
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to fluorinated dienes through hydrofluorination of 2-En-4-ynoates†
Yue Xia‡
,
Aaron D. Charlack‡,
Rui Guo,
Nicholas W. Wade and
Yi-Ming Wang *
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: ym.wang@pitt.edu
First published on 17th December 2024
Abstract
The hydrofluorination of enynoates has been developed for the synthesis of fluorinated dienoates. Using a pyridinium tetrafluoroborate salt that is easily prepared on large scale, this approach enabled the direct conversion of these substrates to fluorinated targets through a vinyl cation mediated process. This approach was applied to a range of aryl-substituted enynoates to deliver the (Z)-configured products with high levels of stereo- and regioselectivity. Mechanistic studies were conducted to provide insights into the stereochemical outcome and reaction efficiency under different reaction conditions.
Introduction
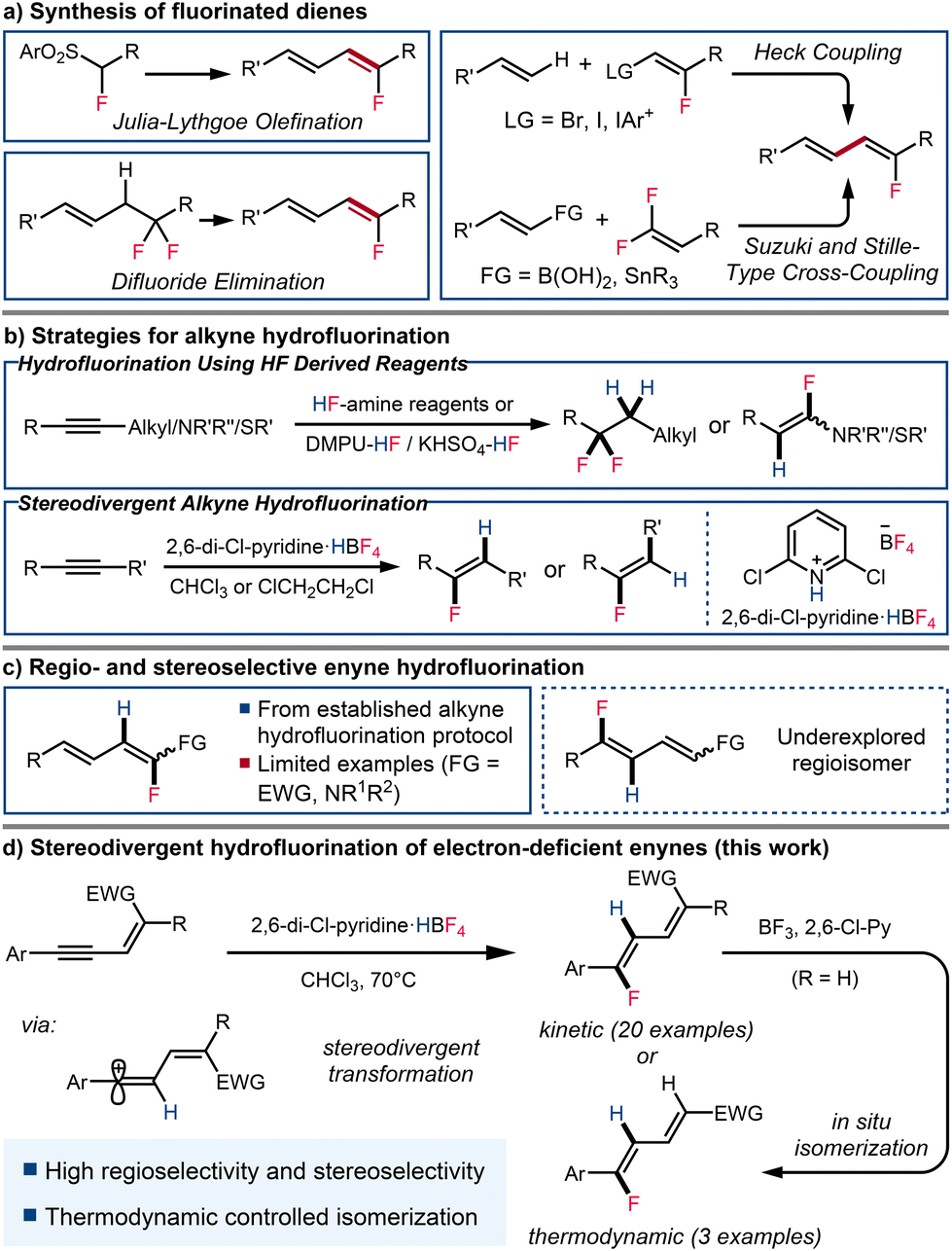
Fluorinated molecules have gained particular interest in the fields of medicinal and agricultural chemistry in recent decades.1 The introduction of a fluorine atom into organic molecules has become an important approach to modulate their physicochemical properties, lipophilicity, and metabolic stability. Fluorinated dienes and polyenes that serve as analogues of bioactive lipids have received particular attention.2 Among methods for the introduction of fluorine atoms in a selective manner, hydrofluorination of unsaturated bonds is one of the most significant and exploited pathways, but this strategy is rarely applied to the synthesis of fluorinated dienes and polyenes.3Various methods have been developed to synthesize C(sp2)-fluorinated dienes, allowing for the selective synthesis of products of various substitution patterns and stereochemistry (Scheme 1a). One of the most common ways to synthesize fluorinated dienes is the coupling of pre-functionalized vinyl fluorides with olefins.4 However, this approach typically requires less accessible starting materials. Geminal difluorides are also common precursors to monofluorodienes. Mono-elimination of fluorine in aliphatic geminal difluorides has led to the synthesis of conjugated vinyl fluorides.5 gem-Difluoroalkenes have also been shown to be good coupling partners, either under Stille conditions or with boronic acids under rhodium catalysis, resulting in the selective formation of the desired monofluorodiene.6 α-Fluorinated aryl sulfones, reacting with α,β-unsaturated carbonyls via Julia olefination, have also been demonstrated to give rise to fluorinated dienes in good yields.7 However, this method tends to result in modest to moderate stereoselectivity. While most of the existing methods for the synthesis of fluorinated dienes require the utilization of fluorine containing fragments and/or multiple synthetic steps from non-fluorinated starting materials, direct hydrofluorination of enynes could provide a reliable synthetic pathway and expand the toolbox for the construction of fluorinated dienes.
 | ||
| Scheme 1 Enyne hydrofluorination for accessing fluorinated dienes. | ||
Recent advances have led to hydrofluorination emerging as an efficient and effective pathway for the incorporation of fluorine into unsaturated compounds, including the synthesis of alkenyl fluorides from alkynes.8 Numerous amine-modulated hydrogen fluoride sources have been investigated to achieve this transformation.9 However, reports by both Olah and Hammond indicate that generally, even with careful control of the reaction conditions, these HF derived reagents deliver the gem-difluoride product without allowing isolation of the presumed alkenyl fluoride intermediate, except in special cases where the alkyne bears a heteroatomic substituent (Scheme 1b, top).10
Considering that tetrafluoroborate salts are safe, stable, and affordable fluoride sources,11 our group has developed a novel pyridinium tetrafluoroborate based reagent for hydrofluorination (Scheme 1b, bottom).12 This reagent enabled the stereoselective hydrofluorination of alkynes, delivering vinyl fluoride products with good to excellent regio- and stereoselectivity under mild conditions, with the stereochemical outcome dependent on both substrate structure and reaction conditions. We aimed to broaden the application of this novel hydrofluorinating reagent to the synthesis of a wider array of fluorinated compounds, particularly fluorinated dienes through the hydrofluorination of enynes. Although alkyne hydrofluorination has been extensively studied, the synthesis of fluorinated dienes through the formation of C(sp2)–F bonds remain significantly underreported, with only few isolated examples of enyne hydrofluorination reported as special cases following established protocols for alkyne hydrofluorination.13 These methodologies have primarily yielded fluorinated dienes with specific regioselectivity, placing fluorine atoms α to an existing functional group. To the best of our knowledge, the construction of other regioisomers through enyne hydrofluorination remains unknown (Scheme 1c). While approaches toward fluorinated dienes via the formation of carbon-carbon bonds has enabled access to these regioisomers, the synthesis usually involves multiple steps, and in some cases proceeds through unstable intermediates.14 Hence, the synthesis of fluorinated dienes bearing alternate selectivity via enyne hydrofluorination could present a useful alternative (Scheme 1d).
Results and discussion
We initiated our investigation using 1d as our standard substrate and 2,6-dichloropyridinium tetrafluoroborate salt as both the hydrogen and fluoride source (Table 1). Upon testing the optimal conditions we applied in our previous alkyne hydrofluorination reaction,12 we found that product 2d could be obtained in a moderate yield of 45% with excellent regio- and stereoselectivity. We found that 2.0 equiv. of pyridinium reagent was most efficient for this transformation (50% yield, entry 2), while less or more pyridinium salt led to lower yields. Various weakly coordinating solvents were tested (entries 2, 5–8) and chloroform was found to give the best result (entry 2). Variation of concentration revealed that diluted reaction conditions gave better yields (see ESI†). We also tested various reaction temperatures (entries 9–12) and found that conducting the reaction at 70 °C provided the highest yields. Various additives were also tested, however we found that in this reaction the additives either lowered the yields or otherwise had little impact. Moreover, in some cases, cyclization of the substrate was competitive with hydrofluorination, while prolonged reaction times were also found to cause decomposition of the target product (see the ESI† for discussions of product stability and side product formation). The synthesis of 2,6-dichloropyridinium tetrafluoroborate was reoptimized and reproducibly scaled to 150 mmol (see the ESI†).
|
||||
|---|---|---|---|---|
| Entry | m | Solvent | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 1d (0.1 mmol) and 2,6-dichloropyridinium tetrafluoroborate was added solvent (2.0 mL), stirred at indicated temperature for 6 h.b NMR yields, determined by 1H NMR using 1,3-dinitrobenzene as internal standard.c Isolated yield. 1,2-DCE = 1,2-dichloroethane. | ||||
| 1 | 1.0 | CHCl3 | 70 | 45 |
| 2 | 2.0 | CHCl3 | 70 | 50 (43c) |
| 3 | 3.0 | CHCl3 | 70 | 41 |
| 4 | 4.0 | CHCl3 | 70 | 39 |
| 5 | 2.0 | PhMe | 70 | 21 |
| 6 | 2.0 | PhCl | 70 | 34 |
| 7 | 2.0 | PhCF3 | 70 | 25 |
| 8 | 2.0 | 1,2-DCE | 70 | 0 |
| 9 | 2.0 | CHCl3 | 20 | Trace |
| 10 | 2.0 | CHCl3 | 40 | 16 |
| 11 | 2.0 | CHCl3 | 60 | 39 |
| 12 | 2.0 | CHCl3 | 80 | 45 |
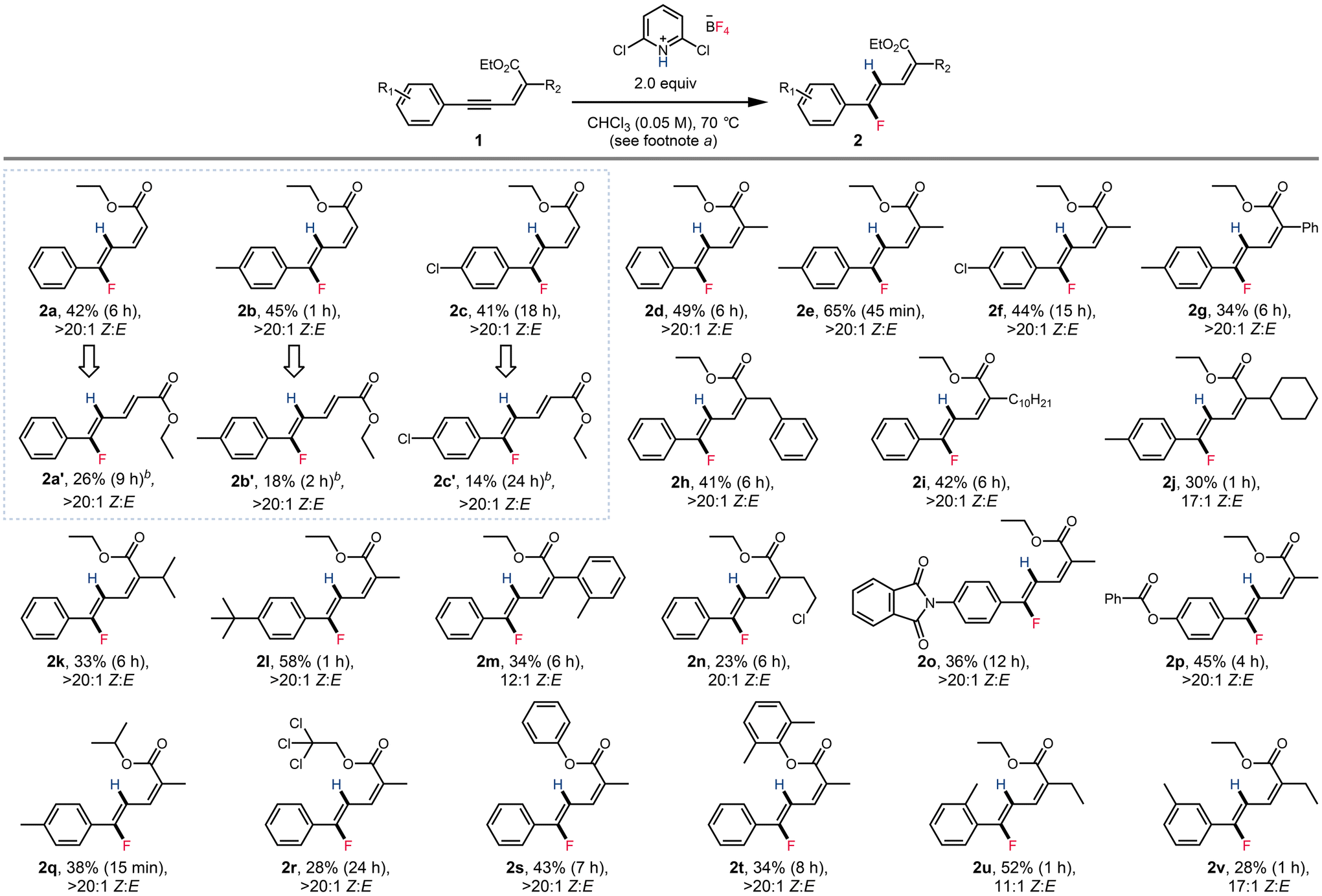
With the optimized conditions established, we set out to explore the scope of this transformation (Scheme 2). Aryl enynoates bearing mildly electron donating substituents (2e, 2l) and mildly electron withdrawing substituents (2f) were tolerated. Enynoates with heteroatomic substituents including phthalimide (2o) and benzoyl (2p) could also be tolerated. 2,5-Diaryl enynoates (2g) were also reactive. Reaction conditions also tolerate various aliphatic and aromatic substitutes, such as benzyl (2h), decyl (2i), cyclohexyl (2j), isopropyl (2l), o-methylphenyl (2m) and 2-chloroethyl (2n). Various esters (2q–2t) could also be applied in this transformation. Substitution at the ortho (2u) and meta (2v) positions of the aryl group were also tolerated. Unfortunately, when the aryl group of the substrate was replaced by an alkyl group, the hydrofluorination protocol was unsuccessful, and no fluorinated products were observed by 19F NMR (see the ESI† for examples attempted). While the yields obtained were generally found to be modest (vide infra), the products were readily purified by column chromatography. In addition, we note that in all cases, the product dienoates were obtained in high regio- and stereoselectivity.
 | ||
Scheme 2 Substrate scope for the formation of fluorinated dienoates 2. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1 (0.25 mmol), 2,6-dichloropyridinium tetrafluoroborate (0.50 mmol), chloroform (5.0 mL), stirred at 70 °C for indicated duration. b1,2-DCE (0.05 M) was used as solvent. Reaction conditions: 1 (0.25 mmol), 2,6-dichloropyridinium tetrafluoroborate (0.50 mmol), chloroform (5.0 mL), stirred at 70 °C for indicated duration. b1,2-DCE (0.05 M) was used as solvent. | ||
Interestingly, we found that when the enynoate C2 position was unsubstituted, isomerization of the product dienoate was observed when the more polar solvent 1,2-dichloroethane was applied (Scheme 2, boxed examples). Enynoates 1a–1c could form both stereoisomers under distinct reaction conditions. However, the (E)-configured products were obtained in considerably lower yields than the (Z) isomer. C2-substituted enynoates were also subjected to similar conditions, but only led to full decomposition of the starting enynoate and dienoate products without the formation of other isomers.
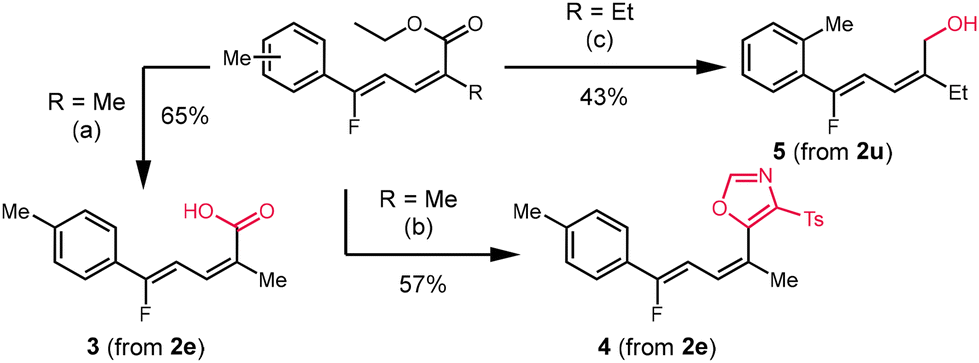
We performed three further derivatizations to demonstrate the synthetic utility of the fluorinated dienoate products (Scheme 3). The saponification of 2e afforded the α,γ-unsaturated carboxylic acid 3 in 65% yield. Cyclization with p-toluenesulfonylmethyl isocyanide (TosMIC) afforded oxazole 4 in 57% yield.15 Reduction of the ester moiety with DIBAL-H afforded the alcohol 5 in 43% yield. We also performed two reactions on mmol scale to demonstrate the scalability of this protocol. Substrate 1u was subjected to conditions at 1.25 mmol scale to successfully synthesize 2u in 49% yield. Similarly, 1t was reacted at 2.5 mmol scale to provide 2t in 35% yield.
 | ||
| Scheme 3 Divergent transformations of products. (a) LiOH (4.0 equiv.), THF (0.5 M), 50 °C, 24 h. (b) TosMIC (1.0 equiv.), nBuLi (2.0 equiv.), THF (0.5 M), 0 °C, 4 h. (c) DIBAL-H (3.0 equiv.), THF (0.5 M), 0 °C, 2 h. | ||
The observation that reactions of substrates 1a, 1b, and 1c in the less polar solvent chloroform (ε = 4.81) or under lower temperatures (see the ESI†) favored the formation of stereo-retention products while reactions conducted in the more polar solvent 1,2-dichloroethane (ε = 10.36) provided the dienoate product with reversed stereochemistry prompted us to investigate the sequence of events in this isomerization and the origin of the divergent stereoselectivity. Two potential pathways for the formation of 2-trans isomers were proposed, which primarily concerned whether the olefin isomerization occurs before or after the enynatee hydrofluorination. To validate the potential reaction pathway, 1a′ was synthesized and subjected to the reaction conditions (Scheme 4). However, even at elevated temperatures, 1a′ remained unreactive, and all material was recovered after 24 h. Additionally, no 1a′ was observed during the hydrofluorination of 1a, instead we observe near quantitative starting material recovery, which strongly suggested that the olefin isomerization occurred following the fluorinated dienoate formation.
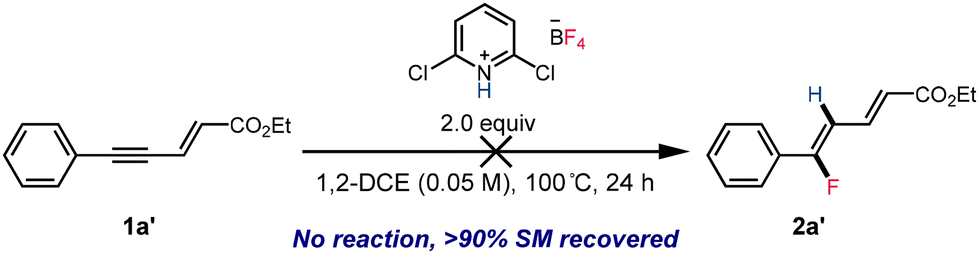 | ||
| Scheme 4 Attempted hydrofluorination of 1a′. | ||
The mechanism of diene isomerization was investigated (Table 2). Upon heating 2a under reaction temperature, no isomerization was observed. However, as Et2O·BF3 was added, a 12% isomerization product was observed, suggesting that the isomerization is a Lewis acid-promoted process. The addition of 2,6-dichloropyridine further enhanced conversion, indicating the possibility of an addition-elimination type mechanism for E/Z isomerization (Scheme 5).
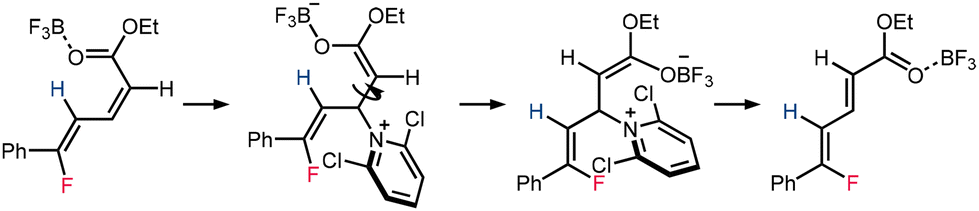 | ||
| Scheme 5 Proposed mechanism for isomerization of dienoate 2a. | ||

The increasing proportion of 2a′ over 12 hours is consistent with gradual equilibration that favors the thermodynamically preferred stereoisomer over prolonged reaction times (Scheme 6, top). The increase in reaction temperature was also found to favor the formation of 2a′ (Scheme 6, bottom).
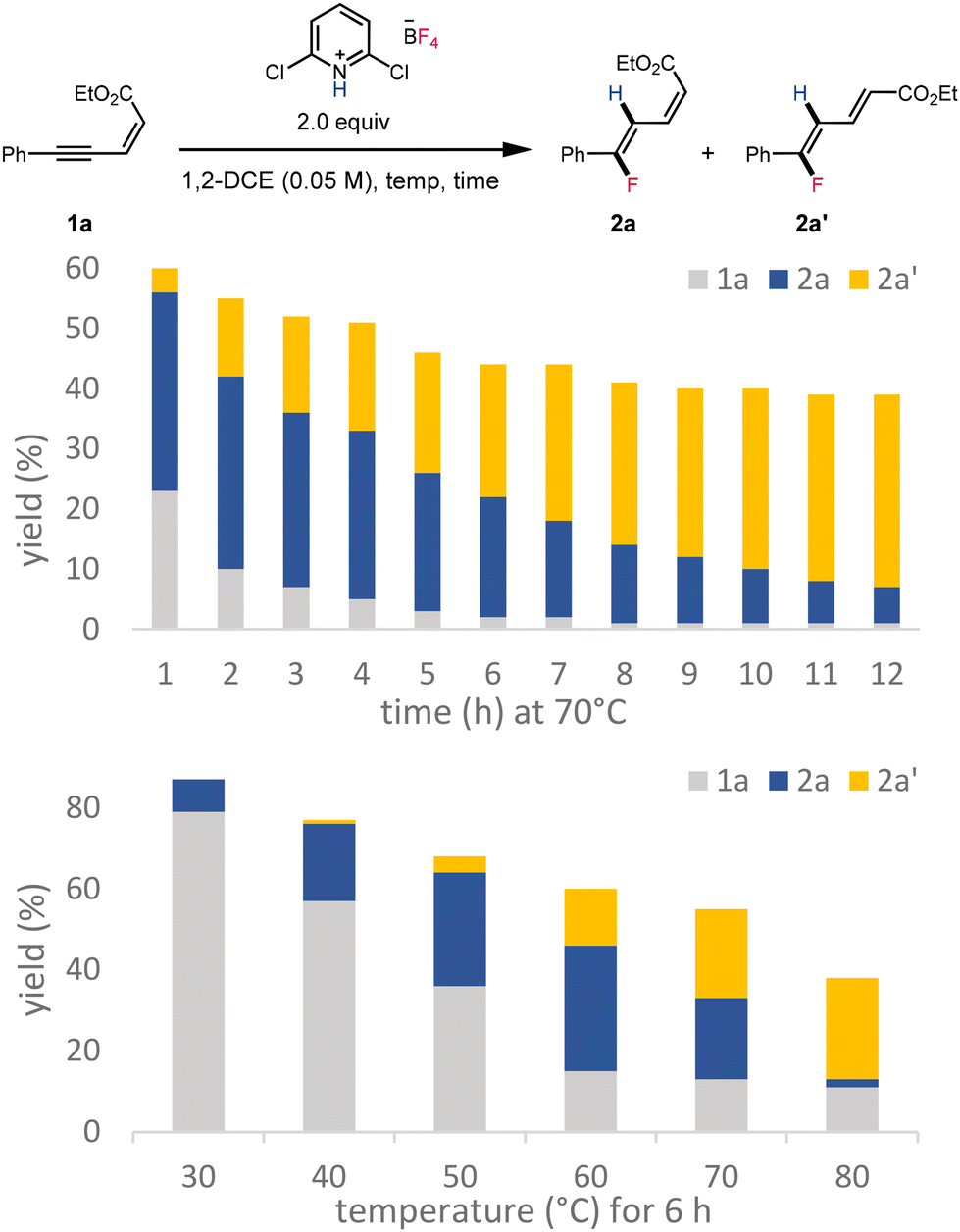 | ||
| Scheme 6 Formation of fluorinated dienoates 2a and 2a′ over time and at various reaction temperatures. | ||
Further experimental study of the dielectric constant of the solvent suggested that more 2a′ could be obtained when increasing the dielectric constant of the solvent or increasing the high dielectric component in the solvent mixture (Scheme 7). These observations further supported the hypothesis that formation of both fluorinated dienoate isomers was under thermodynamic control via polar intermediates.
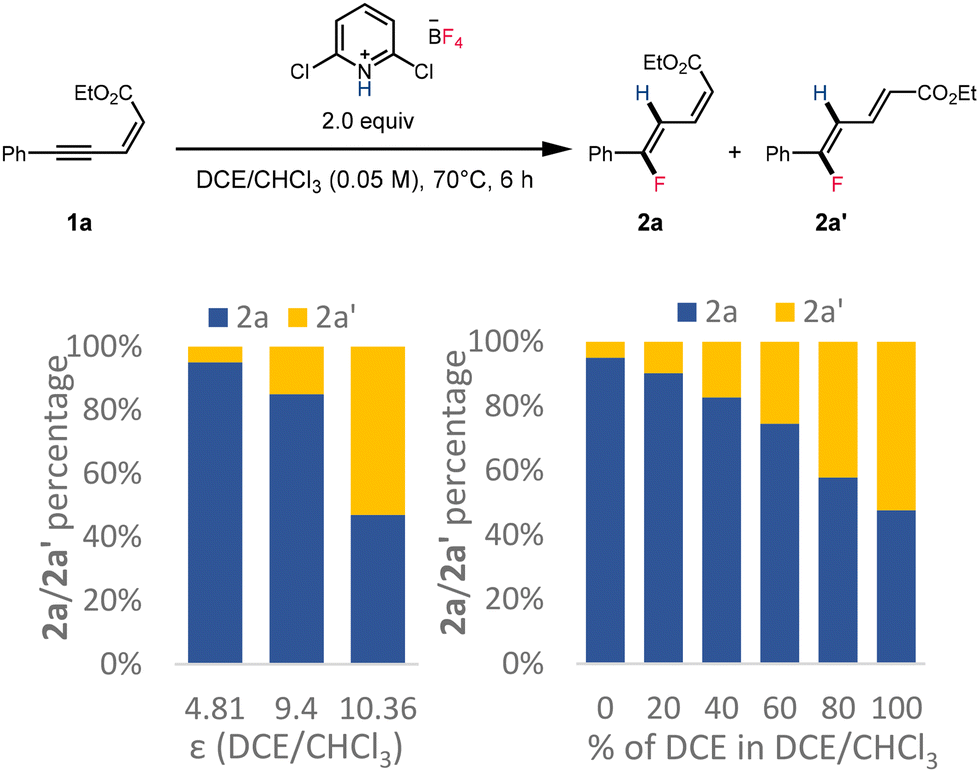 | ||
| Scheme 7 Isomerization of fluorinated dienoate 2a in various solvents. | ||
Conclusions
In conclusion, we have developed straightforward conditions for the hydrofluorination of enynoates using 2,6-dichloropyridinium tetrafluoroborate as fluorinating reagent. This protocol could be applied in the hydrofluorination of a range of enynoates, and stereodivergent protocols were developed for C2-unsubstituted substrates. Mechanistic studies suggested a pathway for the E/Z isomerization of C2-unsubstituted fluorinated dienoates that occurs after initial hydrofluorination of the Z-configured enynoate, allowing for the rationalization of the observed stereodivergence.Data availability
All data supporting the findings of this study, including experimental details and spectroscopic characterization data for all compounds, are available within the article and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences, National Institutes of Health (R35GM142945).References
- For selected reviews on significance of fluorine in material, pharmaceutical, and agricultural science, see: (a) K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in Medicinal Chemistry, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Organic Fluorine Compounds: a Great Opportunity for Enhanced Materials Properties, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (d) T. Fujiwara and D. O'Hagan, Successful Fluorine-Containing Herbicide Agrochemicals, J. Fluor. Chem., 2014, 167, 16–29 CrossRef; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev., 2014, 114, 2432–2506 CrossRef PubMed; (f) V. F. Cardoso, D. M. Correia, C. Ribeiro, M. M. Fernandes and S. Lanceros-Méndez, Fluorinated Polymers as Smart Materials for Advanced Biomedical Applications, Polymers, 2018, 10, 161 CrossRef PubMed; (g) N. A. Meanwell, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design, J. Med. Chem., 2018, 61, 5822–5880 CrossRef; (h) Q. Wang, H. Song and Q. Wang, Fluorine-Containing Agrochemicals in the Last Decade and Approaches for Fluorine Incorporation, Chin. Chem. Lett., 2022, 33, 626–642 CrossRef; (i) G. Shabir, A. Saeed, W. Zahid, F. Naseer, Z. Riaz, N. Khalil, Muneeba and F. Albericio, Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016-2022), Pharmaceuticals, 2023, 16, 1162 CrossRef PubMed.
- (a) M. B. Sporn, N. M. Dunlop, D. L. Newton and W. R. Henderson, Relationships between structure and activity of retinoids, Nature, 1976, 263, 110–113 CrossRef; (b) B. A. Pawson, K.-K. Chan, J. DeNoble, R. J. L. Han, V. Piermattie, A. C. Specian, S. Srisethnil, P. W. Trown and O. Bohoslawec, Fluorinated Retinoic Acids and Their Analogues. 1. Synthesis and Biological Activity of (4-Methoxy-2,3,6-trimethylphenyl)nonatetraenoic Acid Analogues, J. Med. Chem., 1979, 22, 1059–1067 CrossRef PubMed; (c) M. G. Pomper, K. G. Pinney, K. E. Carlson, H. Van Brocklin, C. J. Mathias, M. J. Welch and J. A. Katzenellenbogen, Target tissue uptake selectivity of three fluorine-substituted progestins: Potential imaging agents for receptor-positive breast tumors, Int. J. Radiat. Appl. Instrum., Part B, 1990, 17, 309–319 CrossRef; (d) Y. Zhu and R. S. H. Liu, Divergent Pathways in Photobleaching of 7,9-dicis-Rhodopsin and 9,11-dicis-12-Fluororhodopsin: One-Photon-Two-Bond and One-Photon-One-Bond Isomerization, Biochemistry, 1993, 32, 10233–10238 CrossRef PubMed; (e) D. Hoischen, L. U. Colmenares, J. Liu, C. J. Simmons, G. Britton and R. S. H. Liu, Fluorinated Analogs of the Carotenoprotein, α-Crustacyanin, Bioorg. Chem., 1998, 26, 365–374 CrossRef; (f) M. Prakesch, D. Grée, S. Chandrasekhar and R. Grée, Synthesis of Fluoro Analogues of Unsaturated Fatty Acids and Corresponding Acyclic Metabolites, Eur. J. Org. Chem., 2005, 1221–1232 CrossRef; (g) R. S. H. Liu and J. Liu, Fluorinated Retinoids and Carotenoids, J. Nat. Prod., 2011, 74, 512–517 CrossRef.
- (a) J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, Reversible C−F Bond Formation and the Au-Catalyzed Hydrofluorination of Alkynes, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef PubMed; (b) B. C. Gorske, C. T. Mbofana and S. J. Miller, Regio- and Stereoselective Synthesis of Fluoroalkenes by Directed Au(I) Catalysis, Org. Lett., 2009, 11, 4318–4321 CrossRef; (c) O. E. Okoromoba, J. Han, G. B. Hammond and B. Xu, Designer HF-Based Fluorination Reagent: Highly Regioselective Synthesis of Fluoroalkenes and gem-Difluoromethylene Compounds from Alkynes, J. Am. Chem. Soc., 2014, 136, 14381–14384 CrossRef PubMed; (d) F. Nahra, S. R. Patrick, D. Bello, M. Brill, A. Obled, D. B. Cordes, A. M. Z. Slawin, D. O'Hagan and S. P. Nolan, Hydrofluorination of Alkynes Catalysed by Gold Bifluorides, ChemCatChem, 2015, 7, 240–244 CrossRef PubMed; (e) X. Zeng, S. Liu, G. B. Hammond and B. Xu, Divergent Regio- and Stereoselective Gold-catalyzed Synthesis of α-Fluorosulfones and β-Fluorovinylsulfones from Alkynylsulfones, Chem. – Eur. J., 2017, 23, 11977–11981 CrossRef; (f) R. Gauthier, M. Mamone and J.-F. Paquin, Gold-Catalyzed Hydrofluorination of Internal Alkynes Using Aqueous HF, Org. Lett., 2019, 21, 9024–9027 CrossRef.
- (a) S. Eddarir, H. Mestdagh and C. Rolando, Synthesis of fluorinated enynes and dienes via 1-bromo 2-fluoro alkenes, Tetrahedron Lett., 1991, 32, 69–72 CrossRef; (b) M. Yoshida, D. Nagahara, T. Fukuhara, N. Yoneda and S. Hara, Regio- and stereoselective synthesis of fluoroalkadienes using β-fluoroalkenyliodonium salt, J. Chem. Soc., Perkin Trans. 1, 2001, 2283–2288 RSC; (c) M. Sakai, T. Guan and S. Hara, Stereoselective synthesis of 4-fluoro-1,3-alkadienylboronates and their application in the stereoselective synthesis of fluoropolyenes, J. Fluor. Chem., 2007, 128, 1444–1448 CrossRef.
- Y.-Q. Mei and J.-T. Liu, Synthesis and selective carbocupration reaction of fluorine-containing enynic esters, enynylphosphine oxides, and enynylphosphates, Tetrahedron, 2008, 64, 8801–8806 CrossRef.
- (a) M. Li, Y. Wang and G. C. Tsui, Palladium-Catalyzed Stereoselective C−F Bond Vinylation and Allylation of Tetrasubstituted gem-Difluoroalkenes via Stille Coupling: Synthesis of Monofluorinated 1,3- and 1,4-Dienes, Org. Lett., 2021, 23, 8072–8076 CrossRef PubMed; (b) H. Tan, Y. Zong, Y. Tang and G. C. Tsui, Stereoselective Rhodium(I)-Catalyzed C–F Bond Arylation of Tri- and Tetrasubstituted gem-Difluoroalkenes with Boronic Acids, Org. Lett., 2023, 25, 877–882 CrossRef PubMed.
- R. Kumar and B. Zajc, Stereoselective Synthesis of Conjugated Fluoro Enynes, J. Am. Chem. Soc., 2012, 77, 8417–8427 Search PubMed.
- (a) G. Landelle, M. Bergeron, M.-O. Turcotte-Savard and J.-F. Paquin, Synthetic Approaches to Monofluoroalkenes, Chem. Soc. Rev., 2011, 40, 2867–2908 RSC; (b) H. Yanai and T. Taguchi, Synthetic Methods for Fluorinated Olefins, Eur. J. Org. Chem., 2011, 5939–5954 CrossRef; (c) R. Gauthier and J.-F. Paquin, Hydrofluorination of Alkynes: From (E) to (Z), Chem. – Eur. J., 2023, 29, e202301896 CrossRef PubMed; (d) M. Drouin, J.-D. Hamel and J.-F. Paquin, S ynthesis of Monofluoroalkenes: A Leap Forward, Synthesis, 2018, 881–955 Search PubMed; (e) P. H. S. Paioti, S. A. Gonsales, S. Xu, A. Nikbakht, D. C. Fager, Q. Liu and A. H. Hoveyda, Catalytic and Stereoselective Transformations with Easily Accessible and Purchasable Allyl and Alkenyl Fluorides, Angew. Chem. Int. Ed., 2022, 61, e202208742 CrossRef PubMed.
- (a) G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, Synthetic, Methods and Reactions. 63. Pyridinium Poly(Hydrogen Fluoride) (30% Pyridine-70% Hydrogen Fluoride): a Convenient Reagent for Organic Fluorination Reactions, J. Org. Chem., 1979, 44, 3872–3881 CrossRef; (b) N. Yoneda, S. Nagata, T. Fukuhara and A. Suzuki, A Melamine/HF-Carbon Tetrachloride Liquid–Liquid Two-Phase Mixture. A Highly Versatile Hydrofluorinating Agent for Alkenes to Facilitate Continuous Operations, Chem. Lett., 1984, 13, 1241–1242 CrossRef; (c) G. Haufe, Triethylamine Trishydrofluoride in Synthesis, J. Prakt. Chem./Chem.- Ztg., 1996, 338, 99–113 CrossRef; (d) Z. Lu, B. S. Bajwa, S. Liu, S. Lee, G. B. Hammond and B. Xu, Solventless and Metal-Free Regioselective Hydrofluorination of Functionalized Alkynes and Allenes: An Efficient Protocol for the Synthesis of Gem-Difluorides, Green Chem., 2019, 21, 1467–1471 RSC.
- (a) D. Bello and D. O'Hagan, Lewis Acid-Promoted Hydrofluorination of Alkynyl Sulfides to Generate α-Fluorovinyl Thioethers, Beilstein J. Org. Chem., 2015, 11, 1902–1909 CrossRef PubMed; (b) Y. Murata, K. Hada, T. Aggarwal, J. Escorihuela and N. Shibata, Transition-Metal-Free Approach for Z-Vinyl Fluorides by Hydrofluorination of Alkynes Bearing SF4 and SF5 Groups, Angew. Chem., Int. Ed., 2024, 63, e202318086 CrossRef.
- S. G. Davies and P. M. Roberts, Tetrafluoroborate Salt Fluorination for Preparing Alkyl Fluorides, in Fluorination, ed. J. Hu and T. Umemoto, Springer Singapore, Singapore, 2020, pp. 613–629 Search PubMed.
