
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanophoric hydrogels: when mechanical stress produces useful responses
Franciela
Arenhart Soares
 and
Pol
Besenius
*
and
Pol
Besenius
*
Department of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10–14, 55128 Mainz, Germany. E-mail: besenius@uni-mainz.de
First published on 18th February 2025
Abstract
Mechanophoric hydrogels undergo visible color changes or alterations in optical properties in response to mechanical stimuli. Achieving mechanophoric features in hydrogel materials involves careful control of the polymer microstructure, as well as the incorporation of mechanoresponsive molecular entities known as mechanophores. Upon mechanical activation, mechanophores undergo reversible or irreversible molecular rearrangements, leading to a visual response. This review serves as a practical reference for selecting and designing mechanophores, and classifies their mode of action. By consolidating key information on molecular and materials design criteria, it focuses specifically on mechanophoric responses within hydrogel networks and provides insight into pioneering contributions, as well as some of the most recent advances and literature examples. The multidisciplinary nature of this rapidly growing research field requires expertise in organic synthesis, physical polymer chemistry and photophysics, making this review a valuable resource for researchers interested in the fundamental developments or applications of mechanophoric hydrogel materials.
 Franciela Arenhart Soares | Franciela Arenhart Soares received her Ph.D. degree in Chemistry from the Universidade Federal do Rio Grande do Sul in 2020. Currently she is a post-doctoral fellow in the Besenius group where she is developing organic fluorescent materials for applications within hydrogel networks. She has experience with organic synthesis and analytical methods involving UV-vis and fluorescence spectroscopy in solution and solid state. Her research interests include the synthesis of Excited State Intramolecular Proton Transfer dyes, as well as other luminescent materials. |
 Pol Besenius | Pol Besenius was born and raised in Luxemburg and studied Chemistry in Vienna and Glasgow. He received his PhD from the University of Strathclyde in Glasgow (2008) and undertook postdoctoral studies at the Eindhoven University of Technology, as a Marie-Curie Fellow (2008–2011). He started his independent research group at the Organic Chemistry Institute at the University of Münster supported by a Liebig Fellowship (2011) and moved to the Johannes-Gutenberg University Mainz in 2015 to take up a W2-Professorship. In 2022 he was promoted to W3-Professor of Macromolecular Chemistry and Supramolecular Biomaterials. Pol was awarded an ERC Consolidator Grant (2018), a visiting Faculty Program Fellowship at the Weizmann Institute of Science (2020), and the Forcheur Jean-Marie Lehn Prize (2022). |
Introduction
Hydrogels are three-dimensional networks of hydrophilic polymers that can absorb and retain large amounts of water, typically from 90 wt% to 99 wt%. This unique characteristic makes them resemble biological tissues, and they are used in various applications, including biomedical, pharmaceutical, and environmental fields.1 Hydrogels are formed through either covalent bonds, supramolecular interactions or a mixture of both depending on the nature of the forces between the polymeric chains and the intended applications. While covalent hydrogels possess higher stability and better mechanical properties, they also show limited adaptability and response to external stimuli.2–4 Supramolecular hydrogels are build based on the “self-assembly” of the molecular units.5,6 In contrast to covalent hydrogels, supramolecular hydrogels are known to be more dynamic due to the reversible interactions of the polymeric network, which led to increased external stimuli responsiveness, and adaptable behavior.7 Due to this characteristic, their scope of applications is directly related to the engineering of those individual units8,9 and their mode of crosslinking.10Mechanophoric hydrogels are designed to be sensitive to mechanical stress, i.e. compression, tension, shear, bending or torsion.4,11 The usefulness of the response can be attributed to specific mechanisms within the hydrogel network, like a visible color change, alteration in its optical or catalytic properties, and release of cargo molecules as a response to the mechanical stimuli. Mechanophoric hydrogels have a broad scope of applications in wearable electronics, biomedical devices, sensors, robotics, or controlled release of therapeutic agents making them a promising field of research.12 Mechanophoric hydrogels can be either covalent or supramolecular hydrogel types, depending on the applications. They are obtained through careful control of the microstructure, i.e. by the arrangement of the polymer chains with specific structural features (mechanophoric structural-colored materials) or by the incorporation of inorganic or organic mechanoresponsive molecular entities called mechanophores.13,14 The activation of mechanophores leads to changes in their molecular structure, electronic configuration, or chemical properties.15–17 The ability to undergo a mechanical response makes them valuable tools in the field of materials science, providing a way to visualize and quantify mechanical pressure, stress or damage not only at the macroscopic, but also at the microscopic level.18
Although the development of hydrogels with mechanophoric properties is still challenging due to the hydrophobicity mismatch between most mechanophores and the hydrophilic network, it occurs in a multidisciplinary context, requiring expertise from organic and polymer synthesis, combined with photophysical and macromolecular chemistry. The considerable amount of information available is often challenging for newcomers to the topic, and the demand for the compilation of strategies combining such aspects is critical for successful designing of new materials. This review aims to serve as a practical reference for incorporation of organic mechanophores into the synthesis of hydrogels specifically, and we refer to more extensive reviews for mechanophores in polymer networks wherever appropriate.1,19–21 It achieves this by consolidating information on molecules, materials, and methodologies that have been explored and utilized in this context. The scope of the review specifically focuses on the utilization of organic molecular mechanoresponsive units within covalent and supramolecular hydrogels. The examples discussed are limited to these classes of soft materials containing mechanophores and where the mechano-response was studied systematically. Mechanochromism within other polymeric materials was only indicated as a guide to the state of the art of specific mechanophores. Mechanophores based on metal exchange and inorganic nanomaterials as well as photonic hydrogels, i.e. hydrogels with periodically ordered structures were not included and have been recently reviewed elsewhere.12,15,21,22
The present review is structured following the classical grouping of mechanophores depending on the nature of their activation.23 Before the introduction of the different types of mechanophores, a brief section with conceptual design features will be presented. The second part of the review focusses on type I mechanophores, i.e. materials where the rupture of covalent bonds occurs, including reversible and bond scission materials. The third part will be dedicated to type II mechanophores, materials that do not require bond scission to feature a color change or fluorescence emission. The final section will conclude the review by presenting an outlook and future perspectives.
Response types for mechanophores
Mechanophoric probes or mechanophores are molecular units specially designed to respond with visual color changes to mechanical forces like compression, tension, bending, torsion or shearing forces. As a response to these forces, the material exhibits a visual color change that is often related to alterations at the molecular or supramolecular level. At the molecular level, mechanophores can respond to mechanical stress through the cleavage of bonds, conformational changes, shifts in the fluorescence emission, metal complexation or formation of radicals.13,17,24 Within a hydrogel network, mechanophores can act as molecular reporters for damage, chain mobility, swelling ratios, structural integrity or environmental changes. Beyond the more general classification of type I and II for mechanophores mentioned above, mechanophores can also be categorized according to their response into mechanochromic, mechanoluminescent or mechanofluorescent probes (Fig. 1). | ||
| Fig. 1 Schematic classification of mechanophores and summary of main characteristics of each group. | ||
Mechanochromic mechanophores are molecules which show changes in the absorption spectrum upon mechanical activation, i.e. an intense color change is observed. In general, the changes are caused by expansion or compression of conjugation systems which can also be reflected in molar absorptivity coefficients. Most type I mechanophores show a mechanochromic type response and spiropyran is the most studied mechanophore of this class.25 In contrast, mechanochemical activation of chemiluminescent mechanophores are compounds that emit light upon covalent bond scission, i.e. when light is emitted due to an irreversible chemical reaction in a process known as chemiluminescence.26 An important parameter for these mechanophores is the threshold force required for activation. It represents the force necessary to activate one single molecular mechanophore. Experimentally, this can be measured using optical tweezers or single-molecule force spectroscopy (SMFC).27–29 The threshold forced can also be predicted theoretically with theoretical calculations like Constrained Geometry simulating External Force methods.30,31
Mechanofluorescent/-phosphorescent probes are specific types of mechanophores designed to exhibit a change in their photoluminescence properties in response to mechanical forces. Note, that these are processes where light emission occurs as a relaxation process upon photon absorption from an external light excitation source, which is fundamentally different from chemiluminescence. The terms mechanoluminescent and mechanofluorescent are sometimes used synonymously in the literature; however, for didactic purposes, we distinguish between the two to emphasize to the reader that the origins of their light emissions are fundamentally different.
Mechanofluorescent probes are often incorporated into materials or molecules to serve as sensors for detecting mechanical stress or damage with much faster response compared to traditional mechanophores.20 The change in fluorescence can be visually observed and quantified, providing a convenient and non-destructive means of monitoring mechanical forces at the molecular level.32 One of the advantages of using fluorescent mechanophores relies on the possibility to monitor stress and mechanical constraints with high spatiotemporal resolution and high sensitivity. Molecularly, the fluorescence changes can originate from the disruption of conjugated systems, heterolytic or concerted cleavage/formation of chemical bonds that can either release fluorophores or that will directly result in changes of the electronic structure of the fluorophore.33
Fluorescence changes can arise from a series of different mechanisms like the formation of exciplexes, i.e. heterodimeric complexes in the excited state,34 or based on the restriction of large amplitude molecular motions. In this case, the mechanophores exhibit enhanced fluorescence upon reduction of internal motions in an opposite behavior to typical fluorophores that tend to quench the fluorescence emission when the π-systems are tightly packed.35 More complex mechanisms of fluorescence like the Förster Resonance Energy Transfer (FRET) or more recently the Excited State Intramolecular Proton Transfer (ESIPT) are also encountered. The FRET process involves the transfer of energy between an excited state donor to a nearby ground state acceptor. To efficiently occur, the FRET pair needs to be located around 10 to 100 Å in proximity to each other.36 In contrast, the ESIPT process involves the transfer of a proton from one part of a molecule to another within the excited state, leading to changes in molecular structure and optical properties.37 The ESIPT mechanism induces a strong structural reorganization of the electronic structure of the material, leading to a fluorescence emission largely red-shifted compared to the absorption. This way, ESIPT fluorophores emission is not impaired by self-absorption and inner filter effects, resulting in good photostability over time.38 Although most of ESIPT-related research focuses mainly on pure organic materials, some examples of ESIPT and supramolecular chemistry are emerging.39 Throughout the next sections several examples exploring the diverse types of mechanophores will be discussed and thereby the key aspects of each mechanophore, like activation mode, photoluminescence mechanism and structural design will be highlighted.
Type I: covalent mechanophores
Covalent mechanophores or type I are a large group of mechano-responsive materials in which the response arises from the reversible or non-reversible scission of covalent bonds. These types of sacrificial bonds largely determine the applications of the mechanophores. Reversible mechanophores where the covalent bonds are restored upon light irradiation or heat can be found in self-healing materials40 and in smart systems like write-and-erase or encryption systems.41,42 Although nonreversible mechanophores are preferred for some specific monitoring,1 the loss of activity after the activation might also result in changes of the material properties.Reversible bond scission
SP was also largely explored the preparation of soft materials like hydrogels, either as a “in chain” component56,57 or, more recently, as a crosslinker. Following this second approach, Zheng and coworkers developed a multi-stimuli-responsive hydrogel based on hydrophobic poly(methyl acrylate) (PMA) and hydrophilic polyacrylamide (PAAm).58 The hydrogel was designed to respond to photo-, thermo- and mechanostimuli. The double functionalized SP moiety was placed in the crosslinks through micellar copolymerization due to the low water solubility of the SP molecules (Fig. 2). Under mechanical stress, i.e. by applying tensile strain, the hydrogel changed from yellow to purple due to the conversion of the SP into the MC. The SP-crosslinked hydrogels could reversibly alternate between force-induced network damage state (MC state) and light-induced network recovery state (SP state). Moreover, the final material containing from 0.09 to 0.36 mol% of SP in relation to MA polymer, showed appreciable mechanical properties with tensile strength of 1.45 MPa, tensile strain of approximately 600%, and fracture energy around 7300 J m−2. The high strength and toughness of the SP-crosslinked hydrogels was mainly attributed to the covalent bond breakage of SP mechanophore via SP to MC conversion.
