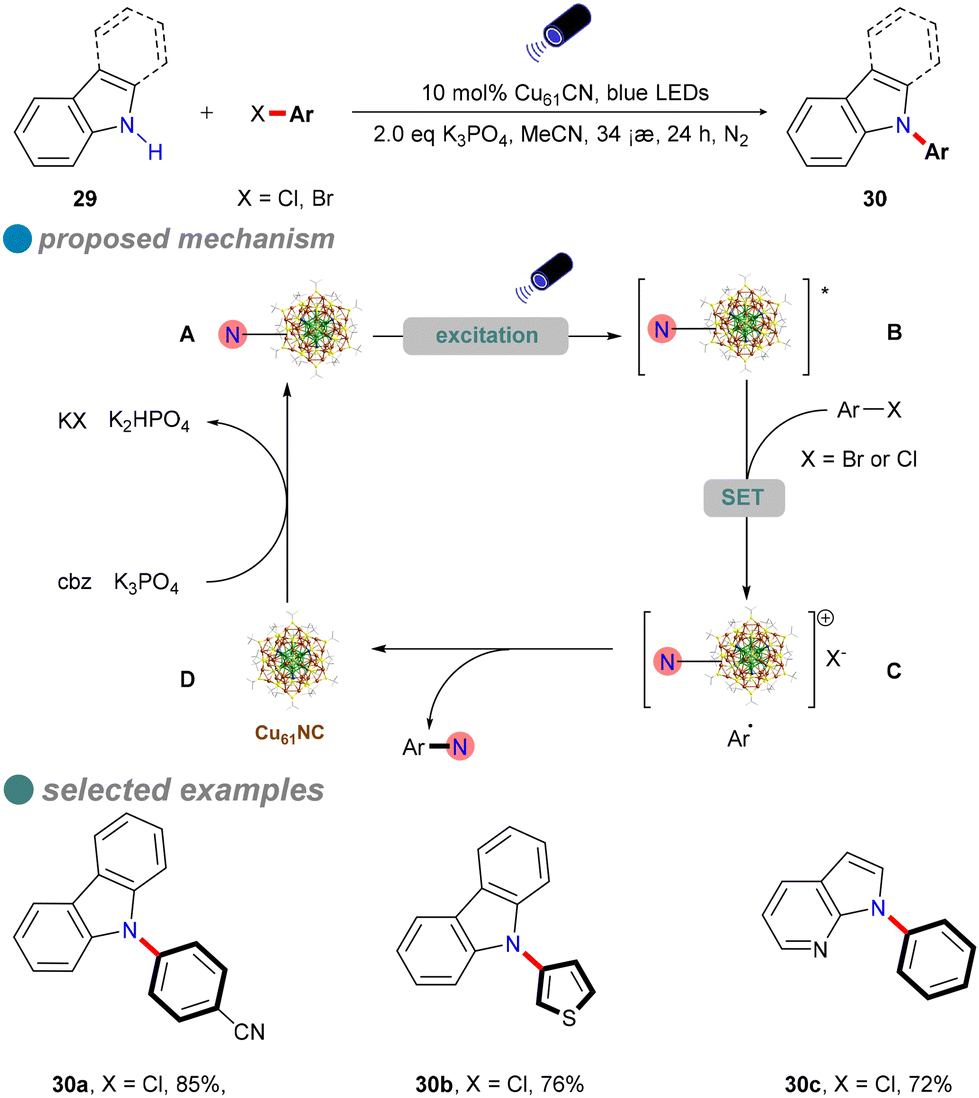 | ||
| Scheme 18 Visible-light-induced N-arylation catalyzed by copper nanocluster. | ||
Mechanistic studies, including radical clock experiments, radical trapping experiments, radical competition experiments, cyclic voltammetry (CV) analyses, and various spectroscopic techniques (absorption, excitation, emission spectra, photoluminescence, photoluminescence quenching, and photoluminescence lifetime measurements), indicate that a single-electron transfer (SET) mechanism underpins the aryl chloride amination reaction, as illustrated in Scheme 18.
In 2023, Xu's group developed copper oxide nanocrystals (CuO NCs) as a heterogeneous photocatalyst for the visible light-promoted Goldberg reaction, showcasing their unique ability to efficiently produce N-arylated amides 32 (Scheme 19).53 The group dissolved a copper salt in deionized water, followed by the addition of a NaOH solution. The mixture was then stirred at a specific temperature. Once cooled to room temperature, the product was subjected to centrifugation, washing, and drying in an oven to yield the desired copper oxide nanoparticles (CuO NCs). The morphology of CuO, particularly the nanoplate structure, significantly influences its electronic properties, demonstrating superior photogenerated electron capabilities and stronger photooxidation ability compared to other morphologies like nanosheets and nanorods. Eosin Y, serving as both a photosensitizer and an electron donor/acceptor, plays a crucial role in enhancing the reaction efficiency, highlighting its potential for effective photocatalytic processes. The catalytic performance of CuO(II) nanocrystals is further improved by optimizing surface adsorption energy and reaction heat, resulting in broad substrate versatility and favorable outcomes in the cross-coupling of electron-rich aryl halides with both aryl and aliphatic amides. This method is particularly significant for the synthesis of pharmaceuticals and agrochemicals, including imatinib, nefiracetam, propanil, and mebenil, achieving moderate to good yields. The reaction mechanism is based on a single-electron transfer (SET) process (Scheme 20), where eosin Y modulates the oxidation states of the CuO nanoplates, facilitating the challenging CuII-catalyzed photoinduced couplings of aryl halides with amides 31. The novel mechanistic paradigm developed in this study shows promising applications for challenging visible light-promoted CuII-catalyzed Ullmann-type couplings.
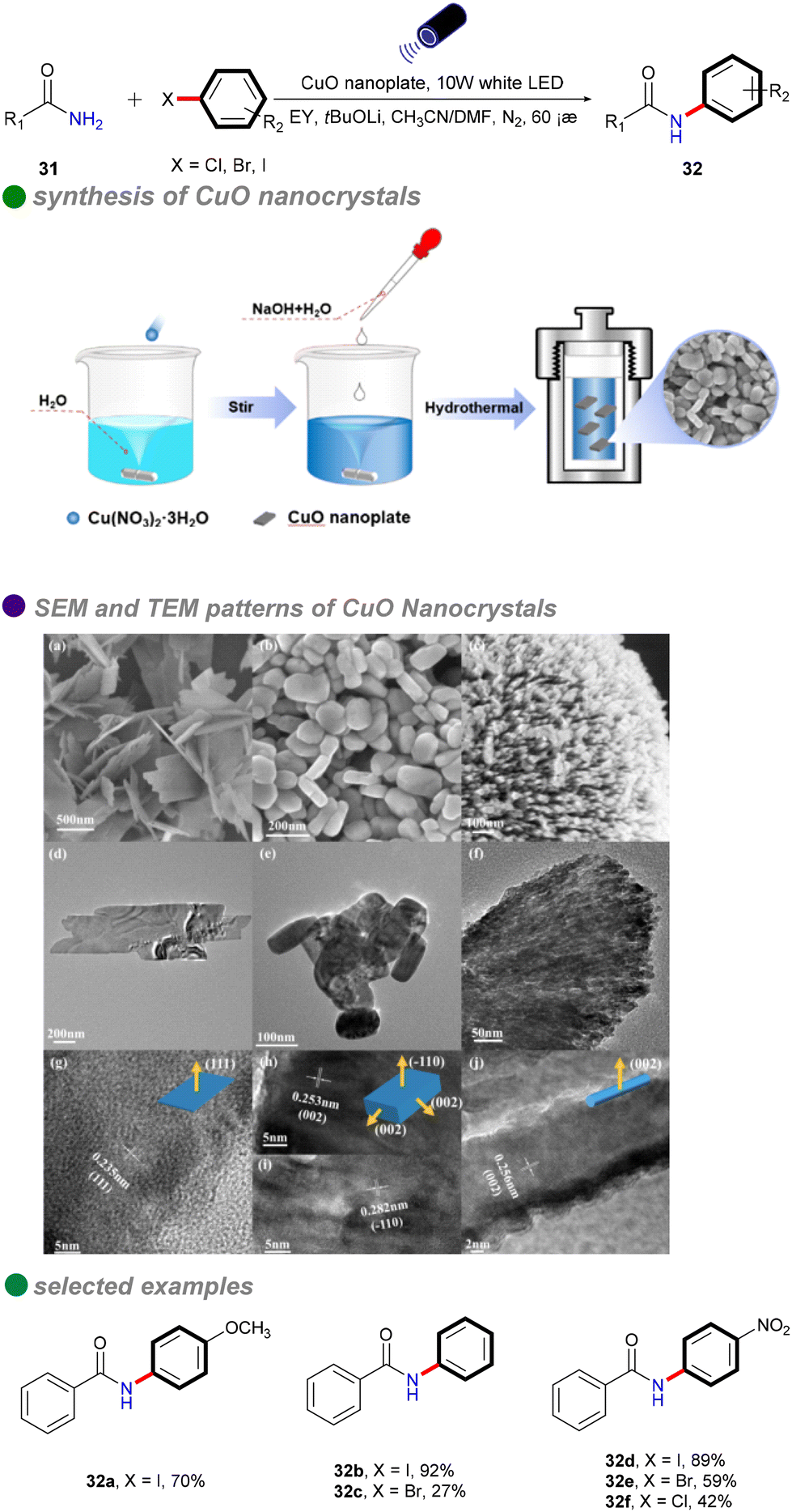 | ||
| Scheme 19 Visible-light-induced N-arylation of amides catalyzed by CuO nanoplate. SEM images show (a) CuO nanosheet, (b) CuO nanoplate, and (c) CuO nanorod. TEM images depict (d) the CuO nanosheet, (e) CuO nanoplate, and (f) CuO nanorod. HRTEM images illustrate (g) the CuO nanosheet, (h) CuO nanoplate with a (111) facet, (i) CuO nanoparticle with a (−110) facet, and (j) CuO nanorod with a (002) facet. | ||
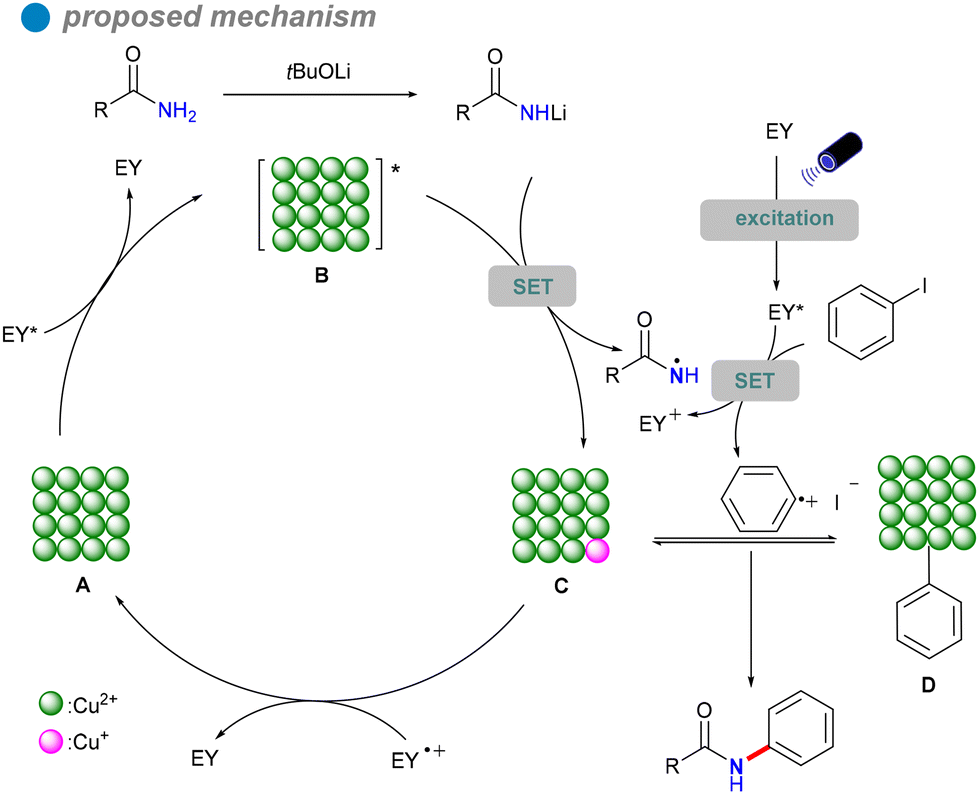 | ||
| Scheme 20 Proposed mechanism for CuO nanoplate photocatalysis. | ||
The development of homogeneous and heterogeneous catalysts has expanded the range of substrates and reaction conditions, making this method an important tool for the synthesis of nitrogen-containing compounds. Advances in photoinduced copper-catalyzed Ullmann C–N coupling reactions have significantly addressed the limitations of traditional Ullmann coupling reactions, which require high-temperature heating or the use of ligands, providing an economical and environmentally friendly approach for the synthesis of nitrogen-containing compounds.
