
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(BO)2-doped tetrathia[7]helicenes: synthesis and property-change induced by “BO bond inversion”†
Luigi
Menduti
 *ab,
Clara
Baldoli
c,
Simone
Manetto
d,
Claudio
Villani
d,
Marta
Penconi
ef,
Sara
Grecchi
a,
Serena
Arnaboldi
a,
Giuseppe
Mazzeo
g,
Giovanna
Longhi
g,
Michael
Bolte
b,
Alexander
Virovets
b,
Hans-Wolfram
Lerner
b,
Matthias
Wagner
*b and
Emanuela
Licandro
a
*ab,
Clara
Baldoli
c,
Simone
Manetto
d,
Claudio
Villani
d,
Marta
Penconi
ef,
Sara
Grecchi
a,
Serena
Arnaboldi
a,
Giuseppe
Mazzeo
g,
Giovanna
Longhi
g,
Michael
Bolte
b,
Alexander
Virovets
b,
Hans-Wolfram
Lerner
b,
Matthias
Wagner
*b and
Emanuela
Licandro
a
aDipartimento di Chimica, Università degli Studi di Milano, Via C. Golgi 19, 20133, Milano, Italy. E-mail: luigi.menduti@unimi.it
bInstitut für Anorganische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Strasse 7, 60438, Frankfurt am Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
cCNR Istituto di Scienze e Tecnologie Chimiche Giulio Natta, Via C. Golgi 19, 20133, Milano, Italy
dDipartimento di Chimica e Tecnologie del Farmaco, Sapienza Università di Roma, P.le A. Moro 5, 00185, Roma, Italy
eCNR Istituto di Scienze e Tecnologie Chimiche Giulio Natta, Via G. Fantoli 16/15, 20133 Milano, Italy
fSmartMatLab Center, via C. Golgi 19, 20133 Milano, Italy
gDipartimento di Medicina Molecolare e Traslazionale, Università di Brescia, Viale Europa 11, 25123, Brescia, Italy
First published on 20th November 2024
Abstract
Helical distortion of polyaromatic hydrocarbons gives rise to a special class of π-conjugated systems, namely helicenes. Owing to their configurational stability and easily tunable optoelectronic properties (via heteroatom-doping), helicenes have recently come to the fore as building blocks for applications in materials science (CP-OLEDs, chiroptical switches). In this context, boron-doped helicenes are particularly promising. Herein, we report the synthesis of the new (BO)2-doped tetrathia[7]helicene 2, derived by the formal inversion of (Mes)B–O moieties in the (previously reported) isomer 1. Theoretical characterization of 2, and comparison with 1, revealed that the inversion of the BO vectors promotes the extension of the LUMO via the central thiophene–benzene–thiophene fragment (and not via the terminal thiophene rings, as in 1), resulting in a considerable lowering of the LUMO energy (ELUMO(2) = −2.22 eV vs. ELUMO(1) = −1.65 eV). Spectroscopic studies revealed that the “BO bond inversion” also contributes to the narrowing of the energy gap (Eoptg (2) = 2.90 eV vs. Eoptg (1) = 3.16 eV) and causes a significant red-shift of the absorption/emission bands (≈40 nm). Interestingly, besides low fluorescence quantum yield (ΦPL(2) = 7%), 2 shows detectable circularly polarized luminescence (glum = 0.8 × 10−3) and pronounced phosphorescence at low temperature (77 K). (P)-/(M)-enantiomers of 2 were successfully separated by CSP-UHPLC and proved to be stable (ΔG‡enant = 29.4 ± 0.1 kcal mol−1 at 353 K). Racemization studies combined with theoretical calculations confirmed that BO-doping is an extremely perturbative tool for tuning the mechanical rigidity of tetrathia[7]helicenes (ΔG‡enant (2) is 10 kcal mol−1 lower than ΔG‡enant (7TH)).
Introduction
Helicenes, which are non-planar, screw-shaped chiral structures composed of ortho-fused aromatic or heteroaromatic rings, represent a unique class of polycyclic aromatic hydrocarbons (PAHs).1 Configurationally stable helicenes show peculiar chiroptical and optoelectronic features suitable for their application in the field of materials science.2,3Recently, the so-called heteroatom doping, based on the replacement of selected carbon atoms by other p-block elements,4 opened new perspectives in helicene chemistry, allowing the modulation of their electronic structures and optoelectronic properties. The most commonly used doping elements to date are Si, N, P, O and S, but recently several examples of B-atom introduction have been described.5 Only a limited number of purely B(sp2)-doped helicenes and chiral PAHs incorporating helicene subunits have been reported so far,6 while in most of the examples, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds are replaced by B(sp2)–N/B(sp2)–O7,8 or B(sp3)–N bonds.9 Our groups have recently contributed to this field by establishing the synthesis and investigating the optoelectronic and chiroptical properties of the first doubly BO-doped tetrathia[7]helicene 110 (Fig. 1).
C double bonds are replaced by B(sp2)–N/B(sp2)–O7,8 or B(sp3)–N bonds.9 Our groups have recently contributed to this field by establishing the synthesis and investigating the optoelectronic and chiroptical properties of the first doubly BO-doped tetrathia[7]helicene 110 (Fig. 1).
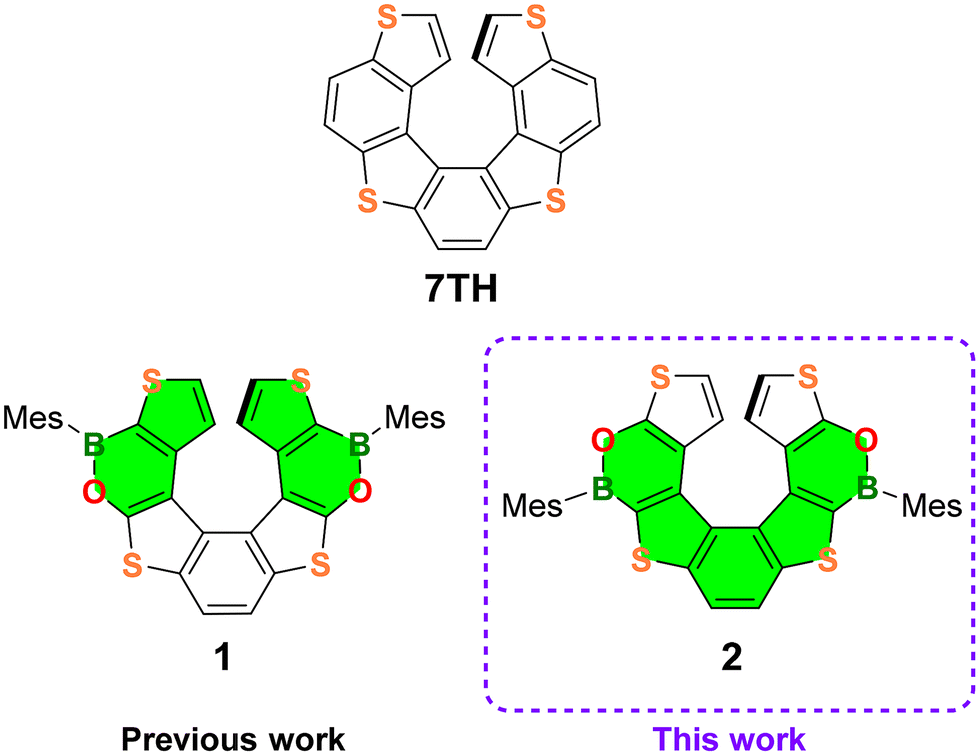 | ||
| Fig. 1 Structures of the parent tetrathia[7]helicene (7TH), the previously reported 1 and the newly developed (BO)2-helicene 2. Mes = 2,4,6-trimethylphenyl (mesityl). The LUMO extension in the structures of 1 and 2 is highlighted in green. | ||
Although 1 showed poor optical performance (ΦPL = 6%, Eoptg = 3.16 eV), our studies have demonstrated that the incorporation of two BO fragments into the tetrathiahelicene (7TH; Fig. 1) scaffold provides a powerful tool to manipulate the mechanical rigidity, the chiroptical properties, and the electron distribution across the helix.
With the aim of thoroughly investigating the effect of BO doping of the 7TH framework to obtain helicenes with improved photophysical features, we designed the new (BO)2-helicene 2, an isomer of 1, derived by the formal inversion of the BO bonds in the two oxaborine rings (Fig. 1).
(BO)2-helicene 2 was tailored based on our previous results, which showed that the LUMO of 1 (highlighted in green in Fig. 1) is mainly localized on the B-substituted terminal thiophene rings,10 while the HOMO, as in 7TH,11 is spread over the entire helix (for DFT calculations, see Fig. 6 and Fig. S49†).
These results suggested that the position of the B atoms could be crucial for the extension of the LUMO across the helix, with only minor effects on the HOMO distribution/energy (as in the case of boron-doped planar polyaromatics).4c The inverted positions of the BO bonds in 2 were therefore expected to promote the extension of the LUMO via the central thiophene–benzene–thiophene fragment rather than via the terminal thiophene rings only (Fig. 1). This could provide a helicene with an extended and energetically lowered LUMO, a narrowed HOMO–LUMO energy gap, and improved optoelectronic performance.
Herein, we report the synthesis of the (BO)2-helicene 2, along with its optoelectronic and chiroptical properties and a comparison with those of the previously reported (BO)2-helicene isomer 1.10 Moreover, we describe attempts to synthesize the (BO)-doped helicene 3 (Scheme 2a), formally derived by the replacement of the central CC bond in 7TH with a (Mes)B–O fragment. Although the target BO-helicene 3 was not obtained, the investigation of two different synthetic approaches provided important information for the design of new derivatives and allowed us to isolate the penta-fused intermediate 11 (see Results and discussion).