- R. Guo, X. Qi, H. Xiang, P. Geaneotes, R. Wang, P. Liu and Y.-M. Wang, Stereodivergent Alkyne Hydrofluorination Using Protic Tetrafluoroborates as Tunable Reagents, Angew. Chem., Int. Ed., 2020, 59, 16651–16660 CrossRef.
- (a) G. Compain, K. Jouvin, A. Martin-Mingot, G. Evano, J. Marrot and S. Thibaudeau, Stereoselective Hydrofluorination of Ynamides: A Straightforward Synthesis of Novel α-Fluoroenamides, Chem. Commun., 2012, 48, 5196–5198 RSC; (b) B. Métayer, G. Compain, K. Jouvin, A. Martin-Mingot, C. Bachmann, J. Marrot, G. Evano and S. Thibaudeau, Chemo- and Stereoselective Synthesis of Fluorinated Enamides from Ynamides in HF/Pyridine: Second-Generation Approach to Potent Ureas Bioisosteres, J. Org. Chem., 2015, 80, 3397–3410 CrossRef; (c) J. Che, Y. Li, F. Zhang, R. Zheng, Y. Bai and G. Zhu, Silver-Promoted trans-Hydrofluorination of Ynamides: a Regio- and Stereoselective Approach to (Z)-α-Fluoroenamides, Tetrahedron Lett., 2014, 55, 6240–6242 CrossRef; (d) G. He, S. Qiu, H. Huang, G. Zhu, D. Zhang, R. Zhang and H. Zhu, Cu(I)- or Ag(I)-Catalyzed Regio- and Stereocontrolled trans-Hydrofluorination of Ynamides, Org. Lett., 2016, 18, 1856–1859 CrossRef; (e) G. Zhu, S. Qiu, Y. Xi, Y. Ding, D. Zhang, R. Zhang, G. He and H. Zhu, (IPr)CuF-Catalyzed α-Site Regiocontrolled trans-Hydrofluorination of Ynamides, Org. Biomol. Chem., 2016, 14, 7746–7753 RSC; (f) T. J. O'Connor and F. D. Toste, Gold-Catalyzed Hydrofluorination of Electron-Deficient Alkynes: Stereoselective Synthesis of β-Fluoro Michael Acceptors, ACS Catal., 2018, 8, 5947–5951 CrossRef PubMed.
- (a) A. J. Lovey and B. A. Pawson, Fluorinated Retinoic Acids and Their Analogs. 3. Synthesis and Biological Activity of Aromatic 6-Fluoro Analogs, J. Med. Chem., 1982, 25, 71–75 CrossRef CAS PubMed; (b) S. Eddarir, H. Mestdagh and C. Rolando, Synthesis of Fluorinated Enynes and Dienes via 1-Bromo 2-Fluoro Alkenes, Tetrahedron Lett., 1991, 32, 69–72 CrossRef CAS; (c) M. Yoshida, A. Komata and S. Hara, Stereoselective Synthesis of Fluoroalkenes via (Z)-2-Fluoroalkenyliodonium Salts, Tetrahedron, 2006, 62, 8636–8645 CrossRef CAS; (d) T. Guan, M. Yoshida and S. Hara, Stereoselective Synthesis of (E)- and (Z)-Fluoroalkenylboronates Using 2-Fluoroalkylideneiodonium Ylides Generated from (2-Fluoro-1-alkenyl)iodonium Salts, J. Org. Chem., 2007, 72, 9617–9621 CrossRef CAS; (e) M. Sakai, T. Guan and S. Hara, Stereoselective Synthesis of 4-Fluoro-1,3-Alkadienylboronates and Their Application in the Stereoselective Synthesis of Fluoropolyenes, J. Fluor. Chem., 2007, 128, 1444–1448 CrossRef CAS; (f) Y.-Q. Mei and J.-T. Liu, Synthesis and Selective Carbocupration Reaction of Fluorine-Containing Enynic Esters, Enynylphosphine Oxides, and Enynylphosphates, Tetrahedron, 2008, 64, 8801–8806 CrossRef CAS; (g) D. Mandal, R. Gupta and R. D. Young, Selective Monodefluorination and Wittig Functionalization of gem-Difluoromethyl Groups to Generate Monofluoroalkenes, J. Am. Chem. Soc., 2018, 140, 10682–10686 CrossRef; (h) R.-Y. Yang and B. Xu, Chemo-, Regio- and Stereoselective Synthesis of Monofluoroalkenes via a Tandem Fluorination–Desulfonation Sequence, Chem. Commun., 2021, 57, 7802–7805 RSC; (i) M. Li, Y. Wang and G. C. Tsui, Palladium-Catalyzed Stereoselective C−F Bond Vinylation and Allylation of Tetrasubstituted gem-Difluoroalkenes via Stille Coupling: Synthesis of Monofluorinated 1,3- and 1,4-Dienes, Org. Lett., 2021, 23, 8072–8076 CrossRef; (j) Y. Wang and G. C. Tsui, Stereoselective Palladium-Catalyzed C–F Bond Alkenylation of Tetrasubstituted gem-Difluoroalkenes via Mizoroki–Heck Reaction, Org. Lett., 2023, 25, 6217–6221 CrossRef PubMed.
- S. P. J. M. van Nispen, C. Mensink and A. M. van Leusen, Use of dilithio-tosylmethyl isocyanide in the synthesis of oxazoles and imidazoles, Tetrahedron Lett., 1980, 21, 3723–3726 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for the substrates and products. See DOI: https://doi.org/10.1039/d4qo02049a |
| ‡ Y.X. and A.D.C. contributed equally. |
| This journal is © the Partner Organisations 2025 |