3.3 Photoinduced C–O cross-couplings
Ullmann-type cross-coupling products containing C–O subunits are prevalent in various organic compounds, playing essential roles in the development of pharmaceuticals such as the nonsteroidal anti-inflammatory agent FK3311 (I)62 and CRF antagonists (II and III),63,64 as shown in Scheme 21. These compounds also have significant applications in natural products,9 agrochemicals,10–12 and functional materials.13 Additionally, the Ullmann coupling reaction provides an easy approach to converting industrial wastes often composed of phenols from sectors such as petrochemicals, pharmaceuticals, coking, printing, and dyeing into valuable products.65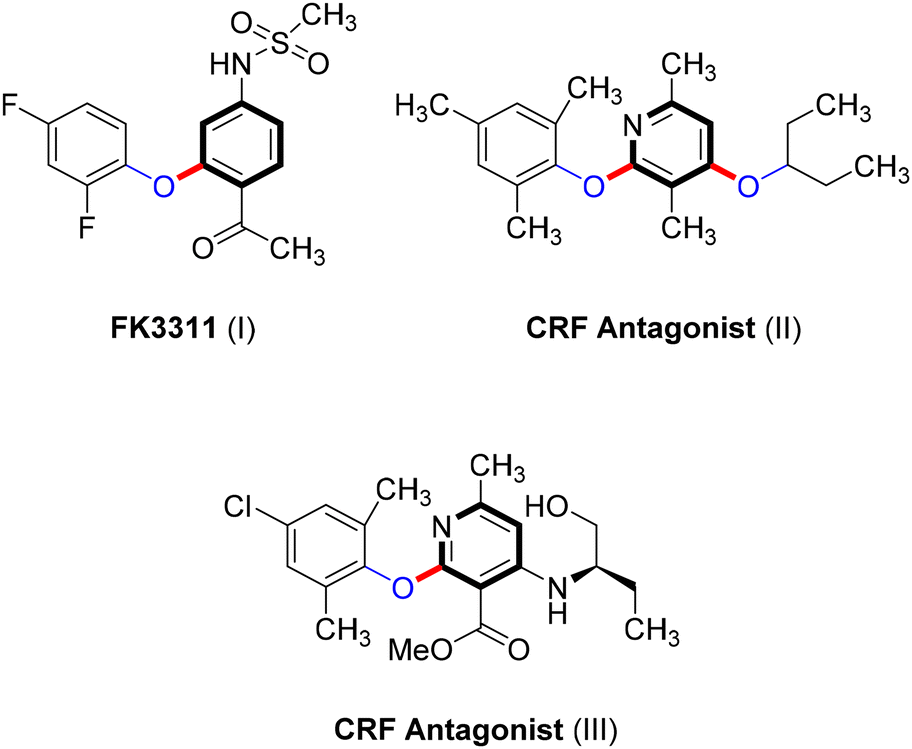 | ||
| Scheme 21 Pharmaceutical agents based on Ullmann O-arylations. | ||
However, the conventional synthesis of C–O Ullmann-type compounds faces challenges, including environmental concerns due to catalyst leaching, the formation of homocoupling byproducts, scalability issues, and the need for harsh conditions.3,65,66 The development of new catalytic systems, such as Cu-photocatalysts, has broadened the utility of the Ullmann coupling reaction, offering environmentally friendly conditions and accommodating a wider range of substrates.
This section discusses the role of homogeneous and heterogeneous Cu-photocatalysts in C–O bond formation via Ullmann-type cross-coupling reactions. Notably, during the single-electron transfer process in photoinduced Ullmann coupling reactions, O-nucleophiles, due to their higher electronegativity, are more resistant to electron loss compared to C-, N-, and S-nucleophiles. This resistance results in higher energy barriers, making these photoreactions challenging and less frequently reported.
The plausible cyclic mechanism of this reaction is illustrated in Scheme 22. Within this mechanism, the pre-prepared nucleophile (phenoxide) engages with the copper complex, resulting in the formation of the CuI-phenoxide complex [LnCu(Nu)] B. Upon photoexcitation by absorbing incident light at 254 nm, this complex transitions to [LnCu(Nu)]* C. Subsequently, a single electron transfer (SET) from the excited state to the aryl halide, leading to the cleavage of the C–X bond and the generation of a chemically stable intermediate [LnCu(Nu)]+ X− containing an aryl radical (Ar˙) D. The aryl radical then undergoes coupling with the nucleophile to furnish the desired diaryl ethers 34. The endurance of the CuI-phenoxide complex is validated through a controlled experiment of [Cu(OPh)2] [N(n-Bu)4] (1) with an aryl iodide derivative, resulting in the target product with a satisfactory yield of 62%. Furthermore, they proposed an alternative reaction mechanism scenario, where direct photolysis of the C–X bond takes place under light exposure, resulting in the generation of Ar˙. This species subsequently interacts with the CuI-phenoxide complex like 1 (instead of CuII-phenoxide [LnCu(Nu)]+ X−) to afford the formation of a C–O bond and Cu0, as a CuI/Cu0/CuI mechanism. So, further controlled experiments were conducted to challenge this hypothesis by examining the reaction of complex 1 with diazonium salt solutions [PhN2] [BF4] as the source of phenyl radicals, and [Ru(bpy)3] [PF6]. The goal was to determine if a phenyl free radical could react with the CuI complex (1) to produce a C–O coupling product. However, the main product observed was benzene, with only a trace amount of diaryl ether (<5%), suggesting that the hypothesis excludes alternative radical-based pathways different from that shown in Scheme 21.
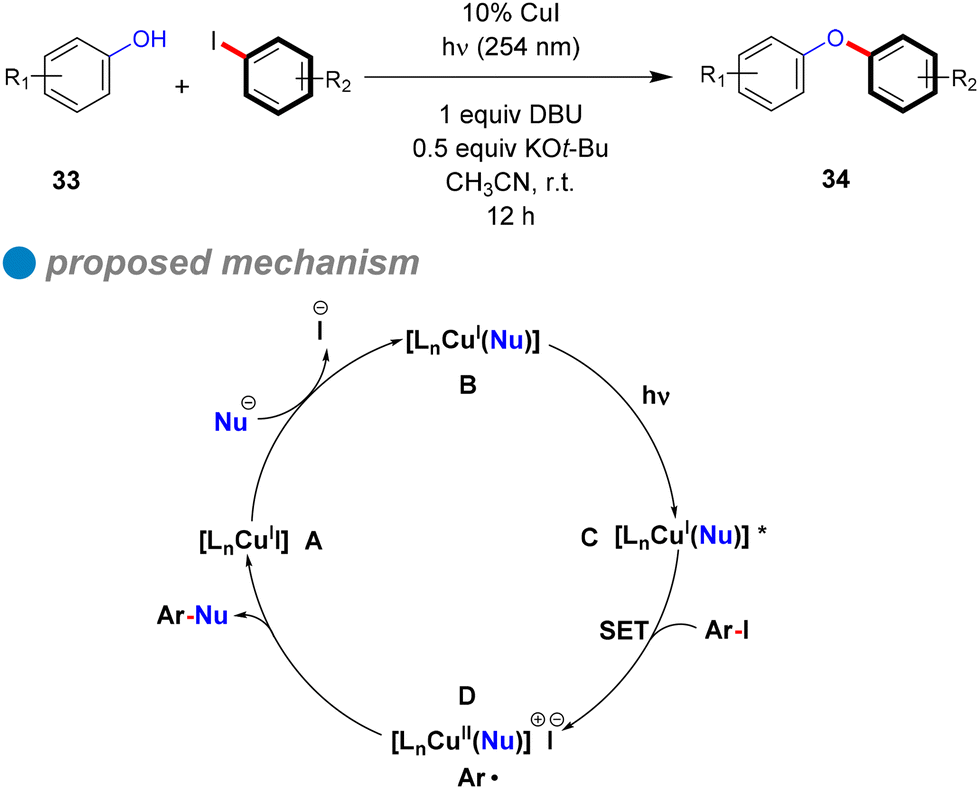 | ||
| Scheme 22 Photoinduced O-arylation of phenols catalyzed by CuI. | ||
In 2018, Ha and his colleagues synthesized a Cu-MOF photocatalyst (Cu2(BDC)2(DABCO)) with a narrow energy band gap of 2.7 eV, determined via the empirical Tauc plot formula.73 They hypothesized that increasing the resonance effect in the organic linker would narrow the energy band gap, which was obvious on (Cu2(BDC)2(DABCO)) band gap (2.7 eV) compared to the Cu-BDC catalyst, which has a wider band gap of 3.6 eV. They then evaluated the photocatalytic activity of the synthesized Cu-MOFs for C–O cross-couplings between phenols 35 and aryl halide, affording the corresponding diaryl ethers 36 (Scheme 23), achieving high to moderate yields with various substrates at ambient temperature using 365 nm light, rather than the optimal 254 nm wavelength for homogeneous C–O bond formation. This reaction required a base, ligand, solvent, catalyst, and illumination to maximize photoactivity, as determined through optimized experimentation. Furthermore, leaching assessments confirmed the catalyst's heterogeneity, maintaining 95% of its activity after seven cycles, indicating its strong stability.