Results and discussion
Synthesis, NMR spectra, and X-ray characterization of 2
The (BO)2-helicene 2 was prepared in four steps, starting from 4-bromothiophene-2-carbaldehyde12 (Scheme 1), which was converted into the alkene 4 in 89% yield (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis = 84:16) via a McMurry coupling. The trans isomer was isolated in 82% yield by washing the crude product with Et2O.
:cis = 84:16) via a McMurry coupling. The trans isomer was isolated in 82% yield by washing the crude product with Et2O.
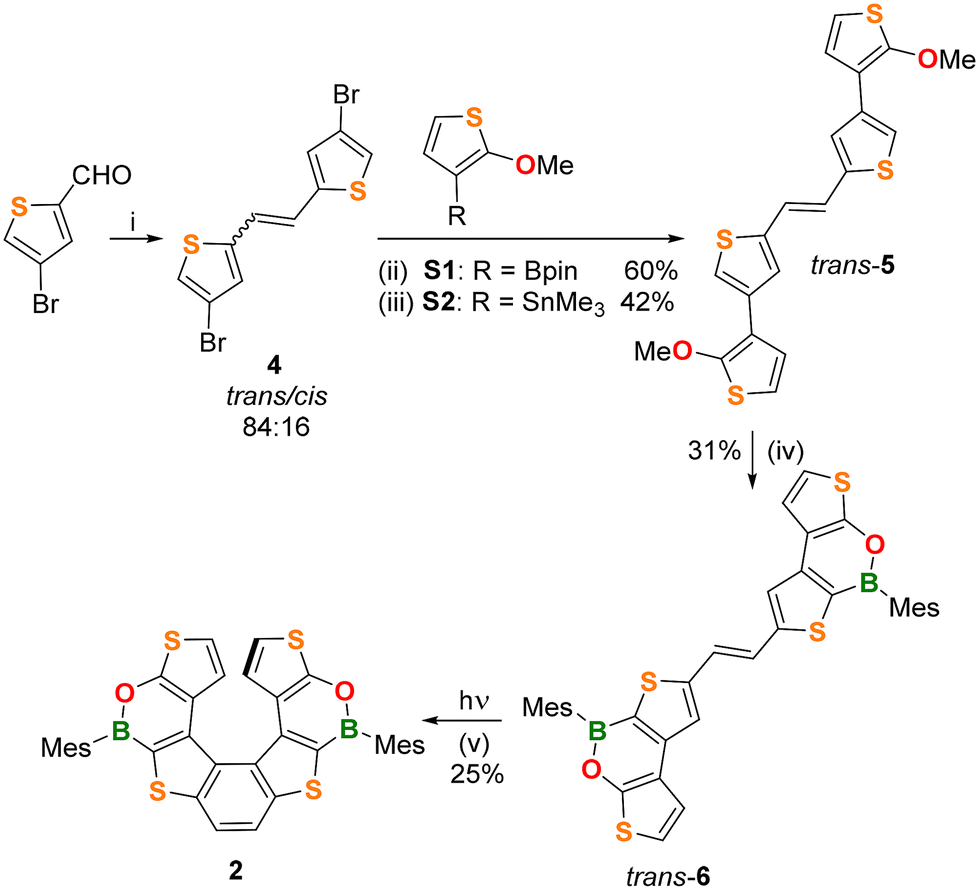 | ||
| Scheme 1 (i) 2.6 equiv. Zn, 1.2 equiv. TiCl4, THF, 0–15 °C; (ii) 2.4 equiv. S1 (R = Bpin), Pd(PPh3)4 10 mol%, 4 equiv. NaOH (3 M), THF, 80 °C, 15 h; (iii) 2.1 equiv. S2 (R = SnMe3), Pd(Pt-Bu3)2 10 mol%, toluene, 80 °C, 17 h; (iv) 2.2 equiv. BCl3, 2 equiv. Et3N, 2.4 equiv. [nBu4N]I, chlorobenzene, 135 °C, then THF, 6 equiv. MesMgBr; and (v) 405 nm LED, C6D6, r.t., 1.5 h. Mes = 2,4,6-trimethylphenyl (mesityl). | ||
The next step was performed on trans-4 and involved Suzuki or Stille coupling with the corresponding partners 2-(2-methoxythiophen-3-yl)-1,3,2-dioxaborolane S1 or (2-methoxythiophen-3-yl)trimethylstannane S2 and furnished the alkene trans-5 in 60% and 42% yields, respectively. Although Suzuki coupling gave a better yield, our preferred route for trans-5 remains Stille coupling, due to the easier preparation and purification of the stannane S2. The subsequent two-fold borylation of trans-5 was performed by demethylative borylation with BCl3/[nBu4N]I/Et3N13 in chlorobenzene at 135 °C, followed by the addition of MesMgBr. In this way, the alkene trans-6 was obtained in 31% yield after purification.
The structures and the trans-configurations of the double bonds in intermediates 4, 5, and 6 were confirmed by X-ray crystallography (Fig. S36–S38†). The last reaction to obtain the helicene structure was a Mallory photocyclization14 involving as the first step the isomerization of trans-6 to cis-6, which afterwards cyclized to give 2.
We first investigated the trans → cis isomerization. To this end, an NMR tube charged with a non-degassed C6D6 solution of trans-6 was irradiated with a 405 nm LED lamp and the reaction progress was monitored by 1H NMR. Surprisingly, this test showed that not only does trans-6 smoothly isomerize to cis-6 but also that cis-6 undergoes photocyclization to give 2, even in the absence of an added oxidant (e.g. I2; differently from what was required for the synthesis of 1).10
In light of these results, we then performed the synthesis of 2 under the same conditions: four NMR tubes charged with trans-6 and C6D6 (10 mg mL−1) were simultaneously irradiated with the 405 nm LED lamp until almost complete disappearance of the alkene precursor (1.5 h; NMR-spectroscopic control). The NMR solutions were then combined and, after chromatographic purification, (BO)2-helicene 2 was isolated in 25% yield.
NMR characterization of the new (BO)2-helicene 2 was done along with a comparison with the chemical shifts of the isomeric helicene 1 (Fig. 2a and b). The inversion of the BO-bonds in the oxaborine rings causes a substantial upfield shift of the diagnostic 1H and 13C signals of the terminal thiophene rings.
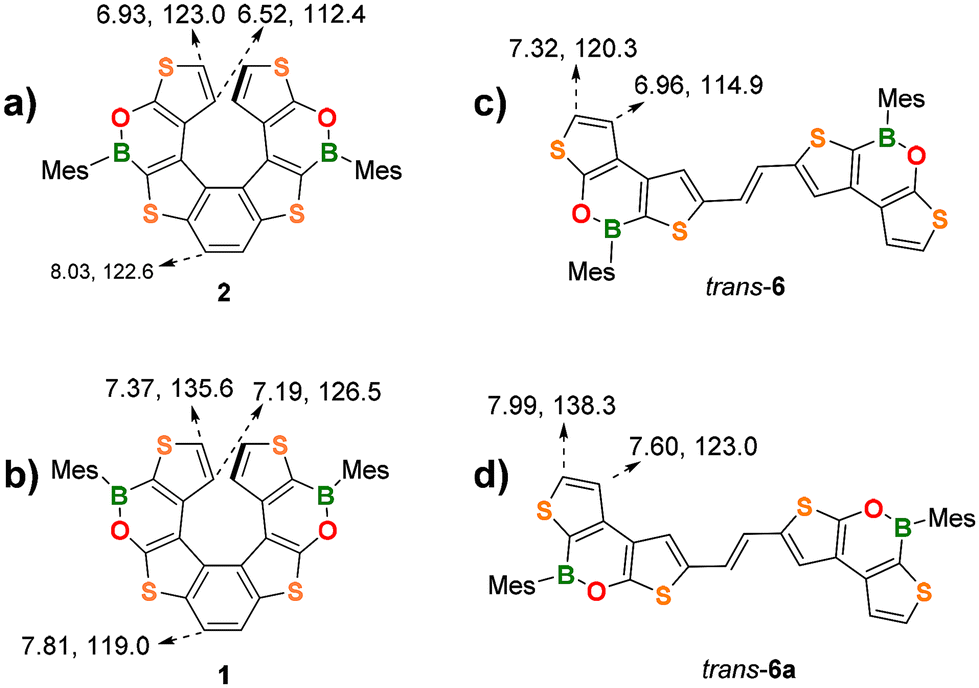 | ||
| Fig. 2 Comparison of diagnostic 1H and 13C NMR resonances (CDCl3) of the two (BO)2-helicenes (a) 2 and (b) 1 and of the trans-alkenes (c) 6 and (d) 6a. | ||
For instance, in 2, the protons at the α and β positions of the terminal thiophene rings substituted with an electron-donating O atom (O-thiophene) resonate at 6.93 and 6.52 ppm, respectively, whereas in the corresponding isomer 1, they are significantly deshielded due to the electron-withdrawing effect of the B atom (B-thiophene) and resonate at 7.37 and 7.19 ppm (cf. parent 7TH: 6.83, 6.67 ppm). The same trend is observed for the 13C shifts: 2 C(α) and C(β) resonate at 123.0 and 112.4 ppm, respectively, but are significantly deshielded in 1 (C(α) = 135.6 ppm, C(β) = 126.5 ppm).
A similar behavior is observed for the corresponding alkene precursors trans-6 and trans-6a for both the H(α)/C(α) and H(β)/C(β) resonances (Fig. 2c and d). Due to the proximity of the electron-withdrawing B atoms, the protons/carbons of the central benzene ring in 2 are deshielded (8.03/122.6 ppm) with respect to those at the same positions in 1 (7.81/119.0 ppm).