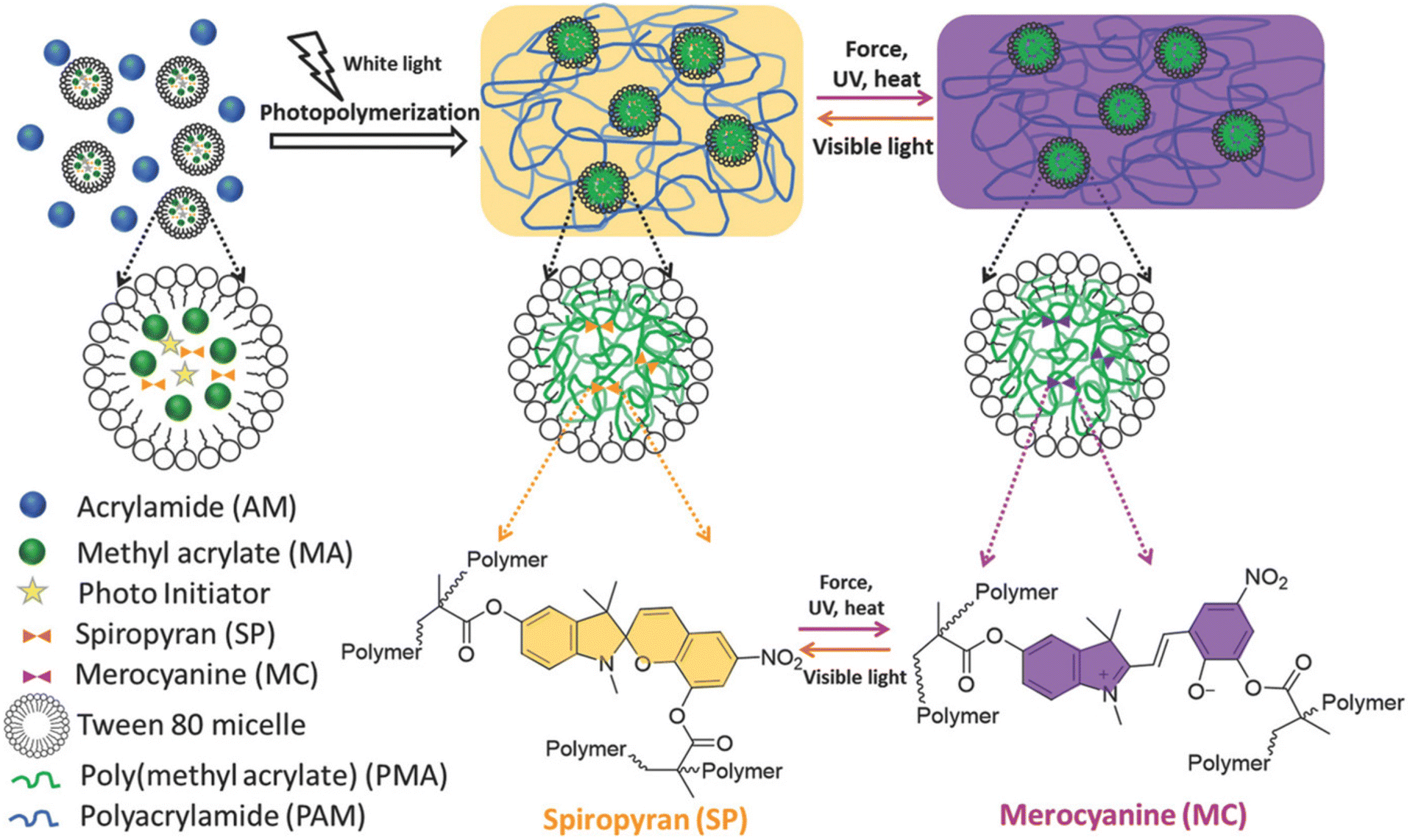 | ||
| Fig. 2 Synthetic procedure for mechanoresponsive poly(AM-co-MA/SP) hydrogels via micellar copolymerization of acrylamide (AM), methyl acrylate (MA) and a spiropyran dimethacrylate crosslinker. The hydrogel exhibits color change from yellow to purple upon force, UV light, and heat stimuli and color reversion from purple to yellow upon white light. Both color change and reversion are triggered by a reversible structural transformation between spiropyran (SP, yellow) and merocyanine (MC, purple) states. Reproduced with permission.58 Copyright 2017, John Wiley and Sons 2017. | ||
In a similar approach, Qiao and coworkers reported on a multi component network organogel elastomer consisting of double (DN) and triple polymeric network (TN) layers containing a SP moiety cross-linked in the first network to investigate their mechanochromic performance.59 For the preparation of the first network, the SP was trifunctionalized with methylacrylate groups and polymerized with butyl acrylate (BA). The second network of acrylate polymer was obtained through photopolymerization of the monomers swelled into the first network. The first network was pre-stretched during the second polymerization, to facilitate the orientation of the SP mechanophores. The triple network was introduced following the same swelling-curing procedure. To characterize the color change during the mechanical tests, the red-green-blue color intensity analysis was used. The tensile strength of the DN material was 7.57 MPa, while the first network alone showed a tensile strength of 0.24 MPa. For the TN hydrogel, the tensile strength was not measured due to necking phenomenon, i.e. when the tensile strain is not dispersed uniformly through the sample, generating heterogenous areas.
With relation to the mechanochromic properties of the samples, the authors observed that the effect of the concentration of the SP on the onset strain of the DN hydrogel was negligible. As expected, under a given deformation, the color intensity increased with the SP concentration. The cyclic tension tests indicated a notable hysteresis loop during the initial loading, and the intensity of the blue color gradually decreased with an increase in the number of cycles. A comparison between the conversion of SP to MC induced by stretching and UV irradiation revealed a significant enhancement in mechanical activation compared to simple-structure polymers containing mechano-active SP. While the DN hydrogel showed a color reversibility even after rupture, the same did not happen to the TN hydrogel, suggesting a rupture of the first network.59
As mentioned before, one of the challenges related to the incorporation of SP mechanophores is the low hydrophilicity of the SP moiety. In order to increase the compatibility between SP and water, Jeong and coworkers recently reported on the preparation of chameleon like hydrogels employing SP functionalized with acrylate terminated polyethylene glycol (PEG) chains.60 This SP containing crosslinker was copolymerized with sodium acrylate. The resulting material containing 10 wt% of crosslinks transitioned from the SP form to the MC form in response to internal stress during the swelling process, enabling monitoring through UV-Vis spectroscopy. The increase of the absorption peak at λabs = 549 nm corresponding to the open MC form, indicated the ring opening of the SP due to swelling of the material.
The SP is considered by many as an efficient mechanophore, whereby the reversibility is conditioned to the exposure of the MC to white light. In the context of mechanophoric hydrogels, the reversibility is a unique feature of the SP mechanophore which can be determining for its applications, like precise control of hydrogel shrinking and swelling and the possibility to reset the system back to its original state.60 Depending on the polymeric matrix, the exposure times can be up to 6 hours.25 Moreover, the formation of aggregates of SP, although slow, can also impact the kinetics of the SP/MC interconversions.61 Despite the SP popularity as a mechanophore, a range of compounds with faster reversibility to the initial state was explored as will be discussed in the following sections.
Based on the advantages of xanthene compounds, Yang and coworkers reported on the development of a double network (DN) hydrogel containing a rhodamine derivative.69 DN hydrogels, like the name suggest, are hydrogels composed by two intercalating polymeric networks, which, in general, yields a material with improved mechanical properties when compared with micellar hydrogels.70 The rhodamine derivative HPR (Fig. 3a) was combined with poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS) to form the first hydrogel network. The second hydrogel network was composed of PAAm which was formed under UV irradiation in the swelled PAMPS. The resulting material is transparent, however, when stretched a pinkish color appears. The mechanochromic effect is much more pronounced under UV light (λexc = 365 nm) since the fluorescence changes from blue (λem = 426 nm) to orange (λem = 566 nm). The system was reported to revert to the initial state 2 hours after being relaxed. When submitted to uniaxial tensile stress, the DN showed a fluorescence emission at λem = 574 nm. The redshift compared to the emission on the tensile tests was attributed to a higher planar conformation of the mechanophore. To investigate the effect of the pH on the mechanochromic response, the authors prepared hydrogels with different pH values. The variation of pH showed negligible effects on the mechanical properties of the hydrogel, but at lower pH the fluorescence emission at 566 nm in the pristine material becomes more intense. This variation implies that a shift between the ring close and the ring open forms of the HPR mechanophore takes place. In addition, at lower pH the authors also observed that the fluorescence emission increased faster upon uniaxial stretching.69
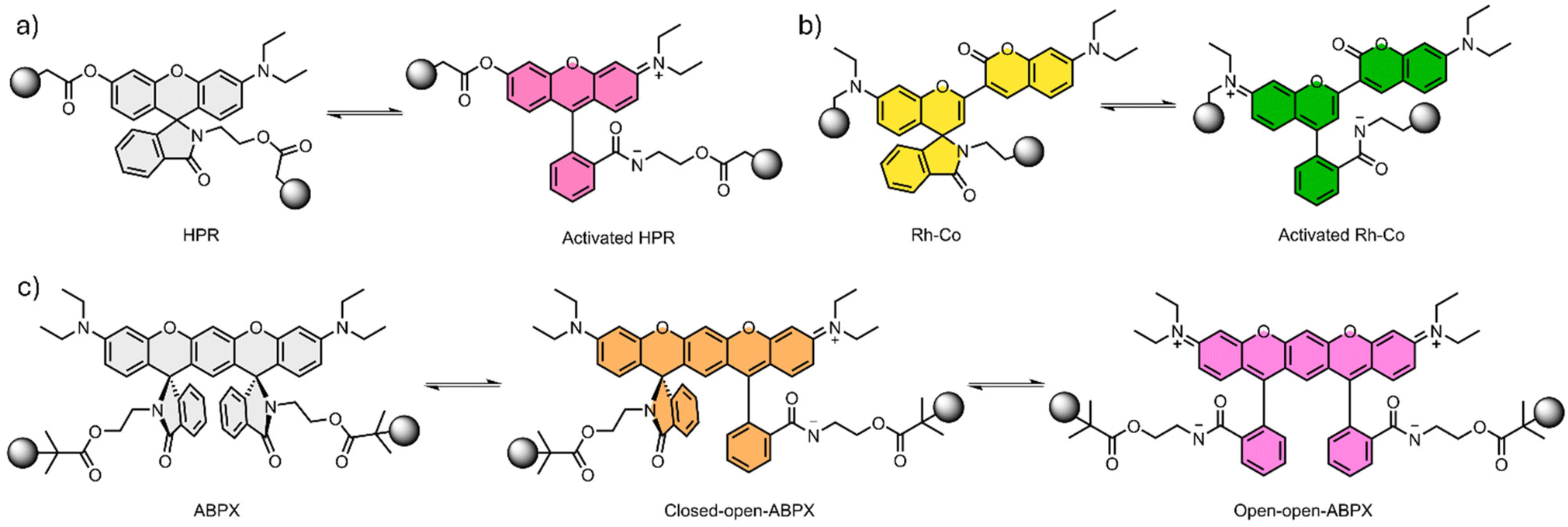 | ||
| Fig. 3 Xanthenes derivates and their respective closed and open forms. (a) HPR derivative, (b) semirhodamine coumarin (Rh–Co) and (c) aminobenzopyranoxanthene (ABPX). | ||
Another example of HPR incorporation as a mechanophore into hydrogels was recently described by Wang and coworkers.71 Inspired by the cephalopods ability to change their skin colors and patterns, the author produced DN hydrogels with arbitrary shapes by using direct ink writing as printing method. The printing ink was composed of the microgels previously prepared containing the mechanophores and swelled with polymerizable monomer, like AM, solution. UV irradiation while printing yielded materials with different shapes, like stars or shells. The printed hydrogel showed a tensile strength of 0.56 MPa and the HPR mechanophores were activated after compression. The samples showed simultaneously mechanochromic and mechanofluorescent responses, a red color and increase of fluorescence emission at λem = 566 nm, respectively. By varying the structure of the mechanophore in the ink, samples showing different colors upon mechanical stress were also prepared (Fig. 4).
 | ||
| Fig. 4 Natural and UV-light photographs of the star shaped hydrogel before and after compression. Scale bars: 1 cm. Reproduced with permission.71 Copyright 2023, American Chemical Society. | ||
Still based on DN hydrogels, Zhang and coworkers employed a rhodamine derivative to prepare a suitable material based on poly(N-isopropylacrylmide) (PNIPAAm) hydrogels for application as multifunctional smart windows.72 Smart windows are designed to reversibly regulate the amount of light passage in response to external stimuli or a combination of responses like transmittance regulation ability, freezing-/compression-induced mechanochromic and mechanofluorescent properties. These hydrogels are known to have a lower critical solution temperature near room temperature and can reversibly allow light passage or light reflection, which are suitable characteristics for designing transparent materials like windows. The rhodamine derivative was placed as a crosslinker in the first polymeric network containing NIPAAm and 2-acrylamido-2-methylpropanesulfonic acid sodium (NaAMPS). The second polymeric network was immersed in an aqueous solution containing NIPAAm and Irgacure 2959 as photoinitiator. The second polymerization under UV light was carried into a glass mold. The incorporation of rodhamine was confirmed by infrared and UV. The hydrogels containing the highest amount of NaAMPS showed the best mechanical properties and higher transparency. The hydrogels showed reversible color change from transparent to pink when frozen to −25 °C and heated up to 25 °C. Upon increasing compression, the hydrogels concomitantly showed color change from transparent to pink and redshift of the fluorescence emission from λem = 460 nm to λem = 575 nm. Although the material showed good reversibility after heating/cooling, the reversibility after compression was conditioned to the preservation of the first network containing rhodamine.72
Although most examples discussed so far are related to DN or TN hydrogels, micellar hydrogels displaying mechanochromic properties containing rhodamine derivatives are also found in the literature. Chen and coworkers developed a micellar hydrogel using a semirhodamine and coumarin (Rh–Co) hybrid (Fig. 3b) in order to explore both mechanochromic and mechanofluorescence response.73 The ring open form of the Rh–Co shows simultaneously high molar extinction coefficient (9.43 × 104 L mol−1 cm−1) and intense near infra-red (NIR) emission (λem = 722 nm) with high quantum yield in dichloromethane (ΦF = 73.45%). The mechanochromic behavior was initially evaluated with sonication experiments. For these, the Rh–Co was incorporated in the center of PMA chains using single-electron transfer living radical polymerization resulting in a PMA-Rh-Co-PMA polymer. As a control, a single end functional PMA-Rh-Co polymer was also prepared. As the solution changed from slight yellow to green during the sonication, the fluorescence at λem = 722 nm gradually increased, revealing the mechanochromic response.
To obtain the micellar hydrogels, the Rh–Co, methyl acrylate (MA) and an initiator were dispersed into an emulsion with Tween 80 and acrylamide was added. After polymerization, the resulting composite was swelled and the mechanochromic properties of the hydrogels were evaluated through mechanical tests including tensile strain, compression and friction. As the strain increased, the chromaticity coordinates of the hydrogel film followed almost a linear pathway from the yellow region to the green one. At the same time, a new NIR emission band around λem = 700 nm gradually increased. The initial yellow state was recovered after approximately 6 hours of the strain release. The compression tests showed similar behavior to the stretched sample. The fluorescence responded accordingly, although the emission was around λem = 728 nm, probably due to a more planar conformation of the ring open Rh–Co. A plot of the change in the red channel against the compression stress showed a sigmoidal shape. Similar results were obtained from the friction tests.73
Although the vast majority of the mechanophores are designed to generate one mechanochemical product, which means one color response, some examples displaying more responsive modes in the same mechanophore are beginning to be designed.74,75 Even if the polymeric system employed was not a hydrogel, it is worth mentioning two examples. First, Ma and coworkers reported on the development of a dual-color mechanophore based on a double-spiro-ring structure (Fig. 3c).76 The aminobenzopyranoxanthene (ABPX) was functionalized to afford a bifunctional initiator in order to covalently link the mechanophore to a PMA network. As a control, a one-sided ABPX initiator was also prepared. The mechanochromism of the resulting systems was evaluated with sonication experiments, which showed a dependence with the molecular weight of the PMA for the same ultrasonic conditions. The system with an average molecular weight of 245 kDa changed from transparent to yellow and finally to reddish color after 30 min of sonication. For lower molecular weights systems, the mechanochromic response was not significant.76 Second, Chen and coworkers concomitantly employed the same strategy to build four ABPX dyes with different configurations (trans-ABPX; cis-ABPX; iso-trans-ABPX; iso-cis-ABPX).77 The isomeric mechanophores were employed to evaluate the geometry-controlled ring opening reactivity using sonication, SMFS and CoGEF calculations. The materials displayed greatly varied mechanochromic and mechanofluorescence behavior, which was attributed mainly to the different force-coupled geometry changes alongside the polarity enhancement. The results from these two studies indicated the significance of exerting control over both the geometric and polarity aspects, emphasizing the role of thermodynamic equilibrium in mechano-responsive polymers.
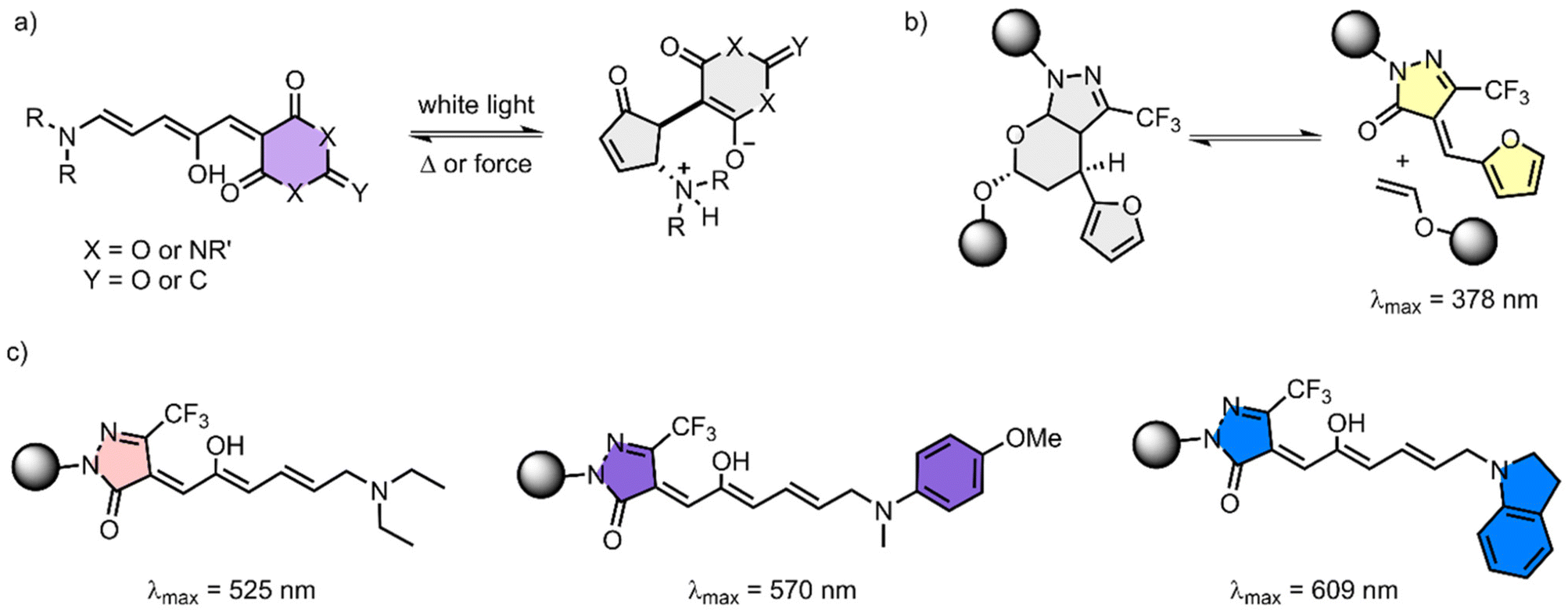 | ||
| Fig. 5 DASA examples. (a) general structure of the DASA adduct, (b) activation of the DASA adduct by mechanical force and (c) distinctly colored products obtained after contact with different secondary amines. | ||
DASA compounds were used in several applications, like targeted drug release, photothermal actuators and as photo-switches in polymeric materials.78,82 Although the applications in hydrogels are still limited a few important concepts can already be highlighted. Recently, Overholts et al. published the first example of DASA derivatives as mechanophores embed in a PMA network.83 Since the properties of the DASA compounds are highly controlled by the amino substitution, the authors used one single DASA precursor as a mechanophore which could be differentiated after its activation, in order to produce a wide range of functionally diverse DASA (Fig. 5b). After the mechanochemical activation of the DASA precursor, different amines were added, which upon reaction with the activated DASA precursor resulted in distinct colored products (Fig. 5c).
The mechanochemical activation of the DASA precursors was firstly confirmed in DCM/HFIP solution. For this, PMA functionalized with DASA adducts was prepared by polymerization of MA with a chain centered DASA precursor bearing two α-bromoisobutyryl ester groups. As a control, a PMA containing a DASA precursor at the end of the chain was also prepared. After sonication, a new absorption peak appeared at λabs = 378 nm, indicating the formation of the furan activated product. The addition of different secondary amines resulted in new absorption peaks and the solution changed from slight yellow to pink (λem = 525 nm), purple (λem = 570 nm) or blue (λem = 609 nm), depending on the nature of the amine. As expected, the control showed no color change after sonication and subsequent addition of the amines.83
To demonstrate the mechanophore activation and subsequent formation of the DASA in bulk, the DASA precursor functionalized with two or one terminal vinyl groups was covalently linked to polydimethylsiloxane (PDMS) chains. The resulting colorless films were subjected to tensile force and no color change was observed. However, after immersion of the mechanically activated films in a solution containing the secondary amine, the blue color was visible. The system was reverted to the initial state by irradiation with white light. The authors also successfully applied the films for multicolor soft lithography.83
The main advantages of the DASA building blocks as mechanophore units are related to the possibility of fine tuning their optical properties via the amino substitution. Depending on the acceptor group, the absorption of the open DASA isomer is tunable between 450–750 nm. Moreover, the DASA photoswitches can be activated with visible or near infrared light, which enhances their compatibility in biological applications. Despite their many advantages, the main challenges related to the use of DASA units as mechanophores is related to their incorporation into the polymeric chains. DASA precursors typically show limited stability in the presence of radicals and strong nucleophiles. Moreover, depending on the synthesis conditions, different ratios of the open and closed isomer can be produced, requiring careful control of the polymerization conditions.78
Non-reversible mechanophore motifs
The bis(adamantyl)-1,2-dioxetane mechanophore undergoes an irreversible homolytic bond scission with production of luminescence (λlum = 420 nm).26 The luminescence results from the rupture of the four-membered ring and subsequent relaxation through radiative emission of the excited adamantanone to the ground state. Bis-Ad was successfully employed as a mechanophore to a series of polymeric materials like elastomers,85 composites68 and more recently into hydrogels.86
Chen and coworkers produced a micellar hydrogel containing the bis-Ad as crosslinker agent. Since one of the biggest challenges related to the application of bis-Ad as a mechanophore in hydrogels is related to the weak luminescence of the adamantanone in aqueous media, the authors used TWEEN 80 micelles loaded with bis-Ad to overcome this issue. In order to characterize the bond scission in real time, a fluorescent dye was also physically incorporated to the micelles previously to the polymerization. After polymerization under UV light, the resulting material MA-bis-Ad-AM formed a transparent film which was subsequently swelled for different times. The confirmation of the mechanophore activation in the hydrogels was done using a rheometer equipped with a high-speed camera. All the three different dyes employed with absorption around λabs = 420 nm acted as FRET pairs, showing fluorescence emission at different regions like red (λem = 614 nm), green (λem = 502 nm), and blue (λem = 430 nm). The same behavior was also observed during stretching experiments. In addition, the damage distribution and bond-breakage mapping of the deformed hydrogel and the notched sample was described with high spatial–temporal resolution.86
Unlike the bis-Ad the homolytic cleavage of the C–C bond in DFSN results in a relatively stable pink cyanofluorenyl radical.84,87 In addition to the mechanochromic response, the DFSN was also reported to improve the mechanical properties when used as a crosslinker.88 However, to the date of preparation of this review, no examples were found in the literature of DFSN applications in hydrogels.
 | ||
| Fig. 6 Force-induced elimination of π-extended anthracenes through retro Diels–Alder reaction. | ||
Employing the same mechanophoric system, the group of Göstl and coworkers reported on a series of microgels and their associated physicochemical transformations under shear force.93–95 The poly(N-vinylcaprolactam) (PVCL) microgels (d ∼ 400 nm) were cross-linked with a force-responsive mechanofluorophore, based on the Diels–Alder adduct of a 9-π-extended anthracene and maleimide. The authors employed fluorescence spectroscopy and CLSM to analyze the extent of covalent bond scission within the microgels after different times of sonication (0–120 min). An increase of the fluorescence was observed with progressing sonication time, indicating that the shear force reached the dense mechanophore rich core. Moreover, upon shearing, the microgel size decreased rapidly and the number of cleaved covalent bonds in the network increased.95 In a similar approach, the group investigated the effects of supramolecular bonds between the network and (+)-catechin (+C) hydrate on the microgels. As the content of +C increased, resistance against shear force exerted by ultrasonication also increased, becoming comparable to the resistance of covalent microgels. The presence or absence of mechanical energy dissipation through the sacrificial +C bonds was visualized by the absence or presence of a fluorescence signal from the mechanophore. While the +C H-bonds were cleaved easily and dissipated the mechanical energy, they protected the covalent bonds of the colloidal network as sacrificial bonds. In this sense, the microgels containing +C showed no significant fluorescence at λem = 430 nm, indicating the efficiency of +C in dissipating the sonication energy, while microgels without +C showed intense fluorescence emission at λem = 430 nm after 10 seconds of sonication.94
Robb and coworkers reported a strategy using a mechanically triggered cascade reaction to release a cargo molecule.96 The release a hydroxycoumarin over time could be monitored via fluorescence (λem = 380 nm). To achieve this, a furan–maleimide Diels–Alder adduct was included at the center of PMA polymeric chains. The mechanoactivation with ultrasound pulses (1 s on/2 s off, 0 °C, 20 kHz, 8.2 W cm−2) induced a retro Diels–Alder reaction, revealing a metastable furfuryl carbonate that quickly decomposed in polar protic media (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN:MeOH) to release carbon dioxide and the hydroxycoumarin.
:1 MeCN:MeOH) to release carbon dioxide and the hydroxycoumarin.
The mechanoactivation for the release of small molecules was also successfully reported by Herrmann, Göstl and coworkers using ultrasound to activate drugs from inactive macromolecules or nano-assemblies through the controlled scission of mechanochemically labile covalent bonds.97 Herrmann and Göstl also expanded the applications of sonochemistry for the release of bigger cargo molecules like DNA- and RNA-based oligonucleotides.98 More recently, the authors reviewed the applications of mechanochemistry for biomacromolecular systems and genetics.99 Lately, the authors reported on the ultrasound sonication of network core-structured star polymers (NCSPs) containing disulfide bridges as mechanophores.100 The NCSPs of poly(ethylene glycol) methyl ether methacrylate (PEGMEMA) with a molecular weight of ≈109 kDa were submitted to sonication. The efficiency of the system was initially quantified by ‘turn on’ sensor molecules leveraging the Michael addition of the mechanochemically generated thiol groups and subsequent retro Diels–Alder reaction to release a fluorophore, in a similar approach as described above in the work of Robb and coworkers.96 By comparing linear polymer functionalized with the disulfide mechanophore and NCSPs, the latter enabled faster mechanophore scission and were responsive to lower sonication powers compared to linear chains. The result highlights the importance of polymer architecture in the mechanoactivation. In addition, as a proof-of-concept the authors prepared a furylated derivative of Doxorubicin (Dox), as anticancer agent. The release of furylated Dox after mechanoactivation was confirmed with in vitro experiments. The IC50 of the sonicated and defurylated Dox-loaded NCSPs was significantly lower compared to the control samples and similar to pristine Dox. These systems highlight the potential of mechanophore strategies in biomedical applications and represent significant milestones in the research field of mechanochemistry and more specific to sonopharmacology, which was also recently reviewed.101,102
Type II mechanophores: supramolecular mechanophores
So far, we have discussed examples of mechanophores that require reversible/non-reversible covalent bond rupture to show a response. In this section, we collected some examples of mechanophores that do not require such sacrificial bonds. This second class of mechanophores encompasses a wide range of molecules that can undergo conformational changes, form/disassemble aggregates or respond to changes of intermolecular interactions and secondary structures. Those supramolecular interactions are important for the detection of low mechanical stress and strain. In general, these kinds of mechanophores show good reversibility and high sensitivity. In the following sections, some examples of supramolecular mechanophores which can form supramolecular assembly of π-systems, FRET and NRET pairs, and ESIPT are discussed.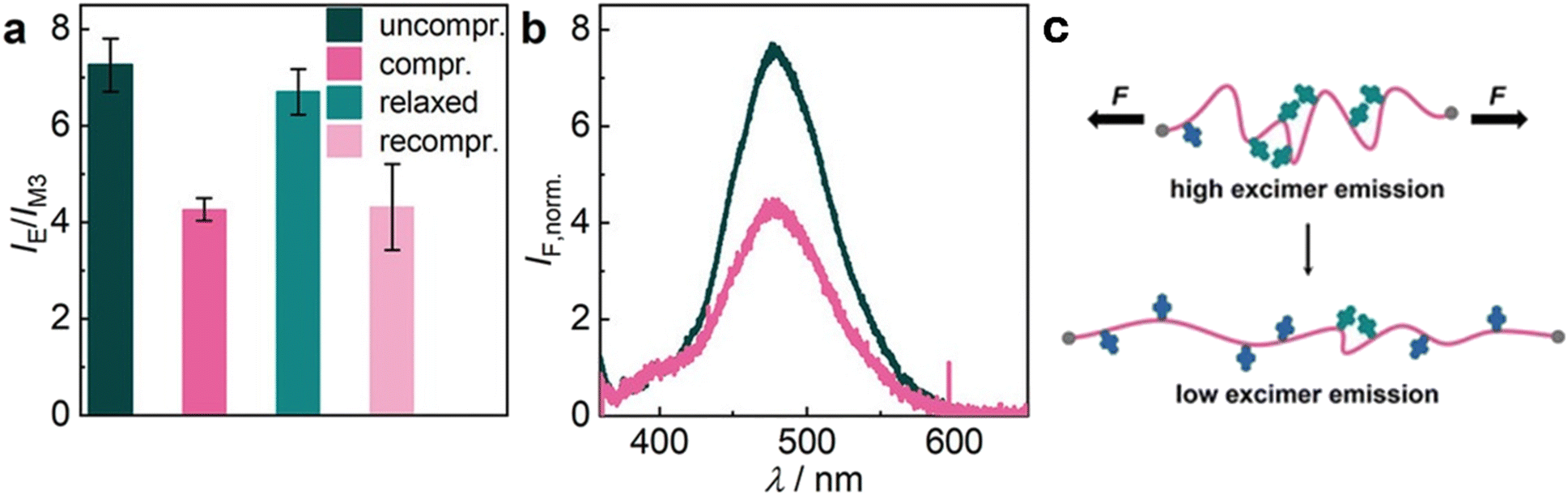 | ||
| Fig. 7 Reversible alteration of PyMC32-crosslinked hydrogel H3.2 emission upon uniaxial compression. (a) IE/IM3 ratio. Mean values ± SD from the mean. N = 5 independent fluorescence measurements. (b) Individual, normalized (to M3) emission spectra color-coded to the bars of panel a. λexc = 340 nm. (c) The hypothesized associated mechanophore transition upon mechanical hydrogel deformation as observed by in situ measurements. Reproduced with permission.104 Copyright 2022, Georg Thieme Verlag KG Stuttgart-New York. | ||
Following a similar strategy, Sagara and coworkers prepared a cyclophane based mechanophore where the aromatic portion consisted of a pyrene derivative core.105 The cyclophane mechanophore was expected to show excimer emission in the pristine state and monomeric emission under mechanical stress. In fact, the photophysical characterization of the material showed a single emission band at λem = 530 nm which is characteristic of the excimer emission of 1,6-bis(phenylethynyl)pyrene. The fluorescence spectra of the monomeric species display clear vibronic structures with peaks at λem = 449 and λem = 469 nm. The hydrogels of AM, ethyl acrylate (EA), and N,N-methylenebisacrylamide with the cyclophane mechanophore as crosslinkers showed dependence of the excimer/monomer emission rate with the content of water. When the ratio of water increases from 10% to 50% the fluorescence spectra showed a blue-shift and the samples become opaque, as an indicative of a phase separation phenomenon in the hydrogel. During the mechanical tests the excimer emission gradually decreased, but was restored after release, confirming that the distances between the pyrene derivative cores increased during strain.
Another excimer forming dye is the perylene diimide (PDI). Depending on the chemical substituents, PDI can emit fluorescence in a wide range of wavelengths making them attractive platforms for the design new supramolecular mechanophores.106 Recently, Draper and coworkers prepared a series of ten water soluble PDI derivatives containing amino acids.107 The authors investigated the PDI derivatives monomers and aggregates in water and the formation of hydrogels. Despite the amino acid substituent, the fluorescence emission was around λem = 550 nm. Although the goal of the study was not related to the mechano-response of the PDI derivates, the authors found a clear relationship between the quantum yield and aggregation/monomer emissions. These observations directly point to the PDI derivatives as promising supramolecular mechanophores in hydrogels.
Some mechanophores are designed with molecular structures that prevent efficient non-radiative decay pathways when in the aggregated state, i.e. when in close interaction with other mechanophores in the surrounding. In the dispersed (or in solution) state, these molecules may have limited fluorescence, but in more viscous environments or due to supramolecular interactions, the non-radiative decay pathways like intramolecular motions and twisted intramolecular charge transfer (TICT), for example, are restricted, playing a constructive effect on the emission process and therefore leading to enhanced fluorescence.35,108,109 From the supramolecular perspective, the enhancement of fluorescence can be achieved by supramolecular polymerizations of individual weakly fluorescent monomeric units.110 In practice, the same principles of molecular motion restriction and TICT are applied.
Tetraphenylethene (TPE) derivatives are commonly used as mechanophores where the combination of phenomena like molecular rigidification and TICT play an important role in the aggregate state.20 A TPE-based mechanophore may have a molecular structure that allows intramolecular rotation in the dispersed state, generally leading to weak fluorescence in solution. Upon aggregation, it becomes more planar and the intramolecular motions are restricted, leading to increased fluorescence intensities. Although TPE was extensively explored as a mechanophore in diverse polymeric systems,20 its mechanofluorescent behavior remains quite unexplored within hydrogels. Recently, two studies have employed TPE derivatives to monitor the aggregation of polymeric chains in hydrogels. The molecular motions were induced either by cationic surfactants111 or by variations on the pH and temperature.112 In both studies the TPE derivatives acted as crosslinkers and were covalently linked to the polymeric matrix during the polymerization step. The fluorescence intensity of the TPE derivatives increased as the mechanophores showed increased proximity, i.e. where the polymeric chains showed small amplitude of molecular motions or shrinkage of the hydrogel network.
In contrast to type I mechanophores that need to be covalently link to the polymeric matrix, type II mechanophores do not require this covalent attachment and can be incorporated in polymeric matrix via physical interactions to form composites. As an example of this strategy, Saengdet and Ogawa developed a bionanocomposite hydrogel based on gelatin and a cyanine dye, 1,1′-diethyl-2,2′-cyanine (PIC), which was adsorbed in smectite (a layered clay mineral).113 The tensile strength of the bionanocomposite hydrogel was 0.18 MPa, with reversible mechano-response through the fluorescence emission from red to blue. The PIC aggregates showed a fluorescence emission around λem = 570 nm. When the samples were stretched, this fluorescence signal decreased and a peak at λem = 520 nm increased, suggesting that the aggregates were broken into the monomeric species. More recently, the same authors reported the mechanofluorescent response of the same bionanocomposite hydrogel to different swelling ratios.114 To investigate the effect of swelling ratio, the authors employed hydrogel beads which possess higher interfacial area per unit compared to hydrogels with anisotropic shape. The swelling in water and deswelling in ethanol triggered the mechanofluorescence behavior between monomers and J-aggregate of the cyanine dyes due to solvent-induced polymer chains untangling (Fig. 8a). In addition, the hydrogel beads showed mechanofluorescent response upon uniaxial stretching, and the phenomena was explained by the formation and disruption of PIC J-aggregates on the smectite surface (Fig. 8b).
 | ||
| Fig. 8 (a) Schematic representation of the mechanofluorescent behavior of the bionanocomposite hydrogel and (b) Photographs of the gel bead during compression under daylight (left) and UV 365 nm (right) and the absorbance and fluorescence spectra form the same samples. Reproduced with permission.114 Copyright 2024, American Chemical Society. | ||
Although in the literature a wide variety of supramolecular π-systems and materials were developed, their applications as supramolecular mechanophores into hydrogels remain sparse. Several examples of mechanophores able to monitor the rigidity of the environment and polymer chain flexibility like the push–pull papillons,115 norborn-2-en-7-one derivatives,116 naphthalimide derivatives,117 among others,118 has already been studied along other polymeric systems than hydrogels. Besides the limited examples, mechanophores based on molecular motion restrictions have the advantages of reversibility and intense fluorescence emission, with moderate to high quantum yields, which facilitates the use of high precision techniques. Moreover, the photoluminescence spectrum can cover a broad spectral range, going from blue emitters, like the pyrene based mechanophores, to near infrared regime using perylene diimide derivatives. However, one of the main challenges to bring together this group of mechanophores and hydrogels, is still related to their low solubility in aqueous media due to their highly hydrophobic structures.
 | ||
| Fig. 9 Strain and temporal response of the mechanofluorescent DNA hydrogels with different force-sensing modules. (a) Sequence and structure of the different modules investigated. Note that the rest of the modules, fluorophores (Atto488, Atto565), quencher (Iowa Black RQ), and attachment strands are identical in all experiments. The T and N modules are controls in which the fluorophore and quencher strands are (T) connected via a T6 covalent link without sacrificial duplex or not connected (N) at all. (b) Fluorescence imaging at different elongations of a 0.7 wt% hydrogel functionalized with D1. Scale bar = 1 mm. (c) Evolution of the red/green fluorescence ratio R/G(ε) after subtraction of R/Gini for the different DNA hydrogels functionalized with the mechanofluorescent modules. (d) Temporal recovery of the fluorescence ratio after hydrogel failure (normalized to R/Gmax and R/Gini). Reproduced with permission.119 Copyright Springer Nature 2019. | ||
Although most of the known FRET pairs are composed of organic dyes, fluorescent proteins (FP) can also actively participate in the FRET process.36 The ability of FP fold and unfold under mechanical stress can be useful to monitor, for example, cell-generated mechanical forces in living systems. Vogel and coworkers developed a hybrid hydrogel where the proteins were covalently linked to the polymeric network through click-reactions.121 Dimeric fibronectin (Fn) was functionalized with several donors (Alexa488) and acceptor (Alexa546) fluorophores, in order that sufficient conformational changes could be read out by FRET. After the functionalization, the Fn was covalently attached to the polymeric chain of PNIPAAm through strain-promoted azide–alkyne cycloaddition (SPAAC) reaction. The four arm star-shaped polyethylegycol functionalized with dibenzocyclooctyne was used as a crosslinker. The conformation dependent FRET was primarily confirmed by protein denaturation where the secondary structure of the protein is lost, therefore generating the FRET signal. The final material was gently stretched over a PDMS surface, and the fluorescence was monitored by confocal microscopy. At a stretching strain around 90% the hydrogel fluorescence emission changed from green to blue, confirming the activation of protein chains.
Another exciting class of supramolecular compounds able to show mechano activation using FRET as probe are rotaxanes. Rotaxanes are molecular interlaced structures where the relative position of the modules can be reversible controlled by an external stimulus.122 Recently, Sagara and coworkers developed a rotaxane-based supramolecular mechanophore with reversible on/off FRET emission.123 The rotaxane was composed by a cyclic molecule equipped with a green light emitting fluorophore which is connected through a short spacer to a red acceptor BODIPY fluorophore (Fig. 10). Due to its good photophysical properties, the donor was 9,10-bis(phenylethynyl)anthracene (An) and the quenching pair was 1,4,5,8-naphthalenetetracarboxylic diimide (NpI). The red fluorophore was a BODIPY derivative (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) which also possessed high emission efficiency. The rotaxane mechanophore was covalently incorporated in a polyurethane-urea (PUU) matrix to form the hydrogel. In contrast with common polyurethanes, the presence of carboxyl groups on the main chain of the PUU polymer improves the hydrophilicity of the material. In the pristine state, the green emitter is positioned in close proximity to the quencher. This spatial arrangement hinders green emission and inhibits FRET from the green emitter to the red emitter due to charge transfer interactions or photo-induced electron transfer between the two moieties. Following the deformation of the PUU hydrogels, the intensity of the red fluorescence (λem = 660 nm) rises due to the force-induced separation of the emitter from the quencher, in a turn on of FRET. Upon subsequent removal of the force, the fluorescence intensity gradually decreases, showing the reversibility of the system. As showed by Sagara and coworkers, rotaxanes as supramolecular mechanophores are powerful tools, since by modification of the acceptor, the emission color can be tuned, and a wide range of colors can be achieved. Moreover, the development of redshifted emitting mechanophores is highly desirable for biological applications.123
 | ||
| Fig. 10 (a) Schematic illustration of FRET control by forces on the rotaxane-based supramolecular mechanophore in stretchable hydrogels. In the force-free state (left), the green emission is quenched because of CT complex formation or PeT. On the right, during stretching the distance between the 9,10-bis(phenylethynyl)anthracene (green) and the 1,4,5,8-naphthalenetetracarboxylic diimide increases (blue), resulting in energy transfer from the anthracene to the red-emitting BODIPY dye. (b) Molecular structures of cyclic compound rotaxane-based supramolecular mechanophore. Reproduced with permission.123 Copyright 2023, American Chemical Society. | ||
Similarly to FRET, NRET is a distance dependent nonradiative process. It is employed as potent spectroscopic nanometric ruler to measure the distance between a NRET fluorophore pair. With the use of a (9-phenanthryl)methyl methacrylate (Ph), and (9-anthryl)methacrylate (An) as a NRET pair, Saunders and coworkers developed a nanogel probe for the quantitative fluorescence sensing of pH and strain in hydrogel matrices.124 The nanogel particles containing Ph and An were prepared by emulsion polymerization and showed pH response. The fluorescence emission of the Ph donor band (ID) at λem = 366 nm increased, whereas the intensity of the An acceptor band (IA) at λem = 414 nm decreased when the pH increased from 4.5 to 9.0. By incorporating the nanogel particles in a host hydrogel, the ratio ID/IA was also employed to monitor the swelling rate. The decrease of the An emission was a direct result of the separation between the donor and acceptor pair during the swelling indicating the chain movement inside the hydrogel.
By changing the NRET pair, the same group reported the development of a nanogel probe which could sense a series of environmental stimuli affecting the swelling of the hydrogel.125 The (9-anthryl)methacrylate and BODIPY FL amine NRET pair showed reversible and quantitative response to pH, temperature, presence of divalent cations, gel degradation and tensile strain. A third study aiming biological applications was also reported.126 Due to the high complexity of the local environment in living cells, the author chooses a NRET pair able to emit fluorescence at NIR. To prepare the microgels the commercially available sulfo-cyanine fluorophores Cy5 (donor) and Cy5.5 (acceptor) were employed. Compared to the previously reported systems, the new probes showed few major advantages: they enable ratiometric fluorescence intensity detection of particle swelling over much higher volume ratios (from 1–90). The excitation and the emission occur at NIR which represent an important feature for applications in living systems where the background absorption is high. In addition, the fluorescence emission maxima provided a second microgel size-dependent parameter to monitor changes in microgel swelling.
As highlighted in the beginning of the section, the main advantages of employing FRET pairs as mechanophores are the combination of photoluminescent detection of the individual components as well as the force probe, the high spatiotemporal resolution of the response and tuning of the emission color.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– or –CO). The two species are usually referred as enol (E) and keto (K) forms and have distinct photophysical properties. The proton transfer between the E and K is dependent on a series of factors, like the polarity of the environment, the presence of substituents and the close proximity and orientation between the acceptor and donor groups. The control over the distinct emission of the E and K species is well understood in solution and solid state.37 Several examples of hydrogels exploring the fluorescence properties of ESIPT molecules can also be found in the literature.127–131 However, when it comes to mechanocromism, only a few studies are available in the literature, suggesting that this control is much more difficult to achieve in swollen polymeric matrices. In fact, only two studies were published by Ma, Sijbesma and coworkers proposing the control of the dihedral angle between the donor and acceptor, so the ESIPT compounds could act as mechanophores.23,132 In both cases, although it was possible to detect variations between the ratios of E and K emission, these changes were not intense enough to produce an intense variation in the emission color with the naked eye. The pre-existence of intermolecular hydrogen bonds with the polymeric matrix strongly supported the E emission instead of the K emission in the pristine state.132
N– or –CO). The two species are usually referred as enol (E) and keto (K) forms and have distinct photophysical properties. The proton transfer between the E and K is dependent on a series of factors, like the polarity of the environment, the presence of substituents and the close proximity and orientation between the acceptor and donor groups. The control over the distinct emission of the E and K species is well understood in solution and solid state.37 Several examples of hydrogels exploring the fluorescence properties of ESIPT molecules can also be found in the literature.127–131 However, when it comes to mechanocromism, only a few studies are available in the literature, suggesting that this control is much more difficult to achieve in swollen polymeric matrices. In fact, only two studies were published by Ma, Sijbesma and coworkers proposing the control of the dihedral angle between the donor and acceptor, so the ESIPT compounds could act as mechanophores.23,132 In both cases, although it was possible to detect variations between the ratios of E and K emission, these changes were not intense enough to produce an intense variation in the emission color with the naked eye. The pre-existence of intermolecular hydrogen bonds with the polymeric matrix strongly supported the E emission instead of the K emission in the pristine state.132
In order to overcome such issues, another study employing an ESIPT-rhodamine mechanophore was recently published by the same group.133 The ESIPT-rhodamine derivative was designed to show a multicolor mechano-response in addition to mechanochromic behavior. The samples showed no activation of the rhodamine spiro-lactam and the mechanofluorescence response was solely related to the conformational changes on the ESIPT unit.
Also founded on ESIPT, but using a slightly different strategy, Weder and coworkers reported on a mechanofluorophore based on an aliphatic ester of 2-(2′-hydroxyphenyl)benzoxazole.134 In the mechanofluorophore proposed by the authors, the hydroxyl group able to perform ESIPT is masked with an ester group, which was then polymerized into PMA chains as a crosslinker. Upon sonication, the functionalized polymer that presented a blue fluorescence emission, showed not only a significant decrease in the molecular weight, but also pronounced changes in the fluorescence emission. The fluorescence emission at λem = 500 nm showed a mirroring behavior of the molecular weight decrease as a function of the sonication time, indicating the activation of the mechanofluorophore by breaking the ester bond masking the hydroxyl group and restoring the 2-(2′-hydroxyphenyl)benzoxazole motif. Although the ratiometric signal produce is advantageous, the system is irreversible, since the ester bond cannot be restored after the force is removed.
From the examples above, it is clear that ESIPT materials still need further development to be successfully employed as mechanophores. Although systems showing ratiometric activation of ESIPT mechanophores were already described, the main challenges are related to intermolecular interactions between the matrix and the ESIPT molecules and the E/K emission ratio, as well as to the achievement of a more intense fluorescence color variation. This leaves room for synthetic chemists and polymer scientists to investigate new structures and polymeric materials in which the ratio between E and K tautomers can be controlled, or systems were the hydroxyl group can be reversibly masked and unmasked.
Perspectives
Although most of the examples presented in this revision are related to mechanophores embedded in covalent hydrogels networks, we further highlight two examples based on supramolecular hydrogels. First, Li and Du reported on the use of SP as a crosslinker in a host–guest system with cucurbit[8]uril (CB[8]).42 For the preparation of the hydrogel, the authors employed two SP derivatives containing a polymerizable methacrylate group (Fig. 11). After attaching the SP derivates to the acrylamide (AM) and 2-hydroxyethyl methacrylate (HEMA) copolymer, 0.5 equivalents of CB[8] were added and the system went through a fast sol–gel transition indicating the complexation. The formed supramolecular hydrogel was found to recover to a fluidic solution upon irradiation with blue light, indicating that the uncharged and ring closed SP species left the CB[8] complexation decrosslinking the system. For the rheological properties the authors prepared samples with solid contents of 10 wt% to 30 wt% and in all cases observed fast relaxation. The network relaxation time consistently decreased across hydrogels with variable solid contents upon irradiation with λem = 450 nm. The hydrogels with 30 wt% exhibited an elongation of 790% and 910% before breaking upon irradiation with blue and UV light, resulting in a toughness of 43.805 kJ m−3 and 70.867 kJ m−3, respectively. In addition to the mechanical properties the authors also evaluated the shape remolding and self-healing properties of the supramolecular hydrogels. The photoswitching properties were also explored for color-changeable patterning and information storage applications, preparation of photoresponsive inks and light-assisted fabrication of conductive supramolecular hydrogel fibers. These properties would indeed be relevant for future applications and evaluation in mechanoresponsive hydrogels. In addition, we highlight one example aiming for applications of mechanochemical release of encapsulated small molecules. The group of Schmidt in collaboration with Göstl reported on a supramolecular coordination cage PdII6(TPT)4 for the release of its nanoconfined guests.135 To build the modular cage, the authors employed the 4-bromomethyl-4′-methyl-2,2′-bipyridine which was further functionalized with poly(ethylene glycol) methyl ether (PEG, Mn = 10 kDa) and complexed to PdII. The walls of the cage were build using four equivalents of 2,4,6-tris(4-pyridyl)-1,3,5-triazine (TPT) and the loading of the system was performed with progesterone or ibuprofen, by adding excess drug to an aqueous solution of the cage. The release of the cargo was induced by sonication and monitored by 1H NMR. The major advantage of the strategy presented by Schmidt, Göstl and coworkers is related to the universality of the supramolecular encapsulation system. This system is the first reported example of a supramolecular coordination cage able to form star-shaped, water-soluble polymer structure which responds to ultrasonication-induced shear forces in solution, enabling the use of its hydrophobic cavity for drug release.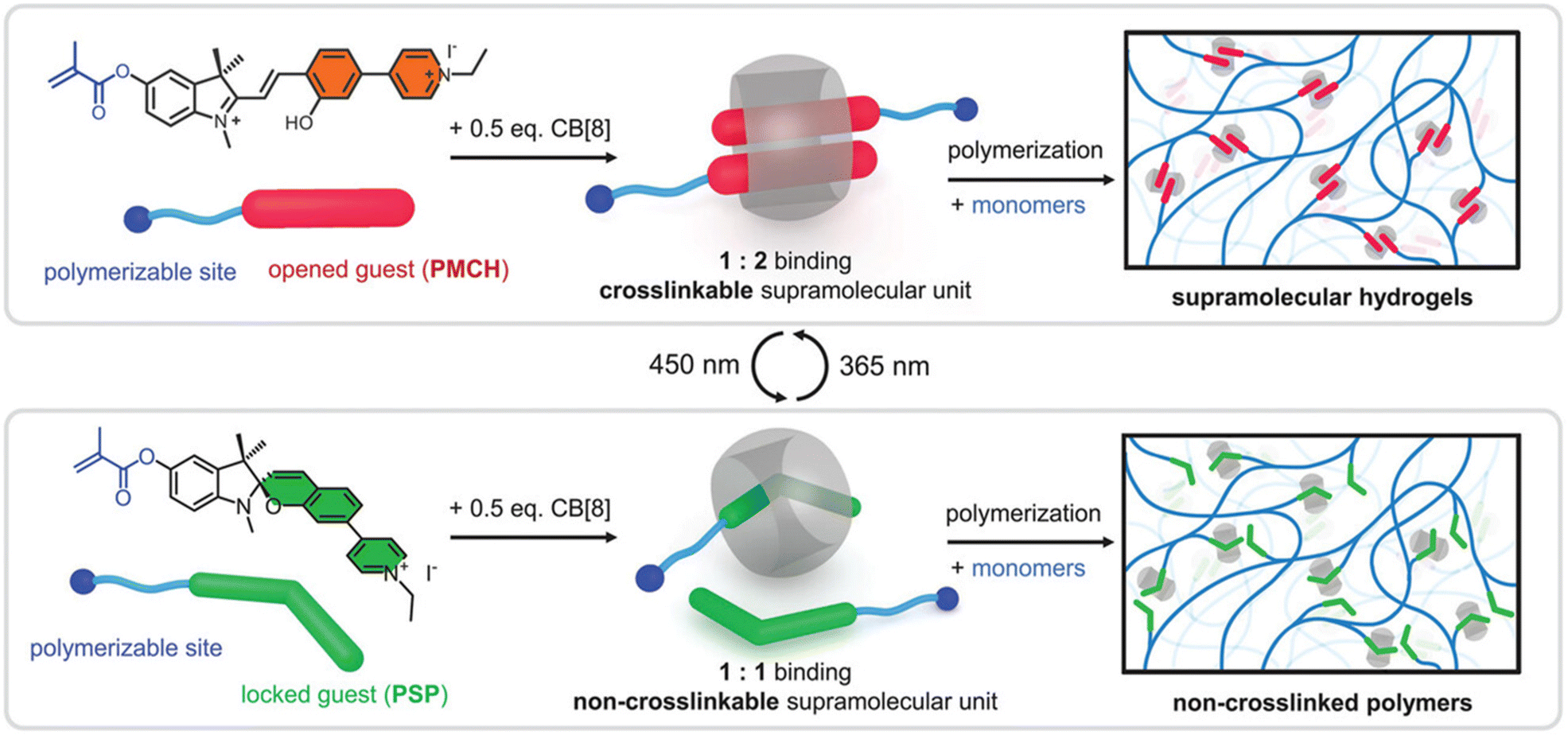 | ||
| Fig. 11 Schematic representation of reversible switching of the host–guest binding ratio between 1:2 (CB[8]/PMCH) and 1:1 (CB[8]/PSP) in response to blue and UV light for the construction of supramolecular hydrogels (top) and non-crosslinked polymers (bottom), respectively. Reproduced with permission.42 Copyright Wiley-VCH GmbH 2024. | ||
Conclusions
Mechanophoric hydrogels represent an exciting area of research, and ongoing efforts are focused on developing new materials with enhanced performance and tailored responses for specific applications. However, many challenges still need to be overcome like the durability, biocompatibility and the sensitivity of the materials. In addition, it is worth mentioning that the incorporation of mechanophores that have a predominantly hydrophobic nature into the hydrophilic network of hydrogels remains a big challenge. Even if the desired network structure is successfully achieved, the network's rupture force is reduced because the mechanophore introduces a weaker bond that breaks before the other covalent bonds in the network.136 Ensuring the durability and repeatability of the mechanophoric response is essential for practical applications. In the case of biomedical applications, ensuring biocompatibility is crucial to avoid adverse effects and only a few studies are addressing this issue during the preparation of the material. Only few examples propose practical applications.72,119,120,137 Further progress on shaping and printing the hydrogels is still necessary in order to overcome this gap and the development of printable inks is appealing.42,71Regarding the combination of hydrogels and mechanophores, some generalizations can be made so far. First, type I mechanophores, i.e. bond scission mechanophores are more often employed in hydrogels with higher mechanical properties, like DN hydrogels.69 In contrast, type II mechanophores are often combined with hydrogels that do not require improved mechanical properties, but require higher sensitivity to stress and/or strain. This is also reflected in the applications. While mechanchromic and mechanoluminescent mechanophores are more often employed to monitor the propagation of the external mechanical forces effects in the polymeric matrix, supramolecular fluorescent mechanophores are employed to monitor molecular movements with precision.119 Second, the mode of introduction of the mechanophore into the polymeric matrix is crucial. In most of the cases discussed throughout this review, mechanophores functionalized either with methylacrylate or bromoisobutyrate are polymerized together with the monomers to yield either the linear backbone chain or a crosslinked material. In general, mechanophores positioned at the crosslinks tend to show more intense color changes. Solvent exchange methods and micellar polymerizations are often used as a strategy to overcome the compatibility barriers between hydrophobic mechanophores and hydrophilic polymeric networks.57,72 And third, still most of the examples encountered in the literature refer to covalent hydrogels. To date, very few studies have pointed out mechanochromism in supramolecular hydrogels.
So far, most of the examples encountered in the literature rely on rhodamine derivatives as mechanophores. Not only the chromic and emissive response of rhodamine is well known, but also its biocompatibility and hydrophilicity are detrimental for applications in highly polar media like hydrogels. Designing new mechanophores with specific chromic and/or emissive responses can be a complex task, requiring a deep understanding of both photochemical and mechanical principles. The use of multi-activation systems and rotaxanes, for example, can inspire chemists to develop new structural motives.76,123 Changes in the fluorescence emission upon mechanical activation are the most frequent type of response. In some cases these changes are not intense or easy to detect without rigorous analytical procedures.23 Moreover, achieving precise control over the activation of mechanophores is essential for their successful application.119 Supramolecular mechanophores which are able to perform FRET-type emission are among the most successful mechanophores due to the modular response, spatiotemporal resolution and sensitivity. It is worth noting that the specific choice of a mechanophore or the development of a full new mechanoresponsive molecule highly depends on the magnitude of the forces to be monitored and on the polymeric context that it is inserted.
Mechanochromic hydrogels are a blooming area of research which require a multidisciplinary approach, involving expertise in organic and physical polymer chemistry, materials science, and photophysics. Throughout this review we pointed to some key aspects beyond the types of mechanophores, their mode of action, the architecture of mechanochromic hydrogels, strategies of incorporation of the mechanophores and some useful characterizations that might guide new researchers into this topic. While the design of mechanochromic hydrogels presents challenges, it also offers opportunities for creating versatile materials with tailored luminescent properties and applications.
Author contributions
F. A. S. writing, research and figures preparation. P. B. conceptualization, structuring and revision.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the DFG (Deutsche Forschungsgemeinschaft) is acknowledged: F. A. S. and P. B. are members of the SFB 1552 (Project No. 465145163).References
- S. Shahi, H. Roghani-Mamaqani, R. Hoogenboom, S. Talebi and H. Mardani, Stimuli-Responsive Covalent Adaptable Hydrogels Based on Homolytic Bond Dissociation and Chain Transfer Reactions, Chem. Mater., 2022, 34, 468–498 CrossRef CAS.
- C. M. A. Leenders, T. Mes, M. B. Baker, M. M. E. Koenigs, P. Besenius, A. R. A. Palmans and E. W. Meijer, From supramolecular polymers to hydrogel materials, Mater. Horiz., 2014, 1, 116–120 RSC.
- R. Eelkema and A. Pich, Pros and Cons: Supramolecular or Macromolecular: What Is Best for Functional Hydrogels with Advanced Properties?, Adv. Mater., 2020, 32(20), 1906012 CrossRef CAS PubMed.
- C. Xu, Y. Chen, S. Zhao, D. Li, X. Tang, H. Zhang, J. Huang, Z. Guo and W. Liu, Mechanical Regulation of Polymer Gels, Chem. Rev., 2024, 124, 10435–10508 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Supramolecular Polymerization, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Supramolecular Polymers in Aqueous Media, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- Y. Shao, H. Jia, T. Cao and D. Liu, Supramolecular Hydrogels Based on DNA Self-Assembly, Acc. Chem. Res., 2017, 50, 659–668 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Supramolecular polymeric hydrogels, Chem. Soc. Rev., 2012, 41, 6195 RSC.
- D. Spitzer, V. Marichez, G. J. M. Formon, P. Besenius and T. M. Hermans, Surface–Assisted Self–Assembly of a Hydrogel by Proton Diffusion, Angew. Chem., Int. Ed., 2018, 57, 11349–11353 CrossRef CAS PubMed.
- O. S. Stach, K. Breul, C. M. Berač, M. Urschbach, S. Seiffert and P. Besenius, Bridging Rigidity and Flexibility: Modulation of Supramolecular Hydrogels by Metal Complexation, Macromol. Rapid. Commun., 2022, 43, 2100473 CrossRef CAS PubMed.
- J. Chen, Q. Peng, X. Peng, L. Han, X. Wang, J. Wang and H. Zeng, Recent Advances in Mechano-Responsive Hydrogels for Biomedical Applications, ACS Appl. Polym. Mater., 2020, 2, 1092–1107 CrossRef CAS.
- J. Wang, K. Zhao, C. Ye and Y. Song, Emerging interactively stretchable electronics with optical and electrical dual-signal feedbacks based on structural color materials, Nano Res., 2023, 1–19 Search PubMed.
- Q. Zhu, K. Van Vliet, N. Holten-Andersen and A. Miserez, A Double–Layer Mechanochromic Hydrogel with Multidirectional Force Sensing and Encryption Capability, Adv. Funct. Mater., 2019, 29, 1808191 CrossRef.
- W. Yang, S. Yamamoto, K. Sueyoshi, T. Inadomi, R. Kato and N. Miyamoto, Perovskite Nanosheet Hydrogels with Mechanochromic Structural Color, Angew. Chem., Int. Ed., 2021, 60, 8466–8471 CrossRef PubMed.
- Q. Mu and J. Hu, Polymer mechanochemistry: from single molecule to bulk material, Phys. Chem. Chem. Phys., 2024, 26, 679–694 RSC.
- A. Pucci and G. Ruggeri, Mechanochromic polymer blends, J. Mater. Chem., 2011, 21, 8282 RSC.
- G. Zanchetta, Control and optical mapping of mechanical transitions in polymer networks and DNA-based soft materials, Curr. Opin. Colloid Interface Sci., 2019, 40, 1–13 CrossRef CAS.
- T. Watabe and H. Otsuka, Enhancing the Reactivity of Mechanically Responsive Units via Macromolecular Design, Macromolecules, 2024, 57, 425–433 CrossRef CAS.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Mechanochemical tools for polymer materials, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, From Molecules to Polymers—Harnessing Inter– and Intramolecular Interactions to Create Mechanochromic Materials, Macromol. Rapid. Commun., 2021, 42, 2000573 CrossRef CAS PubMed.
- F. Ahmed, M. M. Hussain, W. U. Khan and H. Xiong, Exploring recent advancements and future prospects on coordination self-assembly of the regulated lanthanide-doped luminescent supramolecular hydrogels, Coord. Chem. Rev., 2024, 499, 215486 CrossRef CAS.
- M. Lei, Q. Wang, R. Gu and D. Qu, Stimuli–responsive fluorescent hydrogels: Strategies and applications, Responsive Mater., 2024, 2, e20230027 CrossRef.
- H. Hu, X. Cheng, Z. Ma, R. P. Sijbesma and Z. Ma, Polymer Mechanochromism from Force-Tuned Excited-State Intramolecular Proton Transfer, J. Am. Chem. Soc., 2022, 144, 9971–9979 CrossRef CAS PubMed.
- G. Kim, Q. Wu, J. L. Chu, E. J. Smith, M. L. Oelze, J. S. Moore and K. C. Li, Ultrasound controlled mechanophore activation in hydrogels for cancer therapy, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2109791119 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Force-induced activation of covalent bonds in mechanoresponsive polymeric materials, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Y. Chen and R. P. Sijbesma, Dioxetanes as Mechanoluminescent Probes in Thermoplastic Elastomers, Macromolecules, 2014, 47, 3797–3805 Search PubMed.
- K. C. Neuman and A. Nagy, Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy, Nat. Methods, 2008, 5, 491–505 Search PubMed.
- H. A. Heus, E. M. Puchner, A. J. van Vugt-Jonker, J. L. Zimmermann and H. E. Gaub, Atomic force microscope-based single-molecule force spectroscopy of RNA unfolding, Anal. Biochem., 2011, 414, 1–6 CrossRef CAS PubMed.
- W. Ott, M. A. Jobst, C. Schoeler, H. E. Gaub and M. A. Nash, Single-molecule force spectroscopy on polyproteins and receptor–ligand complexes: The current toolbox, J. Struct. Biol., 2017, 197, 3–12 CrossRef CAS PubMed.
- M. K. Beyer, The mechanical strength of a covalent bond calculated by density functional theory, J. Chem. Phys., 2000, 112, 7307–7312 Search PubMed.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, Validation of the CoGEF Method as a Predictive Tool for Polymer Mechanochemistry, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- Z. Qiu, W. Zhao, M. Cao, Y. Wang, J. W. Y. Lam, Z. Zhang, X. Chen and B. Z. Tang, Dynamic Visualization of Stress/Strain Distribution and Fatigue Crack Propagation by an Organic Mechanoresponsive AIE Luminogen, Adv. Mater., 2018, 30, 1803924 CrossRef PubMed.
- J. R. Hemmer, C. Rader, B. D. Wilts, C. Weder and J. A. Berrocal, Heterolytic Bond Cleavage in a Scissile Triarylmethane Mechanophore, J. Am. Chem. Soc., 2021, 143, 18859–18863 CrossRef CAS PubMed.
- G. Zhu, T. Yu, J. Chen, R. Hu, G. Yang, Y. Zeng and Y. Li, Dipyrene-Terminated Oligosilanes Enable Ratiometric Fluorescence Response in Polymers toward Mechano- and Thermo-Stimuli, ACS Appl. Mater. Interfaces, 2023, 15, 11033–11041 CrossRef CAS PubMed.
- F. Würthner, Aggregation–Induced Emission (AIE): A Historical Perspective, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Materials for Fluorescence Resonance Energy Transfer Analysis: Beyond Traditional Donor–Acceptor Combinations, Angew. Chem., Int. Ed., 2006, 45, 4562–4589 CrossRef CAS PubMed.
- F. A. Soares, G. Martinez-Denegri, L. A. Baptista, P. Ślęczkowski and A. Steinbüchel, Balancing the Push–Pull Effect on the Synthesis and Fluorescent Properties of New ESIPT Dyes for Thin Film Applications, J. Phys. Chem. C, 2023, 127, 17624–17636 CrossRef.
- T. Stoerkler, G. Ulrich, A. D. Laurent, D. Jacquemin and J. Massue, Interplay between Dual-State and Aggregation-Induced Emission with ESIPT Scaffolds Containing Triphenylamine Substituents: Experimental and Theoretical Studies, J. Org. Chem., 2023, 88, 9225–9236 CrossRef CAS PubMed.
- P.-Y. Fu, S.-Z. Yi, M. Pan and C.-Y. Su, Excited-State Intramolecular Proton Transfer (ESIPT) Based Metal–Organic Supramolecular Optical Materials: Energy Transfer Mechanism and Luminescence Regulation Strategy, Acc. Mater. Res., 2023, 4, 939–952 Search PubMed.
- J. Han, Y. Yuan and Y. Chen, Visible Light-driven Self-healable Mechanochromic Polyurethanes, Chem. Res. Chin. Univ., 2023, 39, 757–762 CrossRef CAS.
- Z. Wang, Z. Ding, Y. Yang, L. Hu, W. Wu, Y. Gao, Y. Wei, X. Zhang and G. Jiang, Time-resolved encryption via photochromic and mechanochromic system based on silane-substituted spiropyran, Chem. Eng. J., 2023, 457, 141293 Search PubMed.
- M. Du and C. Li, Engineering Supramolecular Hydrogels via Reversible Photoswitching of Cucurbit[8]uril–Spiropyran Complexation Stoichiometry, Adv. Mater., 2024, 36(40), 2408484 Search PubMed.
- Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, Dynamic Behavior of Molecular Switches in Crystal under Pressure and Its Reflection on Tactile Sensing, J. Am. Chem. Soc., 2015, 137, 931–939 CrossRef CAS PubMed.
- M. Kang, O. Oderinde, X. Han, G. Fu and Z. Zhang, Development of oxidized hydroxyethyl cellulose-based hydrogel enabling unique mechanical, transparent and photochromic properties for contact lenses, Int. J. Biol. Macromolecules, 2021, 183, 1162–1173 CrossRef CAS PubMed.
- A. K. Chibisov and H. Görner, Photoprocesses in Spiropyran-Derived Merocyanines, J. Phys. Chem. A, 1997, 101, 4305–4312 CrossRef CAS.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, Cephalopod-inspired design of electro-mechano-chemically responsive elastomers for on-demand fluorescent patterning, Nat. Commun., 2014, 5, 4899 CrossRef PubMed.
- C. K. Lee, D. A. Davis, S. R. White, J. S. Moore, N. R. Sottos and P. V. Braun, Force-Induced Redistribution of a Chemical Equilibrium, J. Am. Chem. Soc., 2010, 132, 16107–16111 Search PubMed.
- Z. Cao, Highly Stretchable Tough Elastomers Crosslinked by Spiropyran Mechanophores for Strain–Induced Colorimetric Sensing, Macromol. Chem. Phys., 2020, 221, 2000190 CrossRef CAS.
- X. Song, Y. Song, X. Cui, J.-P. Wang, Y. Luo, T. Qi and G. L. Li, Intrinsic healable mechanochromic materials via incorporation of spiropyran mechanophore into polymer main chain, Polymer, 2022, 250, 124878 CrossRef CAS.
- C. M. Kingsbury, P. A. May, D. A. Davis, S. R. White, J. S. Moore and N. R. Sottos, Shear activation of mechanophore-crosslinked polymers, J. Mater. Chem., 2011, 21, 8381 RSC.
- R. Janissen and G. A. Filonenko, Mechanochemistry of Spiropyran under Internal Stresses of a Glassy Polymer, J. Am. Chem. Soc., 2022, 144, 23198–23204 Search PubMed.
- T. A. Kim, B. A. Beiermann, S. R. White and N. R. Sottos, Effect of Mechanical Stress on Spiropyran-Merocyanine Reaction Kinetics in a Thermoplastic Polymer, ACS Macro Lett., 2016, 5, 1312–1316 CrossRef CAS PubMed.
- L.-J. Wang, X.-J. Zhou, X.-H. Zhang and B.-Y. Du, Enhanced Mechanophore Activation within Micelles, Macromolecules, 2016, 49, 98–104 CrossRef CAS.
- Y. Chen, A. Kovalenko, A. Brûlet, B. Bresson, A. Lantheaume, L. Olanier and C. Creton, Spiropyran Mechano-Activation in Model Silica-Filled Elastomer Nanocomposites Reveals How Macroscopic Stress in Uniaxial Tension Transfers from Filler/Filler Contacts to Highly Stretched Polymer Strands, Macromolecules, 2023, 56, 5336–5345 CrossRef CAS.
- H. G. Jang, J. Y. Jo, H. Park, Y. C. Jung, Y. Choi, S. Jung, D. C. Lee and J. Kim, Mechano–Responsive Spiropyran Microbeads: A Facile Fabrication Strategy for Self–Reporting Materials, Adv. Mater. Technol., 2023, 8, 2200566 Search PubMed.
- G. Li, Z. Pan, Z. Jia, J. Wang, J. Wang, N. Zhang, M. Pan and J. Yuan, An effective approach for fabricating high-strength polyurethane hydrogels with reversible photochromic performance as a photoswitch, New J. Chem., 2021, 45, 6386–6396 RSC.
- Y. Zhang, B. Ren, F. Yang, Y. Cai, H. Chen, T. Wang, Z. Feng, J. Tang, J. Xu and J. Zheng, Micellar-incorporated hydrogels with highly tough, mechanoresponsive, and self-recovery properties for strain-induced color sensors, J. Mater. Chem. C, 2018, 6, 11536–11551 Search PubMed.
- H. Chen, F. Yang, Q. Chen and J. Zheng, A Novel Design of Multi–Mechanoresponsive and Mechanically Strong Hydrogels, Adv. Mater., 2017, 29, 1606900 CrossRef PubMed.
- W. Qiu, P. A. Gurr and G. G. Qiao, Regulating Color Activation Energy of Mechanophore-Linked Multinetwork Elastomers, Macromolecules, 2020, 53, 4090–4098 CrossRef CAS.
- H. Jeong, S. Cho, E. Heo, C. Woo, S. G. Shin, M. H. Kim and J. H. Jeong, Mechano-responsive chameleon-gel integrated with dumbbell-shaped spiropyran cross-linker, Mater. Lett., 2024, 361, 136141 Search PubMed.
- A. Meeks, M. M. Lerch, T. B. H. Schroeder, A. Shastri and J. Aizenberg, Spiropyran Photoisomerization Dynamics in Multiresponsive Hydrogels, J. Am. Chem. Soc., 2022, 144, 219–227 Search PubMed.
- S. Kamino and M. Uchiyama, Xanthene-based functional dyes: towards new molecules operating in the near-infrared region, Org. Biomol. Chem., 2023, 21, 2458–2471 RSC.
- X. Song, J. Wang, Y. Song, T. Qi and G. L. Li, Bioinspired Healable Mechanochromic Function from Fluorescent Polyurethane Composite Film, ChemistryOpen, 2020, 9, 272–276 CrossRef CAS PubMed.
- H. Gross, H. Dürr and W. Rettig, Emission spectra of photochromic spiro[1,8-a]dihydroindolizines and mechanism of the electrocyclic ring opening reaction, J. Photochem., 1984, 26, 165–178 CrossRef CAS.
- L.-J. Wang, K.-X. Yang, Q. Zhou, H.-Y. Yang, J.-Q. He and X.-Y. Zhang, Rhodamine Mechanophore Functionalized Mechanochromic Double Network Hydrogels with High Sensitivity to Stress, Chin. J. Polym. Sci., 2020, 38, 24–36 CrossRef CAS.
- C.-H. Wu, C.-W. Tu, J. Aimi, J. Zhang, T. Chen, C.-C. Wang and C.-F. Huang, Mechanochromic double network hydrogels as a compression stress sensor, Polym. Chem., 2020, 11, 6423–6428 RSC.
- G. Lin, M. Si, L. Wang, S. Wei, W. Lu, H. Liu, Y. Zhang, D. Li and T. Chen, Dual–Channel Flexible Strain Sensors Based on Mechanofluorescent and Conductive Hydrogel Laminates, Adv. Opt. Mater., 2022, 10, 2102306 Search PubMed.
- Y. Shen, Y. Yuan, X. Ma, W. Yang and Y. Chen, Mechanically induced chemiluminescence of xanthene-modified 1,2-dioxetane in polymers, Polym. Chem., 2023, 14, 4148–4152 Search PubMed.
- B. Xu, Z. Luo, R. Xiao, Z. Wang and J. Yang, Hybrid Phenol–Rhodamine Dye Based Mechanochromic Double Network Hydrogels with Tunable Stress Sensitivity, Macromol. Rapid. Commun., 2022, 43, 2200580 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Double–Network Hydrogels with Extremely High Mechanical Strength, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- B. Xu, H. Wang, Z. Luo, J. Yang and Z. Wang, Multi-material 3D Printing of Mechanochromic Double Network Hydrogels for On-Demand Patterning, ACS Appl. Mater. Interfaces, 2023, 15, 11122–11130 CrossRef CAS PubMed.
- L. Wang, J. Wang, Y. Wang and X. Zhang, Rhodamine-containing double-network hydrogels for smart window materials with tunable light transmittance, low-temperature warning, and deformation sensing, React. Funct. Polym., 2022, 181, 105408 CrossRef CAS.
- F. Yang, X. Li and Y. Chen, A Chromic and Near–Infrared Emissive Mechanophore Serving as a Versatile Force Meter in Micelle–Hydrogel Composites, Adv. Opt. Mater., 2022, 10, 2102552 CrossRef CAS.
- W. He, Y. Yuan, M. Wu, X. Li, Y. Shen, Z. Qu and Y. Chen, Multicolor Chromism from a Single Chromophore through Synergistic Coupling of Mechanochromic and Photochromic Subunits, Angew. Chem., Int. Ed., 2023, 62, e202218785 Search PubMed.
- J. R. Hemmer, V. Bauernfeind, C. Rader, M. Petroselli, C. Weder and J. A. Berrocal, Triarylmethane Mechanophores Enable Full-Visible Spectrum Mechanochromism, Macromolecules, 2023, 56, 8614–8622 Search PubMed.
- H. Hu, X. Cheng, Z. Ma, Z. Wang and Z. Ma, A double-spiro ring-structured mechanophore with dual-signal mechanochromism and multistate mechanochemical behavior: non-sequential ring-opening and multimodal analysis, Polym. Chem., 2022, 13, 5507–5514 RSC.
- M. Wu, Y. Li, W. Yuan, G. De Bo, Y. Cao and Y. Chen, Cooperative and Geometry-Dependent Mechanochromic Reactivity through Aromatic Fusion of Two Rhodamines in Polymers, J. Am. Chem. Soc., 2022, 144, 17120–17128 Search PubMed.
- M. Clerc, S. Sandlass, O. Rifaie-Graham, J. A. Peterson, N. Bruns, J. R. de Alaniz and L. F. Boesel, Visible light-responsive materials: the (photo)chemistry and applications of donor–acceptor Stenhouse adducts in polymer science, Chem. Soc. Rev., 2023, 52, 8245–8294 RSC.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. R. de Alaniz, Photoswitching Using Visible Light: A New Class of Organic Photochromic Molecules, J. Am. Chem. Soc., 2014, 136, 8169–8172 Search PubMed.
- S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. R. de Alaniz, Design and Synthesis of Donor–Acceptor Stenhouse Adducts: A Visible Light Photoswitch Derived from Furfural, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS PubMed.
- M. M. Lerch, S. J. Wezenberg, W. Szymanski and B. L. Feringa, Unraveling the Photoswitching Mechanism in Donor–Acceptor Stenhouse Adducts, J. Am. Chem. Soc., 2016, 138, 6344–6347 CrossRef CAS PubMed.
- N. Mallo, P. T. Brown, H. Iranmanesh, T. S. C. MacDonald, M. J. Teusner, J. B. Harper, G. E. Ball and J. E. Beves, Photochromic switching behaviour of donor–acceptor Stenhouse adducts in organic solvents, Chem. Commun., 2016, 52, 13576–13579 Search PubMed.
- A. C. Overholts, W. G. Razo and M. J. Robb, Mechanically gated formation of donor–acceptor Stenhouse adducts enabling mechanochemical multicolour soft lithography, Nat. Chem., 2023, 15, 332–338 CrossRef CAS PubMed.
- H. Sakai, D. Aoki, K. Seshimo, K. Mayumi, S. Nishitsuji, T. Kurose, H. Ito and H. Otsuka, Visualization and Quantitative Evaluation of Toughening Polymer Networks by a Sacrificial Dynamic Cross-Linker with Mechanochromic Properties, ACS Macro Lett., 2020, 9, 1108–1113 Search PubMed.
- X. Li, J. Li, W. Wei, F. Yang, M. Wu, Q. Wu, T. Xie and Y. Chen, Enhanced Mechanochemiluminescence from End-Functionalized Polyurethanes with Multiple Hydrogen Bonds, Macromolecules, 2021, 54, 1557–1563 CrossRef CAS.
- Q. Li, Q. Wang, Y. Yuan and Y. Chen, Mechanochemiluminescent Hydrogels for Real-Time Visualization of Chemical Bond Scission, Synlett, 2022, 879–884 CAS.
- K. Yanada, D. Aoki and H. Otsuka, Mechanochromic elastomers with different thermo- and mechano-responsive radical-type mechanophores, Soft Matter, 2022, 18, 3218–3225 RSC.
- T. Watabe and H. Otsuka, Swelling–induced Mechanochromism in Multinetwork Polymers, Angew. Chem., 2023, 135, e202216469 CrossRef.
- F. Yang, T. Geng, H. Shen, Y. Kou, G. Xiao, B. Zou and Y. Chen, Mechanochemical Release of Fluorophores from a “Flex–activated” Mechanophore, Angew. Chem., Int. Ed., 2023, 62, e202308662 CrossRef CAS PubMed.
- T. Ouchi, B. H. Bowser, T. B. Kouznetsova, X. Zheng and S. L. Craig, Strain-triggered acidification in a double-network hydrogel enabled by multi-functional transduction of molecular mechanochemistry, Mater. Horiz., 2023, 10, 585–593 RSC.
- R. Göstl and R. P. Sijbesma, π-extended anthracenes as sensitive probes for mechanical stress, Chem. Sci., 2016, 7, 370–375 RSC.
- M. Stratigaki, C. Baumann, L. C. A. van Breemen, J. P. A. Heuts, R. P. Sijbesma and R. Göstl, Fractography of poly(N -isopropylacrylamide) hydrogel networks crosslinked with mechanofluorophores using confocal laser scanning microscopy, Polym. Chem., 2020, 11, 358–366 RSC.
- S. He, S. Schog, Y. Chen, Y. Ji, S. Panitz, W. Richtering and R. Göstl, Photoinduced Mechanical Cloaking of Diarylethene–Crosslinked Microgels, Adv. Mater., 2023, 35, 2305845 CrossRef CAS PubMed.
- E. Izak-Nau, S. Braun, A. Pich and R. Göstl, Mechanically Resistant Poly(N-vinylcaprolactam) Microgels with Sacrificial Supramolecular Catechin Hydrogen Bonds, Adv. Sci., 2022, 9, 2104004 CrossRef CAS PubMed.
- E. Izak-Nau, D. E. Demco, S. Braun, C. Baumann, A. Pich and R. Göstl, Shear-Induced Structural and Functional Transformations of Poly(N-vinylcaprolactam) Microgels, ACS Appl. Polym. Mater., 2020, 2, 1682–1691 CrossRef CAS.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, Mechanically Triggered Small Molecule Release from a Masked Furfuryl Carbonate, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- S. Huo, P. Zhao, Z. Shi, M. Zou, X. Yang, E. Warszawik, M. Loznik, R. Göstl and A. Herrmann, Mechanochemical bond scission for the activation of drugs, Nat. Chem., 2021, 13, 131–139 CrossRef CAS PubMed.
- A. Ishaqat, J. Hahmann, C. Lin, X. Zhang, C. He, W. H. Rath, P. Habib, S. E. M. Sahnoun, K. Rahimi, R. Vinokur, F. M. Mottaghy, R. Göstl, M. Bartneck and A. Herrmann, In Vivo Polymer Mechanochemistry with Polynucleotides, Adv. Mater., 2024, 36, 2403752 CrossRef CAS PubMed.
- J. Hahmann, A. Ishaqat, T. Lammers and A. Herrmann, Sonogenetics for Monitoring and Modulating Biomolecular Function by Ultrasound, Angew. Chem., Int. Ed., 2024, 63, e202317112 CrossRef CAS PubMed.
- J. Fan, M. Xuan, K. Zhang, R. Vinokur, L. Zheng, R. Göstl and A. Herrmann, Accelerated Mechanophore Activation and Drug Release in Network Core–Structured Star Polymers Using High–Intensity Focused Ultrasound, Small Sci., 2024, 4, 2400082 CrossRef CAS.
- D. Yildiz, R. Göstl and A. Herrmann, Sonopharmacology: controlling pharmacotherapy and diagnosis by ultrasound-induced polymer mechanochemistry, Chem. Sci., 2022, 13, 13708–13719 RSC.
- R. Küng, R. Göstl and B. M. Schmidt, Release of Molecular Cargo from Polymer Systems by Mechanochemistry, Chem. – Eur. J., 2022, 28, e202103860 CrossRef PubMed.
- F. Cellini, L. Block, J. Li, S. Khapli, S. D. Peterson and M. Porfiri, Mechanochromic response of pyrene functionalized nanocomposite hydrogels, Sens. Actuators, B, 2016, 234, 510–520 CrossRef CAS.
- D. Rasch and R. Göstl, Pyrene-Based Macrocrosslinkers with Supramolecular Mechanochromism for Elastic Deformation Sensing in Hydrogel Networks, Org. Mater., 2022, 4, 170–177 CrossRef CAS.
- S. Shimizu, H. Yoshida, K. Mayumi, H. Ajiro and Y. Sagara, Mechanochromic luminescence of phase-separated hydrogels that contain cyclophane mechanophores, Mater. Chem. Front., 2023, 7, 4073–4079 RSC.
- H. Traeger, Y. Sagara, J. A. Berrocal, S. Schrettl and C. Weder, Strain-correlated mechanochromism in different polyurethanes featuring a supramolecular mechanophore, Polym. Chem., 2022, 13, 2860–2869 RSC.
- T. A. Welsh, J. G. Egan, B. Dietrich, N. Rafferty, R. E. Ginesi, J. Doutch, R. Schweins and E. R. Draper, The effects of amino acid functionalisation on the optoelectronic properties and self-assembly of perylene bisimides, J. Phys.: Mater., 2024, 7, 015004 CAS.
- J. Z. Sun, W. Bai, Z. Wang and B. Z. Tang, Host–Guest Supramolecular Systems Containing AIE-Active Building Blocks, in Comprehensive Supramolecular Chemistry II, Elsevier, 2017, vol. 8, pp. 89–105 Search PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission: phenomenon, mechanism and applications, Chem. Commun., 2009, 4332 RSC.
- P. Ahlers, C. Götz, S. Riebe, M. Zirbes, M. Jochem, D. Spitzer, J. Voskuhl, T. Basché and P. Besenius, Structure and luminescence properties of supramolecular polymers of amphiphilic aromatic thioether–peptide conjugates in water, Polym. Chem., 2019, 10, 3163–3169 RSC.
- L.-L. Zhang, Y. Zhao, K.-X. Li, S.-S. Yu, R.-Z. Dong, S.-H. Ma, H. Liu, L.-B. Xing and F. Zhou, Bioinspired simultaneous regulation in fluorescence of AIEgen-embedded hydrogels, Soft Matter, 2023, 19, 7093–7099 RSC.
- B. Li, B. Yan, J. Wang, Y. Zhang, Z. Qiu, J. Liang and Q. Zhou, A multistimuli-responsive fluorescent hydrogel based on a fluorescence response to macromolecular segmental motion, Nano Res., 2023, 16, 12098–12105 CrossRef CAS.
- P. M. Saengdet and M. Ogawa, Mechanochromic luminescence of a bionanocomposite hydrogel, Chem. Commun., 2022, 58, 3278–3281 RSC.
- P. M. Saengdet and M. Ogawa, Swelling-Induced Chromotropism of Bionanocomposite Hydrogel Beads, Langmuir, 2024, 40, 1016–1023 CrossRef CAS PubMed.
- H. V. Humeniuk, G. Licari, E. Vauthey, N. Sakai and S. Matile, Mechanosensitive membrane probes: push-pull papillons, Supramol. Chem., 2020, 32, 106–111 CrossRef CAS.
- R. W. Barber and M. J. Robb, A mechanochemical two for one: Mechanical activation of norborn–2−en–7−one releases carbon monoxide and switches on aggregation–induced emission, Aggregate, 2022, 3, e196 CrossRef.
- X. Yu, X. Ge, H. Lan, Y. Li, L. Geng, X. Zhen and T. Yi, Tunable and Switchable Control of Luminescence through Multiple Physical Stimulations in Aggregation-Based Monocomponent Systems, ACS Appl. Mater. Interfaces, 2015, 7, 24312–24321 CrossRef CAS PubMed.
- S. Ghosh, H. Bhambri, A. K. Singh, S. K. Mandal, L. Roy and P. S. Addy, A convenient route to a vinylogous dicyano aryl based AIEgen with switchable mechanochromic luminescence properties, Chem. Commun., 2023, 59, 4463–4466 RSC.
- R. Merindol, G. Delechiave, L. Heinen, L. H. Catalani and A. Walther, Modular Design of Programmable Mechanofluorescent DNA Hydrogels, Nat. Commun., 2019, 10, 528 CrossRef CAS PubMed.
- S. Sethi, T. Xu, A. Sarkar, C. Drees, C. Jacob and A. Walther, Nuclease–Resistant L–DNA Tension Probes Enable Long–Term Force Mapping of Single Cells and Cell Consortia, Angew. Chem., Int. Ed., 2024, 2024, e202413983 Search PubMed.
- M. Taki, T. Yamashita, K. Yatabe and V. Vogel, Mechano-chromic protein–polymer hybrid hydrogel to visualize mechanical strain, Soft Matter, 2019, 15, 9388–9393 RSC.
- D. Aoki, A rational entry to cyclic polymers via spontaneous and selective cyclization reactions, Polym. J., 2021, 53, 257–269 CrossRef CAS.
- T. Muramatsu, S. Shimizu, J. M. Clough, C. Weder and Y. Sagara, Force-Induced Shuttling of Rotaxanes Controls Fluorescence Resonance Energy Transfer in Polymer Hydrogels, ACS Appl. Mater. Interfaces, 2023, 15, 8502–8509 CrossRef CAS PubMed.
- M. Zhu, D. Lu, S. Wu, Q. Lian, W. Wang, A. H. Milani, Z. Cui, N. T. Nguyen, M. Chen, L. A. Lyon, D. J. Adlam, A. J. Freemont, J. A. Hoyland and B. R. Saunders, Responsive Nanogel Probe for Ratiometric Fluorescent Sensing of pH and Strain in Hydrogels, ACS Macro Lett., 2017, 6, 1245–1250 CrossRef CAS PubMed.
- M. Zhu, D. Lu, S. Wu, Q. Lian, W. Wang, L. A. Lyon, W. Wang, P. Bártolo and B. R. Saunders, Using green emitting pH-responsive nanogels to report environmental changes within hydrogels: a nanoprobe for versatile sensing, Nanoscale, 2019, 11, 11484–11495 RSC.
- M. Zhu, D. Lu, Q. Lian, S. Wu, W. Wang, L. A. Lyon, W. Wang, P. Bártolo, M. Dickinson and B. R. Saunders, Highly swelling pH-responsive microgels for dual mode near infra-red fluorescence reporting and imaging, Nanoscale Adv., 2020, 2, 4261–4271 RSC.
- S. Maity, A. Chatterjee, N. Chakraborty and J. Ganguly, A dynamic sugar based bio-inspired, self-healing hydrogel exhibiting ESIPT, New J. Chem., 2018, 42, 5946–5954 RSC.
- S. Maity, S. S. Ray, A. Chatterjee, N. Chakraborty and J. Ganguly, Sugar–Based Self–Assembly of Hydrogel Nanotubes Manifesting ESIPT: Theoretical Insight and Application in Live Cell Imaging, ChemistrySelect, 2018, 3, 6575–6580 CrossRef CAS.
- B. Jana, D. Pan, A. Chatterjee, N. Parshi, S. Ghorai, N. Chakraborty and J. Ganguly, Chitosan@4,6-Dihydroxyisophthalaldehyde Microgels with Hydrazine-Induced Fluorescence for Cell Imaging Applications, ACS Appl. Polym. Mater., 2022, 4, 4208–4218 CrossRef CAS.
- S. Ghorai, B. Jana, D. Pan, T. Ramasamy, N. Parshi, G. Arumugam and J. Ganguly, Evaluation of nanofibril chitosan@8−formyl–7−hydroxy–coumarin hydrogel having distinct auto–fluorescence efficiency: Structure–properties relation, improved antioxidant, and cellular imaging, J. Appl. Polym. Sci., 2022, 139, e52908 CrossRef CAS.
- D. Chen, C. Bao, L. Zhang, Q. Zhang, Z. Wu, Z. Li, X. Sun, L. Wang and T. Xiao, Dynamic Time–Dependent Emission in Solution and Stable Dual Emission in Solid Matrix Exhibited by a Single–Component Fluorescence System, Adv. Funct. Mater., 2024, 2314093 CrossRef CAS.
- Y. Wu, X. Cheng, H. Hu, S. Hu, Z. Ma and Z. Ma, Impact of Polymer Matrix on Polymer Mechanochromism from Excited State Intramolecular Proton Transfer, Chin. J. Chem., 2023, 42, 611–616 CrossRef.
- H. Hu, X. Cheng, Y. Wu, M. Chen, Z. Ma, Q. Yang, R. P. Sijbesma and Z. Ma, Spirolactam and ESIPT in a Double-Network Elastomer: Multicolor Mechanochromism and Dual-Ratiometric Strain Sensing in Low and High Tensile Stress, Macromolecules, 2024, 57, 3368–3375 CrossRef CAS.
- M. Karman, E. Verde-Sesto and C. Weder, Mechanochemical Activation of Polymer-Embedded Photoluminescent Benzoxazole Moieties, ACS Macro Lett., 2018, 7, 1028–1033 CrossRef CAS PubMed.
- R. Küng, T. Pausch, D. Rasch, R. Göstl and B. M. Schmidt, Mechanochemical Release of Non–Covalently Bound Guests from a Polymer–Decorated Supramolecular Cage, Angew. Chem., Int. Ed., 2021, 60, 13626–13630 CrossRef PubMed.
- T. Matsuda, R. Kawakami, T. Nakajima and J. P. Gong, Crack Tip Field of a Double-Network Gel: Visualization of Covalent Bond Scission through Mechanoradical Polymerization, Macromolecules, 2020, 53, 8787–8795 CrossRef CAS.
- X. Yao, J. A. Vishnu, C. Lupfer, D. Hoenders, O. Skarsetz, W. Chen, D. Dattler, A. Perrot, W. Wang, C. Gao, N. Giuseppone, F. Schmid and A. Walther, Scalable Approach to Molecular Motor–Polymer Conjugates for Light–Driven Artificial Muscles, Adv. Mater., 2024, 36, 2403514 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2025 |