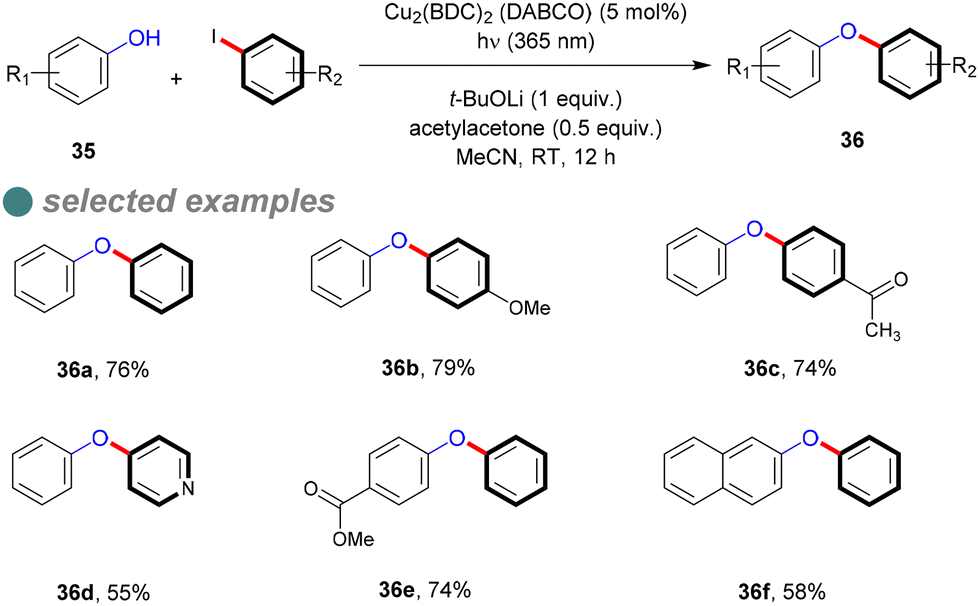 | ||
| Scheme 23 Photoinduced Cu-MOF catalyzed O-arylation of phenols. | ||
In 2022, Ji and his team reported the photo-thermal synthesis of diaryl ethers through Ullmann coupling of phenols and aryl halides under visible-light irradiation (Scheme 24),74 using plasmonic Cu nanoparticles (Cu NPs) supported on CNTs derived from various plastic wastes, such as foam (polystyrene), bags (polyethylene), and food wraps (polypropylene), via microwave-initiated dehydrogenation approaches. XRD patterns indicated no change in the surface of the CNT support. The heterogeneous Cu-catalyst structure was confirmed through various analyses, including TEM, HRTEM (Scheme 23), UV, BET, and XPS. Furthermore, optimization experiments revealed that light-excited Cu NPs and a base are essential for the reaction, as traces of diaryl ethers were produced only in the absence of Cu NPs, light, or a base. Additionally, Cu NPs inhibit the degradation or mineralization of phenols and proceed with the cross-coupling reaction products 38 between aryl halides and phenols 37. Notably, the highest yield was achieved using a broad range of visible light (470–630 nm), without the need for UV light which consisted with the green chemistry principles.
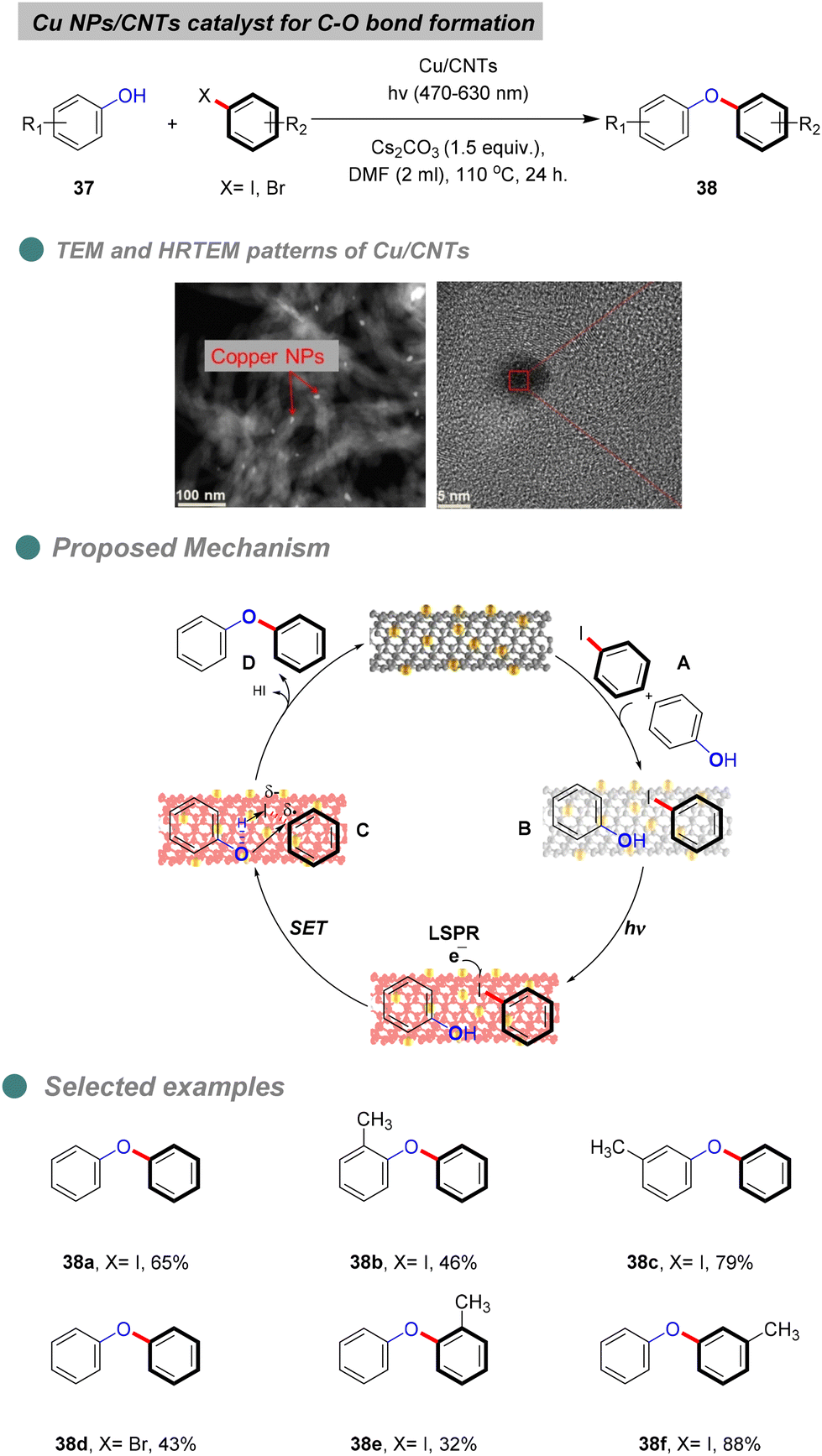 | ||
| Scheme 24 Visible-light-induced O-arylation catalyzed by Cu/CNTs. | ||
Controlled experiments involving radical trapping and electron scavenging demonstrate that the electrons generated by the localized surface plasmon resonance (LSPR) effect of copper nanoparticles (Cu NPs) initiate the reaction. The proposed mechanism entails the adsorption of both reactants I onto the surface of carbon nanotubes (CNTs) II (Scheme 24). Under visible light irradiation, the copper catalyst is excited, facilitating electron transfer to the aryl halide substrates, which leads to the formation of aryl radicals C that couple with the adsorbed phenols to yield the desired diaryl ether D, along with hydrogen iodide and the regeneration of the copper catalyst. Furthermore, the stability and recyclability of the synthesized catalyst were evaluated, revealing that the product yield remained relatively constant after the third cycle, although the copper content diminished due to the oxidation of the Cu NPs. The catalyst's structure was confirmed to be unchanged by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) analyses after four cycles. It is noteworthy that the reaction cannot proceed at room temperature, as the energy provided by light is insufficient to surpass the reaction's energy barrier.