Interestingly, the α and β protons of the terminal thiophene rings of 2 are more shielded than those of its planar precursor trans-6 (6.93/6.52 ppm vs. 7.32/6.96 ppm). This effect can be attributed to the helical 3D-structure, in which each terminal thiophene ring falls into the shielding region of the corresponding ring in the opposite helicene wing. (BO)2-helicene 1 and its precursor trans-6a show the same trend.
The crystal structure of 2 was determined by single-crystal X-ray crystallography using the high-flux DESY Petra III synchrotron source. 2 crystallizes as a racemic mixture of the (P)- and (M)-enantiomers in the monoclinic crystal system (Fig. 3a). 2 shows a dihedral angle between the two terminal thiophene rings of 58.36(6)° (Fig. 3b), significantly larger than those of 1 (50.26(9)°)10 and 7TH (48.6(1)°),15 while the associated intramolecular centroid⋯centroid distances have comparable values (4.299(4) Å for 2; 4.153(6) Å for 1).
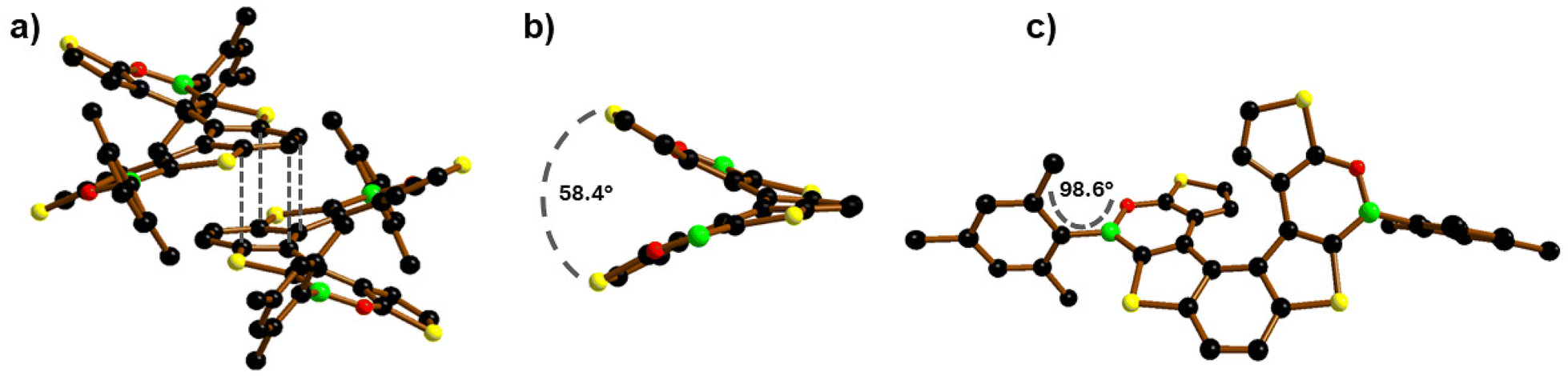 | ||
| Fig. 3 (a) π-stacking interaction between a (P)/(M)-pair of 2 with H atoms omitted for clarity;18 grey dashed lines indicate the shortest intermolecular C⋯C contacts. (b) Dihedral angle between the terminal thiophene rings of (P)-2; Mes rings are omitted for clarity. (c) Front view of (P)-2. B: green, C: black, O: red, and S: yellow spheres. | ||
As for 1, the central benzene ring of 2 is twisted (Fig. 3c), as evidenced by the torsion angle of −10.5° (9.7° for 1) between the (H)CC(H) vector and the opposite CC bond.
Contrary to 1 and similar to most of the Mes-protected B-doped polycyclic aromatic hydrocarbons (PAHs),16 the Mes substituents of 2 are nearly orthogonal to the oxaborine rings (Fig. 3c), as evidenced by the Mes//O–B–C dihedral angle (98.6(2)° for 2; 59.0(5)° and 58.6(6)° for 1). In both 2 and 1, the ortho-CH3 groups of the Mes substituents are positioned at approximately 4 Å to the CH(β) atoms of the opposite thiophene rings, thus excluding that the different Mes orientations in the two (BO)2-helicenes could be caused by steric factors (e.g. H/H clashes). The reason for the two different orientations apparently lies in the crystal packing of 1 and 2.
The B–O bond lengths of 2 (1.393(3) Å) are slightly shorter than those of 1 (1.402(4) and 1.401(5) Å) but longer than those of the reference compound 9-mesityl-10,9-oxaboraphenanthrene (av. B–O = 1.378 Å; av. Mes//O–B–C = 104.1(3) and 84.8(3)° for two crystallographically independent molecules in the asymmetric unit).17 In the crystal, alternating molecules of (P)- and (M)-2 form infinite rods by means of π-stacking interactions between the central benzene rings (Fig. 3a; similar to 7TH).15 The corresponding intramolecular centroid⋯centroid distances are 3.666(4) Å, and the shortest intermolecular C⋯C contacts are 3.552(4) and 3.666(4) Å (grey lines in Fig. 3a). As for 1, the shortest intermolecular S⋯S distances are smaller than the intramolecular ones (3.665(5) and 6.140(5) Å, respectively, for 2; 3.9911(14) and 5.8156(2) Å, respectively, for 1).
Synthesis attempts for 3
Next, we attempted the synthesis of the (BO)-helicene 3 (Scheme 2a), formally derived by the replacement of the central CC bond of 7TH with a (Mes)B–O fragment.
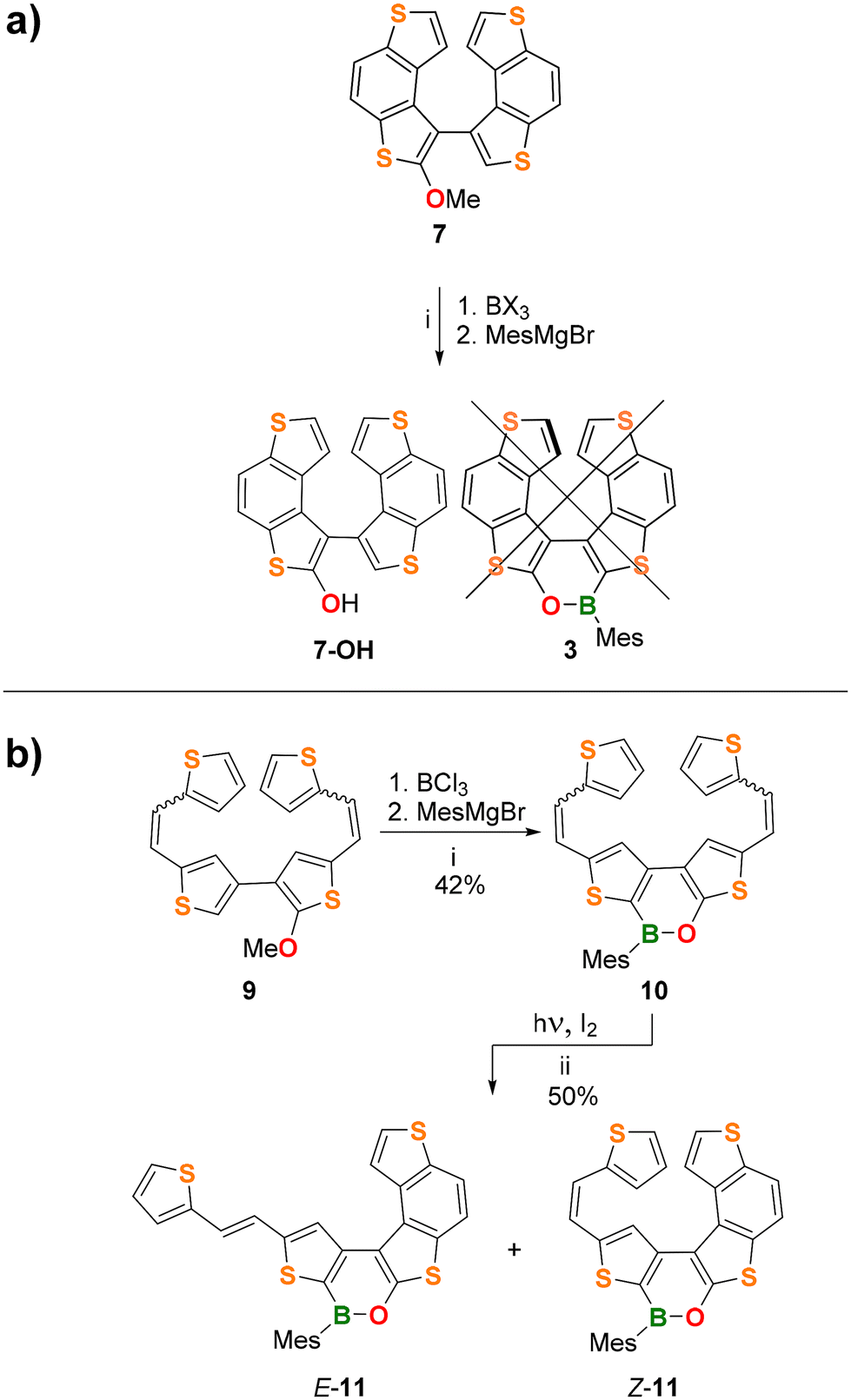 | ||
| Scheme 2 (a) (i) 1.5 equiv. BCl3 or BBr3, 1 equiv. Et3N, 1.2 equiv. [nBu4N]I, chlorobenzene, 135 °C, then THF, 3 equiv. MesMgBr; (b) (i) 1.1 equiv. BCl3, 1 equiv. Et3N, 1.2 equiv. [nBu4N]I, chlorobenzene, 135 °C, then THF, 3 equiv. MesMgBr; (ii) 385 nm LED, 2.1 equiv. I2, 200 equiv. propylene oxide, C6H6, r.t., 3.5 h. | ||
To this end, we pursued two different synthetic strategies (Scheme 2a and b). The first approach involved the late-stage borylation of 7 (Scheme 2a; see the ESI† for details of its synthesis). In the second approach, the oxaborine ring was constructed first (via borylative cyclization 9 → 10; see the ESI† for the synthesis of 9), and the obtained bis-olefinated oxaborine 10 was then subjected to Mallory photocyclization (Scheme 2b).