This section provides a comprehensive review of the literature on photoinduced Cu-catalyzed Ullmann C–O cross-coupling reactions, focusing on both homogeneous and heterogeneous catalysts, including CuI, Cu2(BDC)2(DABCO), and Cu/CNTs. It emphasizes the pivotal role of Cu-photocatalyst systems in expanding the scope of Ullmann coupling reactions, positioning them as indispensable tools in modern synthetic chemistry. Additionally, it delves into the potential mechanisms for these reactions, employing the SET cycle mechanism (CuI/CuI*/CuII/CuI) to illustrate the advantages of photocatalysis (Scheme 22), including enhanced efficiency and lower energy requirements when compared to traditional thermal methods. These innovative methods and catalyst systems not only inspire chemists but also facilitate the exploration of sustainable approaches to enhance C–O bond formation, offering valuable insights into reaction mechanisms supported by both experimental and theoretical evidence.
3.4 Photoinduced C–S cross-couplings
Carbon–sulfur subunits are vital in various natural products and pharmaceuticals. For example, Thymitaq (nolatrexed), a thymidylate synthase inhibitor used to treat hepatocellular carcinoma and nasopharyngeal cancer, is synthesized through a ligand-free Ullmann C–S cross-coupling method (Scheme 25I).75,76 Likewise, AZD7545, a pyruvate dehydrogenase kinase (PDK) inhibitor developed as an oral treatment for type II diabetes, is produced using Ullmann coupling with a catalytic amount of Cu2O under thermal conditions (Scheme 25II).77 Traditionally, C–S bond formation involves cross-coupling reactions between aryl halides and sodium or potassium arenethiolates, generally using copper catalysts at high temperatures. However, this method often requires extended reaction times and toxic solvents.78 Recent advances in photocatalytic technology have significantly improved this process, enabling C–S bond formation under milder conditions. Light irradiation efficiently activates copper catalysts, allowing for selective bond formation without harsh reaction conditions. This section explores the application of photoinduced homogeneous and heterogeneous copper catalysts in Ullmann C–S cross-coupling reactions.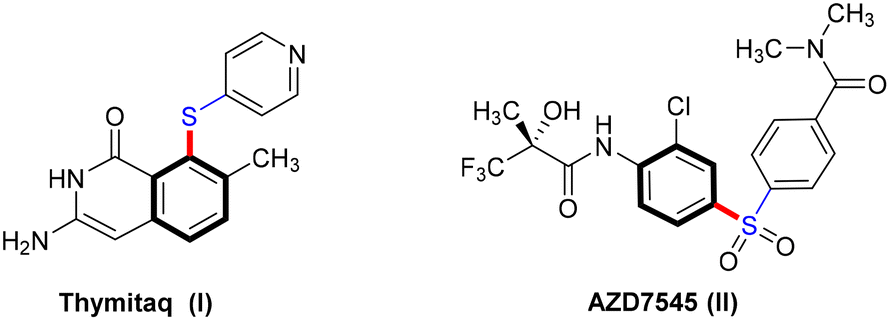 | ||
| Scheme 25 Pharmaceutical application of Ullmann S-arylation. | ||
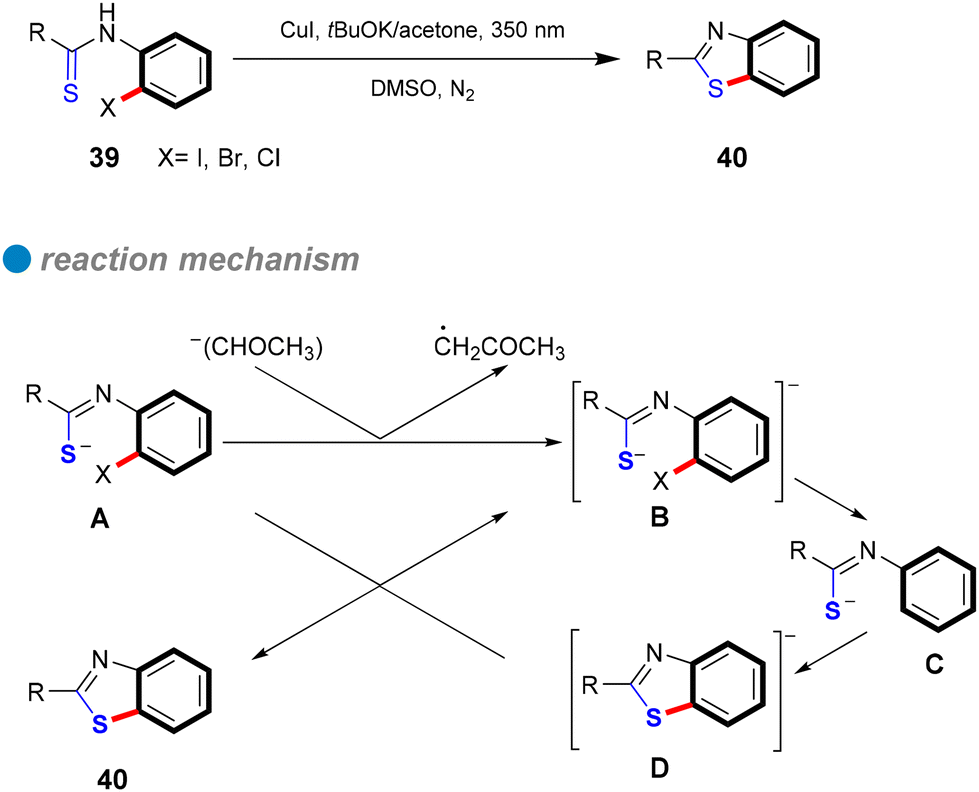 | ||
| Scheme 26 Intramolecular S-arylation by SRN1 mechanism. | ||
In 1984, Bowman's research group conducted a detailed study of the copper(I)-catalyzed C–S cross-couplings, highlighting its catalytic mechanism in contrast to the SRN1 mechanism.80 Notably, strong electron acceptors or scavengers did not inhibit either the catalytic or mediated CuI intramolecular nucleophilic substitution for synthesizing 2-phenyl and 2-methyl-1,3-benzothiazoles, suggesting the mechanism is unlikely SRN1. Several experiments, including the use of chloro-iodobenzene as a test for aryl radical anions typical in SRN1, were conducted. In these tests, phenyl thiolate 41 reacted with 1-chloro-4-iodobenzene under SRN1 conditions to yield a disubstituted product 42, while only a monosubstituted product was obtained under catalytic or mediated CuI conditions (Scheme 27a). Similarly, the reaction of p-chlorophenyl thiolate 43 with iodobenzene produced only the monosubstituted product 44 (Scheme 27b). These findings indicate that aryl radical anions are absent from the mechanism. Additionally, a radical clock test for aryl radicals and aryl radical anions further corroborated the di-halobenzene test results (Scheme 27). Consequently, Bowman and his colleagues concluded that the mechanism involves inner electron transfer from CuI complex to the aryl halide, forming an adduct of CuII complex with aryl radical anion that leads to a CuIII intermediate, followed by reductive elimination of the product and the regeneration of the CuI catalyst. Their research confirmed that copper(I) catalysis offers an efficient coupling method for forming C–S bonds, especially in challenging reaction scenarios that traditional SRN1 conditions struggle to achieve.
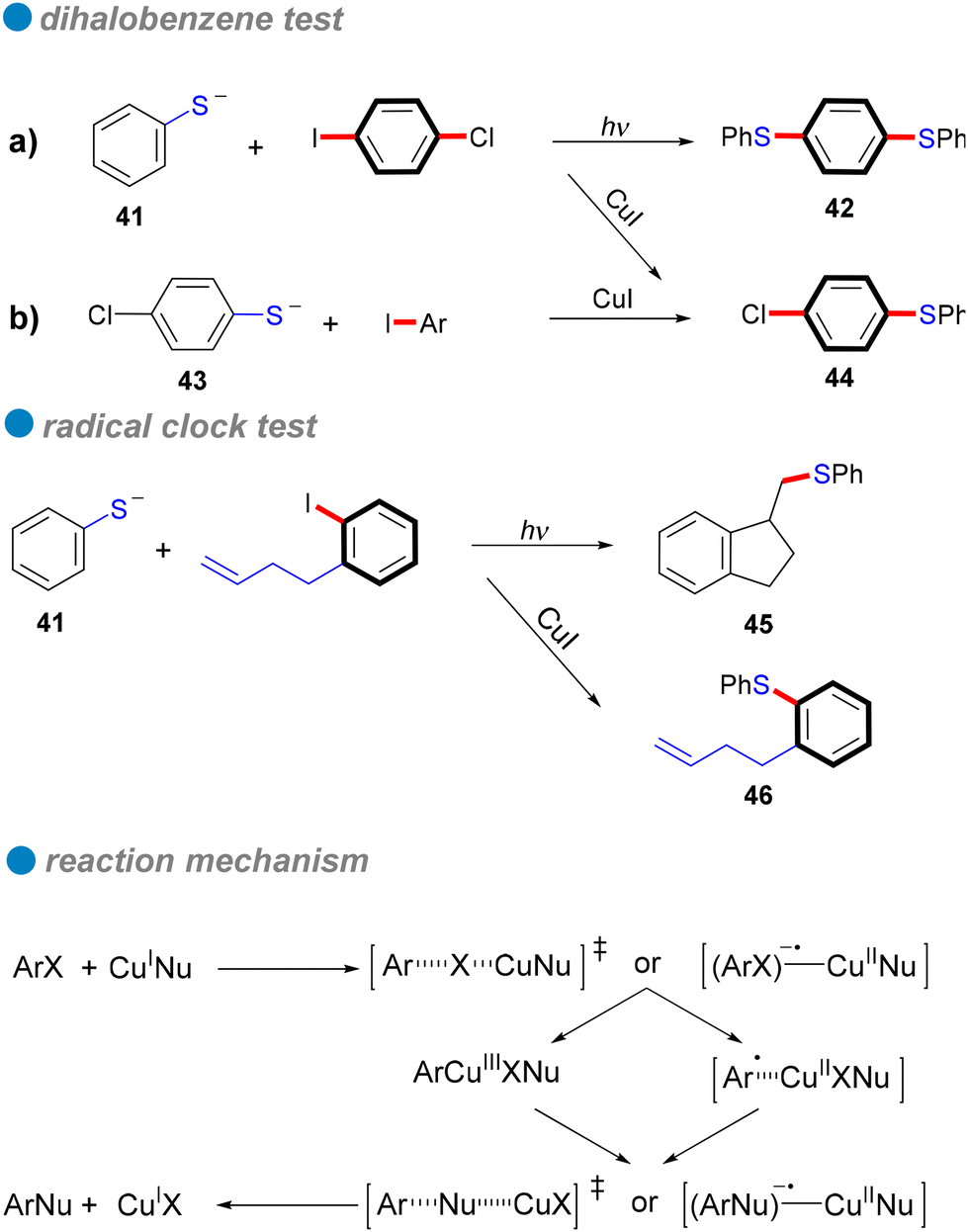 | ||
| Scheme 27 Controlled experiments conducted to eliminate the haloaryl radical-anion intermediate and the plausible mechanism. Only the monosubstituted product 44 was obtained under two sets of CuI conditions: a) catalytic CuI conditions utilizing phenyl thiolate 41, and b) mediated CuI conditions employing p-chlorophenyl thiolate 43. | ||
In 2013, Fu and Peters expanded their method for photoinduced, copper-catalyzed cross-couplings from N-phenylation and N-alkylation of carbazoles to include the coupling of aryl thiols with aryl halides at low temperatures (0 °C), yielding diaryl thioethers (Scheme 28).19 Using copper(I) iodide as a pre-catalyst without additional ligands, their approach efficiently formed C–S bonds via a single-electron transfer (SET) mechanism (Scheme 28). In this process, photoexcited copper(I)-thiolate complexes B react with aryl halides, generating copper(II) and aryl radicals C through electron transfer from copper. The aryl radicals then react with thiols to form thioethers, regenerating the CuI catalyst D. This method accommodates a broad range of hindered, electron-rich, electron-poor, and heterocyclic thiols 47, as well as aryl halides (iodides and bromides), delivering C–S products 48 with good yields (Scheme 28). Notably, the method enables C–S cross-couplings with inactive aryl iodides at temperatures as low as −40 °C. The successful photoinduced coupling of activated aryl chlorides with thiols at 0 °C and below represents a significant advancement over previous methods that required temperatures above 60 °C. These findings further confirm that the SET mechanism offers a lower energy barrier compared to the concerted oxidative addition/reductive elimination (OA/RE) mechanism, underscoring the efficiency and practicality of this approach.
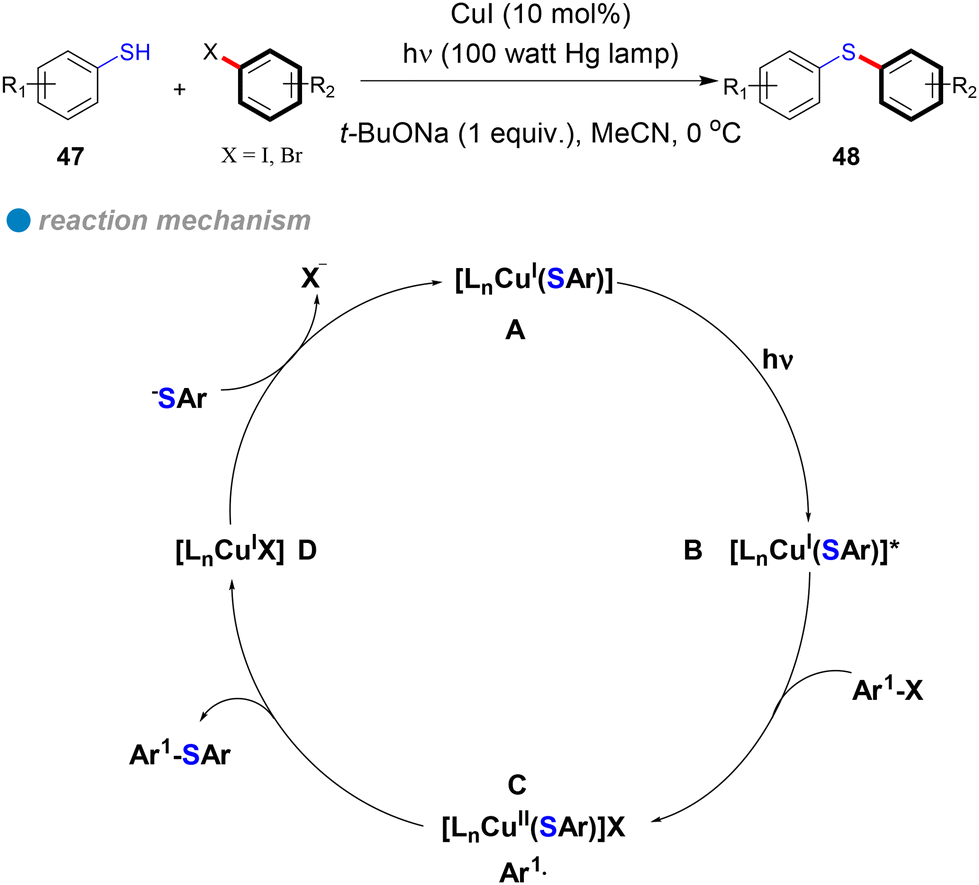 | ||
| Scheme 28 Photoinduced Cu-catalyzed S-arylation of aryl thiols. | ||
In 2016, Fu and Peters conducted a detailed investigation into the photoinduced copper-catalyzed cross-coupling of aryl thiols with aryl halides (Scheme 28).28 Their research demonstrated that the reaction follows a single-electron transfer (SET) mechanism (Scheme 28), ruling out alternative pathways such as iodine atom transfer (IAT), nucleophilic substitution radical (SRN1), and concerted oxidative addition. This conclusion was strongly supported by a combination of controlled radical cycle experiments, electrochemical measurements (cyclic voltammetry), and photophysical studies, including optical spectra, Stern–Volmer kinetic analysis, quantum yield, and chain length. Additionally, spectroscopic techniques, such as EPR and UV, further confirmed the mechanistic pathway. The core innovation of this study is the demonstration of the photoinduced SET mechanism, which was validated through carefully designed control experiments and comprehensive photophysical analyses.
In 2017, the research group led by Mo and Ackermann unveiled a groundbreaking methodology for photoinduced Cu-catalyzed C–S bond formation through cascade S-arylation and C–H chalcogenation under ambient temperature conditions (see Scheme 29).81 This three-component reaction facilitated the interaction between aryl iodides and bromides with elemental sulfur and benzothiazoles 49, exhibiting remarkable functional group tolerance across both electron-rich and electron-deficient aryl iodides (Scheme 29). This innovative approach represents a substantial advancement compared to conventional methods, which frequently rely on precious metals and harsh reaction conditions. Mechanistic investigations indicate a single-electron transfer (SET) mechanism, with glavinoxyl employed as a radical scavenger to elucidate the presence of aryl radical intermediates; notably, its incorporation inhibited the reaction's efficiency by as much as 0% when three equivalents of glavinoxyl were utilized (refer to Scheme 29). Furthermore, the study assessed the influence of the thiolating reagent on C–H chalcogenation, revealing that thiophenol did not produce the anticipated product, whereas diphenyl disulfide yielded a high synthesis yield of the target compound (Scheme 29). Additionally, the reaction between iodobenzene and elemental sulfur generated both diphenyl disulfide and diphenyl sulfide, signifying that diaryl disulfides are crucial intermediates formed in situ throughout the process.
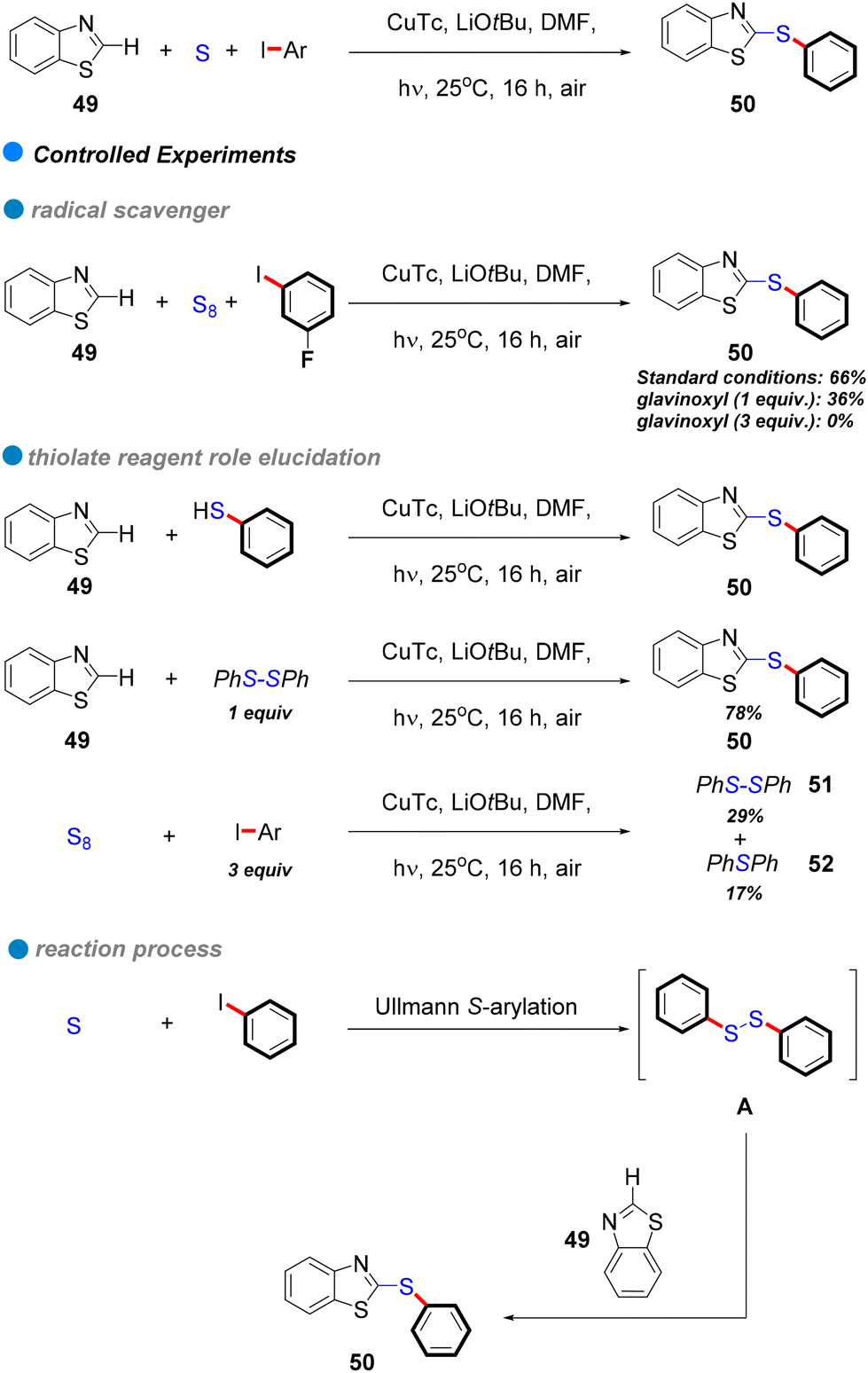 | ||
| Scheme 29 Photoinduced Cu-catalyzed domino synthesis of thioethers via successive S-arylation and C–H chalcogenation. | ||
In 2021, Yang and colleagues developed an efficient method for the sulfonylation of aryl halides (iodides and bromides) with sulfinate salts under visible light and copper catalysis (Scheme 30).82 Using an in situ-generated single-ligand CuI photocatalyst, they achieved the synthesis of various organic sulfones with high yields. This environmentally friendly approach avoids the need for costly and toxic catalysts, while demonstrating excellent functional group compatibility and scalability for large-scale synthesis. Furthermore, the reaction was found to be particularly effective for electron-deficient aryl halides under mild visible light conditions. Mechanistic studies, including electron paramagnetic resonance (EPR) spectroscopy and fluorescence quenching assays, confirmed that a single-electron transfer (SET) mechanism played a crucial role in the reaction. These findings provide valuable tools for advancing synthetic chemistry and open new avenues for future research in photocatalysis and sulfonylation reactions.