Unfortunately, the borylative cyclization 7 → 3 failed for BCl3 and BBr3; only the hydroxy derivative 7-OH was isolated besides a small amount of unidentified products (Scheme 2a; see the ESI for details, Table S1†). The crystal structure of the precursor compound 7 (Fig. S39†) indicates that the final B–C bond-forming step must overcome adverse steric factors and is therefore unfavorable.
In contrast, borylation of the more flexible bis-olefin 9 (see the ESI† for details of its synthesis) gave the bis-olefinated oxaborine 10 in a satisfactory yield (42%) as a mixture of four E/Z isomers after chromatographic purification. Treatment of the mixture with n-pentane allowed the isolation of crystals of E,E-10, the configuration of which was confirmed by single-crystal X-ray diffraction (Fig. 4a and Fig. S42†). Photocyclization of 10 was carried out on the mixture of isomers in benzene solution by irradiation with a 385 nm LED lamp for 3.5 h at room temperature in the presence of 2 equiv. of I2 and excess propylene oxide as an HI scavenger (Scheme 2b).19 In this way, the penta-fused product E/Z-11 was obtained in 50% yield as an almost equimolar mixture of the E and Z-isomers. We found no indication for the formation of 3. 1H NMR analysis of E/Z-11 allowed the assignment of most of the proton resonances of both structures (see the ESI†). An X-ray diffraction study of single crystals of E-11 confirmed that the photocyclization had occurred at the “O-thiophene” double bond (Fig. 4b and Fig. S43†).
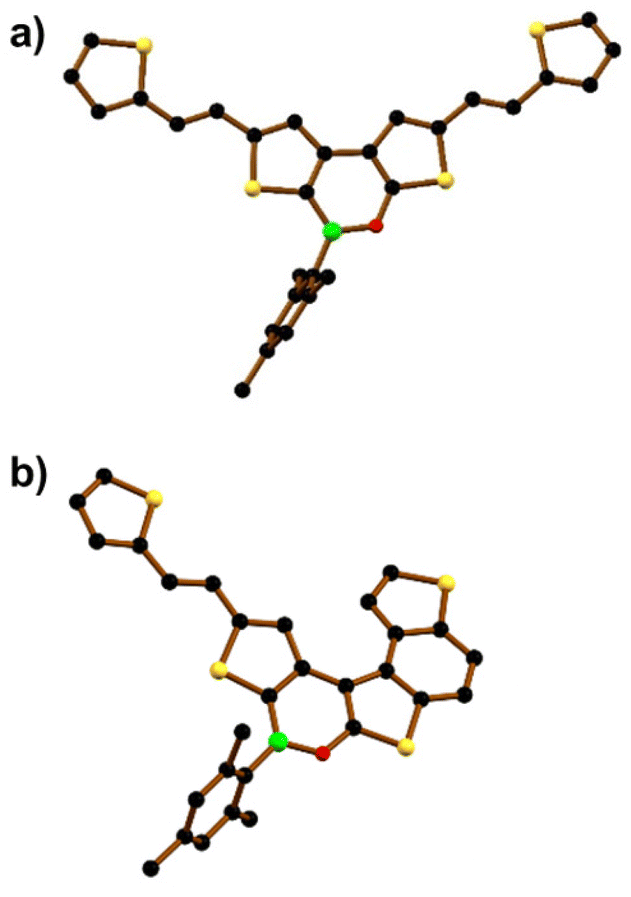 | ||
| Fig. 4 Solid-state structures of (a) E,E-10 and (b) E-11. Hydrogen atoms are omitted for clarity. B: green, C: black, O: red, and S: yellow spheres. | ||
Spectroscopic, electrochemical, theoretical, and chiroptical characterization of 2
The electrochemical, spectroscopic, and chiroptical properties of the new (BO)2-helicene 2 were investigated by cyclic voltammetry, UV/Vis and fluorescence spectroscopy, and chiral HPLC analysis and compared with the corresponding properties of isomer 1.Cyclic voltammetry measurements on 2 (THF, [nBu4N][PF6]; referenced against the FcH/FcH+ couple) showed two irreversible reduction processes with peak potentials of EIpc = −1.66 V and EIIpc = −2.36 V in the cathodic scan and an irreversible oxidation peak in the anodic scan (Epa = 0.82 V; Fig. S28, S29, and Table S3†). Helicene 2 is therefore easier to reduce than its isomer 1 (EIpc(1) = –2.83 V). Consequently, the electrochemically determined LUMO energy of 2 (ELUMO = –3.14 eV) is also significantly lower than that of 1 (ELUMO = –1.97 eV), thus confirming that the B–O-bond inversion contributes markedly to the stabilization of the LUMO.
The molar absorptivity and emission spectra of 2 and 1 (c-hexane) are shown in Fig. 5a; the data are summarized in Table 1. Both compounds show an absorption band below 300 nm and a second, more structured band at lower energy, which is red-shifted in 2 and characterized by a lower absorption coefficient relative to 1. Consequently, the energy gap between the frontier orbitals is narrowed (Eoptg(2) = 2.90 eV vs. Eoptg(1) = 3.16 eV). The emission spectrum of 2 shows a band with a well-resolved vibrational fine structure and a maximum at 446 nm which, similar to the absorption band, is bathochromically shifted with respect to the spectrum of 1. The photoluminescence quantum efficiency of 2 remains unchanged, ΦPL = 7% (c-hexane; cf.1: λem = 411 nm, ΦPL = 6%; 7TH: λem ≈ 405 nm, ΦPL = 6%),20 and the lifetime of the excited state is τ = 0.82 ns.
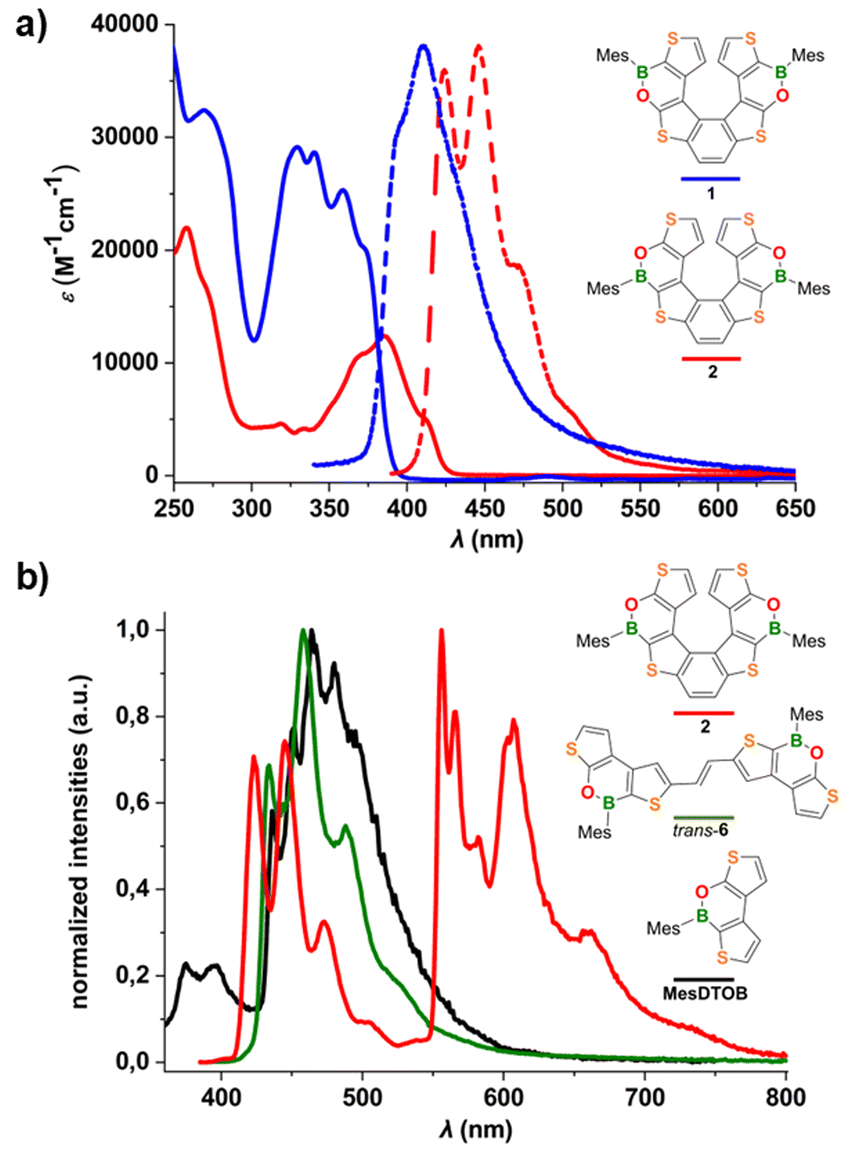 | ||
| Fig. 5 (a) Molar absorptivity (solid lines) and emission (dashed lines) spectra of 2 (red) and 1 (blue) in c-hexane. (b) Low-temperature emission spectra of 2 (red), trans-6 (green) and MesDTOB (black) in a frozen toluene matrix (77 K). | ||
| c-Hexane, r.t.a | Toluene, 77 K | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs [nm] (ε [M−1 cm−1]) | λ em [nm] | τ [ns] | Φ PL | k r [107] | k nr [109] | λ em,FL [nm] | τ FL [ns] | λ em,PH [nm] | τ PH [ms] | E T [eV] | |
| a Quantum yields and lifetimes determined on N2-saturated solutions. [FL] = fluorescence. [PH] = phosphorescence. sh = shoulder. Rate constants kr and knr are calculated using the equations kr = ΦPL/τ and knr = (1 − ΦPL)/τ. Absorption and emission spectra of trans-6 at r.t. are given in the ESI.† For absorption and emission spectra of MesDTOB at r.t., see ref. 10. | |||||||||||
| 2 | 258 (21900), 271 (sh), 319 (4600), 334 (4200), 368 (10600), 385 (12400), 413 (5000) |
424, 446, 472, 506 (sh) | 0.82 | 7 | 8.5 | 1.13 | 423, 445, 473, 505 (sh) | 1.09 | 556, 566, 582, 603, 607, 660, 735 (sh) | 63.3 | 2.23 |
| trans-6 | 270 (26500), 355 (30700), 370 (30000), 393 (sh), 414 (sh) |
429, 452, 489 (sh) | 0.31 | 6 | 19.3 | 3.03 | 433, 457, 489, 530 (sh) | 0.48 | — | — | — |
| MesDTOB | 269 (32400), 330 (29100), 341 (28600), 359 (25300), 374 (sh) |
366, 380 | 0.30 | 6 | 20.0 | 3.13 | 375, 395 | 0.70 | 436, 452, 464, 480 | 104 | 2.84 |
To gain insight into the origin of the low quantum efficiency of 2, low-temperature photoluminescence spectra (77 K) were measured. To avoid degradation of 2 in 2-MeTHF (which usually contains peroxides), measurements were carried out in a frozen toluene matrix; for comparison, the corresponding spectra of the helicene precursor trans-6 and the mesityl dithienooxaborine (MesDTOB) subunit have also been included in Fig. 5b.
At 77 K, the fluorescence emission of 2 between 400 and 530 nm overlaps with the room temperature photoluminescence in c-hexane and the corresponding average lifetime of the singlet excited state is τ = 1.09 ns. Interestingly, at low temperature, we observed long-wavelength photoluminescence, between 530 and 800 nm, not present at room temperature. This emission corresponds to phosphorescence from the triplet excited state with a typical lifetime of τ = 63.3 ms. Both fluorescence and phosphorescence signals are highly structured and with spacings of about 1300 and 300–700 cm−1, respectively, as observed for similar thiahelicene and carbohelicene structures reported in the literature.11,21 In particular, the low-frequency vibronic progression of the phosphorescence band is associated with breathing vibrational modes, so-called “helix breathing”.11,22 The observed phosphorescence of 2 results from the population of the triplet state by intersystem crossing from the singlet excited state and the subsequent relaxation of the triplet state by radiative processes. In this type of structure, these spin-forbidden singlet–triplet transitions become partially allowed by the enhanced spin–orbit coupling arising from two different contributions: the helically twisted molecular scaffold and the presence of heavy sulfur heteroatoms along the conjugated system. Notably, phosphorescence emission was also observed in our previous studies on α-dimesitylboryl benzodithiophene systems23 (resembling the B-thiophene–benzene–thiophene-B fragment of 2) and in tetrathia[7]helicene derivatives.11
Under the same conditions, no phosphorescence was detected for the more flexible precursor trans-6, likely due to rotational deactivation processes. Interestingly, intense phosphorescence was also observed at 77 K for the dithienooxaborine model compound MesDTOB, albeit at much shorter wavelengths than in the case of 2 (Fig. 5b). Overall, the photophysical characterization of 2 showed that the introduction of two BO moieties into the parent 7TH results in a red-shift of the phosphorescence emission and an enhanced phosphorescence/fluorescence ratio (for low-temperature emission measurements on 7TH (in 2-MeTHF), see ref. 11).
The HOMOs and LUMOs of the (BO)2-helicenes 1 and 2 have been computed at the M06/6-311+G(d,p) level of theory (Fig. 6 and Fig. S49†). A comparison with the HOMO/LUMO of the parent 7TH confirms that the introduction of the BO moieties substantially influences the LUMO delocalization and that the Mes rings make no contribution to the frontier orbitals, in agreement with reports on (BN)-doped carbohelicenes.7e The LUMO of 2 is ≈0.6 eV lower than that of 1 (−2.22 eV vs. −1.65 eV), in line with the trend observed for experimental values (−3.14 eV vs. −1.97 eV). As expected, the LUMO of 2 is mainly delocalized over the thiophene–benzene–thiophene central fragment rather than over the terminal thiophene rings (as in the case of 1). Conversely, the HOMOs of 1, 2, and 7TH present only minor differences, both in terms of energy and degree of delocalization.
 | ||
| Fig. 6 Isovalue surfaces for the LUMO and HOMO of 1, 2, and 7TH, calculated at the M06/6-311+G(d,p) level. | ||
Preliminary racemization tests showed that the enantiomers of 2 are stereochemically stable at room temperature. The (P)-/(M)-enantiomers of 2 were therefore separated by chiral stationary phase ultra-high performance liquid chromatography (CSP-UHPLC) on an (S,S)-Whelk-O 1 column, using n-hexane/CH2Cl2 (95:5) as the eluent (Fig. 7a).24 Later, the analytical HPLC resolution was scaled up to the semipreparative level using the same eluent and a 10 mm internal diameter column. Racemization studies were conducted on the second eluted enantiomer showing an ee = 97 % (Rt ≈ 7 min; Fig. 7a, red trace). The collected fractions containing the second eluted enantiomer were combined and evaporated at room temperature. The solid residue was dissolved in decalin and subjected to thermal racemization at 80 °C.
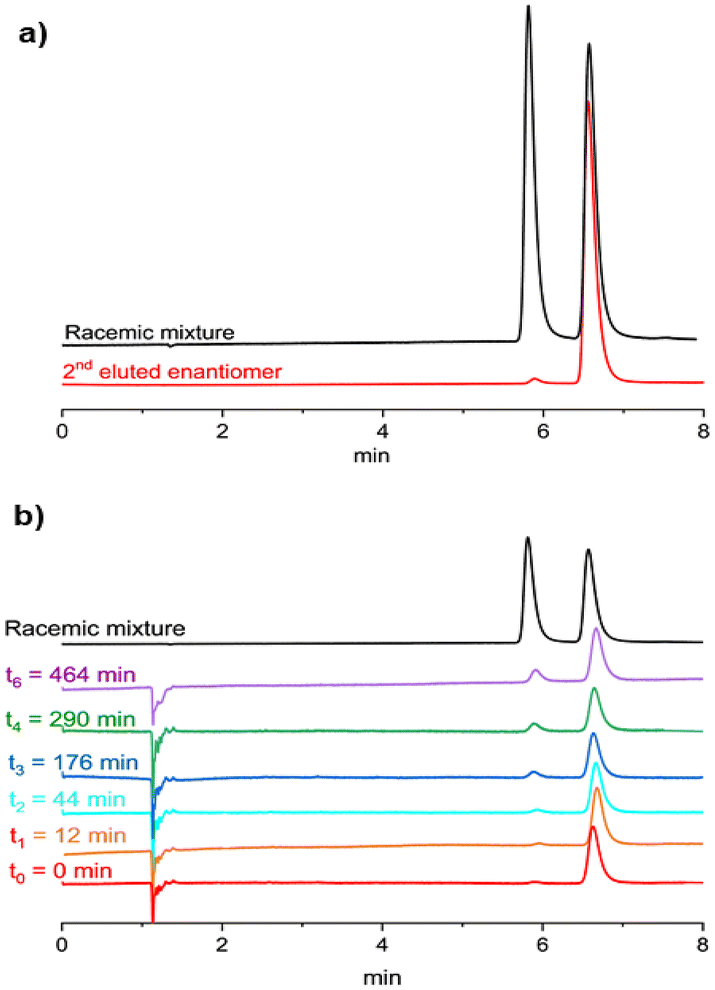 | ||
| Fig. 7 (a) CSP-UHPLC resolution of 2 (stationary phase: (S,S)-Whelk-O 1, mobile phase: n-hexane/CH2Cl2 (95:5), flow rate = 1.0 mL min−1, 25 °C). (b) Thermal racemization study conducted on the second eluted enantiomer in decalin at 80 °C; solvent peak: Rt ≈ 1 min. | ||
The changes in the enantiomeric excess of the sample were monitored by CSP-UHPLC (Fig. 7b). The racemization process was monitored by the decrease in the enantiomeric excess of the sample. Significant interconversion of the two enantiomers was visible after 12 min and the elution peak of the first enantiomer (Rt ≈ 6 min) began to emerge (Fig. 7b).
After approximately 8 h, the ee had dropped to 65% (Fig. 7b, purple trace). Data collected over this time (Fig. S46†) were used to determine the racemization constant krac = 1.48 × 10−5 s−1.24,25 The enantiomerization constant was calculated using the relation kenant = 0.5 × krac. The free energies of activation, ΔG‡rac = 28.6 ± 0.1 kcal mol−1 (at 353 K) and ΔG‡enant = 29.4 ± 0.1 kcal mol−1 (at 353 K), were calculated by using the Eyring equation and setting a transmission coefficient equal to 0.5 (see section 8.1 of the ESI†).26
Both ΔG‡rac and ΔG‡enant of 2 are close to the corresponding values of 1 (ΔG‡rac (1) = 26.9 ± 0.1 kcal mol−1, ΔG‡enant (1) = 27.4 ± 0.1 kcal mol−1 at 353 K), which reveals that the inversion of the B–O bonds does not reduce the configurational stability of the enantiomers.
We then evaluated the energy barrier through DFT calculations, using the same functional and basis set as for compound 1.10 Optimization of the transition state has been performed at the PBE1-PBE/TZVP level with empirical dispersion corrections,27 obtaining one imaginary frequency. This gave a barrier of 36 kcal mol−1, significantly higher (≈8 kcal mol−1) than the one obtained from the experimental data (ΔG‡enant (1) = 27.4 ± 0.1 kcal mol−1). The analysis of a relaxed scan to evaluate the racemization barrier of model compounds without Mes groups (1* and 2*; Fig. S50†) allows increasing the basis set level and comparing 1* and 2* with carbo[6]helicene and 7TH to assess the influence of the heteroatoms.28 However, similar values for the enantiomerization barrier were obtained (≈36 kcal mol−1).
By contrast, a second scan carried out for 1, 2, carbo[6]helicene, and 7TH with a semiempirical method (AM-1 level) produced results that were in better agreement with the experimental observations (27.8 kcal mol−1 for 1 and 28.9 kcal mol−1 for 2 (Fig. S51†)). In any case, the enantiomerization barrier is not much influenced by the presence of the Mes groups. As for 1, a comparison of the minimum and transition-state structures of 2 and 7TH suggests that the smaller double-bond character of the BO pairs allows for greater flexibility of the six-membered heterocycles compared to benzene rings (Fig. S52†).10
Circular dichroism (CD) spectra recorded in n-hexane/CH2Cl2 (95:5) at 20 °C showed a perfect mirror-image relationship between the first and second eluted enantiomers of 2 (Fig. 8a); the optical rotation values are −264° (ee = 98%) and +261° (ee = 97%), respectively, which are in the same order of magnitude as those of 1 (−97.5°; +97.7°).
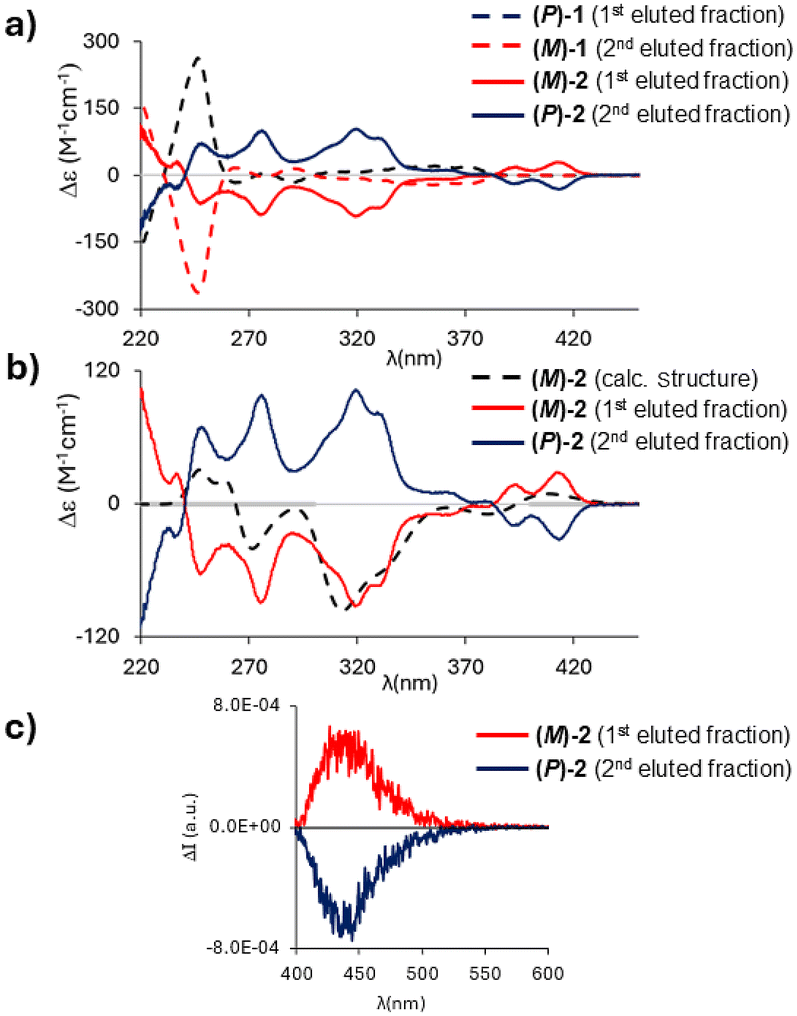 | ||
| Fig. 8 (a) Circular dichroism (CD) spectra of the 1 (dashed lines) and 2 (solid lines) enantiomers in n-hexane/CH2Cl2 (95:5) at 20 °C; red traces correspond to the (M)-structures and blue traces to the (P)-structures. (b) CD spectra of (M)-2 (red trace) and (P)-2 (blue trace) in n-hexane/CH2Cl2 (95:5) at 20 °C; computed CD spectrum of the optimized (M)-2 structure is shown for comparison (brown dashed trace; multiplied by a factor of 1.5). (c) Circularly polarized luminescence (CPL) spectra of (M)-2 (red trace) and (P)-2 (blue trace) in n-hexane/CH2Cl2 (95:5) at 20 °C. | ||
To assign the absolute configuration, the (M)-structure of 2 was optimized at the M06/6-311+G(d,p) level of theory. Similar to compound 1, only one stable conformer was found.
The comparison of calculated and experimental spectra is shown in Fig. 8b; a good correspondence is obtained between the calculated spectrum and the spectrum of the first eluted enantiomer of 2 (opposite to the case of 1, for which the (M)-structure matches the CD spectrum of the second eluted enantiomer). Fig. 8a illustrates how the inversion of the B–O bonds leads to remarkably different CD spectra: while 1 shows only weak spectral features above λ = 260 nm,102 has a more intense and structured CD spectrum with the first feature recorded at 414 nm, in correspondence with the first shoulder observed in the absorption spectrum (Fig. 5a).
Circularly polarized luminescence (CPL) spectra of 2 showed a band at λ = 440 nm (Fig. 8c); the resolution of our CPL apparatus does not allow the reproduction of all the vibronic features observed in the emission spectrum of 2 (Fig. 5a). The dissymmetry ratio (defined as glum = ΔI/I and gabs = ΔA/A)29 in emission is glum = 0.8 × 10−3 and is comparable to those of other organic molecules30 and lower than gabs of the first CD band (gabs = 2 × 10−3; Fig. 8b). The first transition, responsible for the absorption/emission band (in the two optimized geometries), is associated with an electric and magnetic dipole transition moment parallel to the C2-axis. Interestingly, the most intense CD band (gabs = 8.5 × 10−3) is found at 320 nm (Fig. 8b); conversely, in the corresponding region of the absorption spectrum, a low-intensity band is observed (Fig. 5a). This indicates that electric and magnetic dipole moments have the prevalent component along the helical axis (see Table S15†).
Conclusions
We reported the synthesis and full characterization of the (BO)2-helicene 2, which was obtained through a four-step synthesis involving a Mallory photocyclization as the last step. Moreover, the study of the synthesis of the singly-doped BO-helicene (3) was also described.A combined experimental and theoretical effort allowed us to evaluate (1) the property-switch produced by the BO bond inversion 1 → 2, along with (2) the effect of BO-doping on 7TH.
Although the effect on the quantum yield is negligible (ΦPL(2) = 7% vs. ΦPL(1) = 6%), switching of BO vectors proved to be a useful tool to achieve a lowered LUMO, red-shifted absorption/emission bands (≈40 nm), and a smaller energy gap (Eoptg (2) = 2.90 eV vs. Eoptg (1) = 3.16 eV).
As previously observed for 1, comparison of 2 with 7TH highlighted that the incorporation of BO bonds does not significantly improve fluorescence performances (ΦPL (2) = 7% vs. ΦPL (7TH) = 5%) but provides a detectable CPL signal (glum = 0.8 × 10−3).
At low temperature (77 K), 2 showed intense and structured phosphorescence (peaked at 556 nm; τ = 63.3 ms), reflecting noticeable vibronic components from “helix breathing” modes. This is important evidence that the incorporation of B–O bonds enhances the flexibility of the helical scaffold, as also highlighted by kinetics studies (ΔG‡enant (2) is 10 kcal mol−1 lower than ΔG‡enant (7TH)).
These findings now indicate how BO-doping is a means of influencing the rigidity of tetrathia[7]helicenes and demonstrate how it is possible to manipulate the electronic distribution across the helix by changing the position of the B centers within the scaffold. Overall, our results provide essential information for the synthesis and the structural design of new (BO)-helicene derivatives with tailored optoelectronic and mechanical properties.
Author contributions
L.M. and M.W conceived the idea. L.M., M.W., E.L., and C.B. wrote the manuscript. All authors discussed and commented on the manuscript. In detail: L.M. and C.B. synthesized the compounds; L.M. and M.P. performed spectroscopic measurements; S.A. and S.G. performed the electrochemical measurements; S.M. and C.V. performed the enantiomer separation and measured CD spectra; G.L. and G.M. performed the theoretical calculations and measured CPL spectra; A.V. and M.B. performed single crystal X-ray diffraction studies; L.M., M.W., E.L. and C.B. directed and supervised the project.Data availability
The data that support the findings of this study are available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. is grateful to Prof. Todd B. Marder and Prof. Ivo Starý for helpful scientific discussions during the PhD defence. L. M. is grateful to the University of Milan for providing a Ph.D. scholarship and to the Goethe-Universität Frankfurt for enabling a binational joint Ph.D. programme. Parts of this research (project I-20231044) were carried out at PETRA III at DESY (Hamburg), a member of the Helmholtz Association (HGF). We thank Drs M. Tolkiehn and E. Peresypkina for their assistance regarding the use of the beamline P24 at DESY. M. P. gratefully acknowledges Regione Lombardia and Fondazione Cariplo for funding the SmartMatLab Center project.References
- (a) J. Crassous, I. G. Stará and I. Starý, Helicenes: Synthesis, Properties and Applications, John Wiley & Sons, 2022 CrossRef; (b) C.-F. Chen and Y. Shen, Helicene Chemistry, Springer, 2017 CrossRef; (c) P. Ravat, Carbo[n]helicenes Restricted to Enantiomerize: An Insight into the Design Process of Configurationally Stable Functional Chiral PAHs, Chem. – Eur. J., 2021, 27, 3957–3967 CrossRef CAS PubMed; (d) G. Gingras, One hundred years of helicene chemistry. Part 3: applications and properties of carbohelicenes, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC; (e) J. M. Fernández-Garciá, P. J. Evans, S. Filippone, M. Á. Herranz and N. Martín, Chiral Molecular Carbon Nanostructures, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef PubMed; (f) Z. Zhou and M. A. Petrukhina, Adding multiple electrons to helicenes: how they respond?, Chem. Sci., 2024 10.1039/d4sc06062h.
- (a) S. Abbate, G. Longhi and T. Mori, Helicenes: Chiroptical Properties of Helicenes, John Wiley & Sons, 2022, pp. 373–394 Search PubMed; (b) T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed; (c) P. Izquierdo-García, J. M. Fernández-García, S. Medina Rivero, M. Šámal, J. Rybáček, L. Bednárová, S. Ramírez-Barroso, F. J. Ramírez, R. Rodríguez, J. Perles, D. García-Fresnadillo, J. Crassous, J. Casado, I. G. Stará and N. Martín, Helical Bilayer Nanographenes: Impact of the Helicene Length on the Structural, Electrochemical, Photophysical, and Chiroptical Properties, J. Am. Chem. Soc., 2023, 145, 11599–11610 CrossRef.
- (a) S. T. J. Ryan and M. J. Fuchter, Helicenes: Helicenes for Optoelectronic Applications and Devices, John Wiley & Sons, 2022, pp. 473–503 Search PubMed; (b) J. R. Brandt, F. Salerno and M. J. Fuchter, The added value of small-molecule chirality in technological applications, Nat. Rev. Chem., 2017, 1, 17–45 CrossRef; (c) H. Isla and J. Crassous, Helicene-based chiroptical switches, C. R. Chim., 2016, 19, 39–49 CrossRef CAS; (d) K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Achieving high circularly polarized luminescence with push–pull helicenic systems: from rationalized design to top-emission CP-OLED applications, Chem. Sci., 2021, 12, 5522–5533 RSC.
- (a) A. Borissov, Y. K. Maurya, L. Moshniaha, W. S. Wong, M. Żyła-Karwowska and M. Stępień, Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds, Chem. Rev., 2022, 122, 565–788 CrossRef CAS; (b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Structurally Constrained Boron-, Nitrogen-, Silicon-, and Phosphorus-Centered Polycyclic π-Conjugated Systems, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS; (c) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Doping Polycyclic Aromatics with Boron for Superior Performance in Materials Science and Catalysis, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (d) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications, Chem. Rev., 2017, 117, 3479–3716 CrossRef.
- (a) A. Nowak-Król, P. T. Geppert and K. R. Naveen, Boron-containing helicenes as new generation of chiral materials: opportunities and challenges of leaving the flatland, Chem. Sci., 2024, 15, 7408–7440 RSC; (b) K. Dhbaibi, L. Favereau and J. Crassous, Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P), Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS.
- For related B(sp2)-doped helicenes/helically-chiral PAHs, see: (a) M. Schnitzlein, K. Shoyama and F. Würthner, A highly fluorescent bora[6]helicene exhibiting circularly polarized light emission, Chem. Sci., 2024, 15, 2984–2989 RSC; (b) W. Sun, J. Guo, Z. Fan, L. Yuan, K. Ye, C. Dou and Y. Wang, Ribbon-Type Boron-Doped Polycyclic Aromatic Hydrocarbons: Conformations, Dynamic Complexation and Electronic Properties, Angew. Chem., Int. Ed., 2022, 61, e202209271 CrossRef CAS; (c) F. Miyamoto, S. Nakatsuka, K. Yamada, K. I. Nakayama and T. Hatakeyama, Synthesis of Boron-Doped Polycyclic Aromatic Hydrocarbons by Tandem Intramolecular Electrophilic Arene Borylation, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS; (d) J. Radtke, K. Schickedanz, M. Bamberg, L. Menduti, D. Schollmeyer, M. Bolte, H. W. Lerner and M. Wagner, Selective access to either a doubly boron-doped tetrabenzopentacene or an oxadiborepin from the same precursor, Chem. Sci., 2019, 10, 9017–9027 RSC; (e) K. Schickedanz, T. Trageser, M. Bolte, H. W. Lerner and M. Wagner, A boron-doped helicene as a highly soluble, benchtop-stable green emitter, Chem. Commun., 2015, 51, 15808–15810 RSC; (f) Y. Liu, L. Yuan, Z. Fan, J. Yang, Y. Wang and C. Dou, Boron-doped double [6]carbohelicenes: a combination of helicene and boron-doped π-systems, Chem. Sci., 2024, 15, 12819–12826 RSC; (g) A. Ikeno, M. Hayakawa, M. Sakai, Y. Tsutsui, S. Nakatsuka, S. Seki and T. Hatakeyama, π-Extended 9b-Boraphenalenes: Synthesis, Structure, and Physical Properties, J. Am. Chem. Soc., 2024, 146, 17084–17093 CrossRef CAS PubMed.
- For related B(sp2)–N doped helicenes/helically-chiral PAHs, see: (a) Y. Appiarius, S. Míguez-Lago, P. Puylaert, N. Wolf, S. Kumar, M. Molkenthin, D. Miguel, T. Neudecker, M. Juríček, A. G. Campaña and A. Staubitz, Boosting quantum yields and circularly polarized luminescence of penta- and hexahelicenes by doping with two BN-groups, Chem. Sci., 2024, 5, 466–476 RSC; (b) A. Abengózar, P. García-García, D. Sucunza, A. Pérez-Redondo and J. J. Vaquero, Synthesis of functionalized helical BN-benzo[c]phenanthrenes, Chem. Commun., 2018, 54, 2467–2470 RSC; (c) Z. Sun, C. Yi, Q. Liang, C. Bingi, W. Zhu, P. Qiang, D. Wu and F. Zhang, π-Extended C2-Symmetric Double NBN-Heterohelicenes with Exceptional Luminescent Properties, Org. Lett., 2020, 22, 209–213 CAS; (d) F. Zhang, F. Rauch, A. Swain, T. B. Marder and P. Ravat, Efficient Narrowband Circularly Polarized Light Emitters Based on 1,4-B,N-embedded Rigid Donor-Acceptor Helicenes, Angew. Chem., Int. Ed., 2023, 62, 1–7 Search PubMed; (e) K. Yuan, D. Volland, S. Kirschner, M. Uzelac, G. S. Nichol, A. Nowak-Król and M. J. Ingleson, Enhanced N-directed electrophilic C–H borylation generates BN-[5]- and [6]helicenes with improved photophysical properties, Chem. Sci., 2022, 13, 1136–1145 RSC; (f) M. Yang, I. S. Park and T. Yasuda, Full-Color, Narrowband, and High-Efficiency Electroluminescence from Boron and Carbazole Embedded Polycyclic Heteroaromatics, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed; (g) J. K. Li, X. Y. Chen, Y. L. Guo, X. C. X. Y. Wang, A. C. H. Sue, X. Y. Cao and X. C. X. Y. Wang, B,N-embedded double hetero[7]helicenes with strong chiroptical responses in the visible light region, J. Am. Chem. Soc., 2021, 143, 17958–17963 CAS; (h) T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, Azaboradibenzo[6]helicene: carrier inversion induced by helical homochirality, J. Am. Chem. Soc., 2012, 134, 19600–19603 CAS; (i) Y. Si and G. Yang, Photophysical properties of azaboradibenzo[6]helicene derivatives, J. Mater. Chem. C, 2013, 1, 2354–2361 RSC; (j) G. Meng, J. Zhou, X. S. Han, W. Zhao, Y. Zhang, M. Li, C. F. Chen, D. Zhang and L. Duan, B-N Covalent Bond Embedded Double Hetero-[n]helicenes for Pure Red Narrowband Circularly Polarized Electroluminescence with High Efficiency and Stability, Adv. Mater., 2024, 36, 1–11 Search PubMed; (k) Y. Jiao, Z. Sun, Z. Wang, Y. Fu and F. Zhang, Synthesis of Nonsymmetric NBN-Embedded [6]- and [7]Helicenes with Amplified Activities, Org. Lett., 2023, 25, 8766–8770 CrossRef CAS; (l) D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D. T. Yang, Multiple helicenes defected by heteroatoms and heptagons with narrow emissions and superior photoluminescence quantum yields, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS.
- For related B(sp2)–O doped helicenes/helically-chiral PAHs, see: (a) T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, Two-step synthesis of boron-fused double helicenes, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef CAS PubMed; (b) X. Y. Wang, A. Narita, W. Zhang, X. Feng and K. Müllen, Synthesis of stable nanographenes with OBO-doped zigzag edges based on tandem demethylation-electrophilic borylation, J. Am. Chem. Soc., 2016, 138, 9021–9024 CrossRef CAS PubMed; (c) X. Y. Wang, T. Dienel, M. Di Giovannantonio, G. B. Barin, N. Kharche, O. Deniz, J. I. Urgel, R. Widmer, S. Stolz, L. H. De Lima, M. Muntwiler, M. Tommasini, V. Meunier, P. Ruffieux, X. Feng, R. Fasel, K. Müllen and A. Narita, Heteroatom-doped perihexacene from a double helicene precursor: on-surface synthesis and properties, J. Am. Chem. Soc., 2017, 139, 4671–4674 CrossRef CAS PubMed; (d) Z. Zhou, X. Y. Wang, Z. Wei, K. Müllen and M. A. Petrukhina, Charging OBO–Fused Double[5]Helicene with Electrons, Angew. Chem., Int. Ed., 2019, 58, 14969–14973 CrossRef CAS PubMed.
- For related B(sp3)–N doped helicenes/helically-chiral PAHs, see: (a) F. Full, A. Artigas, K. Wiegand, D. Volland, K. Szkodzińska, Y. Coquerel and A. Nowak-Król, Controllable 1,4-Palladium Aryl to Aryl Migration in Fused Systems – Application to the Synthesis of Azaborole Multihelicenes, J. Am. Chem. Soc., 2024, 146, 29245–29254 CrossRef CAS PubMed; (b) D. Volland, J. Niedens, P. T. Geppert, M. J. Wildervanck, F. Full and A. Nowak-Król, Synthesis of a Blue–Emissive Azaborathia[9]helicene by Silicon–Boron Exchange from Unusual Atropisomeric Teraryls, Angew. Chem., Int. Ed., 2023, 62, e202304291 CrossRef CAS; (c) F. Full, M. J. Wildervanck, D. Volland and A. Nowak-Król, Synthesis of Enantioenriched Azaborole Helicenes by Chirality Transfer from Axially Chiral Biaryls, Synlett, 2023, 477–482 CAS; (d) J. Full, S. P. Panchal, J. Götz, A. M. Krause and A. Nowak-Król, Modular synthesis of organoboron helically chiral compounds: cutouts from extended helices, Angew. Chem., Int. Ed., 2021, 60, 4350–4357 CrossRef CAS; (e) Z. Domínguez, R. López-Rodríguez, E. Álvarez, S. Abbate, G. Longhi, U. Pischel and A. Ros, Azabora[5]helicene Charge-Transfer Dyes Show Efficient and Spectrally Variable Circularly Polarized Luminescence, Chem. – Eur. J., 2018, 24, 12660–12668 CrossRef PubMed; (f) C. Shen, M. Srebro-Hooper, M. Jean, N. Vanthuyne, L. Toupet, J. A. G. Williams, A. R. Torres, A. J. Riives, G. Muller, J. Autschbach and J. Crassous, Synthesis and chiroptical properties of hexa–, octa–, and deca–azaborahelicenes: influence of helicene size and of the number of boron atoms, Chem. – Eur. J., 2017, 23, 407–418 CrossRef CAS PubMed.
- L. Menduti, C. Baldoli, S. Manetto, M. Bolte, H. W. Lerner, G. Longhi, C. Villani, E. Licandro and M. Wagner, (BO)2-Doped Tetrathia[7]helicene: A Configurationally Stable Blue Emitter, Angew. Chem., Int. Ed., 2023, 62, e202215468 CrossRef CAS PubMed.
- A. Bossi, P. R. Mussini, G. Farinola, M. Penconi, S. Cauteruccio, M. E. Thompson and E. Licandro, Sparkling Organic Phosphorescence from Fluorinated Tetrathia[7]helicenes: Synthesis and Photophysical, Electrochemical and Computational Studies, Chem. – Eur. J., 2023, 29, e202300339 CrossRef CAS PubMed.
- Commercially available compound: CAS 18791-75-8.
- S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Intramolecular (directed) electrophilic C–H borylation, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
- K. B. Jørgensen, Photochemical oxidative cyclisation of stilbenes and stilbenoids - the Mallory-reaction, Molecules, 2010, 15, 4334–4358 CrossRef PubMed.
- H. Nakagawa, A. Obata, K. I. Yamada, H. Kawazura, M. Konno and H. Miyamae, Crystal and Molecular Structures of Tetrathia[7]heterohelicene: Racemate and Enantiomer, J. Chem. Soc., Perkin Trans. 2, 1985, 20, 1899–1903 RSC.
- C. Hoffend, M. Diefenbach, E. Januszewski, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Effects of boron doping on the structural and optoelectronic properties of 9,10-diarylanthracenes, Dalton Trans., 2013, 42, 13826–13837 RSC.
- A. Budanow, E. Von Grotthuss, M. Bolte, M. Wagner and H. Lerner, 10,9-Oxaboraphenanthrenes as luminescent fluorophores, Tetrahedron, 2016, 72, 1477–1484 CrossRef CAS.
- Deposition numbers CCDC 2384683 (for 2), 2384684 (for 4), 2384685 (for 5), 2384686 (for 6), 2384687 (for 7), 2384688 (for 8), 2384689 (for 9), 2384690 (for 10) and 2384691 (for 11) contain the supplementary crystallographic data for this paper.†.
- Propylene oxide is a known scavenger for the HI developed during photocyclizations carried out using I2 as the oxidant: L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, Improved methodology for photocyclization reactions, J. Org. Chem., 1991, 56, 3769–3775 CrossRef CAS.
- T. Caronna, M. Catellani, S. Luzzati, L. Malpezzi, S. V. Meille, A. Mele, C. Richter and R. Sinisi, Molecular crystal architecture and optical properties of a thiohelicenes series containing 5, 7, 9, and 11 rings prepared via photochemical synthesis, Chem. Mater., 2001, 13, 3906–3914 CrossRef CAS.
- (a) C. Johannessen, E. W. Blanch, C. Villani, S. Abbate, G. Longhi, N. R. Agarwal, M. Tommasini and D. A. Lightner, Raman and ROA spectra of (−)-and (+)-2-Br-Hexahelicene: Experimental and DFT Studies of a π-Conjugated Chiral System, J. Phys. Chem. B, 2013, 117, 2221–2230 CrossRef CAS PubMed; (b) F. Della Sala, V. Vitale, M. Mazzeo, R. Cingolani, G. Gigli, L. Favaretto and G. Barbarella, The effects of oxygen and boron functionalization on the optical properties of dithienothiophenes, J. Non-Cryst. Solids, 2006, 352, 2461–2464 CrossRef CAS; (c) Z. Sun, W. Xu, S. Qiu, Z. Ma, C. Li, S. Zhang and H. Wang, Thia[n]helicenes with long persistent phosphorescence, Chem. Sci., 2024, 15, 1077–1087 RSC; (d) Y. Kondo, Y. Tsutsui, Y. Matsuo, T. Tanaka and S. Seki, Impacts of heteroatom substitution on the excited state dynamics of π-extended helicenes, Nanoscale Adv., 2024, 6, 4567–4571 RSC.
- S. P. McGlynn and T. Azumi, Molecular spectroscopy of the triplet state, Prentice-Hall, 1969 Search PubMed.
- L. Menduti, C. Baldoli, S. Arnaboldi, A. Dreuw, D. Tahaoglu, A. Bossi and E. Licandro, (Dimesityl)boron Benzodithiophenes: Synthesis, Electrochemical, Photophysical and Theoretical Characterization, ChemistryOpen, 2022, 11, 1–11 CrossRef.
- Full details are given in the ESI.†.
- (a) L. Meca, D. Reha and Z. Havlas, Racemization Barriers of 1,1′-Binaphthyl and 1,1′-Binaphthalene-2,2′-diol: A DFT Study, J. Org. Chem., 2003, 68, 5677–5680 CrossRef CAS; (b) D. C. Patel, R. M. Woods, Z. S. Breitbach, A. Berthod and D. W. Armstrong, Thermal racemization of biaryl atropisomers, Tetrahedron: Asymmetry, 2017, 28, 1557–1561 CrossRef CAS.
- Enantiomerization is the reversible conversion of a single enantiomer into the other one. The corresponding rate constant is kenant. Racemization is the irreversible conversion of a sample of single enantiomers or of a non-racemic mixture of enantiomers into the racemate. The corresponding rate constant is krac. A comparison of calculated and experimental energy barriers was done using the enantiomerization barrier obtained from kenant, which is calculated from the experimentally determined krac according to: kenant = 0.5krac = 8.15 × 10−5 s−1. The free energy for the enantiomerization process (ΔG‡enant) was obtained by using the Eyring equation and setting a transmission equal to 0.5.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef; (b) J. Barroso, J. L. Cabellos, S. Pan, F. Murillo, X. Zarate, M. A. Fernandez-Herrera and G. Merino, Revisiting the racemization mechanism of helicenes, J. Chem. Soc., Chem. Commun., 2017, 54, 188–191 RSC.
- (a) For experimental data on 7TH racemization, see: K. Yamada, H. Nakagawa and H. Kawazura, Thermal racemization of thiaheterohelicenes, Bull. Chem. Soc. Jpn., 1986, 59, 2429–2432 CrossRef CAS; (b) For experimental data on carbo[6]helicene racemization, see: R. H. Janke, G. Haufe, E. U. Würthwein and J. H. Borkent, Racemization Barriers of Helicenes: A Computational Study, J. Am. Chem. Soc., 1996, 118, 6031–6035 CrossRef CAS.
- (a) W. Kuhn, The physical significance of optical rotatory power, Z. Phys. Chem., Abt. B, 1929, 4, 14–36 CrossRef CAS; (b) W. Kuhn, Optical rotatory power, Annu. Rev. Phys. Chem., 1958, 9, 417–438 CrossRef CAS.
- (a) Y. Liu, J. Cerezo, G. Mazzeo, N. Lin, X. Zhao, G. Longhi, S. Abbate and F. Santoro, Vibronic coupling explains the different shape of electronic circular dichroism and of circularly polarized luminescence spectra of hexahelicenes, J. Chem. Theory Comput., 2016, 12, 2799–2819 CrossRef CAS; (b) H. Tanaka, Y. Inoue and T. Mori, Circularly polarized luminescence and circular dichroisms in small organic molecules: correlation between excitation and emission dissymmetry factors, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2384683 (for 2), 2384684 (for 4), 2384685 (for 5), 2384686 (for 6), 2384687 (for 7), 2384688 (for 8), 2384689 (for 9), 2384690 (for 10) and 2384691 (for 11). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01897d |
| This journal is © the Partner Organisations 2025 |