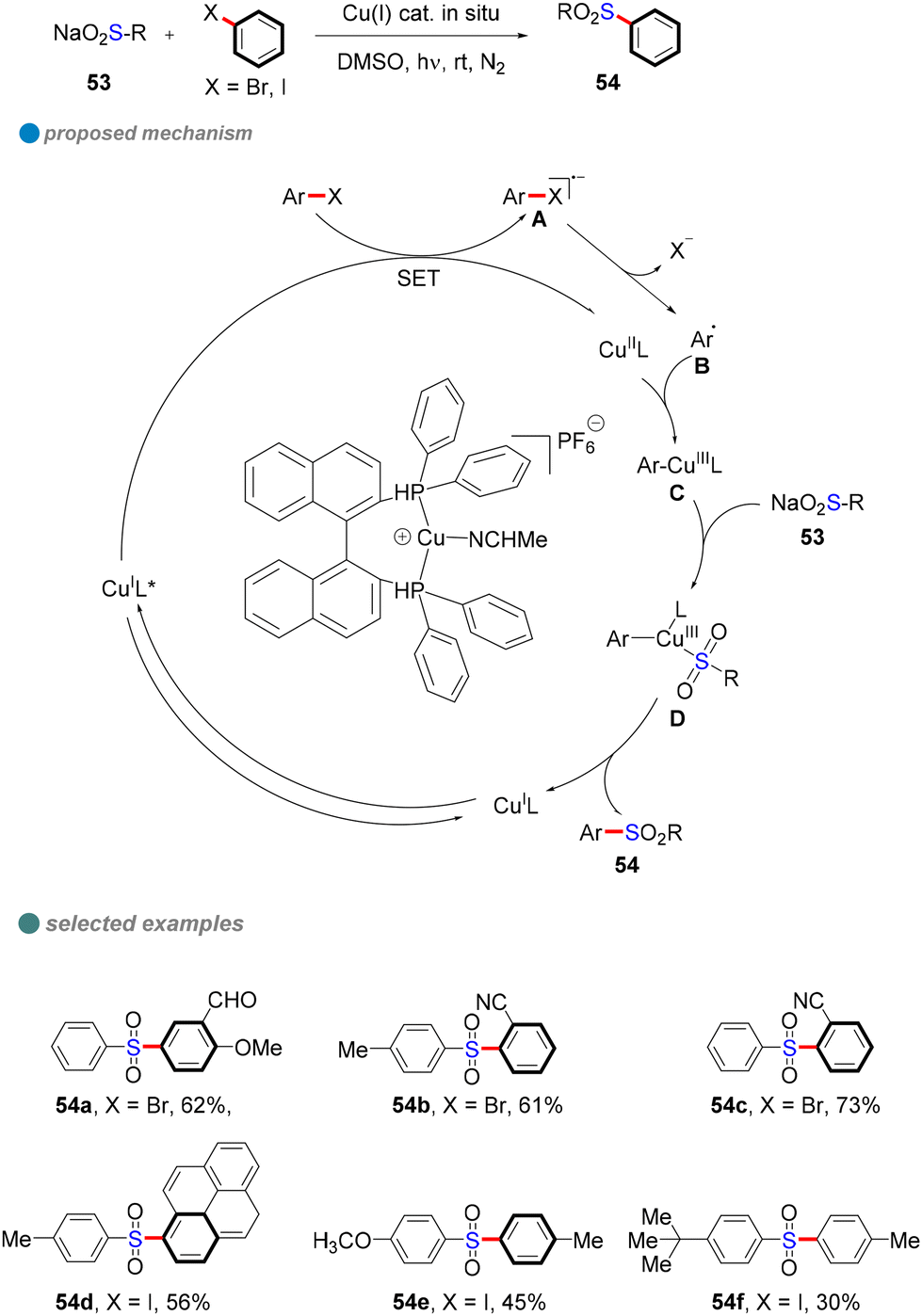 | ||
| Scheme 30 Visible-light-induced Cu-catalyzed S-arylation of sulfinate salts. | ||
This section reviews recent advancements in photo-induced copper-catalyzed C–S bond coupling reactions, focusing exclusively on homogeneous catalysis from Bowman (1982)79 to Mo and Yang (2021).82 It discusses the mechanisms involved, including intramolecular SRN1 and SET pathways. This green, efficient synthesis method employs photoinduction to activate copper catalysts, enabling the selective formation of C–S bonds under mild conditions with good functional group tolerance. Photoinduced copper-catalyzed C–S bond coupling represents a promising approach for green synthesis. While electron-rich nucleophiles have been the primary focus thus far, future research should prioritize enhancing the diversity of substrates and expanding the substrate scope by optimizing the copper catalytic systems.
4 Conclusions and perspectives
This review provides a detailed analysis of the existing literature on photoinduced Cu-catalyzed Ullmann cross-couplings, which encompass the formation of C–C, C–N, C–O, and C–S bonds, focusing on both homogeneous and heterogeneous copper catalysis. These methods tackle significant challenges encountered in traditional Ullmann reactions, such as the need for high temperatures, limited substrate diversity, and low atomic efficiency associated with copper catalysis.We also discuss the evolution of reaction mechanisms, moving from oxidative addition/reductive elimination (OA/RE) to iodine atom transfer (IAT) and radical nucleophilic substitution (SRN1), and ultimately to the single-electron transfer (SET) mechanism, which employs light as an energy source (see Scheme 2I–IV). The SET mechanism has attracted considerable interest due to its ability to address the limitations of oxidative addition in copper-catalyzed reactions, particularly the instability, and challenges in forming trivalent copper intermediates. By using more stable divalent copper intermediates, this approach allows for milder reaction conditions, a broader range of substrates, lower copper concentrations, and improved yields. These advancements highlight the potential of photoinduced Cu-catalyzed Ullmann cross-couplings as a more efficient, environmentally friendly, and economically sustainable method.
However, despite these advancements, several challenges remain that limit the widespread application of these techniques. For instance, the dependence on harmful UV light sources in certain reactions raises safety and environmental issues. Additionally, there is a lack of studies focusing on reactions involving electron-poor nucleophiles and less reactive aryl halides, which restricts the range of possible transformations. Moreover, the energy barriers associated with the iodine atom transfer (IAT) and single-electron transfer (SET) mechanisms are closely aligned, making it difficult to completely eliminate the IAT pathway in many cases, and research into the photocatalytic IAT mechanism is still limited. Catalyst leaching and unwanted side reactions also present significant challenges. The development of heterogeneous catalysts offers a promising solution to these issues. By employing surface adsorption and taking advantage of synergistic interactions among multiple catalytic sites, heterogeneous catalysts can function effectively under visible light, thereby enhancing reaction efficiency and selectivity while addressing the problems linked to traditional catalysis.
Data availability
No new data were created or analyzed in this study. Data sharing is not applicable.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to acknowledge the financial support from the Henan Province Science and Technology Development Plan Project (242102230066), National Natural Science Foundation of China (U1904184 and U1904191), and the Key Scientific Research Project Plan of Higher Education Institutions in Henan Province (24A150004).References
- F. Ullmann and J. Bielecki, Ueber Synthesen in der Biphenylreihe, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- F. Ullmann, Ueber eine Neue Bildungsweise von Diphenylaminderivaten, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- F. Ullmann and P. Sponagel, Ueber die Phenylirung von Phenolen, Ber. Dtsch. Chem. Ges., 1905, 38, 2211–2212 CrossRef CAS.
- I. Goldberg, Ueber Phenylirungen bei Gegenwart von Kupfer als Katalysator, Ber. Dtsch. Chem. Ges., 1906, 39, 1691–1692 CrossRef.
- W. R. H. Hurtley, CCXLIV.—Replacement of Halogen in Orthobromo-Benzoic Acid, J. Chem. Soc. (Resumed), 1929, 1870–1873 RSC.
- A. Hossain, A. Bhattacharyya and O. Reiser, Copper's Rapid Ascent in Visible-Light Photoredox Catalysis, Science, 2019, 364, eaav9713 CrossRef.
- A. D. Kute, R. P. Gaikwad, I. R. Warkad and M. B. Gawande, A Review on the Synthesis and Applications of Sustainable Copper-Based Nanomaterials, Green Chem., 2022, 24, 3502–3573 RSC.
- J. F. Hartwig, Carbon–Heteroatom Bond Formation Catalysed by Organometallic Complexes, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- G. Evano, N. Blanchard and M. Toumi, Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Copper in Cross-Coupling Reactions, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS.
- F. Monnier and M. Taillefer, Catalytic C–C, C–N, and C–O Ullmann-Type Coupling Reactions, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS.
- Q. Yang, Y. Zhao and D. Ma, Cu-Mediated Ullmann-type Cross-Coupling and Industrial Applications in Route Design, Process Development, and Scale-up of Pharmaceutical and Agrochemical Processes, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- A. Kolanowska, A. W. Kuziel, R. G. Jędrysiak, M. Krzywiecki, E. Korczeniewski, M. Wiśniewski, A. P. Terzyk and S. Boncel, Ullmann Reactions of Carbon Nanotubes—Advantageous and Unexplored Functionalization toward Tunable Surface Chemistry, Nanomaterials, 2019, 9, 1619 CrossRef CAS PubMed.
- J. M. Fordham, P. Kollmus, M. Cavegn, R. Schneider and M. Santagostino, A ‘pool and Split’ Approach to the Optimization of Challenging Pd-Catalyzed C–N Cross-Coupling Reactions, J. Org. Chem., 2022, 87, 4400–4414 CrossRef CAS PubMed.
- S. S. Murthy Bandaru, S. Bhilare, N. Chrysochos, V. Gayakhe, I. Trentin, C. Schulzke and A. R. Kapdi, Pd/PTABS: Catalyst for Room Temperature Amination of Heteroarenes, Org. Lett., 2018, 20, 473–476 CrossRef CAS PubMed.
- W. Xu, C. Liu, D. Xiang, Q. Luo, Y. Shu, H. Lin, Y. Hu, Z. Zhang and Y. Ouyang, Palladium Catalyst Immobilized on Functionalized Microporous Organic Polymers For C–C Coupling Reactions, RSC Adv., 2019, 9, 34595–34600 RSC.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Selected Copper-Based Reactions for C–N, C–O, C–S, and C–C Bond Formation, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Photoinduced Ullmann C–N Coupling: Demonstrating the Viability of a Radical Pathway, Science, 2012, 338, 647–651 CrossRef CAS PubMed.
- C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, A New Family of Nucleophiles for Photoinduced, Copper-Catalyzed Cross-Couplings via Single-Electron Transfer: Reactions of Thiols with Aryl Halides under Mild Conditions (O °C), J. Am. Chem. Soc., 2013, 135, 9548–9552 CrossRef CAS.
- B. Michelet, C. Deldaele, S. Kajouj, C. Moucheron and G. Evano, A General Copper Catalyst for Photoredox Transformations of Organic Halides, Org. Lett., 2017, 19, 3576–3579 CrossRef CAS PubMed.
- S. Zhu, H. Li, Y. Li, Z. Huang and L. Chu, Exploring Visible Light for Carbon–Nitrogen and Carbon–Oxygen Bond Formation via Nickel Catalysis, Org. Chem. Front., 2023, 10, 548–569 RSC.
- J.-H. Qin, Y. Wang, J.-Y. Ouyang, M. Liu and X.-H. Ouyang, Recent Progress in the Synthesis of N-Substituted Arylamines by Reductive Cross-Coupling of Nitroarenes, Org. Chem. Front., 2024, 11, 2638–2664 RSC.
- S. S. Talekar, S. Dutta, M. V. Mane and B. Maity, Visible Light–Induced Photoredox and Copper-Catalyzed C–N Cross-Coupling: A Mechanistic Perspective, Eur. J. Org. Chem., 2024, e202301312 CrossRef CAS.
- G. M. Whitesides, W. F. Fischer Jr., J. San Filippo Jr., R. W. Bashe and H. O. House, Reaction of Lithium Dialkyl- and Diarylcuprates with Organic Halides, J. Am. Chem. Soc., 1969, 91, 4871–4882 CrossRef CAS.
- G. M. Whitesides and P. E. Kendall, Stereochemistry of the Conjugate Addition of Derivatives of Endo-2-Norbornylcopper(I) to Mesityl Oxide, J. Org. Chem., 1972, 37, 3718–3725 CrossRef CAS.
- C. R. Johnson and G. A. Dutra, Reactions of Lithium Diorganocuprates(I) with Tosylates. II. Stereochemical, Kinetic, and Mechanistic Aspects, J. Am. Chem. Soc., 1973, 95, 7783–7788 CrossRef CAS.
- T. Cohen, J. Wood and A. G. Dietz, Organocopper Intermediates in the Exchange Reaction of Aryl Halides with Salts of Copper(I). the Possible Role of Copper(III), Tetrahedron Lett., 1974, 15, 3555–3558 CrossRef.
- M. W. Johnson, K. I. Hannoun, Y. Tan, G. C. Fu and J. C. Peters, A Mechanistic Investigation of the Photoinduced, Copper-Mediated Cross-Coupling of an Aryl Thiol with an Aryl Halide, Chem. Sci., 2016, 7, 4091–4100 RSC.
- J. F. Bunnett and C. C. Wamser, Radical Abstraction of Iodine from Aryl Iodides, J. Am. Chem. Soc., 1966, 88, 5534–5537 CrossRef CAS.
- G. O. Jones, P. Liu, K. N. Houk and S. L. Buchwald, Computational Explorations of Mechanisms and Ligand-Directed Selectivities of Copper-Catalyzed Ullmann-type Reactions, J. Am. Chem. Soc., 2010, 132, 6205–6213 CrossRef CAS.
- C. L. Jenkins and J. K. Kochi, Homolytic and Ionic Mechanisms in the Ligand-Transfer Oxidation of Alkyl Radicals by Copper(II) Halides and Pseudohalides, J. Am. Chem. Soc., 1972, 94, 856–865 CrossRef CAS.
- A. Cai, W. Yan, C. Wang and W. Liu, Copper-Catalyzed Difluoromethylation of Alkyl Iodides Enabled by Aryl Radical Activation of Carbon–Iodine Bonds, Angew. Chem., Int. Ed., 2021, 60, 27070–27077 CrossRef CAS.
- N. Kornblum, R. E. Michel and R. C. Kerber, Chain Reactions in Substitution Processes which Proceed via Radical-Anion Intermediates, J. Am. Chem. Soc., 1966, 88, 5662–5663 CrossRef CAS.
- G. A. Russell and W. C. Danen, Coupling Reactions of the 2-Nitro-2-Propyl Anion, J. Am. Chem. Soc., 1966, 88, 5663–5665 CrossRef CAS.
- J. F. Bunnett, Aromatic Substitution by the SRN1 Mechanism, Acc. Chem. Res., 1978, 11, 413–420 CrossRef CAS.
- S. Arai, T. Yamagishi, S. Ototake and M. Hida, Ullmann Condensation Reaction of Haloanthraquinone Derivatives with Amines in Aprotic Solvents, Bull. Chem. Soc. Jpn., 1977, 50, 547–548 CrossRef CAS.
- J. K. Kochi, Organometallic Mechanisms and Catalysis: The Role of Reactive Intermediates in Organic Processes, Academic Press, New York, 1978 Search PubMed.
- E. Sperotto, G. P. M. Van Klink, G. Van Koten and J. G. De Vries, The Mechanism of the Modified Ullmann Reaction, Dalton Trans., 2010, 39, 10338 RSC.
- S. J. Blanksby and G. B. Ellison, Bond Dissociation Energies of Organic Molecules, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Selective, Nickel-Catalyzed Hydrogenolysis of Aryl Ethers, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- M. Krzeszewski, O. Vakuliuk and D. T. Gryko, Color-Tunable Fluorescent Dyes Based on Benzo[c]coumarin, Eur. J. Org. Chem., 2013, 5631–5644 CrossRef CAS.
- Z. T. He and J. F. Hartwig, Palladium-Catalyzed α-Arylation of Carboxylic Acids and Secondary Amides via a Traceless Protecting Strategy, J. Am. Chem. Soc., 2019, 141, 11749–11753 CrossRef CAS PubMed.
- K. Kudo and N. Yamamoto, Scalable Synthesis of 8-Amino-3-Hydroxy-6H-Benzo[c]Chromen-6-One: Key Intermediate for SEGRA via the Hurtley Reaction, Org. Process Res. Dev., 2015, 19, 309–314 CrossRef CAS.
- R. Akhtar, A. F. Zahoor, M. Irfan, T. H. Bokhari and A. ul Haq, Recent Green Synthetic Approaches toward Ullmann Reaction: A Review, Chem. Pap., 2022, 76, 7275–7293 CrossRef CAS.
- F. Yang, J. Koeller and L. Ackermann, Photoinduced Copper-Catalyzed C–H Arylation at Room Temperature, Angew. Chem., Int. Ed., 2016, 55, 4759–4762 CrossRef CAS.
- X. Ma and G. Zhang, Visible Light-Induced Copper-Catalyzed C–H Arylation of Benzoxazoles, Chin. J. Chem., 2020, 38, 1299–1303 CrossRef CAS.
- J. Wang, Y. Liu, X. Zong, A. Lei and Z. Sun, Recent Advances in the Heterogeneous Photochemical Synthesis of C–N Bonds, Green Chem., 2023, 25, 5010–5023 RSC.
- Z. Wang, X. Hu, Z. Liu, G. Zou, G. Wang and K. Zhang, Recent Developments in Polymeric Carbon Nitride-Derived Photocatalysts and Electrocatalysts for Nitrogen Fixation, ACS Catal., 2019, 9, 10260–10278 CrossRef CAS.
- Z. Zhang, Y. Xu, Q. Zhang, S. Fang, H. Sun, W. Ou and C. Su, Semi-Heterogeneous Photo-Cu-Dual-Catalytic Cross-Coupling Reactions Using Polymeric Carbon Nitrides, Sci. Bull., 2022, 67, 71–78 CrossRef CAS PubMed.
- S. U. Son, I. K. Park, J. Park and T. Hyeon, Synthesis of Cu2O Coated Cu Nanoparticles and Their Successful Applications to Ullmann-type Amination Coupling Reactions of Aryl Chlorides, Chem. Commun., 2004, 778 RSC.
- D.-L. Zhu, J. Li, D. J. Young, Y. Wang and H.-X. Li, Visible-Light-Initiated Nickel-Catalyzed Amination of Aryl Halides Using Thioxanthen-9-one as a Photocatalyst, Org. Chem. Front., 2023, 10, 3612–3618 RSC.
- E.-C. Elliott, J. L. Maggs, B. K. Park, P. M. O’Neill and A. V. Stachulski, Convenient Syntheses of Halo–Dibenz[b,f]Azepines and Carbamazepine Analogues via N–Arylindoles, Org. Biomol. Chem., 2013, 11, 8426–8434 RSC.
- Z. Xu, W. Wang, Y. Xu, S. Song, J. Yu, W. Song, H. Xu, H. Fu and Z. Li, Visible Light-Promoted Goldberg Reaction Driven by a Preponderant Facet of CuO Nanocrystals, ACS Catal., 2023, 13, 13920–13930 CrossRef CAS.
- D. T. Ziegler, J. Choi, J. M. Muñoz-Molina, A. C. Bissember, J. C. Peters and G. C. Fu, A Versatile Approach to Ullmann C–N Couplings at Room Temperature: New Families of Nucleophiles and Electrophiles for Photoinduced, Copper–Catalyzed Processes, J. Am. Chem. Soc., 2013, 135, 13107–13112 CrossRef CAS PubMed.
- W. J. Yoo, T. Tsukamoto and S. Kobayashi, Visible Light–Mediated Ullmann–type C–N Coupling Reactions of Carbazole Derivatives and Aryl Iodides, Org. Lett., 2015, 17, 3640–3642 CrossRef CAS PubMed.
- S. Sana, S. Dastari, D. S. Reddy, R. Tokala, M. Sathish, R. Sonti and N. Shankaraiah, Visible-Light-Mediated Photocatalytic Sequential N–Arylation: An Eco-Friendly Synthetic Route to Unsymmetrical Diarylamines and the Imatinib Drug, Org. Chem. Front., 2023, 10, 4573–4580 RSC.
- G. Singh, M. Kumar and V. Bhalla, Ultrafine Hybrid Cu2O–Fe2O3 Nanoparticles Stabilized by Hexaphenylbenzene–Based Supramolecular Assemblies: A Photocatalytic System for the Ullmann–Goldberg Coupling Reaction, Green Chem., 2018, 20, 5346–5357 RSC.
- M. Li, X. Xing, Z. Ma, J. Lv, P. Fu and Z. Li, Synthesis of Composition Tunable and (111)–Faceted Cu/Cu2O Nanoparticles toward Photocatalytic, Ligand–Free, and Solvent–Free C–N Ullmann Coupling Reactions, ACS Sustainable Chem. Eng., 2018, 6, 5495–5503 CrossRef CAS.
- E. Wang, K. Chen, Y. Chen, J. Zhang, X. Lin and M. Chen, Porous Polymeric Ligand Promoted Copper-Catalyzed C–N Coupling of (Hetero)Aryl Chlorides under Visible-Light Irradiation, Sci. China: Chem., 2021, 64, 17–21 CrossRef CAS.
- G.-J. Zha, W. Ji, Z.-H. Qi, W.-J. Qiu, A.-M. Li, D.-R. Zhu and S. Jing, Microenvironment Modulation of Cuprous Cluster Enables Inert Aryl Chlorides Activation in Single–Molecule Metallaphotoredox Amination, J. Catal., 2022, 405, 313–321 CrossRef CAS.
- A. Sagadevan, A. Ghosh, P. Maity, O. F. Mohammed, O. M. Bakr and M. Rueping, Visible–Light Copper Nanocluster Catalysis for the C–N Coupling of Aryl Chlorides at Room Temperature, J. Am. Chem. Soc., 2022, 144, 12052–12061 CrossRef CAS PubMed.
- A. Zanka, A. Kubota, S. Hirabayashi and H. Nakamura, Process Development of a Novel Anti-Inflammatory Agent. The Regiospecific Bromination of 4′-Acetylmethanesulfonanilide, Org. Process Res. Dev., 1998, 2, 71–77 CrossRef CAS.
- S. G. Ruggeri, B. C. Vanderplas, B. G. Anderson, R. Breitenbach, F. J. Urban, I. Stewart, A. Morgan and G. R. Young, Regioselective Addition of Mesitol to a 2,4-Dichloropyridine, Org. Process Res. Dev., 2008, 12, 411–413 CrossRef CAS.
- S. Caron, N. M. Do, J. E. Sieser, D. C. Whritenour and P. D. Hill, Preparation of a Corticotropin-Releasing Factor Antagonist by Nucleophilic Aromatic Substitution and Copper-Mediated Ether Formation, Org. Process Res. Dev., 2009, 13, 324–330 CrossRef CAS.
- M. Wolter, G. Nordmann, G. E. Job and S. L. Buchwald, Copper-Catalyzed Coupling of Aryl Iodides with Aliphatic Alcohols, Org. Lett., 2002, 4, 973–976 CrossRef CAS.
- F. Gorginpour and H. Zali-Boeini, Synergistic Effect of Copper Nanocrystals-Nanoparticles Incorporated in a Porous Organic Polymer for the Ullmann C–O Coupling Reaction, Mol. Catal., 2021, 504, 111460 CrossRef CAS.
- D. R. McMillin, M. T. Buckner and B. T. Ahn, A Light-Induced Redox Reaction of Bis(2,9-Dimethyl-1,10-Phenanthroline) Copper(I), Inorg. Chem., 1977, 16, 943–945 CrossRef CAS.
- Y. Tan, J. M. Muñoz-Molina, G. C. Fu and J. C. Peters, Oxygen Nucleophiles as Reaction Partners in Photoinduced, Copper-Catalyzed Cross-Couplings: O-Arylations of Phenols at Room Temperature, Chem. Sci., 2014, 5, 2831–2835 RSC.
- J. Y. Kim, J. C. Park, A. Kim, A. Y. Kim, H. J. Lee, H. Song and K. H. Park, Cu2O Nanocube-Catalyzed Cross-Coupling of Aryl Halides with Phenols via Ullmann Coupling, Eur. J. Inorg. Chem., 2009, 2009, 4219–4223 CrossRef.
- L. Wang, J. Zhang, J. Sun, L. Zhu, H. Zhang, F. Liu, D. Zheng, X. Meng, X. Shi and F. Xiao, Copper-Incorporated Porous Polydivinylbenzene as Efficient and Recyclable Heterogeneous Catalyst in Ullmann Biaryl Ether Coupling, ChemCatChem, 2013, 5, 1606–1613 CrossRef CAS.
- Sk. M. Islam, N. Salam, P. Mondal, A. S. Roy, K. Ghosh and K. Tuhina, A Highly Active Reusable Polymer Anchored Copper Catalyst for C–O, C–N and C–S Cross-Coupling Reactions, J. Mol. Catal. A: Chem., 2014, 387, 7–19 CrossRef CAS.
- K. Mullick, S. Biswas, C. Kim, R. Ramprasad, A. M. Angeles-Boza and S. L. Suib, Ullmann Reaction Catalyzed by Heterogeneous Mesoporous Copper/Manganese Oxide: A Kinetic and Mechanistic Analysis, Inorg. Chem., 2017, 56, 10290–10297 CrossRef CAS PubMed.
- H. Q. Ha, H. T. D. Nguyen, T. H. M. Pham, V. T. Pham and T. Truong, Towards Optical Application of Metal-Organic Frameworks: Cu-MOFs as Sole Heterogeneous Photocatalyst for Arylation of Phenols at Room Temperature, Catal. Commun., 2018, 117, 79–84 CrossRef CAS.
- R. Ji, X. Jie, Y. Zhou, Y. Wang, B. Li, X. Liu and J. Zhao, Light-Assisted Ullmann Coupling of Phenols and Aryl Halides: The Synergetic Effect between Plasmonic Copper Nanoparticles and Carbon Nanotubes from Various Sources, Chem. – Eur. J., 2022, 28, e202103703 CrossRef CAS PubMed.
- R. G. Gish, C. Porta, L. Lazar, P. Ruff, R. Feld, A. Croitoru, L. Feun, K. Jeziorski, J. Leighton, J. Knox, J. Gallo and G. T. Kennealey, Phase III Randomized Controlled Trial Comparing the Survival of Patients with Unresectable Hepatocellular Carcinoma Treated with Nolatrexed or Doxorubicin, J. Clin. Oncol., 2007, 25, 3069–3075 CrossRef CAS.
- H. Malmgren, B. Bäckström, E. Sølver and J. Wennerberg, A Large Scale Process for the Preparation of Thymitaq, Org. Process Res. Dev., 2008, 12, 1195–1200 CrossRef CAS.
- B. Patel, C. R. Firkin, E. W. Snape, S. L. Jenkin, D. Brown, J. G. K. Chaffey, P. A. Hopes, C. D. Reens, M. Butters and J. D. Moseley, Process Development and Scale-Up of AZD7545, a PDK Inhibitor, Org. Process Res. Dev., 2012, 16, 447–460 CrossRef CAS.
- R. Adams, W. Reifschneider and D. Nair, The Preparation of Aryl Thioethers by Reaction of Aromatic Halogen Compounds with Cuprous Thiophenolate or Cuprous Thiobutylate, Croat. Chem. Acta, 1957, 29, 277–285 CAS.
- W. R. Bowman, H. Heaney and P. H. G. Smith, Intramolecular Aromatic Substitution (SRN1) Reactions, Use of Entrainment for the Preparation of Benzothiazoles, Tetrahedron Lett., 1982, 23, 5093–5096 CrossRef CAS.
- W. R. Bowman, H. Heaney and P. H. G. Smith, Copper(I) Catalysed Aromatic Nucleophilic Substitution: A Mechanistic and Synthetic Comparison with the SRN1 Reaction, Tetrahedron Lett., 1984, 25, 5821–5824 CrossRef CAS.
- P. Gandeepan, J. Mo and L. Ackermann, Photo-induced Copper-Catalyzed C–H Chalcogenation of Azoles at Room Temperature, Chem. Commun., 2017, 53, 5906–5909 RSC.
- Q. Yan, W. Cui, X. Song, G. Xu, M. Jiang, K. Sun, J. Lv and D. Yang, Sulfonylation of Aryl Halides by Visible Light/Copper Catalysis, Org. Lett., 2021, 23, 3663–3668 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |