
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Remodelling molecular frameworks via atom-level surgery: recent advances in skeletal editing of (hetero)cycles
Rubal
Sharma
 ab,
Mitsuhiro
Arisawa
b,
Shinobu
Takizawa
*a and
Mohamed S. H.
Salem
*ac
ab,
Mitsuhiro
Arisawa
b,
Shinobu
Takizawa
*a and
Mohamed S. H.
Salem
*ac
aSANKEN, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan. E-mail: mohamedsalem43@sanken.osaka-u.ac.jp; taki@sanken.osaka-u.ac.jp
bGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamada-oka, Suita, Osaka 565-0871, Japan
cPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Suez Canal University, 4.5 Km the Ring Road, Ismailia 41522, Egypt
First published on 8th January 2025
Abstract
Skeletal editing is an emerging approach in synthetic chemistry that enables precise atom-level modifications within molecular cores, facilitating complex transformations and minimizing resource-intensive synthesis. This review provides a comprehensive overview of the most recent advancements in skeletal editing, with a particular focus on single atom modifications. While skeletal editing can be applied to both cyclic and acyclic compounds, this review centers on carbo- and heterocyclic systems exclusively. By integrating historical context and categorizing key developments, it highlights the major achievements in insertion, deletion, and transmutation, connecting related works and delving into mechanistic insights.
 Rubal Sharma | Rubal Sharma earned her Bachelor's in Pharmacy from Punjabi University, Patiala, India, in 2022. In 2023, she was awarded a MEXT scholarship to study at Osaka University, where she began her Master's program in 2024. Currently, she is still pursuing her Master's, with research interests spanning skeletal editing, electrochemistry, and data chemistry for material and medicinal applications. |
 Mitsuhiro Arisawa | Mitsuhiro Arisawa received his Ph.D. from Graduate School of Pharmaceutical Sciences, Osaka University, Japan (Prof. Yasuyuki Kita). He joined the Faculty of Pharmaceutical Sciences at Chiba University, Japan, as an assistant professor. From 2002 to 2004, he was a visiting scholar at Harvard University, U.S.A. (Prof. Matthew D. Shair). He became an Associate Professor at Graduate School of Pharmaceutical Science, Hokkaido University, in 2005. In 2013, he moved to Graduate School of Pharmaceutical Sciences at Osaka University, an Associate Professor. He was promoted to a professor in 2019. His current research interest is organic chemistry for drug development. |
 Shinobu Takizawa | Shinobu Takizawa earned his Ph.D. in 2000 from Osaka University under the supervision of Professor Yasuyuki Kita. He was a JSPS research fellow from 1999 to 2000. He joined SANKEN, Osaka University, as an Assistant Professor in 2000, later advancing to an Associate Professor. From 2006 to 2008, he served as a Research Associate with Professor Dale L. Boger at the Scripps Research Institute. Since 2024, he has been a Professor at SANKEN, Osaka University. His current research focuses on developing environmentally friendly and data-driven organic synthetic processes. |
 Mohamed S. H. Salem | Mohamed S. H. Salem, earned his master's degree in pharmaceutical chemistry from Suez Canal University, Egypt. He then received the MEXT scholarship to pursue his Ph.D. at Osaka University, Japan, under the guidance of Prof. Hiroaki Sasai and Prof. Takayoshi Suzuki. In 2022, he completed his Ph.D., working on the electrochemical synthesis of polycyclic heteroaromatics and their optical behavior. From 2022 he has been serving as a postdoctoral researcher in the Takizawa group. He rejoined SANKEN, Osaka University, as a Specially Appointed Assistant Professor in 2025. His current research focuses on developing green synthetic approaches for the bottom-up synthesis of functionalized small organic molecules with various material-based applications. |
1. Introduction
Modern synthetic organic chemistry has increasingly focused on the precise manipulation of molecular frameworks to enable more efficient and versatile transformations across diverse fields, including sustainable synthesis and materials science.1–3 Skeletal editing, an emerging approach that allows for atom-level modifications, is garnering significant attention for its potential to remodel molecular cores with precision while reducing the need for resource-intensive de novo synthesis.4,5 By selectively inserting, deleting, or exchanging atoms within a molecule's skeleton, this technique has the potential to facilitate late-stage modifications, streamline synthetic routes, improve sustainability, and provide access to complex systems that are challenging to achieve through traditional methods (Fig. 1A).6,7 In drug discovery, skeletal editing holds promise for the rapid optimization of lead compounds, potentially enhancing their potency and selectivity through efficient diversification.8–10 It also introduces retrosynthetic elegance to the total synthesis of natural products, akin to widely used reactions such as cross-coupling and amide bond formation.11 Beyond pharmaceuticals, skeletal editing has transformative potential in materials science, with the ability to fine-tune electronic, optical, and catalytic properties and unlock new applications in optoelectronics, energy storage, sustainable catalysis, and next-generation technologies.12–15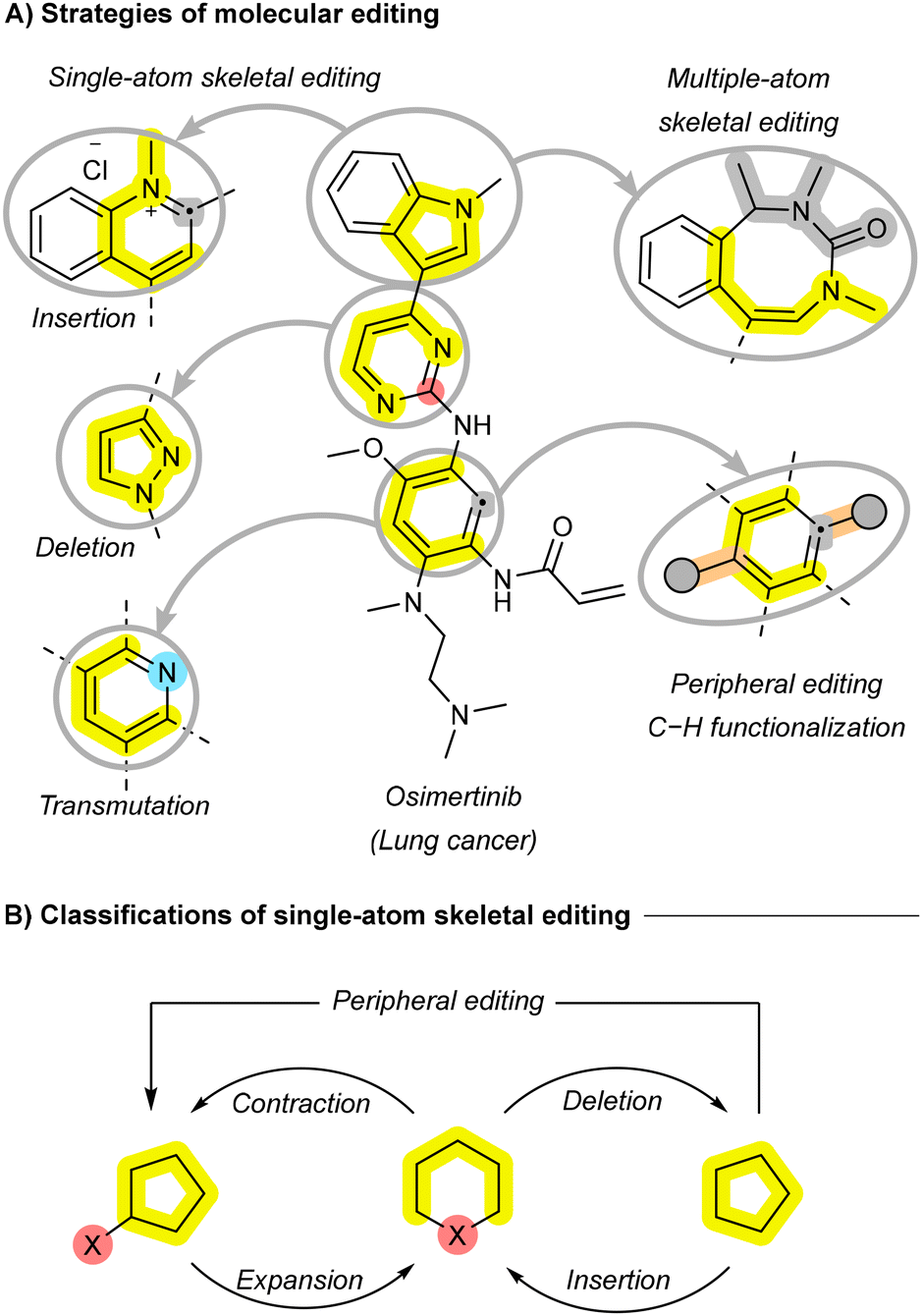 | ||
| Fig. 1 Definitions and categorizations of skeletal editing. | ||
Historically, chemists primarily focused on modifying peripheral functional groups—referred to as “peripheral editing”—to diversify molecules without altering their core skeleton (Fig. 1A). While reactions capable of rearranging molecular cores—like the Buchner ring expansion, Baeyer–Villiger oxidation or the Ciamician–Dennstedt rearrangement, were known by the late 19th century, they were limited in scope and not widely applied to skeletal modifications until recently.16–18 The rise of CRISPR gene editing in the early 2010s inspired similar tools for molecular editing, leading to the conceptualization of “skeletal editing” as a method to precisely edit molecular structures, analogous to CRISPR's role in biology.19 This link is reflected in the shared terminology of “editing”, “mutations”, “transmutations”, “deletions”, and “insertions” across both fields. However, skeletal editing remains a rapidly evolving discipline without standardized terminology, leading to diverse and sometimes inconsistent terms to describe similar processes. Terms such as “skeleton”, “framework”, “scaffold”, and “core” are frequently used interchangeably to refer to molecular skeletons. Moreover, some terms are applied with varying levels of specificity, with researchers distinguish between skeletal editing that includes peripheral modifications and those that do not. In these cases, terms like “deletion” and “insertion” are used to describe skeletal modifications without accompanying peripheral changes, while “contraction” and “expansion” refer to transformations that involve both skeletal and peripheral modifications (Fig. 1B).4 Others, however, treat these terms as synonyms, blurring the distinction between these strategies.5,6,20,21 This inconsistency underscores the need for standardization to promote clearer communication and broader adoption of skeletal editing methodologies. In 2022, Sarpong and Levin published a key review to address these ambiguities, providing clear definitions and categorizations for skeletal editing, supported by key examples.4 A year later, Ball published another key review, focusing on the application of skeletal editing to the interconversion of carbo- and heteroarenes, further advancing the field's scope and practical examples.6
Skeletal editing can be categorized based on the number of atoms involved: single-atom and multiple-atom approaches (Fig. 1A). Single-atom editing involves the insertion, deletion, or substitution of individual atoms within a molecular skeleton, enabling localized modifications. This is particularly valuable for fine-tuning properties in pharmaceuticals and sustainable materials through atom doping. Multiple-atom skeletal editing, on the other hand, involves more extensive alterations, such as the insertion, removal, or exchange of entire fragments, allowing for significant structural reshaping and the design of complex molecular architectures. While single-atom editing has garnered more attention for its potential in applications requiring precision, multiple-atom editing also features prominently, with examples like ring insertion highlighted in recent reviews.22,23 Skeletal editing can include both cyclic and acyclic skeletons. In cyclic systems, the “main skeleton” refers to the central ring(s) that define the molecule's core structure, with modifications often involving ring expansion, contraction, or rearrangement. In acyclic systems, the main skeleton is the longest chain of atoms or the central scaffold, around which modifications occur. In this review, we focus primarily on recent advances in skeletal editing of cyclic systems, particularly arenes and heteroarenes, with an emphasis on single-atom editing. While recent reviews have highlighted key developments in skeletal editing, there is still a pressing need for a comprehensive overview that links these advances to their historical context.4–6,20–22,24–26 Such a review should provide a broader perspective on the evolution of skeletal editing strategies by exploring their mechanisms and the various insertive agents or catalysts used, rather than focusing on a single strategy or specific substrates. Our review addresses this gap by providing a holistic evaluation of the field, discussing key mechanisms across diverse skeletons, and consolidating recent breakthroughs during the last five years scattered across various studies. While some reviews differentiate between skeletal editing with and without peripheral modifications, we adopt a more inclusive approach. Terms like insertion and deletion are used in this review irrespective of whether peripheral modifications are involved or not.
2. Strategies of skeletal editing
Transformations in skeletal editing of cyclic compounds can be broadly classified into three main categories: (1) the insertion of new atom(s) into the main skeleton, leading to ring expansion; (2) the deletion of one or more atoms, resulting in ring contraction; and (3) the exchange of one or more atoms, referred to as transmutation, which alters the atom's identity without changing the overall size of the cyclic system (Fig. 1A). Additional modifications, such as converting monocyclic systems to bicyclic systems, have also been reported.27 This review will focus primarily on these three main categories, further subdividing them based on the nature of the atom(s) being inserted, deleted, or exchanged. For instance, transformations may involve the insertion or deletion of single carbon (C) or nitrogen (N) atoms, each of which can result in distinct structural and functional outcomes.2.1. Ring expansion strategies through atom insertion
Atom insertion is a central strategy in skeletal editing, allowing precise incorporation of new atom(s) into cyclic structures, directly modifying molecular frameworks. Unlike traditional synthetic methods and de novo synthesis, which often require multiple steps, excessive reagents, and generate considerable waste, atom insertion provides a more sustainable and efficient alternative. This strategy can be classified based on the type of atom introduced; while carbon insertion is common, atoms such as nitrogen, oxygen, and boron have also been successfully inserted using various reagents.6 The active species responsible for these insertions, known as insertive agents, exhibit diverse reactivity and selectivity, offering distinct advantages depending on the target transformation. In this review, we focus on the progress made with different insertive agents, exploring their ability to expand the scope of skeletal editing, overcome persistent challenges, and highlight the unique advantages each offers based on substrate and mechanism.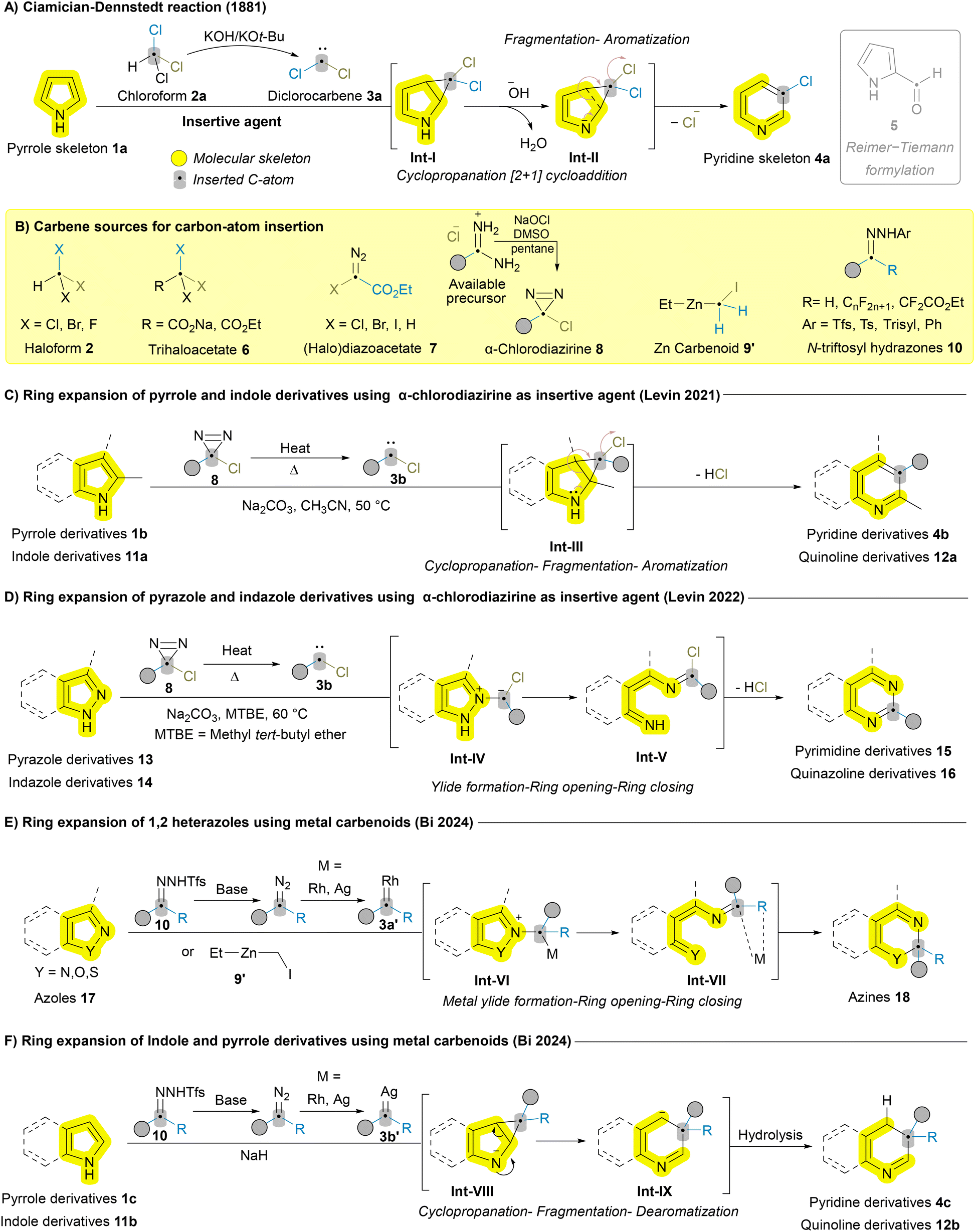 | ||
| Scheme 1 Carbon atom insertion into the main skeleton of azaheterocycles leading to ring expansion. | ||
In 2021, Levin group made a significant breakthrough by applying a novel insertive agent, α-chlorodiazirine 8, to release carbene for the efficient and selective ring expansion of pyrroles 1b and indoles 11a to the corresponding pyridines 4b and quinolines 12a, showcasing the versatility and impact of the cyclopropanation-fragmentation-aromatization mechanistic pathway (Scheme 1C).34 α-Chlorodiazirines 8 can be readily prepared via the oxidation of amidinium salts (Scheme 1B) and exhibit good results across a broad scope for 2-substituted pyrroles 1b and indoles 11a which was not feasible with halodiazoacetates 7.32 This method was further extended by the same group in 2022 to include pyrazoles 13 and indazoles 14via N–N bond cleavage and cyclization. The method involves carbon insertion between the N–N bond through ylide formation Int-IV, initiated by trapping chlorocarbene 3b at the azole N2 terminus, followed by fragmentation (N–N cleavage) affording Int-V and cyclization (Scheme 1D).35 Although α-chlorodiazirine 8 demonstrated good reactivity with various monocyclic azoles (pyrroles 1b and pyrazoles 13) and bicyclic azoles (indoles 11a and indazoles 14), it was less effective for unsubstituted substrates at position 2. In these cases, the products’ nitrogen lone pairs (LPs) interfere with the carbene, forming ylide intermediates and reducing the overall efficiency of the reaction.34 Liam and coworkers recently reported a modified photochemically mediated protocol to overcome this limitation for pyrroles 1, indoles 11, and pyrazoles 13 by introducing N-substitution, which masks the nitrogen's lone pairs, preventing them from reacting with the carbene source.36
Halogenated carbene sources, such as 1,1-dibromoalkanes and bromodifluoroacetate (BrCF2COOEt) 6, have also been employed as insertive agents for azaheterocycles’ skeletal editing.37–39 Metals like rhodium (Rh), silver (Ag), copper (Cu), and zinc (Zn) have been utilized as metal carbenoids in ring expansion, each following substrate-specific pathway. Recently, Bi and coworkers employed Rh- and Ag-catalyzed N-trifluoromethanesulfonyl (N-triftosyl) hydrazones 10 as carbene precursors for the dearomative ring expansion of azoles 17,40 indazoles 14,41 aziridines,42 pyrroles 1,43 and indoles 11.44,45 Despite the use of similar carbene sources and metals, distinct structural features of substrates lead to different pathways.46 Azoles 17, indazoles 14 and aziridines undergo metal ylide formation, ring cleavage, and ring closing (Scheme 1E),40–42 while pyrroles 1c and indoles 11b follow the cyclopropanation-fragmentation-dearomatization pathway (Scheme 1F).43,44 In 2024, the Nakamura group reported the use of zinc carbenoids 9′ for methylene insertion into 1,2-azoles 17, following the metal ylide formation, ring opening, and ring closing pathway (Scheme 1E).47,48 These dearomative transformations elegantly yield diverse valuable products but deviate from the core concept of skeletal editing, which focuses on precise atom-level modifications while retaining the structural class of the scaffold. Transformations like those in Scheme 1E and F, though innovative, involve broader reorganization, such as converting one aromatic system to another. Other examples of metal carbenoids include the formation of spiro compounds from (benzo)isoxazoles.49,50 Carbon insertion into the in situ-generated N-heterocyclic carbenes (NHC) to yield 3,4-dihydroquinoxalin-2(1H)-ones was also reported.51 A few examples of ring expansion in azaheterocycles without a carbene source have been reported, such as the formation of quinolinones from oxindoles,52,53 the ring expansion of pyrazolium ylides to 1,2-dihydropyrimidines,54 and benzoisothiazol-3-ones to 2,3-dihydrobenzothiazin-4-ones.55
In addition to the significant progress made with carbene sources for ring expansion of azaheterocycles, arenes have also been explored for carbene-mediated skeletal editing, notably inspired by the Buchner reaction.56 In 1885, the Buchner reaction was developed to enable the dearomative ring expansion of arenes 19 yielding cycloheptatrienes 20.16 This process involves the formation of norcaradiene intermediates Int-Ivia cyclopropanation at the 1,2-position, followed by 6π electrocyclic ring opening to produce cycloheptatrienes 20 (Scheme 2A).16 While this reaction capitalizes on the inherent reactivity of arenes 19, its applicability to polycyclic (hetero)arenes like naphthalenes 21 was initially limited. The Sarlah group addressed this challenge by introducing the arenophile 4-methyl-1,2,4-triazoline-3,5-dione (MTAD) 22, which redirected cyclopropanation toward the 2,3-position when used in combination with TMS-diazomethane 23 and a palladium (Pd) catalyst (insertive agent), facilitating the formation of benzocycloheptatrienes 24 (Scheme 2B).57 In 2024, the Sarpong group adopted a similar strategy to achieve the total synthesis of the natural diterpenoid harringtolide 27 (Scheme 2C). They first converted cephanolide A 25 into the corresponding p-quinol methylether derivative 26 (benzenoid) following the Kita oxidative dearomatization conditions, and subsequently used diazomethane as an insertive agent to perform a ring expansion, yielding harringtonolide 27 (troponoid) through the Büchner–Curtius–Schlotterbeck reaction.58 In 2024, the Jiang group developed an asymmetric rhodium/boron catalytic system for the single-atom carbon insertion of phenols with cyclopropenes as insertive agents, synthesizing various cycloheptadienones with excellent chemo- and regioselectivity.59
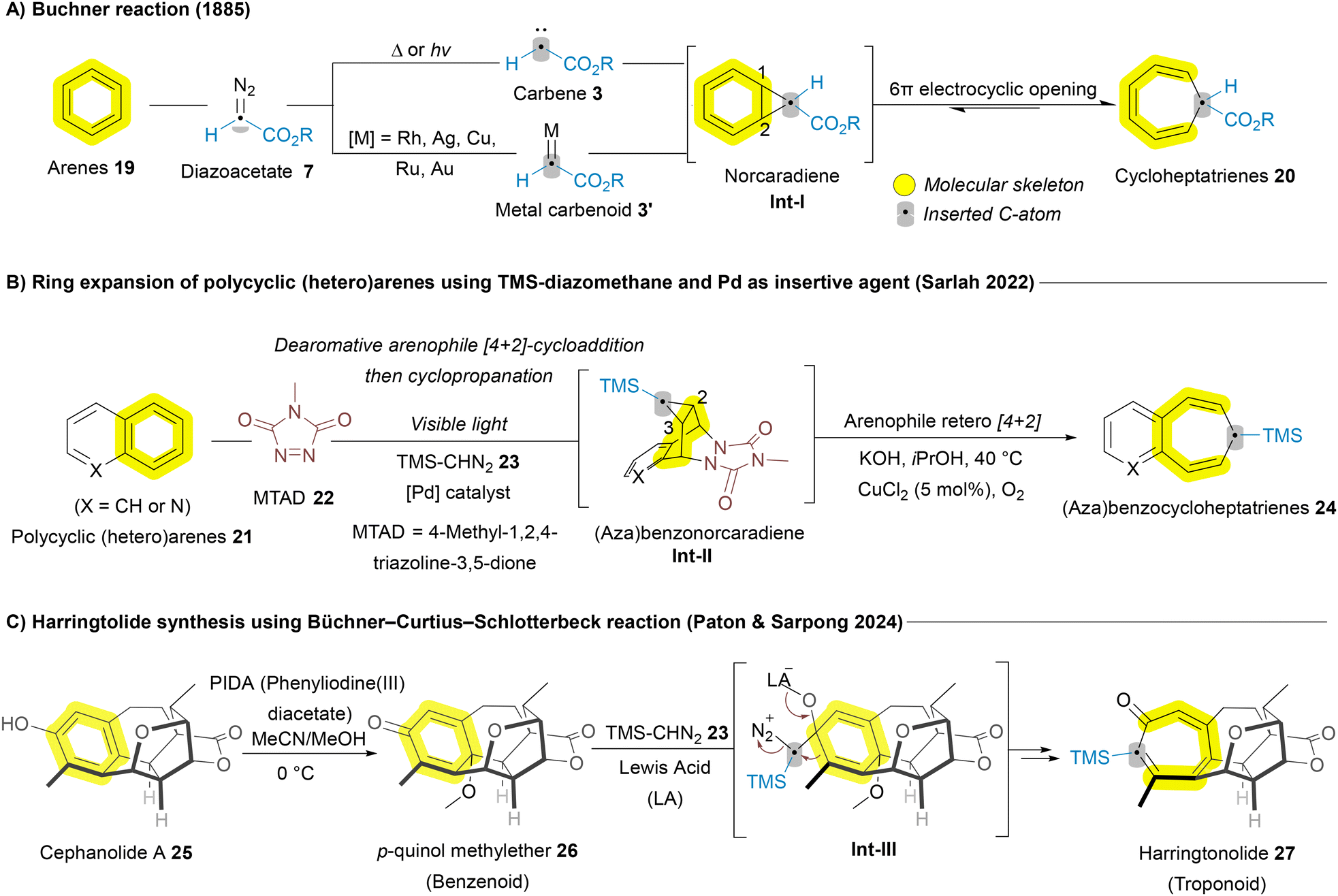 | ||
| Scheme 2 Carbon atom insertion into the main skeleton of arenes and polycyclic (hetero)arenens leading to ring expansion. | ||
As the reaction evolved, various metal carbenoids—including silver (Ag), copper (Cu), rhodium (Rh), and ruthenium (Ru)—have been employed to enable both intra- and intermolecular asymmetric Buchner reactions. In 2019, Qiu and Xu reported a copper-catalyzed carbene/alkyne metathesis (CAM) reaction terminated with the Buchner ring expansion, yielding dihydrocyclohepta[b]indoles 29 (Scheme 3A). This marked the first example of the Buchner ring expansion reaction involving donor/donor-type metal carbene species.60 Donor/acceptor carbenes contain one electron-donating and one electron-withdrawing group, while donor/donor carbenes feature two electron-donating groups.61 Asymmetric versions of these ring-expansion reactions, have also been reported. In 2019, the Iwasa group provided an early example of an efficient enantioselective intramolecular Buchner reaction using diazoacetamides 30 (Scheme 3B). The Ru(II)–Pheox catalyst 32 demonstrated high efficiency in this transformation, achieving both high regioselectivity and enantioselectivity (up to 99% enantiomeric excess ee), yielding various γ-lactam fused 5,7-bicyclic-heptatriene derivatives 31a in quantitative amounts.62 Following this breakthrough, other asymmetric versions of the Buchner reaction, applying various protocols to different substrates and insertive agents, have been reported. In 2021, Nemoto and Harada developed a diazo-free asymmetric intramolecular Buchner reaction for non-activated arenes 33, using ynamides as a carbene source (Scheme 3C). This method enabled asymmetric ring expansion, yielding γ-lactam fused 5,7-bicyclic-cycloheptatrienes 31b in the presence of a chiral phosphoric acid silver salt 34.63 In the same year, Darses and coworkers employed a similar strategy, using a dirhodium (Rh) catalyst to synthesize enantioenriched seven-membered carbocycle-containing bicyclic skeletons.64 In 2022, the Zhu group reported a chiral dirhodium(II) tetracarboxylate-catalyzed enantioselective intramolecular Buchner reaction of donor/donor carbenes, leading to the synthesis of valuable chiral polycyclic products. Both aryloxy enynones and diazo compounds served as efficient carbene precursors, achieving excellent yields and outstanding enantioselectivities.65
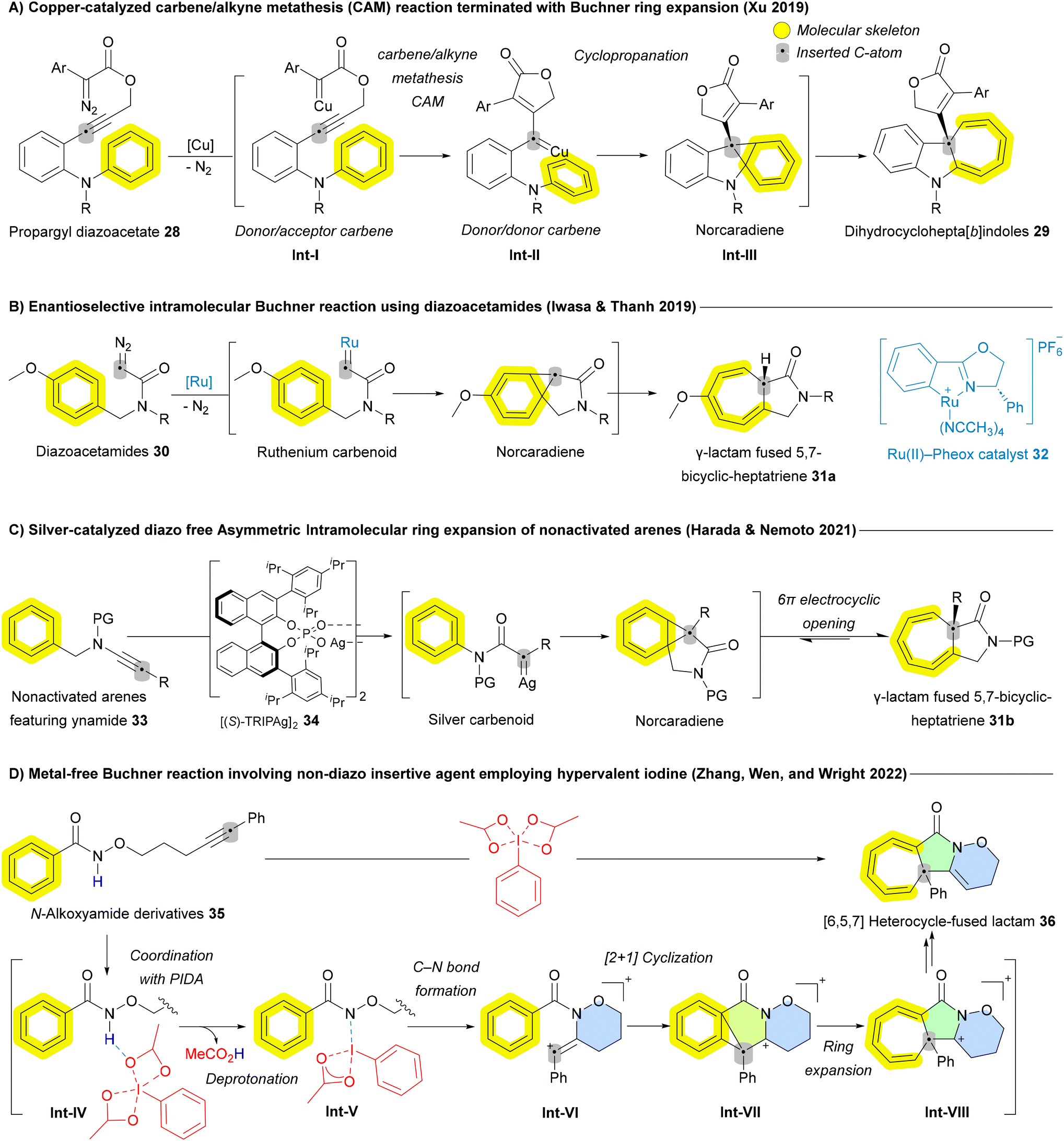 | ||
| Scheme 3 Recent advances of Buchner-mediated carbon atom insertion into the main skeleton of arenes. | ||
Despite all these advances in the Buchner reaction from the perspectives of sustainability, a metal-free Buchner reaction involving non-diazo compounds remains highly desirable. Addressing this need, a new strategy was introduced by Ni, Wen, and Zhang in 2022, employing hypervalent iodine [phenyliodine(III) diacetate (PIDA)] as a promoter for intramolecular Buchner reactions.66 This method utilizes three-carbon-atom tethered N-alkoxyamides 35 as substrates. The proposed mechanism begins with the coordination of the benzamide substrate to phenyliodine(III) diacetate, forming the intermediate Int-IV, followed by a rapid deprotonation process that yields the slightly endergonic species Int-V. From Int-V, the release of the acetate ligand and subsequent C–N bond formation led to a stable vinyl cation, Int-VI. This cation induces a [2 + 1] cyclization reaction, producing a highly stabilized cationic species Int-VII, via a rapid process with a low energy barrier. The ring expansion then proceeds to form the seven-membered ring intermediate Int-VIII. Finally, a deprotonation process results in the formation of a [6,5,7] heterocycle-fused lactam 36, completing the reaction (Scheme 3D).66 All these transformations in Scheme 3 are grounded in well-established reactivity trends, which have been studied extensively for decades. While recent advances allow them to be categorized under the broader umbrella of skeletal editing, they do not represent the forefront of the field and often deviate from the core concept of skeletal editing by converting one aromatic system into another.
In addition to various (polycyclic) arenes, other examples of (polycyclic) heteroarenes, particularly azaheterocycles, follow a sequence similar to the Buchner reaction. This typically involves a dearomative [4 + 2] cycloaddition, cyclopropanation, and subsequent electrocyclic ring opening. In 2019, Mancheño and coworkers developed a metal-free ring expansion protocol for carbon atom insertion into hydroquinolines scaffold 37 employing TMS-diazomethane (TMSCHN2) 23 as the insertive agent to afford a range of benzo[b]azepines 38a (Scheme 4A). The authors proposed two possible pathways for the ring expansion. After the oxidation of 37 to the corresponding intermediate Int-I, nucleophilic attack of TMS-diazomethane 23 on the iminium ion generates diazo intermediate Int-II, which undergoes nitrogen release. This occurs through nucleophilic attack either at the olefinic carbon (3-position) or the nitrogen atom, leading to cyclopropane cationic intermediates Int-III or Int-IV, respectively. Subsequent ring opening gives rise to the 7-membered cationic intermediates Int-V or Int-VI, respectively. Finally, the release of TMS+ leads to the formation of benzo[b]azepine 38a (Scheme 4A).67 In 2021, Beeler and coworkers introduced another approach to access mono- and polycyclic functionalized azepines 38b through the dearomative photochemical rearrangement of aromatic N-ylides 40 (Scheme 4B).68 That same year, the Yoo group developed an unprecedented regioselective silver(I)-catalyzed carbon atom insertion, yielding 4-substituted azepine derivatives 38c through 1,4-dearomative addition of diazoacetates 7 (Scheme 4C).69 Additionally, different azaheterocycles have been reported for carbon atom insertion using various insertive agents and different approaches, including 1,2-dihydropyridines and quinolines using gold-carbenoid as the insertive agent,70 and Cu-iminium catalysis for carbon atom insertion into oxindoles affording quinolinones.53 In 2024, Morandi and coworkers employed the same substrate oxindoles 42 to develop a highly efficient rare example of regiodivergent ring expansion reaction to afford both 3-substituted quinolinones 44 and 4-substiituted quinolinones 45 (Scheme 4D).52 They showed a successful example for the late-stage functionalization of bioactive oxindoles, such as doliracetam (drug for epilepsy) showing the potential of this method in the synthesis of quinoline derivatives and diversification of drugs.
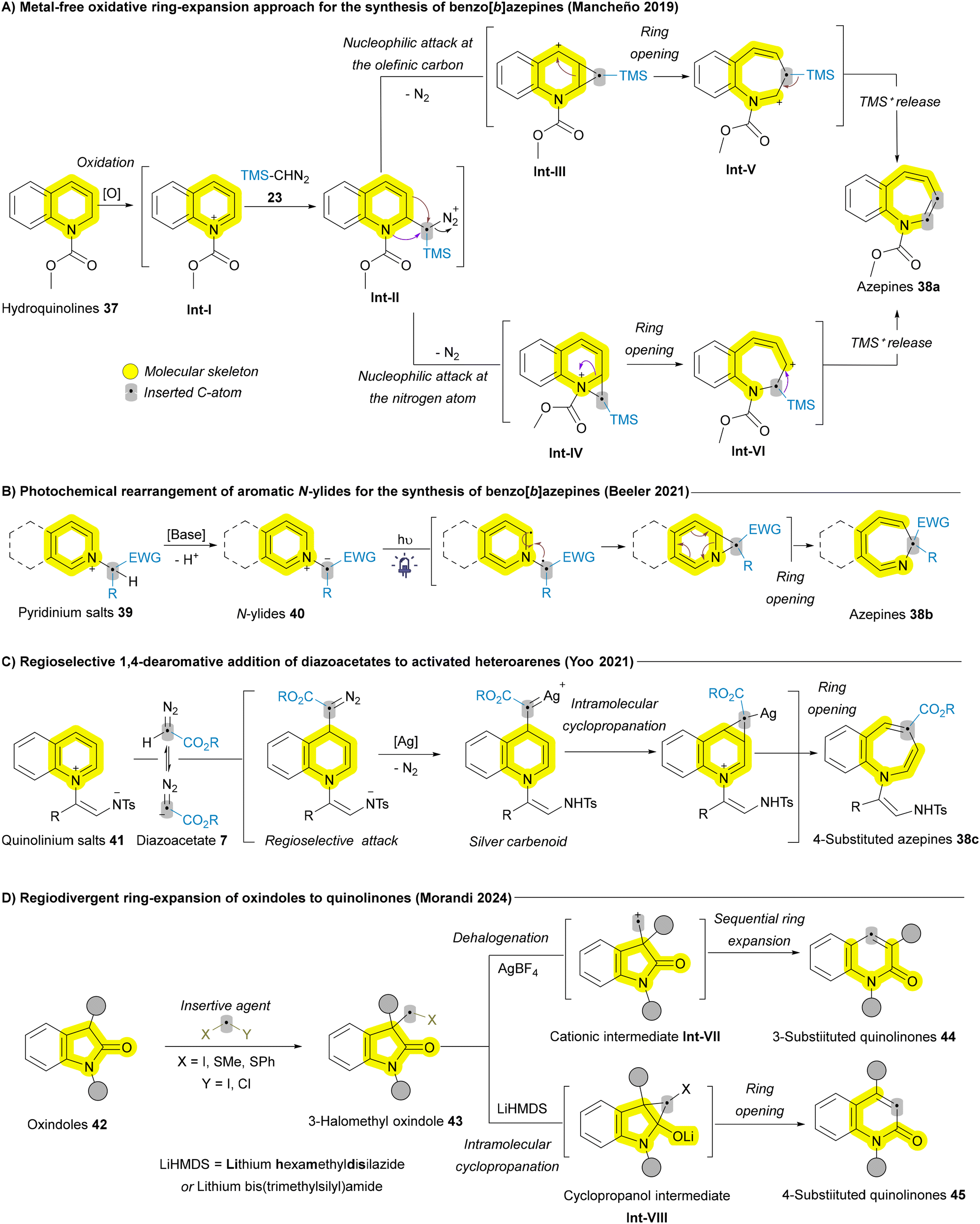 | ||
| Scheme 4 Recent advances of carbon atom insertion into the main skeleton of quinolinium and pyridinium salts and oxindoles. | ||
In 2022, Arnold group, the pioneers of directed evolution to engineer enzymes, reported a landmark example of enantioselective single-carbon atom insertion into aziridines 46 to afford azetidines 47 as a new-to-nature activity of engineered “carbene transferase” enzymes. The iron carbenoid insertive agent could be trapped by nucleophilic aziridine forming aziridinium ylides Int-I, which could undergo intramolecular [1,2]-Stevens rearrangement liberating the desired product 47 (Scheme 5A).71 This pioneering achievement opens the door to bio-catalyzed skeletal editing, enabling unprecedented efficiency and stereocontrol in chemical transformations. Gutierrez and Glorius developed a photoredox-catalyzed ring expansion strategy to efficiently insert functionalized carbon atoms into indenes. The process leverages α-iodonium diazo compounds as masked carbyne equivalents, alongside photoredox catalysis, to achieve carbon insertion under mild conditions.72 Beside all these single atom insertions, some recent reports of multiple atom insertion have been introduced,73,74 including the work of Clayden of the asymmetric deprotonation of N-benzyl urea derivatives of nitrogen heterocycles 48 leads to enantioselective insertion of the benzylic substituent into an aromatic C–N bond via chiral lithium (Scheme 5B).75 Not only azaheterocycles are the heteroarenes that have been reported for the carbon atom insertion, but also other heterocycles for example oxetane and thietane heterocycles have been reported to undergo photo-mediated carbon atom insertion employing diazoacetates 7 as an insertive agent to afford tetrahydrofuran and thiolane heterocycles.76
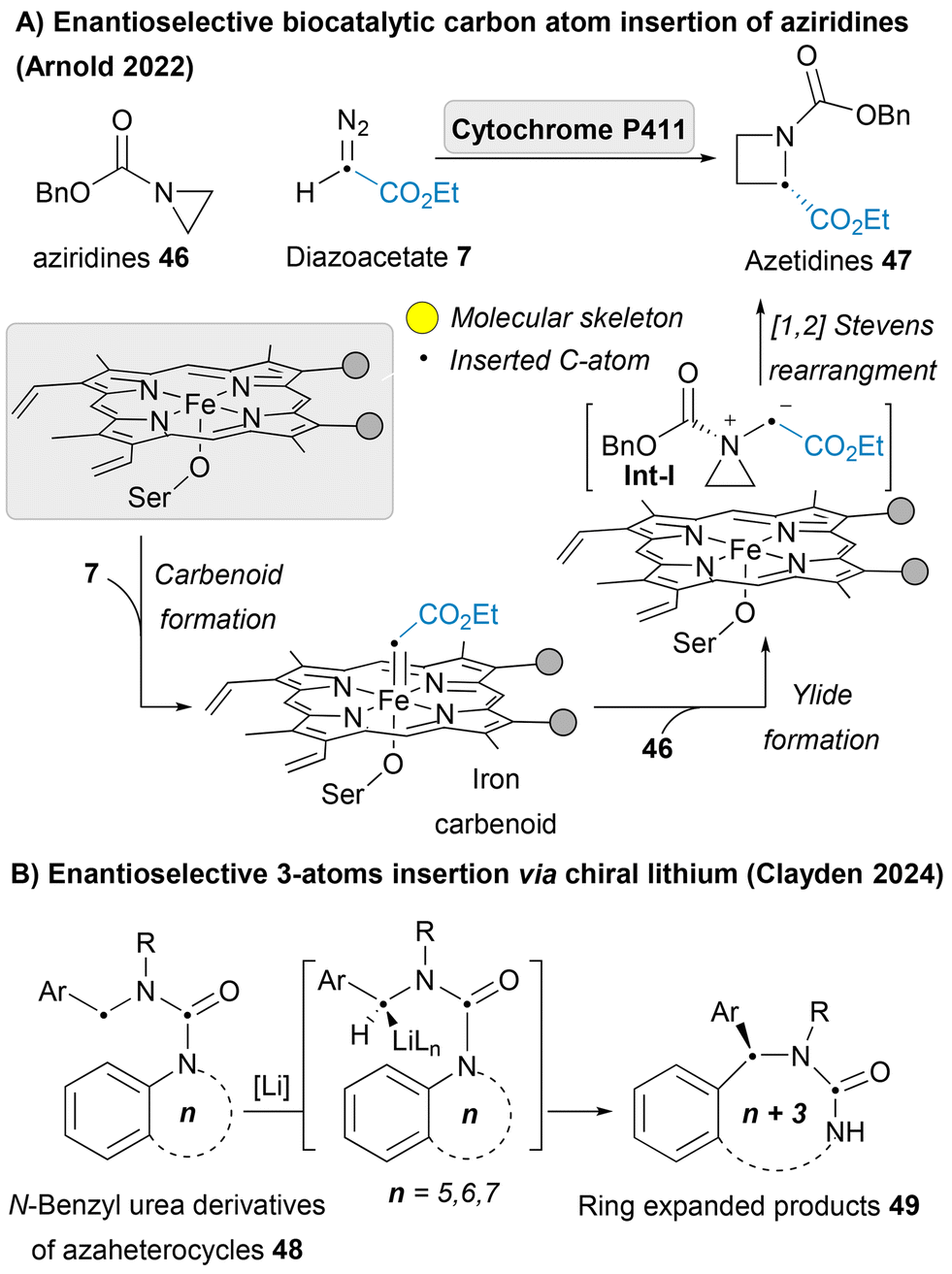 | ||
| Scheme 5 Enantioselective biocatalytic carbon atom insertion and multiple atom insertion. | ||
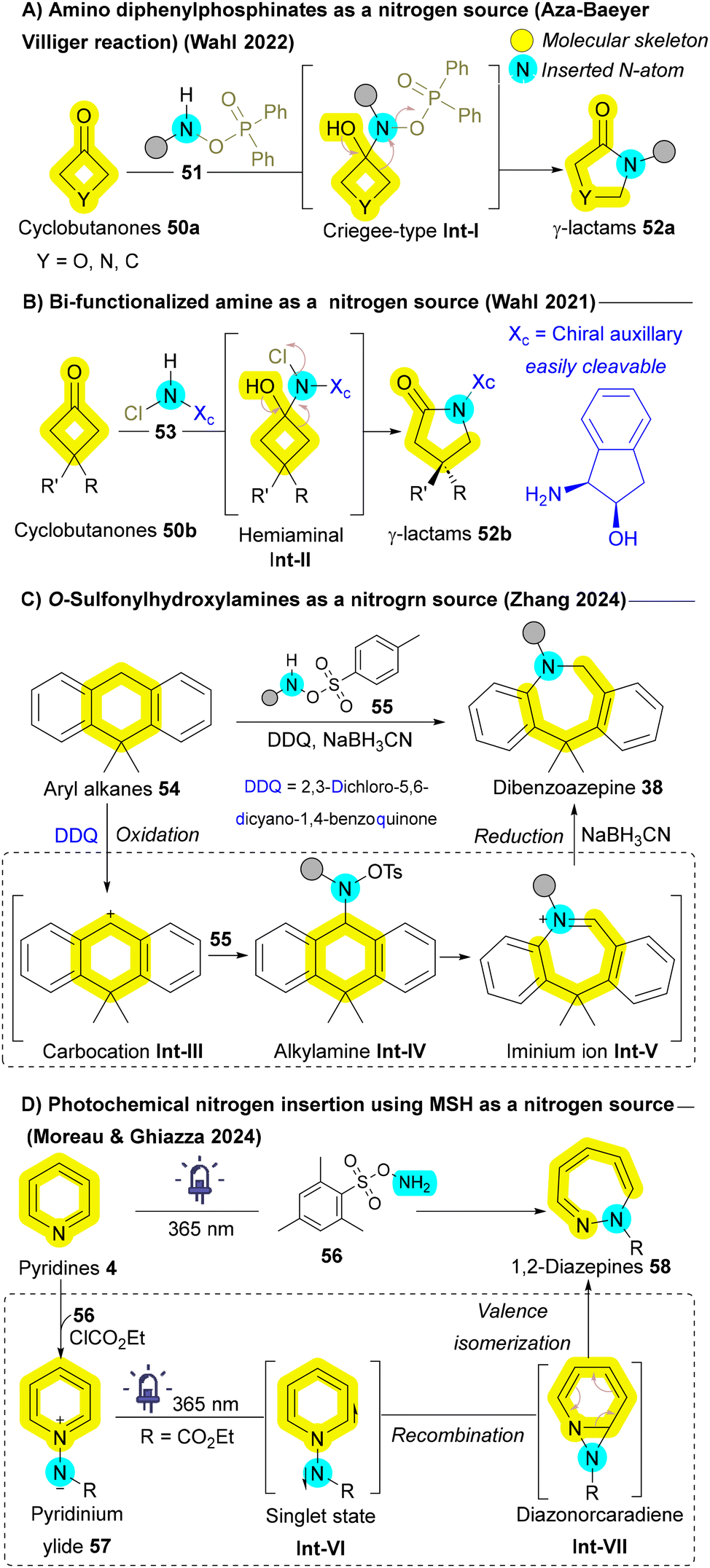 | ||
| Scheme 6 Nitrogen atom insertion employing amine derivatives as insertive agents. | ||
In addition to classical carbonyl-containing substrates, other non-carbonyl substrates have also been explored for nitrogen insertion. O-Sulfonylhydroxylamines 55 have emerged as highly efficient nitrogen sources for the ring expansion of cyclic alcohols and cyclic alkanes 54, as demonstrated by Wahl et al.85 and Zhang et al.,86 respectively. These methodologies typically proceed through the formation of a carbocation intermediate Int-III, which subsequently transforms into an iminium ion Int-V. Upon reduction, this intermediate Int-V affords the desired ring expanded product 38 (Scheme 6C).86 In 2024, Ghiazza and Moreau unveiled an innovative landmark photochemical approach that utilizes O-(mesitylsulfonyl)hydroxylamine (MSH) 56 to induce the ring expansion of pyridines 4 into 1,2-diazepines 58. This skeletal transformation is driven by the in situ formation of 1-aminopyridinium ylides 57, which are then excited to their singlet state Int-VI upon UV irradiation. Subsequently, they undergo rearrangement into diazonorcaradienes Int-VII, followed by sequential fragmentation and dearomatization, ultimately yielding the 1,2-diazepine scaffold 58 (Scheme 6D).87 These pioneering methodologies not only demonstrate the utility of O-sulfonylhydroxylamines 55 and 56 as nitrogen sources but also significantly broaden the substrate scope for nitrogen insertion reactions, highlighting its potential in skeletal editing strategies and representing the forefront of the field.
Another promising insertive agent for N-atom insertion reactions is the nitrene.88 Early investigations into the chemistry of nitrenes were initiated by Huisgen, Doering, Beach and Cotter, which laid the foundation for subsequent research in this area.89–91 Aryl nitrenes have been successfully employed as reactive intermediates to facilitate the synthesis of azepines from corresponding aryl azides 59a which can generate the corresponding nitrenes Int-Ivia thermal decomposition, photochemical activation, or heavy atom tunneling.91–96 Once formed, aryl nitrenes Int-I can undergo aziridination, producing aziridine Int-VI or azirine Int-III intermediates that subsequently undergo oxidative ring opening to yield azacycloheptatetraenes 60. These scaffolds can further convert to the corresponding azepines 38d upon nucleophilic addition (Scheme 7A).92 While this approach yields diverse and valuable skeletons, it sometimes involves converting aromatic rings into non-aromatic analogues and vice versa, which falls short of achieving the optimal goal of atom-level modifications. Wei group expanded the utility of azides as nitrogen sources by employing transition metals such as cobalt and rhodium to mitigate the undesired C–H insertions that are common in traditional nitrene ring expansion reactions.97,98 In 2023, Wei group demonstrated a transformation involving biaryls with peripheral carbamoyl azides 61 that are activated by a rhodium catalyst, allowing direct insertion into the C–C bonds of arene rings to generate fused azepine products 38e (Scheme 7B). Although this transformation is particularly challenging, the employment of a paddlewheel dirhodium complex, Rh2(esp)2, effectively inhibited the unwanted competing C–H amination pathway.98 In addition to azide derivatives 59a and 61, Ji and Wei's groups have explored the incorporation of TMSN3 as an insertive agent in conjunction with metal catalysts for a variety of substrates, including cycloalkenes 62,97 indoles,99 and arenols (more details in section 2.3.1),100 to facilitate nitrogen atom insertion via nitrene intermediates. For example, in reactions involving cobalt, azido radicals generated from TMSN3 by a radical chain reaction selectively attack cycloalkenes 62 to produce carbon radicals Int-VII, which subsequently yield aziridine radicals Int-IX. Through consecutive oxidative ring opening and dehydrogenation, the corresponding pyridine derivatives 4 are formed (Scheme 7C).97 In the case of indoles, a domino reaction occurs that generates azido radicals, leading to diazidation and the formation of quinazolin-4-amine derivatives.99 Conversely, for arenols, dearomatizative azidation followed by aryl migration, afforded the corresponding benzazepine derivatives.100 In 2024, Wang and Luan employed AgOTf as a catalyst and PhI = NTs as insertive agent to induce nitrogen atom insertion of arenols affording azepinone.101 Iron has been utilized as a catalyst by Yu's group to convert α-azidyl phenyl ketones into enamides through nitrogen insertion.102
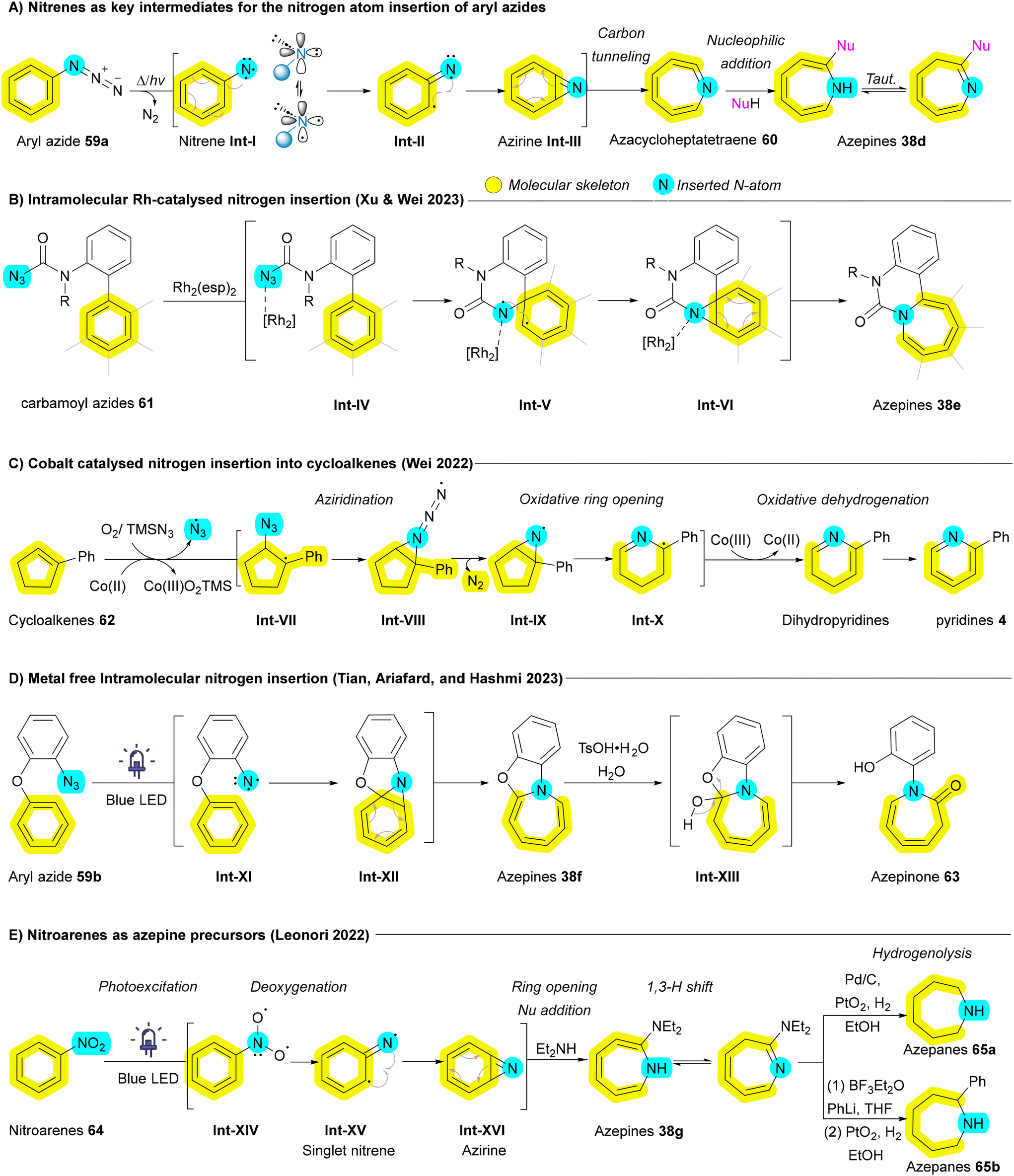 | ||
| Scheme 7 Nitrogen atom insertion into the main skeleton of aryl azides and nitroarenes leading to ring expansion. | ||
In 2023, a metal-free photochemical approach for the intramolecular nitrogen insertion has been reported by Tian, Ariafard and Hashmi, involving a cascade reaction that generates nitrene intermediates Int-XI, aziridination, and subsequent water addition to obtain desired azepinone derivatives 63 (Scheme 7D).103 In 2022, Leonori and his group have explored nitroarenes 64, an unprecedented stable and commercially accessible substrate, to synthesize azepines 38g and azepanes 65 using blue light as an energy source. In this process, nitroarenes 64 are converted to singlet nitrenes Int-XV in the presence of blue light, facilitating azirine Int-XVI formation at C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds followed by a 6π-electrocyclic ring opening, ultimately producing azepines 38g. These azepines 38g can subsequently undergo hydrogenolysis to yield azepanes 65 (Scheme 7E).104
C bonds followed by a 6π-electrocyclic ring opening, ultimately producing azepines 38g. These azepines 38g can subsequently undergo hydrogenolysis to yield azepanes 65 (Scheme 7E).104
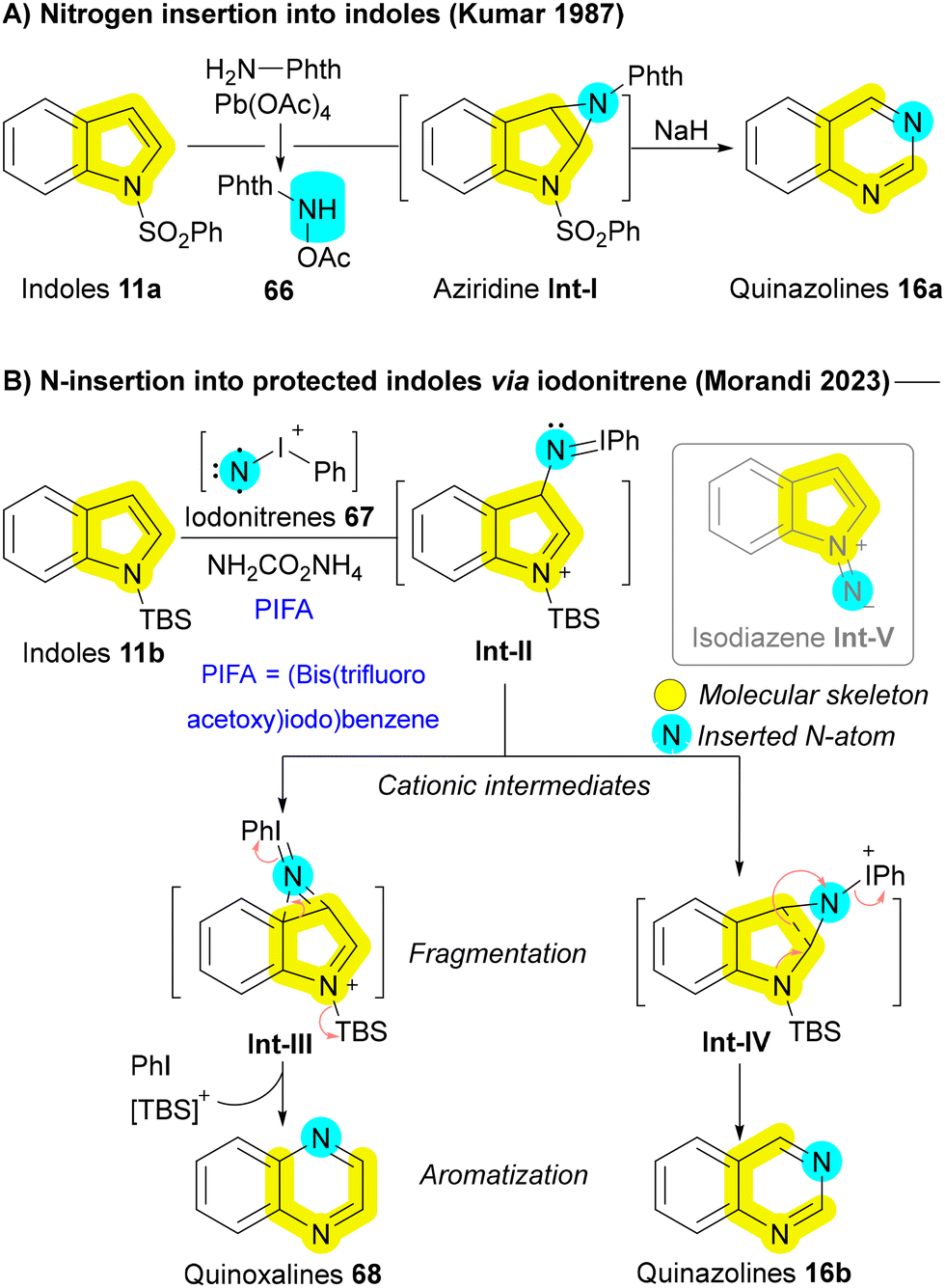
Recently, several innovative methods have been developed to transform indoles 11 and pyrroles 1 into their nitrogen-extended counterparts, including quinazolines 16 and pyrimidines 15. A key example dates back to 1987 when Kumar utilized N-acetoxyaminophthalimide 66, generated via the oxidation of N-amino phthalimide with lead(IV) acetate Pb(OAc)4, as an insertive agent. The reaction followed a pathway including the formation of aziridine Int-I, followed by subsequent ring expansion (Scheme 8A).105 Building on similar principles, the Morandi group later introduced a pioneering strategy for nitrogen insertion into N-protected indoles 11b, enabling access to N,N-heterocycles such as quinazolines 16b and quinoxalines 68, depending on the substitution pattern of the indoles 11b. This reaction exhibits a broad substrate scope, tolerating various functional groups, and thus enabling the bioisosteric diversification of natural products and pharmaceutical agents.106 In 2024, the Alcarazo group applied the same strategy to obtain indoles from cycloalkene substrates, further expanding the versatility of nitrogen insertion chemistry.107 Morandi group's strategy employed iodonitrenes 67, generated in situ from hypervalent iodine and ammonium carbamate (NH4CO2NH2), as highly reactive electrophilic aminating agents. This innovative chemistry not only opened the gate for numerous future developments but also provided elegant solutions to several persistent challenges in nitrogen insertion and amination reactions. The reaction proceeds through the formation of a cationic azirine Int-III and aziridine Int-IV intermediates, followed by the elimination of iodobenzene to afford the desired quinoxaline 68 or quinazoline 16b (Scheme 8B). The use of silyl protecting group (TBS) of indole 11b is critical to avoid side interactions between the nucleophilic nitrogen of the indole 11b and the electrophilic iodonitrene 67, forming an unstable isodiazene intermediate Int-V that can degrade the carbon skeleton.106 Hence, the requirement for a protecting group, coupled with its inefficiency in converting pyrroles to pyrimidines, restricts the applicability of this method to more complex medicinal substrates. In addressing these challenges, the same group improved their protocol one year later with the serendipitous discovery that lithium bis(trimethylsilyl)amide (LiHMDS) could serve a dual function as both a base and a nitrogen atom source. This allowed for the direct insertion of nitrogen atoms into 1H-indoles and 1H-pyrroles, even in complex bioactive molecules, overcoming previous limitations and broadening the synthetic utility of this methodology.108 In 2024, the same Morandi group applied their iodonitrene chemistry for transforming cyclopentenone derivatives into pyridones, through a strategy of silyl enol ether formation, followed by nitrogen insertion, and subsequent aromatization. Their strategy enabled as well to incorporate 15N-labels in various synthetic targets.109
 | ||
| Scheme 8 Nitrogen atom insertion into the main skeleton of indoles leading to ring expansion. | ||
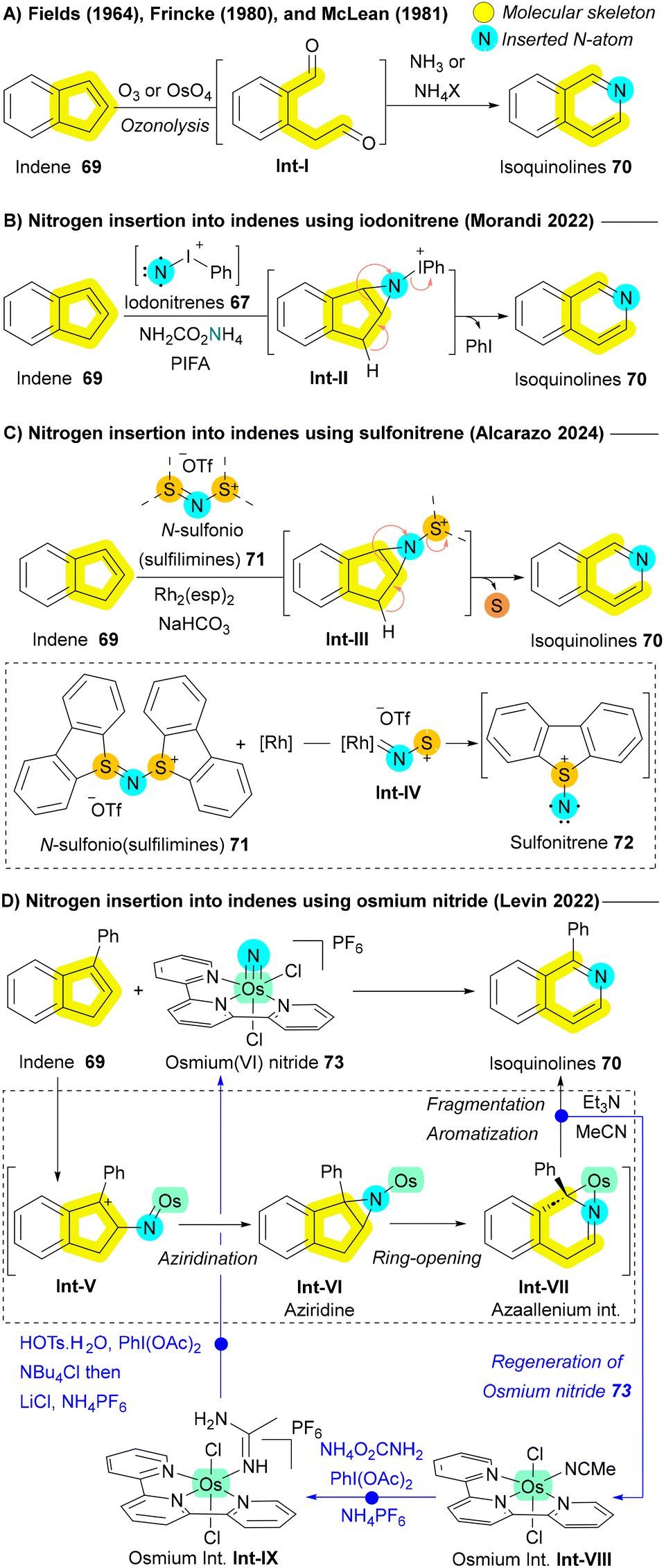
Another significant class of compounds that have been explored for nitrogen insertion reactions is cyclic olefins, particularly indenes 69, which can be transformed into isoquinolines 70—a crucial scaffold in various pharmaceuticals.77 Early studies by Fields, Frincke, and McLean demonstrated this transformation through oxidative cleavage (ozonolysis) of the indene backbone using ozone (O3) or osmium tetroxide (OsO4) in the presence of ammonia or ammonium salts as the nitrogen source (Scheme 9A).110–112 However, the need for these harsh oxidative conditions limited the applicability of these methods. In 2023, The Morandi group advanced this area by applying their iodonitrene 67-based approach for nitrogen insertion in indenes 69, leading to isoquinolines 70 synthesis via an aziridination–fragmentation–aromatization pathway (Scheme 9B).113 However, their protocol required strong oxidizing agents (hypervalent iodine), further constraining its substrate scope.113 In 2024, Alcarazo and colleagues introduced a novel electrophilic nitrogen source, N-(sulfonio)sulfilimine 71 acting as sulfonitrene 72 precursors under rhodium catalysis. These reactive species 72 enabled the same reactivity for indenes 69 without the need for oxidizing agents, via aziridination followed by ring expansion (Scheme 9C). This protocol proved effective for indenes 69, even in the absence of electron-donating groups or aryl rings, but encountered limitations when applied to unprotected indoles or pyrroles due to the inherent stability of iminium cations, which hindered aziridination.107 Levin et al. also contributed to this field by reporting the formation of isoquinolines 70 from indenes 69 through direct nitrogen atom insertion using osmium(VI) nitride 73. The reaction proceeds via an aziridination and ring-opening sequence, leading to azaallenium Int-VII formation. The aromatization of this intermediate Int-VII occurs primarily through base-assisted deprotonation, followed by stepwise regeneration of starting osmium(VI) nitride 73 (Scheme 9D).114
 | ||
| Scheme 9 Nitrogen atom insertion into the main skeleton of indenes leading to ring expansion. | ||
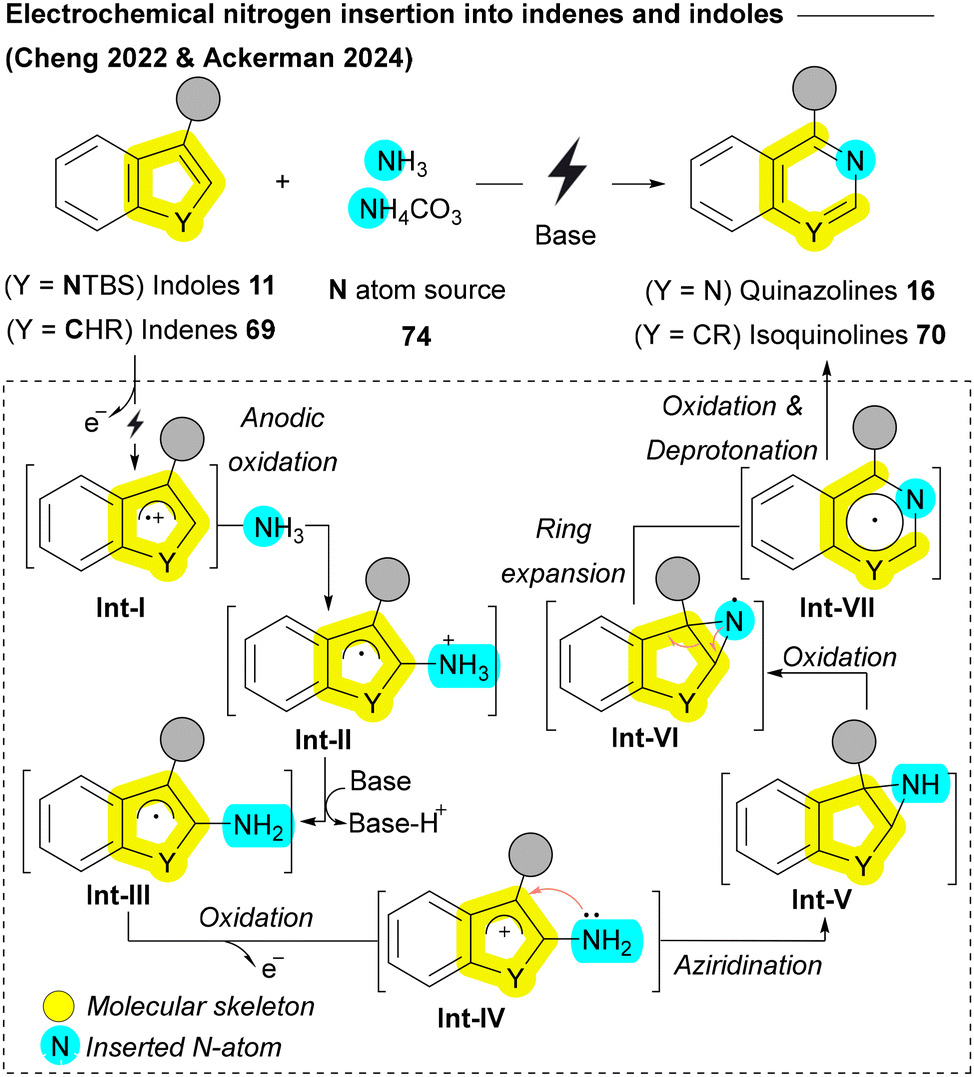
Recently, the Cheng group and Ackerman group achieved a groundbreaking milestone by successfully accomplishing direct ammonia 74 insertion into indenes 69 and indoles 11, respectively, using an electrochemical approach (Scheme 10).115,116 In their methods, a cation radical Int-I is generated through anodic oxidation, which then reacts with ammonia affording Int-II. A subsequent oxidation converts the neutral radical Int-III into a cation Int-IV which undergoes annulation to aziridine Int-V. A third electron transfer oxidation of nitrogen during the conversion of Int-V to Int-VI triggers deprotonation/rearrangement, yielding dihydroisoquinoline radical Int-VII. The fourth electron transfer and deprotonation results in the final products isoquinolines 70 or quinazolines 16 (Scheme 10).115,116
 | ||
| Scheme 10 Electrochemical N-atom insertion into the skeleton of indoles and indenes leading to ring expansion. | ||
 | ||
| Scheme 11 Oxygen atom insertion strategies leading to ring expansion. | ||
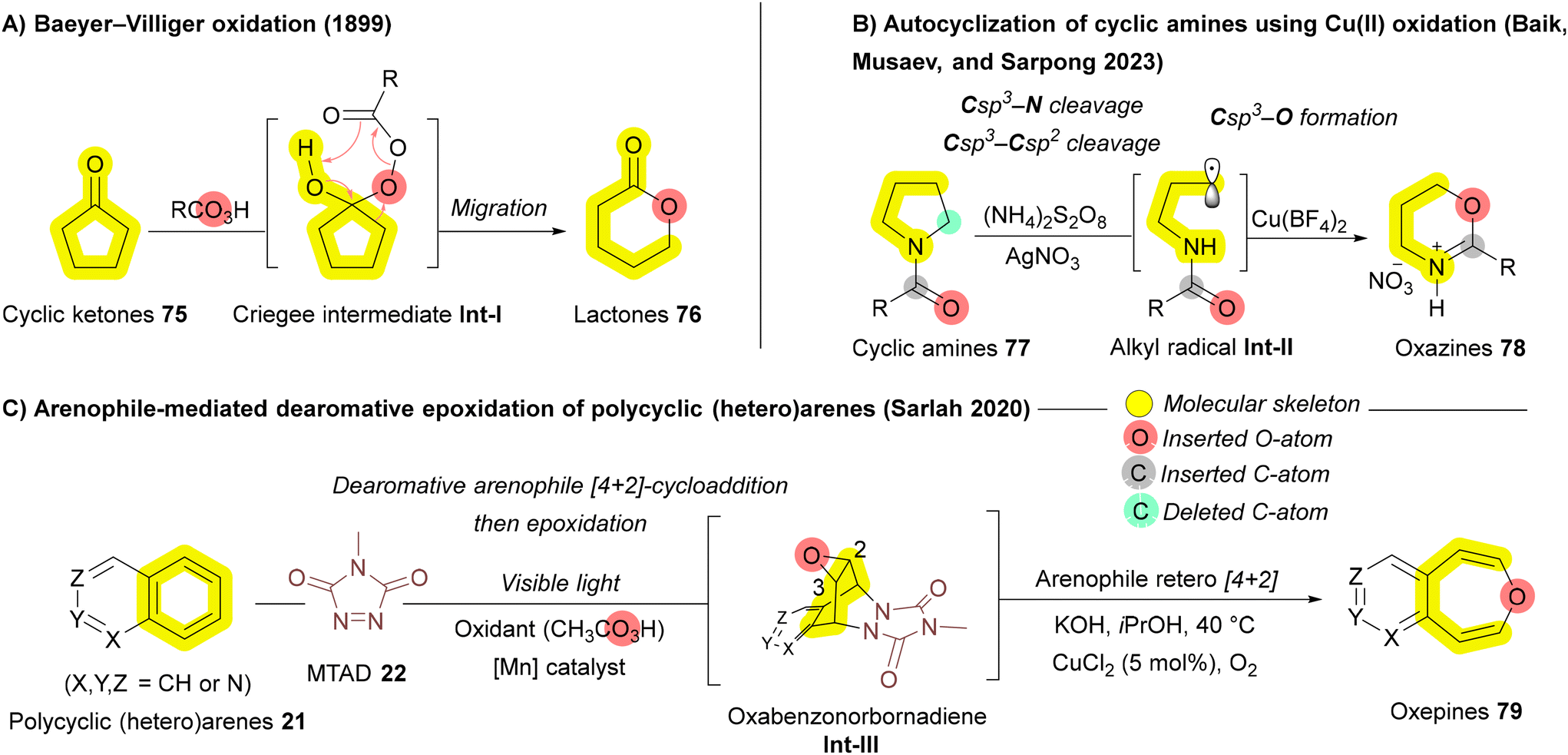
In 2023, the Sarpong group investigated the structural remodeling of cyclic amines 77 through oxidative C–N and C–C bond cleavages, utilizing peroxydisulfate (persulfate) as the oxidant.118 Their proposed mechanism involves the generation of an alkyl radical Int-II from the cyclic amine 77via Ag(I)-mediated activation by persulfate.119 This alkyl radical Int-II subsequently interacts with Cu(II), forming a Cu-complex that undergoes intramolecular cyclization to yield oxazines 78 (Scheme 11B). In 2020, a novel arenophile-based dearomative approach developed by Sarlah and coworkers similar to their work on carbon insertion (Scheme 2B),57 they employed 4-methyl-1,2,4-triazoline-3,5-dione (MTAD) 22 as the reactive arenophile to facilitate oxygen atom insertion. In their strategy, polycyclic (hetero)arenes 21 undergo visible light-induced [4 + 2]cycloaddition, leading to the formation of oxabenzonorbornadiene Int-III, which subsequently undergo a [4 + 2] cycloreversion to yield oxepines 79 (Scheme 11C). This method expands the scope of oxygen insertions, enabling the selective functionalization of polycyclic systems that were previously challenging to modify.120 In 2023, the Liu group achieved the first transformation of pyrrolidine to 1,2-oxazinane via formal oxygen atom insertion, utilizing meta-chloroperbenzoic acid (mCPBA) as the insertive agent to produce complex, medicinally significant bispiro[oxindole–oxazinane] hybrids with remarkable stereocontrol.121
In 2017, Kinjo and coworkers reported the insertion of highly reactive bromoborylene (BrB:) 81 into C–N bonds of substrate 80, leading to N-heterocycle enlargement to afford compound 82 (Scheme 12A).125 Recent approaches to boron insertion primarily involve reductive ring-opening using metals, followed by trapping of the resulting species with organoboronic esters. Yorimitsu's group has employed transition metals such as Ni and Mn to introduce boron into benzofurans.126,127 In 2019, they extended this approach to indoles 11, using lithium metal to achieve reductive ring-opening, followed by trapping of the resulting dianionic species Int-I with organoboronic esters 83, producing 1,2-benzazaborins 84 (Scheme 12B).128 Due to the higher aromatic stabilization energy of pyrrole rings compared to furan, a stronger reductive agent, such as lithium metal, was necessary to facilitate the ring opening.128 In 2024, Jin, Wang, and Wu developed a facile BH3-mediated strategy for boron insertion into indoles and benzimidazoles via the hydroborative cleavage of C–N bonds.129 In 2021, Dong, Liu, and coworkers reported boron insertion into cyclic ethers 85 using tandem zinc/nickel catalysis. Similar to other recent strategies, this process follows a cleavage-then-rebound mechanism, where the ether ring 85 undergoes Zn-enabled reductive ring opening, followed by either radical Int-VII formation or SN2 oxidative addition facilitated by the Ni catalyst. This produces the desired benzoxaborin 87, which can be further transformed to achieve boron-to-carbon transmutations or oxygen/boron-to-nitrogen replacement including one-atom deletion (Scheme 12C).130 Despite these advances, the limited substrates and the need for strong reductants still restricts the broader application of these reactions. In 2024, Yang, Song, and coworkers introduced a more practical boron insertion method for constructing fused BN-heterocycles 90 without strong reducing agents like lithium. This development significantly broadens the scope of these reactions, enhancing their potential in fields such as medicinal chemistry and functional materials. The process begins with boron tribromide (BBr3) 89 inducing the ring opening of N-heterocycles 88 to form intermediate Int-IX, followed by intramolecular C–H borylation that generates the 1,2-benzazaborine Int-X. Finally, the B–N heterocycle 90 is accomplished through intramolecular B–Br/C–Br reductive coupling via Ni catalyst (Scheme 12D).122
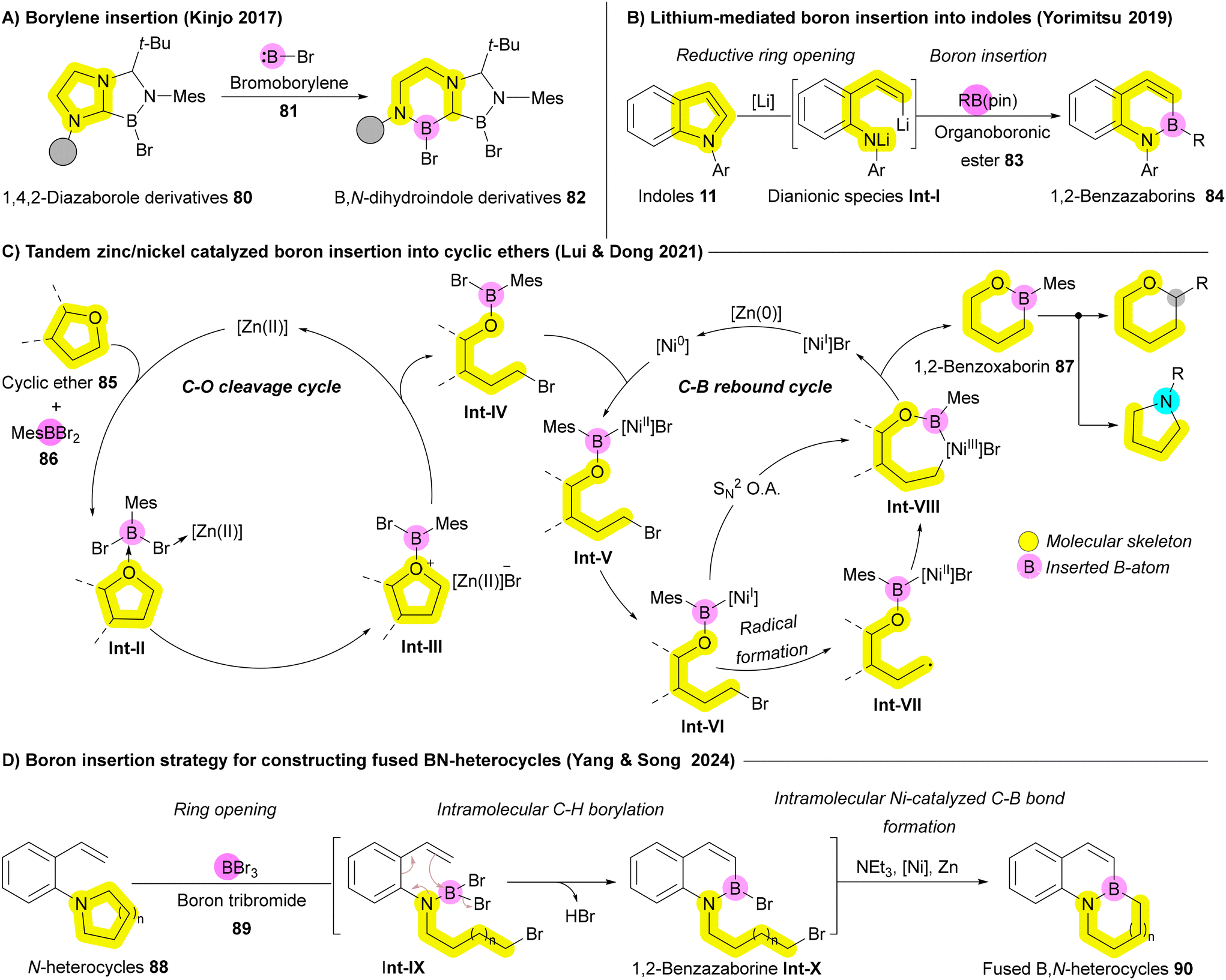 | ||
| Scheme 12 Boron atom insertion strategies leading to ring expansion. | ||
2.2. Ring contraction strategies through atom deletion
Ring contraction via atom deletion or rearrangement of the core skeleton is an efficient strategy in skeletal editing, enabling the selective removal of one or more atoms from the cyclic frameworks to create smaller rings with modified structural and functional features.4 This approach has been well-established for carbocyclic systems, where anionic, carbene, and cationic intermediates have been classically utilized to achieve ring contractions of cyclic ketones.131 Some widely exploited reactions in this context are the Favorskii rearrangement, and the benzilic acid rearrangement. Recent advancements have extended these strategies to non-carbonyl ring systems, including azacyclic compounds, often leveraging photo-induced protocols to achieve the desired transformations.87,132–134 In this review article, we will focus on recent advances subclassifying them based on the type of atom removed; for example, the deletion of carbon or heteroatoms such as boron or nitrogen.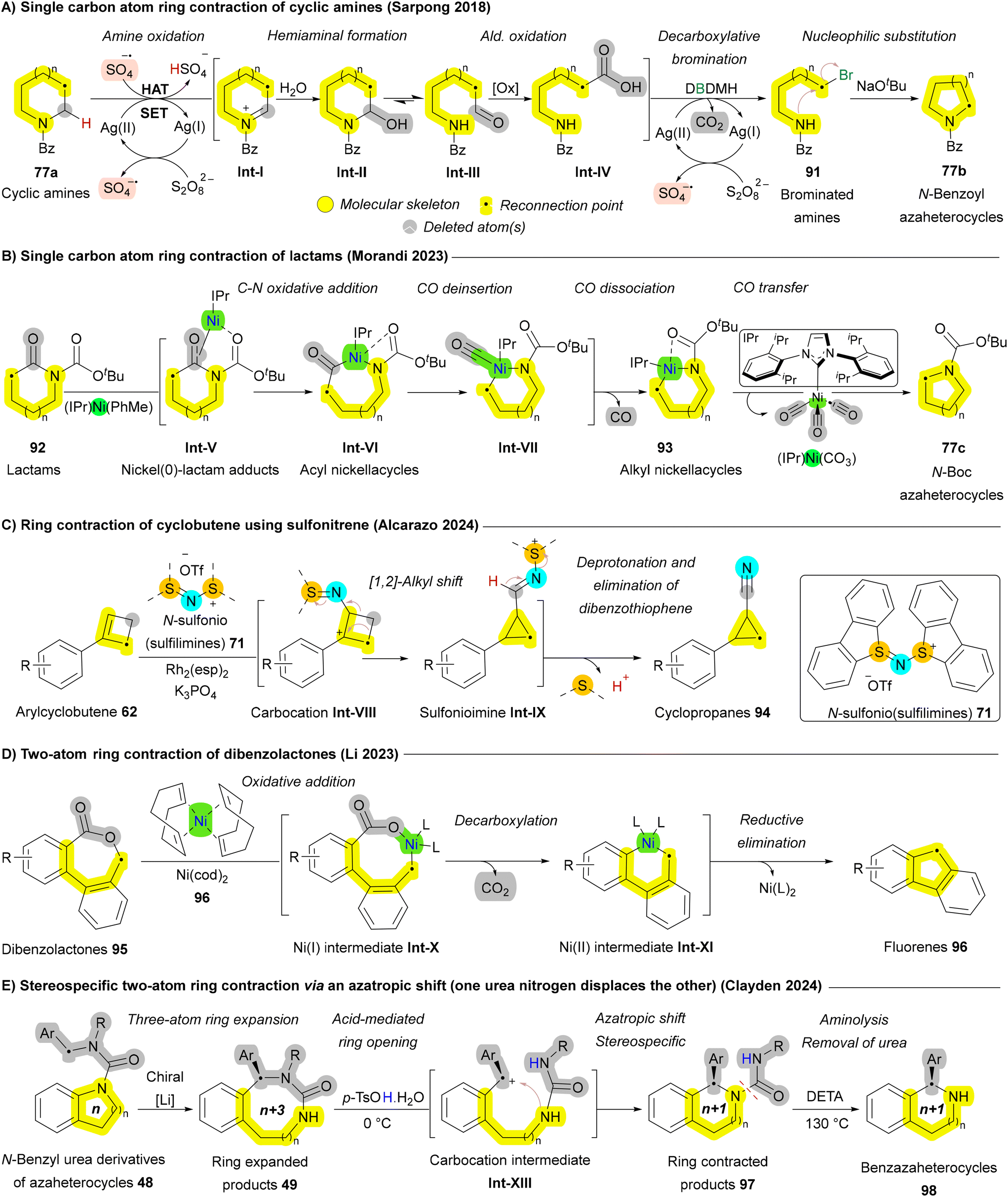 | ||
| Scheme 13 Single and multiple carbon atom deletion of non-classical ring systems. | ||
Besides single-atom deletion approaches, multiple atom deletion has also been reported, though with fewer examples. These strategies involve the removal of two or more atoms from the molecular framework, often resulting in more significant skeletal rearrangements. Dibenzolactone 95 is one of the substrates that has been shown to undergo two-atom ring contraction, yielding the corresponding fluorenes 96 through Ni- or Pd-catalyzed decarboxylative intramolecular coupling. In 2023, the Li group developed a practical approach for the skeletal editing of dibenzolactones 95, which does not require inductively electron-withdrawing ortho substituents on the aryl carboxylate moiety or metal additives. The reaction proceeds via a sequence of oxidative addition, CO2 deinsertion (decarboxylation), and reductive elimination (Scheme 13D).137 As discussed earlier in Scheme 5B, the Clayden group reported an example of multiple carbon atom insertion via the asymmetric deprotonation of N-benzyl urea derivatives of nitrogen heterocycles 48. This process leads to the enantioselective insertion of the benzylic substituent into an aromatic C–N bond via a chiral lithium complex, yielding 49 (Scheme 5B). Subsequent treatment of the ring-expanded (n + 3) ureas 49 with acid triggers a two-atom ring contraction—an “azatropic shift”, in which one urea nitrogen displaces the other—resulting in almost complete retention of stereochemistry. Removal of the urea substituent from 97 was achieved through aminolysis with diethylenetetramine, yielding enantiopure 1-aryl-tetrahydrobenzazaheterocycles 98 (Scheme 13E).75
The potential of ring contraction strategies has been explored to include heteroaromatic systems as well. Early studies catalogued by Buchardt, Kaneko, Streith, and Albini demonstrated that photolysis of azaarenes, such as quinoline N-oxides 99, could result in carbon deletion via the formation of benzoxazepine intermediates Int-III and Int-IV.138,139 However, the use of unselective mercury lamp excitation led to the generation of undesired two-photon byproducts alongside the desired ring contraction products (Scheme 14A). To address this challenge, Levin et al. introduced a selective 390 nm LED light source, significantly enhancing excitation selectivity and improving reaction outcomes. The mechanism involves the formation of a 3,1-benzoxazepine intermediate Int-IIIvia an oxaziridine intermediate Int-I, followed by acid-mediated rearrangement and in situ hydrolysis through two concurrent pathways (Int-V and Int-VI) leading to the formation of N-phenylamides 100. These N-phenylamides 100 then undergo ring closure to form N-acylindoles 11a, which can subsequently undergo deacylation to yield indoles 11b (Scheme 14B).132 In 2021, Tang and Pan et al. reported an oxygen- and visible light-mediated synthesis of pyrroles 1 from pyridinium salts 39 using rhodamine B (RhB) as the photocatalyst. Upon excitation by visible light, RhB facilitates the formation of a pyridinium radical Int-VII, which interacts with molecular oxygen to generate an alkoxy dioxyl radical Int-VIII. Following a proton-coupled electron transfer (PCET) between these intermediates, RhB is regenerated, leading to the formation of an unstable 1,2-dioxetane intermediate Int-X. This intermediate Int-X then undergoes fragmentation and subsequent base-catalyzed aldol condensation, ultimately yielding 3-formylpyrrole 1 (Scheme 14C).133
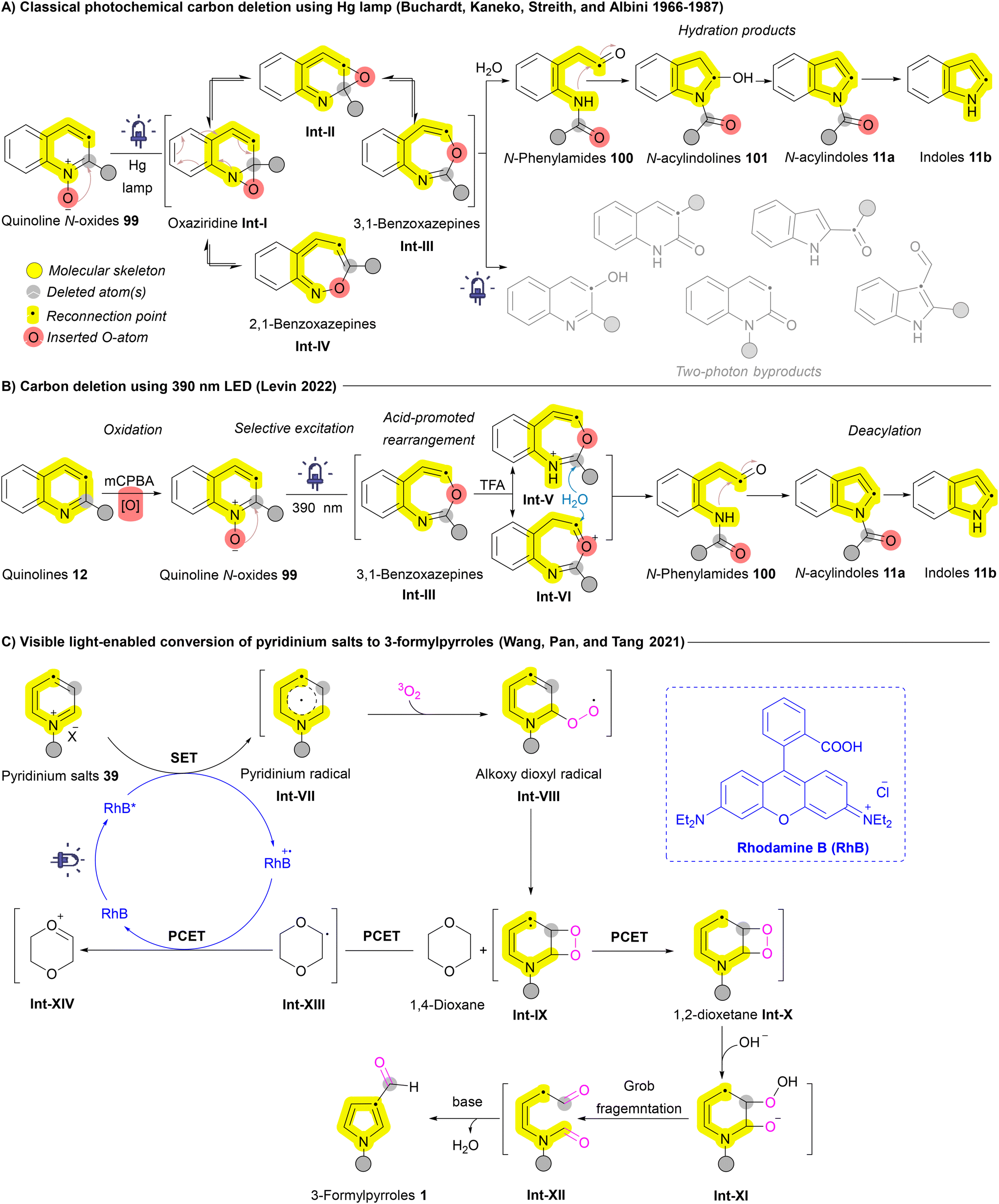 | ||
| Scheme 14 Single carbon atom deletion of azaarenes (quinoline N-oxides and pyridinium salts). | ||
Pyrimidines 15 are the most prevalent diazines in FDA-approved drugs, while pyrazoles 13 are the most common diazoles.140,141 In 1968, Van der Plas and Jongejan pioneered the conversion of pyrimidines 15 to pyrazoles 13 using excess hydrazine (NH2NH2) at extreme temperatures (200 °C). Further investigation revealed that N-methylation with iodomethane enabled successful transformation at a reduced temperature of 100 °C (Scheme 15A).142 Although these methods enabled the conversion of pyrimidines 15 into pyrazoles 13, they typically proceed with low yields and under harsh conditions. In 2022, Sarpong group reported milder conditions, tolerate a wide range of functional groups, and allows for the simultaneous regioselective introduction of N-substituents on the resulting pyrazole 13. The key to the success of this one-carbon deletion method is the N-triflylation of pyrimidines 15, which significantly decreases the LUMO energy of 102b, facilitating hydrazine attack at 23 °C. After nucleophilic attack by hydrazine, intermediate Int-I is formed, followed by Int-IIvia a 3,3-sigmatropic rearrangement, resulting in the ring-opening. Subsequently, the terminal hydrazone nitrogen engages with the ring-opened species Int-III (following tautomerization) at C4, yielding a charge-separated species. Finally, a subsequent proton transfer generates Int-IV, and elimination of N-triflylformamidine through a 1,5-sigmatropic H-shift results in the formation of the pyrazole product 13 (Scheme 15B).143
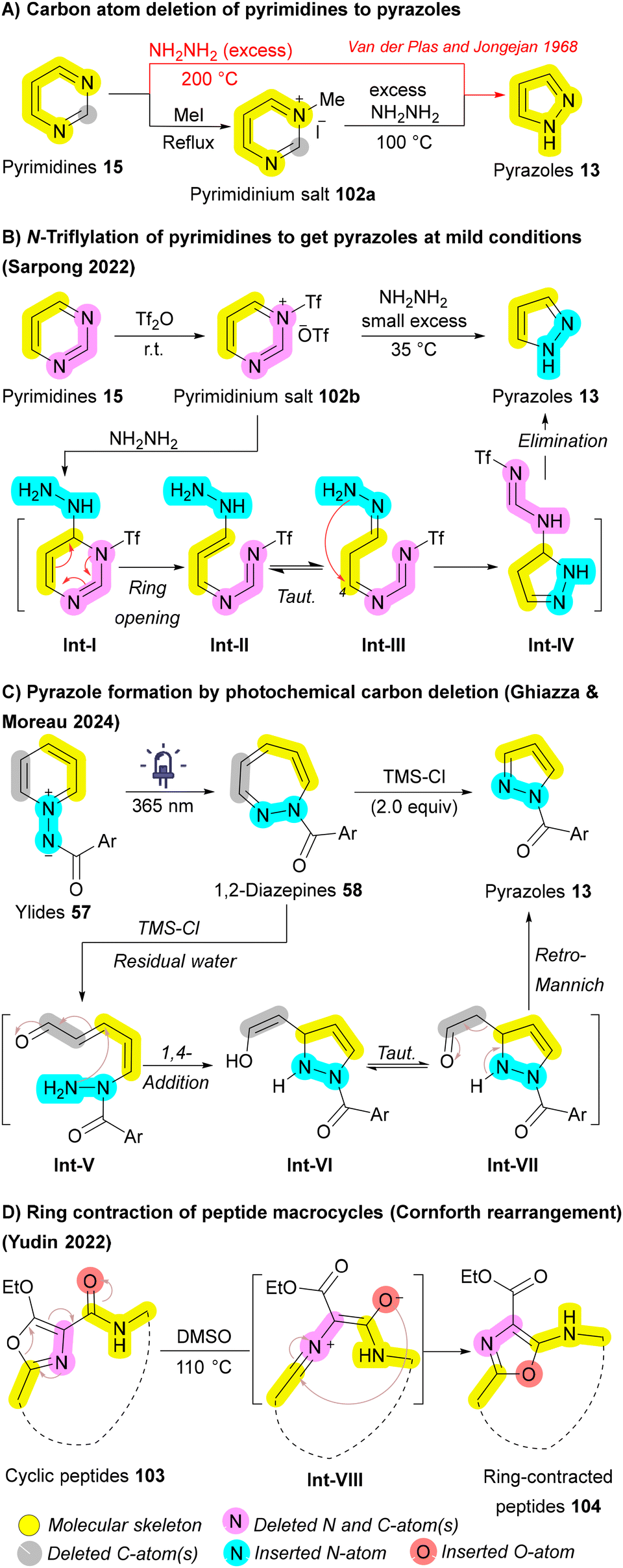 | ||
| Scheme 15 Carbon atom deletion of azaarenes (pyrimidines, pyridinium ylides, and macrocycles). | ||
Another skeletal editing approach for generating pyrazoles 13 was developed in 2024 by Ghiazza and Moreau. While investigating a photochemical method for the ring expansion of pyridinium ylides 57 into 1,2-diazepines 58 (Scheme 6D), they serendipitously discovered a concurrent ring contraction when excess TMS-Cl was applied alongside light, yielding pyrazoles 13. The authors proposed a mechanism in which the Lewis acid TMS-Cl, combined with the residual water present in the medium, facilitates the opening of the 1,2-diazepine ring 58 affording Int-V. Subsequently, a 1,4-addition of the nucleophilic amino group, followed by a retro-Mannich reaction, leads to the formation of the pyrazole ring 13, with the restoration of aromaticity serving as the driving force of the sequence (Scheme 15C).87 Ring contraction in macrocycles, such as cyclic peptides 103, has also been achieved by Yudin et al. through the Cornforth rearrangement. The mechanism involves the formation of a nitrilium intermediate Int-VIII through the opening of the oxazole ring, followed by nucleophilic attack from the adjacent carbonyl group on the nitrilium ion, resulting in the reformation of the oxazole moiety. The final ring-contracted peptide macrocycle 104 exhibited a conformational change, creating more space for studies related to conformation (Scheme 15D).144
 | ||
| Scheme 16 Ring contraction strategies through single nitrogen-atom deletion. | ||
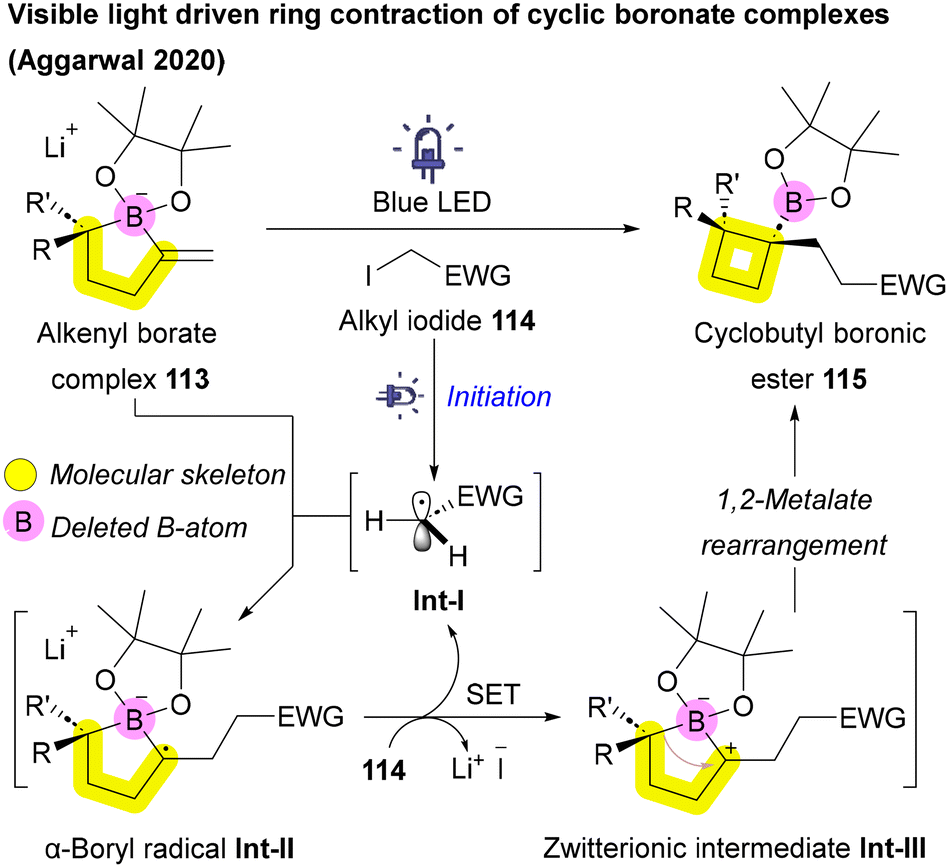 | ||
| Scheme 17 Ring contraction through boron-atom deletion. | ||
2.3. Transmutation strategies through atom exchange
Transmutation in skeletal editing aims to maintain the structural integrity of a ring system while swapping one or more atoms for others, which is especially valuable in medicinal chemistry.4 Achieving these transformations enables a more direct examination of how subtle changes—such as substituting nitrogen for carbon—affect a molecule's biological activity without altering its core shape.158 This process can be further subdivided based on the nature of the atoms being exchanged. | ||
| Scheme 18 Carbon-to-nitrogen (C-to-N) single-atom transmutation of pyridines via nitrene internalization. | ||
Levin et al. extended this expansion/contraction approach to convert quinolines 12a into quinazolines 16. Quinolines 12a were initially transformed into quinoline N-oxides 99, which under LED light (390 nm) undergo rearrangement to 3,1-benzoxazepine intermediates Int-I. Subsequent treatment with ammonium carbamate as a nitrogen source, combined with oxidative conditions (O3 and pyridine), yields an intermediate with two carbonyl termini Int-II. The carboxylate group then acts as a leaving group, allowing the nitrogen source (ammonia) to react with the imidic anhydride, ultimately forming quinazoline 16 (Scheme 19A).164 Xu and Wei applied an iron-mediated ring expansion/contraction strategy for C-to-N atom exchange in arenols 119a. Arenols undergo the addition of nucleophile, ring opening, and ring closing (ANRORC) mechanism. Bromination using N-bromosuccinimide (NBS) produces a brominated ketone intermediate Int-III, which converts to an azido ketone via N(n-Bu)4N3 giving intermediate Int-IV. A 1,2-aryl migration initiates ring expansion affording metal-nitrene intermediate Int-V then, forming an N,O-hemiketal Int-VI upon OH nucleophilic addition. Ring opening produces an amino-ketone intermediate Int-VII, which cyclizes and dehydrates to yield quinolines 12b (Scheme 19B).165 In a parallel strategy, the Wei group employed Cu-catalyzed ring expansion to generate benzo[b]azepines 38l from arenols 119b. This approach was further adapted to achieve a subsequent ring contraction, facilitating a one-carbon-to-nitrogen exchange. Benzazepine 38l, in the presence of mCPBA, forms an oxaziridine intermediate Int-X that undergoes cleavage of N–O bond affording radical intermediate Int-XI. Rearrangement of this radical intermediate Int-XI then produces the desired N-heterocycle 120b (Scheme 19C).100 Wang and Luan developed a silver-catalyzed aminative dearomatization strategy for transmutation of naphthols affording the corresponding isoquinolines.101 Another notable approach introduced by Hrobárik et al. involves the silver-mediated synthesis of benzo[1,2,3]thiadiazoles 124 from benzothiazol-2(3H)-ones 122 and 2-halobenzothiazoles 121. This reaction involves the formation of NO+ facilitated by Ag+, which, through N-nitrosation, converts 2-hydroxybenzothiazole 123 into N-nitrosated benzothiazol-2(3H)-one Int-XIII. Further interaction with Ag+ and NO2− initiates ring opening, followed by ring closure through a nucleophilic sulfur attack on the diazo intermediate Int-XVII, resulting in the formation of the isothiadiazole ring in benzo[1,2,3]thiadiazole 124 (Scheme 19D).166
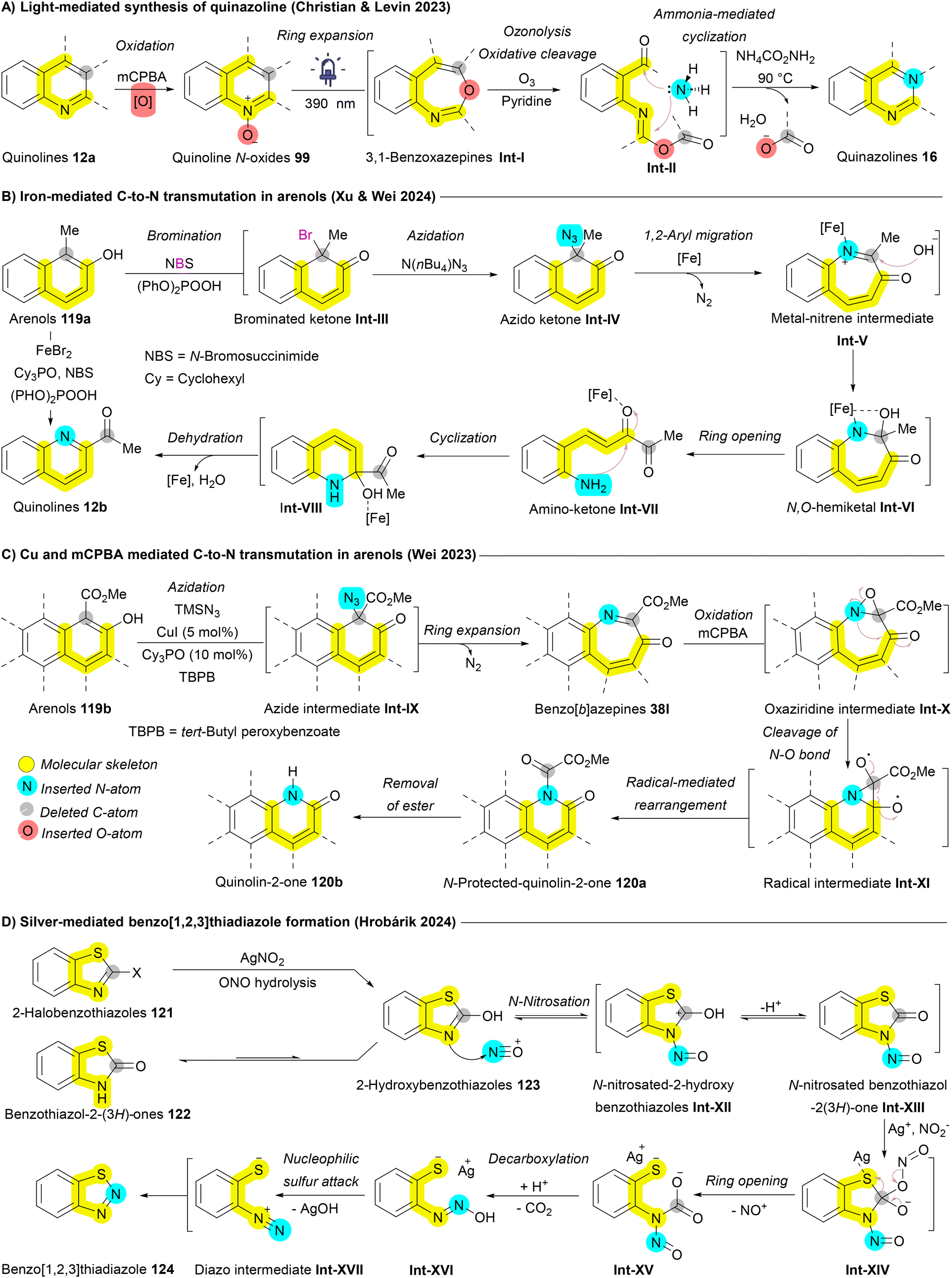 | ||
| Scheme 19 Carbon-to-nitrogen single-atom transmutation of arenes and heteroarenes via expansion/contraction sequence. | ||
 | ||
| Scheme 20 Pyridine to benzene rearrangements (N-to-C transmutation) achieved through Zincke pyridinium chemistry. | ||
In 2024, a great development by Paton and McNally who achieved a deconstruction–reconstruction process to convert pyrimidines 15 into pyridines 4a (Scheme 21A).176 After generating the pyrimidinium salt 133b, pyrrolidine 126d acts as a nucleophile to cleave the pyrimidine, yielding vinamidinium salts 134. Employing Marcoux's protocol with the lithium enolate of commercially available acetyltrimethylsilane 135, followed by a combination of ammonium acetate (NH4OAc) and acetic acid (AcOH), the vinamidinium salts 134 undergo reaction with ketone-derived enolates and ammonium salts, forming substituted pyridines 4a through intermediate Int-V. Under the reaction conditions, the C–Si bond is cleaved after pyridine formation (Scheme 21A).176 Recently, Studer and coworkers described a two-atom switch approach using cycloaddition chemistry. The mechanism involves an initial dearomative cyclization of the pyridine ring 4b, forming an electron-rich diene Int-VI within an oxazino-pyridine structure. In the presence of electron-rich dienophiles, such as alkynes 136, the oxazino-pyridine Int-VI undergoes a [4 + 2] cycloaddition, yielding a bridged intermediate Int-VII. This intermediate Int-VII then retrocyclizes and rearomatizes, affording the target benzene ring 132 (Scheme 21B).177 Boswell et al. introduced a similar two-atom switch approach inspired by water-displacement for the transformation of pyridines to benzenes. A sequence of 1,2-addition, [4 + 2] cycloaddition, and retero-[4 + 2] with alkyne moiety enabled the late stage diversification of various substituted pyridines.178 When such cycloaddition chemistry employ five-membered heterocycles as the 4π-component, two new atoms are installed from the dienophile at the expense of one ring-atom of the substrate achieving ring expansion with atom exchange.6 Specific examples include isoxazoles into pyridines through inverse electron-demand Diels–Alder reaction (IEDDA),179 pyrroles to benzene through Diels–Alder reaction (DA),180 and benzisoxazoles to quinazolines.181 In 2024, Zhou and coworkers introduced the first example of swapping from an S atom to C–O pair atoms, enabling the direct transformation of benzodithiol-3-ones 137 into benzo[d][1,3]oxathiin-4-ones 138 (Scheme 21C). The reaction proceeds in the presence of 1,2-bis(diphenylphosphino)ethane (DPPE) to enable phosphine mediated S–S bond cleavage affording Int-VIII that can undergo nucleophilic addition with formaldehyde giving Int-IX followed by cyclization to the corresponding benzo[d][1,3]oxathiin-4-ones 138.182
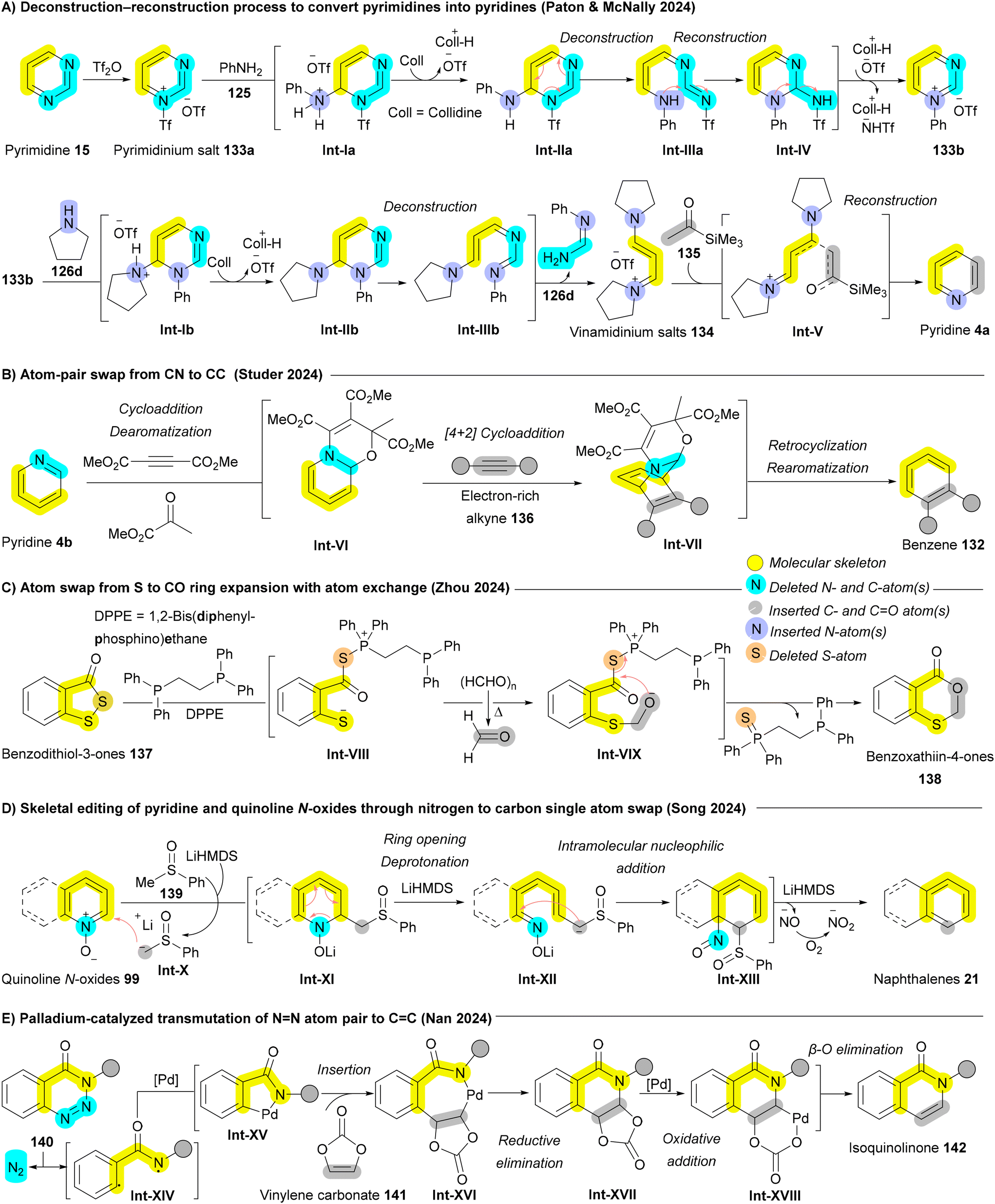 | ||
| Scheme 21 Various strategies for nitrogen -to-carbon (N-to-C) transmutation of heteroarenes. | ||
In 1977, Hamada and Takeuchi discovered that benzo[h]quinoline N-oxide could be transformed into anthracene using DMSO as a carbon source; however, this strategy proved ineffective for broader substrates.183 Recently, the Song and Sorensen groups enhanced this approach by employing n-butyllithium or LiHMDS, significantly increasing conversion efficiency and broadening the substrate scope.184,185 In this method, phenyl methyl sulfoxide (PhSOMe) 139 with LiHMDS generates a methylsulfinyl carbanion Int-X, which acts as a nucleophile for addition, forming an intermediate Int-XI that subsequently undergoes ring opening. Following deprotonation and intramolecular nucleophilic addition, ring closure occurs with nitrite release as NO−, yielding the desired naphthalene 21 (Scheme 21D).184 In 2024, Nan and coworkers reported a rare example of palladium-catalyzed atom-pair exchange in benzotriazinones 140, converting NN to CC and yielding isoquinolinones 142. Benzotriazinone 140 first undergoes in situ denitrogenation, generating radical species Int-XIV, which then forms a five-membered cyclopalladium intermediate, Int-XV. Migratory insertion of vinylene carbonate 141 produces a seven-membered cyclopalladium intermediate, Int-XVI, followed by reductive elimination to afford tricyclic intermediate Int-XVII. This intermediate Int-XVII then undergoes a sequence of oxidative addition and β-O elimination, giving isoquinolinone derivatives 142 (Scheme 21E).186
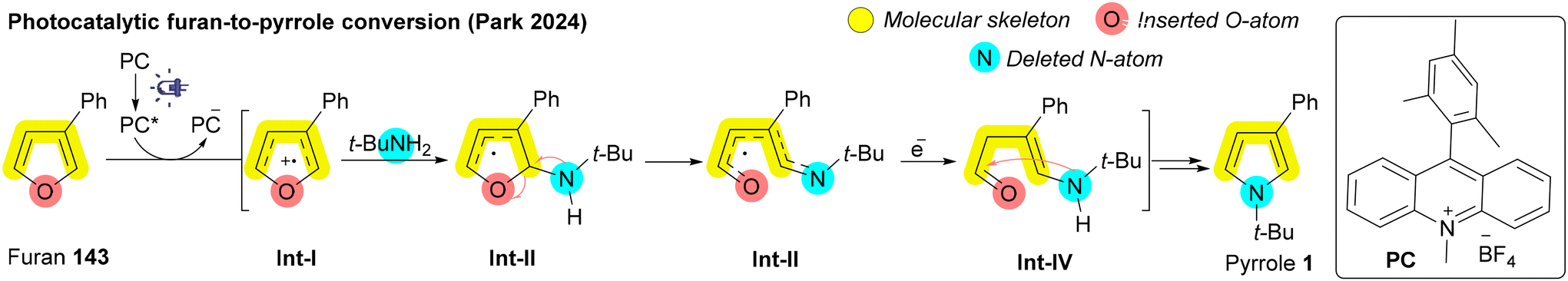 | ||
| Scheme 22 Oxygen-to-Nitrogen (O-to-N) transmutation. | ||
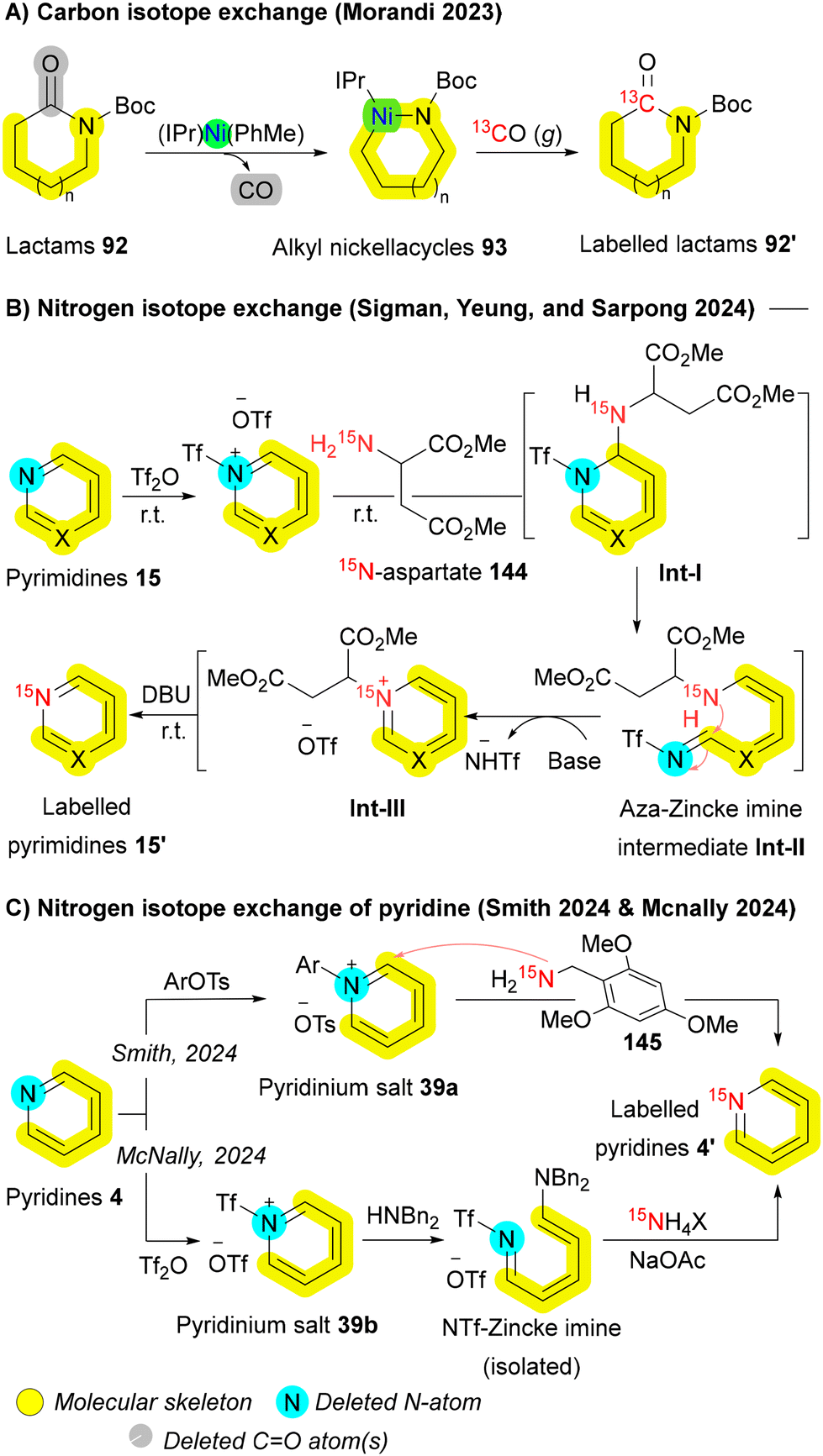 | ||
| Scheme 23 12C-to-13C and 14N-to-15N isotopic exchange via skeletal editing. | ||
Due to nitrogen-15's extensive applications in pharmaceuticals—spanning applications from biomolecular NMR,192,193 structural analysis,194 and mechanism elucidation117,195,196 to bioimaging spin hyperpolarization (e.g., SABRE-SHEATH)197—an efficient 14N to 15N exchange is highly valuable. In 2024, three groups independently reported similar approaches for 14N to 15N exchange in various azaheterocycles198–200 by adapting the Zincke reaction.168 Sigman, Yeung and Sarpong established a protocol that enabling direct 14N → 15N single atom transmutation across diverse nitrogen heteroaromatics, particularly pyrimidines 15, using an easily prepared 15N-enriched aspartate derived diester 144. Central to this transformation is a low-temperature N-triflylation step that activates the heterocyclic nitrogen, allowing a room-temperature Zincke-type ring-opening and ring-closing sequence mediated by 15N-aspartate nucleophile 144. This sequence yields an N-succinyl intermediate Int-III with the nitrogen isotopically swapped. In situ elimination of the succinyl group as fumarate or maleate reveals the labeled heterocycle 15′ (Scheme 23B).198 Similarly, Smith and McNally applied a 15N labeling strategy to pyridines 4. Upon activation via N-triflylation or N-arylation, pyridinium salts 39 undergo ANRORC, ultimately producing labeled pyridines 4′ (Scheme 23C).199,200
2.4. Stereochemical editing
Stereochemical editing is a cutting-edge approach in organic synthesis that enables the direct modification of stereocenters, allowing for the adjustment of relative stereochemistry at a late stage. This technique decouples stereochemistry-defining steps from the main structural assembly, enhancing flexibility and efficiency in generating diverse isomers from a single compound. By utilizing photocatalysis, stereochemical editing can effectively overcome traditional thermodynamic limitations, making it a powerful tool in synthetic chemistry.201,202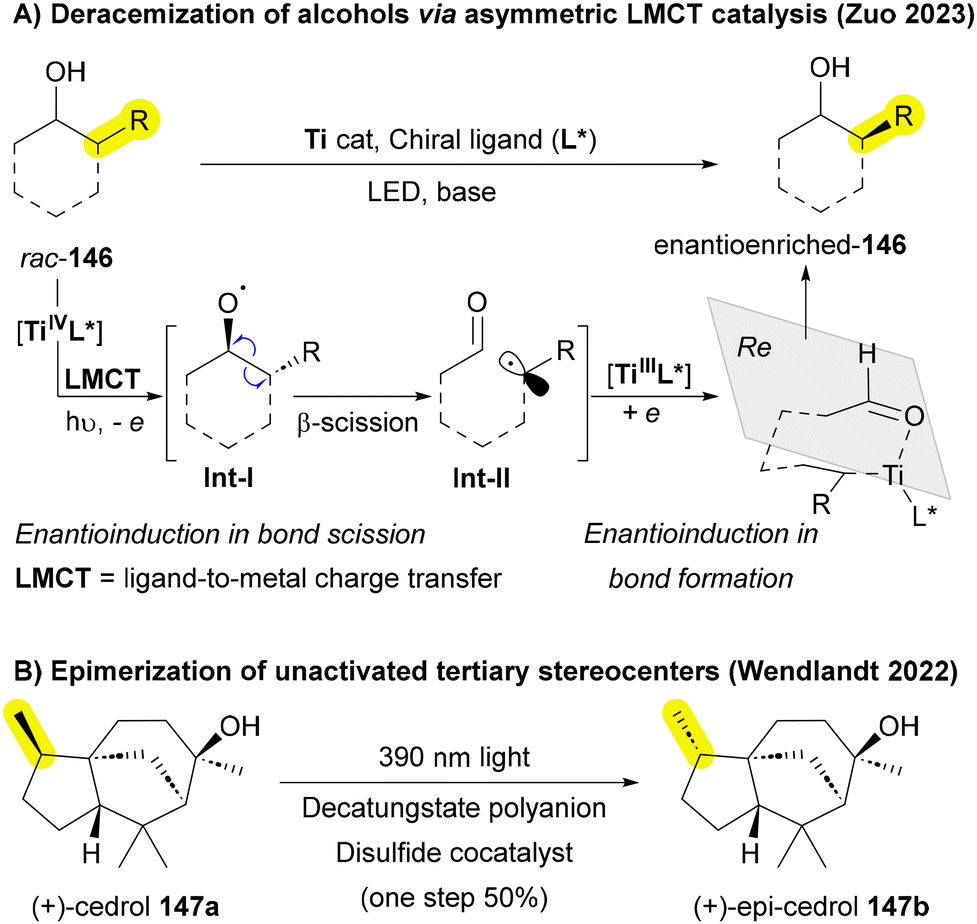 | ||
| Scheme 24 Stereochemical editing. | ||
3. Conclusions and perspectives
The field of skeletal editing has grown rapidly, with impressive advances that allow atom-level modifications in molecular frameworks—offering transformative applications in pharmaceuticals and beyond. Through strategies like insertion, deletion, and transmutation, chemists have successfully reshaped molecular scaffolds with increasing precision and sustainability.4–6 Although significant progress has been made, achieving a level of maturity that enables context-independent deployment remains a challenge. Several limitations hinder further progress and need to be overcome before the field can reach its full potential, and can be summarized as follows:1. Generality: the limited availability of methods that enable diverse transformations by simply altering the insertive agent or reactive species highlights a significant gap in the field of skeletal editing.101 To advance this area of research, it is essential to develop more robust strategies that can reliably achieve various modifications with high selectivity. As discussed in this review, many existing methods share similar underlying mechanisms, suggesting that there is potential for expanding their applicability to meet these objectives.21,56,88
2. Selectivity: high chemo-, regio-, and stereoselectivity remains a significant challenge in skeletal editing, hindering the field's progress. Many current methods exhibit non-selective conditions, resulting in lower yields and unwanted byproducts. Furthermore, the literature on asymmetric skeletal editing is limited, with very few reported examples.75 Notably, Wendlandt and Zuo have made significant contributions to the area of stereochemical editing, highlighting its potential to advance the field.207,208,212 This lack of selectivity complicates product purification and restricts the practical application of these approaches in the synthesis of complex molecules that require high selectivity.213,214
3. Diversity: current methods predominantly focus on carbon and nitrogen, which can be fully understood in the context of drug development. However, there is a need to develop efficient techniques that incorporate a broader range of heteroatoms.130 Expanding diversity will not only enhance the functionality and complexity of synthesized scaffolds but also advance applications in materials science and optoelectronics enabling atom doping and allows for direct comparative studies without de novo synthesis.14
4. Efficiency and complexity: most existing methods for skeletal editing rely on stepwise sequences, which could be enhanced by developing streamlined, single-step processes. Furthermore, many studies focus on simple monocyclic structures lacking other functionalities. The true potential of skeletal editing will be realized when it is applied to the synthesis of complex materials, challenging molecular skeletons, or natural products, as seen in some reports from the Sarpong group,58 and Ng group.188
5. Sustainability: although skeletal editing aligns with sustainability objectives by conserving resources, effort, and time, chemists often compromise these advantages when attempting complex transformations. As a result, many reported methods exhibit low atom economy, limited scalability, and reliance on expensive or hard-to-source metal reagents. Recently, many research groups have recognized these issues and are focusing on more sustainable approaches, employing more practical methods, and greener alternatives such as electrochemistry.115,116
Addressing these limitations and exploring underdeveloped areas is vital for unlocking the full potential of skeletal editing. Future efforts should prioritize ambitious reactions, such as the migration of heteroatoms and enhanced transmutation methods, which would enable straightforward diversification of drug candidates after lead identification. Collaboration between medicinal and synthetic chemists is essential for translating these methods into novel drug designs and structure-oriented approaches, including drug-oriented rational molecular editing (DORME) and structure-guided rational molecular editing (SGRME).9 By tackling these challenges and exploring new avenues, skeletal editing can reach full maturity as a powerful tool for remodelling molecular frameworks with atom-level precision.
Author contributions
R. S. and M. S. H. S. wrote and drafted the manuscript; M. S. H. S. reviewed and edited the manuscript; M. A. and S. T. provided guidance and supervision throughout the work. All authors have approved the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. S. is grateful to Japanese government (MEXT) scholarship for their financial support. The authors acknowledge the support from JSPS KAKENHI Grants 21A204, 21H05217, 22KK0073, 22K06502 and 24K17681 from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), the Japan Society for the Promotion of Science (JSPS), and Core Research for Evolutionary Science and Technology (JST CREST) (No. JPMJCR20R1).References
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, The importance of synthetic chemistry in the pharmaceutical industry, Science, 2019, 363, eaat0805 CrossRef CAS.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Organic synthesis provides opportunities to transform drug discovery, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- Y. Xiao, M. Zhou, M. Zeng and L. Fu, Atomic-scale structural modification of 2D materials, Adv. Sci., 2019, 6, 1801501 CrossRef.
- J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Single-atom logic for heterocycle editing, Nat. Synth., 2022, 1, 352–364 CrossRef CAS PubMed.
- C. Zippel, J. Seibert and S. Bräse, Skeletal editing—nitrogen deletion of secondary amines by anomeric amide reagents, Angew. Chem., Int. Ed., 2021, 60, 19522–19524 CrossRef CAS PubMed.
- B. W. Joynson and L. T. Ball, Skeletal editing: interconversion of arenes and heteroarenes, Helv. Chim. Acta, 2023, 106, e202200182 CrossRef CAS.
- F.-S. Li, X.-Y. Zou, T.-Q. Hu, Q. Sun, Z. Xu, B. Zhou and L.-W. Ye, Asymmetric one-carbon ring expansion of diverse N-heterocycles via copper-catalyzed diyne cyclization, Sci. Adv., 2024, 10, eadq7767 CrossRef CAS PubMed.
- T. Huo, X. Zhao, Z. Cheng, J. Wei, M. Zhu, X. Dou and N. Jiao, Late-stage modification of bioactive compounds: improving druggability through efficient molecular editing, Acta Pharm. Sin. B, 2024, 14, 1030–1076 CrossRef CAS PubMed.
- C. Ma, C. W. Lindsley, J. Chang and B. Yu, Rational molecular editing: a new paradigm in drug discovery, J. Med. Chem., 2024, 67, 11459–11466 CrossRef CAS PubMed.
- E.-Q. Li, C. W. Lindsley, J. Chang and B. Yu, Molecular skeleton editing for new drug discovery, J. Med. Chem., 2024, 67, 13509–13511 CrossRef CAS.
- C. Hui, Z. Wang, S. Wang and C. Xu, Molecular editing in natural product synthesis, Org. Chem. Front., 2022, 9, 1451–1457 RSC.
- R. A. Ditzler, A. J. King, S. E. Towell, M. Ratushnyy and A. V. Zhukhovitskiy, Editing of polymer backbones, Nat. Rev. Chem., 2023, 7, 600–615 CrossRef PubMed.
- M. S. H. Salem, R. Sharma, S. Suzuki, Y. Imai, M. Arisawa and S. Takizawa, Impact of helical elongation of symmetric oxa[n]helicenes on their structural, photophysical, and chiroptical characteristics, Chirality, 2024, 36, e23673 Search PubMed.
- C. Hu, D. Liu, Y. Xiao and L. Dai, Functionalization of graphene materials by heteroatom-doping for energy conversion and storage, Prog. Nat. Sci.: Mater. Int., 2018, 28, 121–132 CrossRef CAS.
- M. Gauthier, J. B. Whittingham, A. Hasija, D. J. Tetlow and D. A. Leigh, Skeletal editing of mechanically interlocked molecules: nitrogen atom deletion from crown ether-dibenzylammonium rotaxanes, J. Am. Chem. Soc., 2024, 146, 29496–29502 Search PubMed.
- E. Buchner and T. Curtius, Ueber die einwirkung von diazoessigäther auf aromatische kohlenwasserstoffe, Ber. Dtsch. Chem. Ges., 1885, 18, 2377–2379 CrossRef.
- G. Ciamician and M. Dennstedt, Ueber die einwirkung des chloroforms auf die kaliumverbindung pyrrols, Ber. Dtsch. Chem. Ges., 1881, 14, 1153–1163 CrossRef.
- A. Baeyer and V. Villiger, Einwirkung des caro'schen reagens auf ketone, Ber. Dtsch. Chem. Ges., 1899, 32, 3625–3633 CrossRef.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity, Science, 2012, 337, 816–821 CrossRef CAS.
- P. Zhang, L. Hua, T. Takahashi, S. Jin and Q. Wang, Recent advances in the dearomative skeletal editing of mono-azaarenes, Synthesis, 2024, 55–70 CAS.
- Z. Liu, P. Sivaguru, Y. Ning, Y. Wu and X. Bi, Skeletal editing of (hetero) arenes using carbenes, Chem. – Eur. J., 2023, 29, e202301227 CrossRef CAS.
- X. Li and Z. Xu, Skeletal editing: ring insertion for direct access to heterocycles, Molecules, 2024, 29, 1920 CrossRef CAS PubMed.
- J. Huang, F. Liu, X. Wu, J.-Q. Chen and J. Wu, Recent advances in the reactions of silacyclobutanes and their applications, Org. Chem. Front., 2022, 9, 2840–2855 RSC.
- T. Zhang and H. Feng, Skeletal editing of isatins for heterocycle molecular diversity, Chem. Rec., 2024, 24, e202400024 CrossRef CAS PubMed.
- S. Manna, J. K. Laha, A. Gupta and P. Bhatti, Skeletal editing by hypervalent iodine mediated nitrogen insertion, Chem. – Eur. J., 2024, 30, e202401993 Search PubMed.
- F. Liu, L. Anand and M. Szostak, Diversification of indoles and pyrroles by molecular editing: new frontiers in heterocycle–to–heterocycle transmutation, Chem. – Eur. J., 2023, 29, e202300096 CrossRef CAS PubMed.
- J. Luo, Q. Zhou, Z. Xu, K. Houk and K. Zheng, Photochemical skeletal editing of pyridines to bicyclic pyrazolines and pyrazoles, J. Am. Chem. Soc., 2024, 146, 21389–21400 CrossRef PubMed.
- T. Yuan and L. Shi, Recent advances in carbon atom addition for ring-expanding single-atom skeletal editing, Org. Chem. Front., 2024, 11, 7318–7332 Search PubMed.
- P. Dowd and S. C. Choi, A new tributyltin hydride-based rearrangement of bromomethyl β-keto esters. A synthetically useful ring expansion to γ-keto esters, J. Am. Chem. Soc., 1987, 109, 3493–3494 CrossRef CAS.
- A. L. Beckwith, D. M. O'Shea, S. Gerba and S. W. Westwood, Cyano or acyl group migration by consecutive homolytic addition and β-fission, J. Chem. Soc., Chem. Commun., 1987, 1987, 666–667 RSC.
- D. Ma, B. S. Martin, K. S. Gallagher, T. Saito and M. Dai, One-carbon insertion and polarity inversion enabled a pyrrole strategy to the total syntheses of pyridine-containing Lycopodium alkaloids: complanadine A and lycodine, J. Am. Chem. Soc., 2021, 143, 16383–16387 CrossRef CAS PubMed.
- M. Mortén, M. Hennum and T. Bonge-Hansen, Synthesis of quinoline-3-carboxylates by a Rh(II)-catalyzed cyclopropanation-ring expansion reaction of indoles with halodiazoacetates, Beilstein J. Org. Chem., 2015, 11, 1944–1949 CrossRef PubMed.
- F.-P. Wu, J. L. Tyler, C. G. Daniliuc and F. Glorius, Atomic carbon equivalent: design and application to diversity-generating skeletal editing from indoles to 3-functionalized quinolines, ACS Catal., 2024, 14, 13343–13351 CrossRef CAS.
- B. D. Dherange, P. Q. Kelly, J. P. Liles, M. S. Sigman and M. D. Levin, Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines, J. Am. Chem. Soc., 2021, 143, 11337–11344 CrossRef CAS PubMed.
- E. E. Hyland, P. Q. Kelly, A. M. McKillop, B. D. Dherange and M. D. Levin, Unified access to pyrimidines and quinazolines enabled by N–N cleaving carbon atom insertion, J. Am. Chem. Soc., 2022, 144, 19258–19264 CrossRef CAS PubMed.
- B. W. Joynson, G. R. Cumming and L. T. Ball, Photochemically mediated ring expansion of indoles and pyrroles with chlorodiazirines: synthetic methodology and thermal hazard assessment, Angew. Chem., Int. Ed., 2023, 62, e202305081 CrossRef.
- H. Guo, S. Qiu and P. Xu, One-carbon ring expansion of indoles and pyrroles: a straightforward access to 3-fluorinated quinolines and pyridines, Angew. Chem., Int. Ed., 2024, 63, e202317104 CrossRef CAS PubMed.
- Y. Zhou, F. Chen, Z. Li, J. Dong, J. Li, B. Zhang and Q. Song, Single-atom skeletal editing of 2H-indazoles enabled by difluorocarbene, Sci. China: Chem., 2023, 66, 1975–1981 CrossRef CAS.
- C. Li, L. Chen, H. Wang, Z. Yan, B. Lyu, W. Lyu, C. Jiang, D. Lu, J. Li and N. Jiao, C−F Bond insertion into indoles with CHBr2F: an efficient method to synthesize fluorinated quinolines and quinolones, Chin. J. Chem., 2024, 42, 1128–1132 CrossRef CAS.
- L. Li, Y. Ning, H. Chen, Y. Ning, P. Sivaguru, P. Liao, Q. Zhu, Y. Ji, G. de Ruiter and X. Bi, Dearomative insertion of fluoroalkyl carbenes into azoles leading to fluoroalkyl heterocycles with a quaternary center, Angew. Chem., Int. Ed., 2024, 63, e202313807 CrossRef CAS.
- L. Li, H. Chen, M. Liu, Q. Zhu, H. Zhang, G. de Ruiter and X. Bi, Silver-catalyzed dearomative skeletal editing of indazoles by donor carbene insertion, Chem. – Eur. J., 2024, 30, e202304227 CrossRef CAS PubMed.
- Y. Ning, H. Chen, Y. Ning, J. Zhang and X. Bi, Rhodium-catalyzed one-carbon ring expansion of aziridines with vinyl-N-triftosylhydrazones for the synthesis of 2-vinyl azetidines, Angew. Chem., Int. Ed., 2024, 63, e202318072 CrossRef CAS PubMed.
- Y. Yang, Q. Song, P. Sivaguru, Z. Liu, D. Shi, T. Tian, G. de Ruiter and X. Bi, Controllable skeletal and peripheral editing of pyrroles with vinylcarbenes, Angew. Chem., Int. Ed., 2024, 63, e202401359 CrossRef CAS PubMed.
- S. Liu, Y. Yang, Q. Song, Z. Liu, Y. Lu, Z. Wang, P. Sivaguru and X. Bi, Tunable molecular editing of indoles with fluoroalkyl carbenes, Nat. Chem., 2024, 16, 988–997 CrossRef CAS PubMed.
- L. Li, H. Chen, X. Zhang, K. Murali, Q. Zhu, M. Liu, H. Zhang, V. Nenajdenko and X. Bi, Silver-catalyzed single-carbon insertion of indoles with acetophenone N-triftosylhydrazones, Org. Lett., 2024, 26, 7207–7211 CrossRef CAS PubMed.
- P. Sivaguru, Y. Pan, N. Wang and X. Bi, Who is who in the carbene chemistry of N-sulfonyl hydrazones, Chin. J. Chem., 2024, 41, 2071–2108 CrossRef.
- M. Tsuda, T. Morita, Y. Morita, J. Takaya and H. Nakamura, Methylene insertion into nitrogen-heteroatom single bonds of 1,2-azoles via a zinc carbenoid: an alternative tool for skeletal editing, Adv. Sci., 2024, 11, 2307563 CrossRef CAS PubMed.
- M. Tsuda and H. Nakamura, Zinc carbenoid-promoted methylene insertion in saturated heterocycles: mechanistic insights and reactivity profiles, Synthesis, 2025, 167–175 CAS.
- M. Qi, M. Suleman, J. Fan, P. Lu and Y. Wang, Cu(I)-catalyzed synthesis of spiro [isoquinoline-4,2′-[1,3] oxazin]-3-ones via ring expansion reactions of isoxazoles with 4-diazoisoquinolin-3-ones, Tetrahedron, 2022, 128, 133092 CrossRef CAS.
- C. Han, W. Wu, Z. Chen and S. Pu, Rhodium-catalyzed [5+1]-cycloaddition reactions to spiro-benzo[e,][1,3] oxazineindoline imines, Asian J. Org. Chem., 2019, 8, 1385–1389 CrossRef CAS.
- J. S. Lamb, F. Koyama, N. Suzuki and Y. Suzuki, Carbon atom insertion into N-heterocyclic carbenes to yield 3,4-dihydroquinoxalin-2(1H)-ones, Org. Chem. Front., 2024, 11, 277–283 RSC.
- H. L. Schmitt, D. Martymianov, O. Green, T. Delcaillau, Y. S. P. Kim and B. Morandi, Regiodivergent ring-expansion of oxindoles to quinolinones, J. Am. Chem. Soc., 2024, 146, 4301–4308 CrossRef CAS PubMed.
- Y.-L. Li, N. Yu, K.-C. He, Y.-Q. Zhou, W.-H. Zheng, K. Jiang and Y. Wei, Skeletal transformation of oxindoles into quinolinones enabled by synergistic copper/iminium catalysis, J. Org. Chem., 2023, 88, 4863–4874 CrossRef CAS.
- R. Tran, D. K. Brownsey, L. O'Sullivan, C. M. Brandow, E. S. Chang, W. Zhou, K. V. Patel, E. Gorobets and D. J. Derksen, Leveraging pyrazolium ylide reactivity to access indolizine and 1,2-dihydropyrimidine derivatives, Chem. – Eur. J., 2024, 30, e202400421 CrossRef CAS PubMed.
- K. Yang, Q. Li, D. Yuan, Y. Luo, C. Qi, Z.-Y. Li, B. Li and X. Sun, Transition-metal-free skeletal editing of benzoisothiazol-3-ones to 2,3-dihydrobenzothiazin-4-ones via a single-carbon insertion, Org. Chem. Front., 2025, 12 10.1039/D4QO01714E.
- G. Ma, K.-F. Wei, M. Song, Y.-L. Dang, Y. Yue, B. Han, H. Su and W.-B. Shen, Recent advances in transition-metal-catalyzed Büchner reaction of alkynes, Org. Biomol. Chem., 2023, 21, 5150–5157 RSC.
- P. Piacentini, T. W. Bingham and D. Sarlah, Dearomative ring expansion of polycyclic arenes, Angew. Chem., Int. Ed., 2022, 61, e202208014 CrossRef CAS PubMed.
- S. Wiesler, G. Sennari, M. V. Popescu, K. E. Gardner, K. Aida, R. S. Paton and R. Sarpong, Late-stage benzenoid-to-troponoid skeletal modification of the cephalotanes exemplified by the total synthesis of harringtonolide, Nat. Commun., 2024, 15, 4125 CrossRef CAS PubMed.
- J. Han, Y. Fan, X. Yang, Y. Zhu, X. Zhang, F. Zhang, G. Hao and Y. Jiang, Synthesis of functionalized cycloheptadienones starting from phenols and using a rhodium/boron asymmetric catalytic system, Angew. Chem., Int. Ed., 2024, 63, e202416468 Search PubMed.
- Q. Zeng, K. Dong, J. Huang, L. Qiu and X. Xu, Copper-catalyzed carbene/alkyne metathesis terminated with the Buchner reaction: synthesis of dihydrocyclohepta[b] indoles, Org. Biomol. Chem., 2019, 17, 2326–2330 RSC.
- D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Recent progress on donor and donor–donor carbenes, Chem. Soc. Rev., 2020, 49, 908–950 RSC.
- N. P. T. Thanh, M. Tone, H. Inoue, I. Fujisawa and S. Iwasa, Highly stereoselective intramolecular Buchner reaction of diazoacetamides catalyzed by a Ru(II)–Pheox complex, Chem. Commun., 2019, 55, 13398–13401 RSC.
- T. Ito, S. Harada, H. Homma, H. Takenaka, S. Hirose and T. Nemoto, Asymmetric intramolecular dearomatization of nonactivated arenes with ynamides for rapid assembly of fused ring system under silver catalysis, J. Am. Chem. Soc., 2021, 143, 604–611 CrossRef CAS PubMed.
- B. Darses, P. Maldivi, C. Philouze, P. Dauban and J.-F. Poisson, Asymmetric intramolecular Buchner reaction: from high stereoselectivity to coexistence of norcaradiene, cycloheptatriene, and an intermediate form in the solid state, Org. Lett., 2021, 23, 300–304 CrossRef CAS.
- D. Zhu, T. Cao, K. Chen and S. Zhu, Rh2(II)-catalyzed enantioselective intramolecular Büchner reaction and aromatic substitution of donor–donor carbenes, Chem. Sci., 2022, 13, 1992–2000 RSC.
- D.-F. Yuan, Z.-C. Wang, R.-S. Geng, G.-Y. Ren, J. S. Wright, S.-F. Ni, M. Li, L.-R. Wen and L.-B. Zhang, Hypervalent iodine promoted the synthesis of cycloheptatrienes and cyclopropanes, Chem. Sci., 2022, 13, 478–485 RSC.
- S. Stockerl, T. Danelzik, D. G. Piekarski and O. G. Mancheño, Mild, metal-free oxidative ring-expansion approach for the synthesis of benzo [b] azepines, Org. Lett., 2019, 21, 4535–4539 CrossRef CAS.
- M. J. Mailloux, G. S. Fleming, S. S. Kumta and A. B. Beeler, Unified synthesis of azepines by visible-light-mediated dearomative ring expansion of aromatic N-ylides, Org. Lett., 2021, 23, 525–529 CrossRef CAS PubMed.
- J. Kim and E. J. Yoo, Catalytic ring expansion of activated heteroarenes enabled by regioselective dearomatization, Org. Lett., 2021, 23, 4256–4260 CrossRef CAS PubMed.
- B. Niu, Q. Nie, B. Huang and M. Cai, Heterogeneous gold(I)-catalyzed oxidative ring expansion of 2-alkynyl-1,2-dihydropyridines or -quinolines towards functionalized azepines or benzazepines, Adv. Synth. Catal., 2019, 361, 4065–4074 CrossRef CAS.
- D. C. Miller, R. G. Lal, L. A. Marchetti and F. H. Arnold, Biocatalytic one-carbon ring expansion of aziridines to azetidines via a highly enantioselective [1,2]-Stevens rearrangement, J. Am. Chem. Soc., 2022, 144, 4739–4745 CrossRef CAS PubMed.
- F.-P. Wu, C. C. Chintawar, R. Lalisse, P. Mukherjee, S. Dutta, J. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Ring expansion of indene by photoredox-enabled functionalized carbon-atom insertion, Nat. Catal., 2024, 7, 242–251 CrossRef CAS.
- X. Dong, Y. Shao, Z. Liu, X. Huang, X. S. Xue and Y. Chen, Radical 6-endo addition enables pyridine synthesis under metal-free conditions, Angew. Chem., Int. Ed., 2024, 63, e202410297 Search PubMed.
- F. Xie, Y. Chen, Y. Li, Z. Wang, J. Zhang and Y. Tang, Photo-induced two-carbon ring expansion of N-alkenyl lactams and N-alkenyl/phenyl benzoazetinones, Org. Chem. Front., 2023, 10, 928–935 RSC.
- R. K. Mallick, M. Žabka and J. Clayden, Benzo-fused nitrogen heterocycles by asymmetric ring expansion and stereochemically retentive re-contraction of cyclic ureas, Angew. Chem., Int. Ed., 2024, 63, e202318417 Search PubMed.
- S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Photochemical ring expansion reactions: synthesis of tetrahydrofuran derivatives and mechanism studies, Chem. Sci., 2019, 10, 10129–10134 RSC.
- C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, An update on the nitrogen heterocycle compositions and properties of US FDA-approved pharmaceuticals (2013–2023), J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS.
- L. Guy Donaruma and W. Z. Heldt, The Beckmann rearrangement, Org. React., 2004, 11, 1–156 Search PubMed.
- A. Wrobleski, T. C. Coombs, C. W. Huh, S. W. Li and J. Aubé, The Schmidt reaction, Org. React., 2004, 78, 1–320 Search PubMed.
- R. Liu, J. Wang, H. Wu, X. Quan, S. Wang, J. Guo, Y. Wang and H. Li, Stereocontrol in an intermolecular Schmidt reaction of equilibrating hydroxyalkyl allylic azides, Chem. Commun., 2024, 60, 4362–4365 RSC.
- M. Ong, M. Arnold, A. W. Walz and J. M. Wahl, Stereospecific nitrogen insertion using amino diphenylphosphinates: an aza-Baeyer–Villiger rearrangement, Org. Lett., 2022, 24, 6171–6175 CrossRef CAS PubMed.
- J. Sietmann, M. Ong, C. Mück-Lichtenfeld, C. G. Daniliuc and J. M. Wahl, Desymmetrization of prochiral cyclobutanones via nitrogen insertion: a concise route to chiral γ-lactams, Angew. Chem., Int. Ed., 2021, 60, 9719–9723 CrossRef CAS.
- Y. Fu, H. Liang, Y. Lu and S. Huang, Photoredox-enabled deconstructive [5+1] annulation approach to isoquinolones from indanones in water, Org. Lett., 2024, 26, 3043–3047 CrossRef CAS PubMed.
- C. Amber, L. T. Göttemann, R. T. Steele, T. M. Petitjean and R. Sarpong, Reductive amination of carbonyl C–C bonds enables formal nitrogen insertion, J. Org. Chem., 2024, 89, 17655–17663 Search PubMed.
- A. Sandvoß and J. M. Wahl, From cycloalkanols to heterocycles via nitrogen insertion, Org. Lett., 2023, 25, 5795–5799 CrossRef PubMed.
- Z. Zhang, Q. Li, Z. Cheng, N. Jiao and C. Zhang, Selective nitrogen insertion into aryl alkanes, Nat. Commun., 2024, 15, 6016 CrossRef CAS PubMed.
- E. Boudry, F. Bourdreux, J. Marrot, X. Moreau and C. Ghiazza, Dearomatization of pyridines: photochemical skeletal enlargement for the synthesis of 1, 2-diazepines, J. Am. Chem. Soc., 2024, 146, 2845–2854 CrossRef CAS PubMed.
- Y. A. Xu, S. H. Xiang, J. T. Che, Y. B. Wang and B. Tan, Skeletal editing of cyclic molecules using nitrenes, Chin. J. Chem., 2024, 42, 2656–2667 CrossRef CAS.
- R. Huisgen, D. Vossius and M. Appl, Die thermolyse des phenylazids in primären aminen; die konstitution des dibenzamils, Chem. Ber., 1958, 91, 1–12 CrossRef CAS.
- W. V. E. Doering and R. Odum, Ring enlargement in the photolysis of phenyl azide, Tetrahedron, 1966, 22, 81–93 CrossRef CAS.
- R. J. Cotter and W. F. Beach, Thermolysis of azidoformates in aromatic compounds. A synthesis of 1H-azepin-1-yl carboxylates, J. Org. Chem., 1964, 29, 751–754 CrossRef CAS.
- Y. Zhang, J.-Y. Xue, X.-C. Su, W.-J. Xiao, J.-Y. Lv, W.-X. Shi, Y. Zou, M. Yan and X.-J. Zhang, Skeletal editing of benzene motif: photopromoted transannulation for synthesis of DNA-Encoded seven-membered rings, Org. Lett., 2024, 26, 2212–2217 CrossRef CAS PubMed.
- C. M. Nunes, A. K. Eckhardt, I. Reva, R. Fausto and P. R. Schreiner, Competitive nitrogen versus carbon tunneling, J. Am. Chem. Soc., 2019, 141, 14340–14348 CrossRef CAS PubMed.
- N. P. Gritsan, I. Likhotvorik, M.-L. Tsao, N. Çelebi, M. S. Platz, W. L. Karney, C. R. Kemnitz and W. T. Borden, Ring-expansion reaction of cyano-substituted singlet phenyl nitrenes: theoretical predictions and kinetic results from laser flash photolysis and chemical trapping experiments, J. Am. Chem. Soc., 2001, 123, 1425–1433 CrossRef CAS.
- H. Inui, K. Sawada, S. Oishi, K. Ushida and R. J. McMahon, Aryl nitrene rearrangements: spectroscopic observation of a benzazirine and its ring expansion to a ketenimine by heavy-atom tunneling, J. Am. Chem. Soc., 2013, 135, 10246–10249 CrossRef CAS.
- A. M. Pandey, S. Mondal, P. Andotra and B. Gnanaprakasam, Solid-state melt rearrangement of azidofluorenes for the synthesis of phenanthridine derivatives, Asian J. Org. Chem., 2024, e202400459 Search PubMed.
- J. Wang, H. Lu, Y. He, C. Jing and H. Wei, Cobalt-catalyzed nitrogen atom insertion in arylcycloalkenes, J. Am. Chem. Soc., 2022, 144, 22433–22439 CrossRef CAS.
- H. Li, N. Li, J. Wu, T. Yu, R. Zhang, L.-P. Xu and H. Wei, Rhodium-catalyzed intramolecular nitrogen atom insertion into arene rings, J. Am. Chem. Soc., 2023, 145, 17570–17576 CrossRef CAS.
- M.-M. Xu, W.-B. Cao, X.-P. Xu and S.-J. Ji, Efficient synthesis of 2-arylquinazolin-4-amines via a copper-catalyzed diazidation and ring expansion cascade of 2-arylindoles, Chem. Commun., 2018, 54, 12602–12605 RSC.
- Y. He, J. Wang, T. Zhu, Z. Zheng and H. Wei, Nitrogen atom insertion into arenols to access benzazepines, Chem. Sci., 2024, 15, 2612–2617 RSC.
- L. Li, D. Wang, Y. Zhang, J. Liu, H. Wang and X. Luan, Diversification of naphthol skeletons triggered by aminative dearomatization, Org. Lett., 2024, 26, 4910–4915 CrossRef CAS.
- T. Yang, X. Fan, X. Zhao and W. Yu, Iron-catalyzed acyl migration of tertiary α-azidyl ketones: synthetic approach toward enamides and isoquinolones, Org. Lett., 2018, 20, 1875–1879 CrossRef CAS PubMed.
- L. Song, X. Tian, K. Farshadfar, F. Shiri, F. Rominger, A. Ariafard and A. S. K. Hashmi, An unexpected synthesis of azepinone derivatives through a metal-free photochemical cascade reaction, Nat. Commun., 2023, 14, 831 CrossRef CAS.
- R. Mykura, R. Sánchez-Bento, E. Matador, V. K. Duong, A. Varela, L. Angelini, R. J. Carbajo, J. Llaveria, A. Ruffoni and D. Leonori, Synthesis of polysubstituted azepanes by dearomative ring expansion of nitroarenes, Nat. Chem., 2024, 16, 771–779 CrossRef CAS PubMed.
- P. R. Kumar, Addition of phthalimidonitrene to substituted indoles, Heterocycles, 1987, 26, 1257–1262 CrossRef CAS.
- J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Late-stage diversification of indole skeletons through nitrogen atom insertion, Science, 2022, 377, 1104–1109 CrossRef CAS.
- T. Heilmann, J. M. Lopez-Soria, J. Ulbrich, J. Kircher, Z. Li, B. Worbs, C. Golz, R. A. Mata and M. Alcarazo, N-(Sulfonio) sulfilimine reagents: non-oxidizing sources of electrophilic nitrogen atom for skeletal editing, Angew. Chem., Int. Ed., 2024, 63, e202403826 CrossRef CAS.
- J. C. Reisenbauer, A.-S. K. Paschke, J. Krizic, B. B. Botlik, P. Finkelstein and B. Morandi, Direct access to quinazolines and pyrimidines from unprotected indoles and pyrroles through nitrogen atom insertion, Org. Lett., 2023, 25, 8419–8423 CrossRef CAS.
- B. B. Botlik, M. Weber, F. Ruepp, K. Kawanaka, P. Finkelstein and B. Morandi, Streamlining the synthesis of pyridones through oxidative amination of cyclopentenones, Angew. Chem., Int. Ed., 2024, 63, e202408230 CrossRef CAS PubMed.
- M. I. Fremery and E. K. Fields, Amozonolysis of cycloolefins, J. Org. Chem., 1964, 29, 2240–2243 CrossRef CAS.
- R. B. Miller and J. M. Frincke, Synthesis of isoquinolines from indenes, J. Org. Chem., 1980, 45, 5312–5315 CrossRef CAS.
- D. S. Dime and S. McLean, Synthesis of isoquinolines from indenes, J. Org. Chem., 1981, 46, 4999–5000 CrossRef CAS.
- P. Finkelstein, J. C. Reisenbauer, B. B. Botlik, O. Green, A. Florin and B. Morandi, Nitrogen atom insertion into indenes to access isoquinolines, Chem. Sci., 2023, 14, 2954–2959 RSC.
- P. Q. Kelly, A. S. Filatov and M. D. Levin, A synthetic cycle for heteroarene synthesis by nitride insertion, Angew. Chem., Int. Ed., 2022, 61, e202213041 CrossRef CAS PubMed.
- S. Liu and X. Cheng, Insertion of ammonia into alkenes to build aromatic N-heterocycles, Nat. Commun., 2022, 13, 425 CrossRef CAS PubMed.
- B. S. Zhang, S. L. Homölle, T. Bauch, J. C. Oliveira, S. Warratz, B. Yuan, X. Y. Gou and L. Ackermann, Electrochemical skeletal indole editing via nitrogen atom insertion by sustainable oxygen reduction reaction, Angew. Chem., Int. Ed., 2024, 63, e202407384 CrossRef CAS.
- M. S. H. Salem, C. Dubois, Y. Takamura, A. Kitajima, T. Kawai, S. Takizawa and M. Kirihara, Light-induced autoxidation of aldehydes to peracids and carboxylic acids, Green Chem., 2024, 26, 375–383 RSC.
- D. M. Soro, J. B. Roque, J. W. Rackl, B. Park, S. Payer, Y. Shi, J. C. Ruble, A. L. Kaledin, M.-H. Baik, D. G. Musaev and R. Sarpong, Photo-and metal-mediated deconstructive approaches to cyclic aliphatic amine diversification, J. Am. Chem. Soc., 2023, 145, 11245–11257 CrossRef CAS PubMed.
- J. B. Roque, Y. Kuroda, L. T. Göttemann and R. Sarpong, Deconstructive diversification of cyclic amines, Nature, 2018, 564, 244–248 CrossRef CAS PubMed.
- Z. Siddiqi, W. C. Wertjes and D. Sarlah, Chemical equivalent of arene monooxygenases: dearomative synthesis of arene oxides and oxepines, J. Am. Chem. Soc., 2020, 142, 10125–10131 CrossRef CAS.
- Q.-H. Shi, Y.-H. Wang, Z.-Y. Chen, X.-R. Wang, W.-H. Zhang, F.-L. Tian, L.-J. Peng, Y. Zhou and X.-L. Liu, Formal oxygen atom insertion as a skeletal-editing step: rapid access natural-product-inspired bispiro[oxindole-oxazinane] hybrids, Org. Chem. Front., 2023, 10, 3307–3312 RSC.
- X. Zhang, W. Su, H. Guo, P. Fang, K. Yang and Q. Song, N-Heterocycle-editing to access fused-BN-heterocycles via ring-opening/C-H borylation/reductive C–B bond formation, Angew. Chem., Int. Ed., 2024, 63, e202318613 CrossRef CAS.
- S. Van der Kerk, J. Boersma and G. Van der Kerk, The generation and some reactions of methylborylene, Tetrahedron Lett., 1976, 17, 4765–4766 CrossRef.
- B. Pachaly and R. West, Photochemical generation of triphenylsilylboranediyl (C6H5)3SiB: from organosilylboranes, Angew. Chem., Int. Ed. Engl., 1984, 23, 454–455 CrossRef.
- B. Su, Y. Li, R. Ganguly and R. Kinjo, Ring expansion, photoisomerization, and retrocyclization of 1,4,2-diazaboroles, Angew. Chem., Int. Ed., 2017, 56, 14572–14576 CrossRef CAS PubMed.
- H. Saito, S. Otsuka, K. Nogi and H. Yorimitsu, Nickel-catalyzed boron insertion into the C2−O bond of benzofurans, J. Am. Chem. Soc., 2016, 138, 15315–15318 CrossRef CAS.
- S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Manganese-catalyzed ring opening of benzofurans and its application to insertion of heteroatoms into the C2−O bond, Org. Lett., 2017, 19, 5557–5560 CrossRef CAS PubMed.
- S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Aromatic metamorphosis of indoles into 1,2-benzazaborins, Org. Lett., 2019, 21, 3855–3860 CrossRef CAS PubMed.
- C. Ren, B. Han, H. Guo, W. Yang, C. Xia, X.-H. Jin, F. Wang and L. Wu, Skeletal editing of aromatic N-heterocycles via hydroborative cleavage of C−N bonds–scope, mechanism, and property, Angew. Chem., Int. Ed., 2024, 63, e202407222 Search PubMed.
- H. Lyu, I. Kevlishvili, X. Yu, P. Liu and G. Dong, Boron insertion into alkyl ether bonds via zinc/nickel tandem catalysis, Science, 2021, 372, 175–182 CrossRef CAS PubMed.
- L. F. Silva Jr, Construction of cyclopentyl units by ring contraction reactions, Tetrahedron, 2002, 58, 9137–9161 CrossRef.
- J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Scaffold hopping by net photochemical carbon deletion of azaarenes, Science, 2022, 376, 527–532 CrossRef CAS PubMed.
- X.-J. Peng, H.-P. He, Q. Liu, K. She, B.-Q. Zhang, H.-S. Wang, H.-T. Tang and Y.-M. Pan, Photocatalyst-controlled and visible light-enabled selective oxidation of pyridinium salts, Sci. China: Chem., 2021, 64, 753–760 CrossRef CAS.
- J. Jurczyk, M. C. Lux, D. Adpressa, S. F. Kim, Y.-H. Lam, C. S. Yeung and R. Sarpong, Photomediated ring contraction of saturated heterocycles, Science, 2021, 373, 1004–1012 CrossRef CAS PubMed.
- M. Takashima, M. Miyoshi, M. Sako, M. Arisawa and K. Murai, Oxidative rearrangement approach for the ring contraction of N−H piperidines to pyrrolidines, Adv. Synth. Catal., 2024, 366, 3325–3331 Search PubMed.
- H. Zhong, D. T. Egger, V. C. Gasser, P. Finkelstein, L. Keim, M. Z. Seidel, N. Trapp and B. Morandi, Skeletal metalation of lactams through a carbonyl-to-nickel-exchange logic, Nat. Commun., 2023, 14, 5273 CrossRef CAS PubMed.
- L.-Y. Chen and J. Li, Skeletal editing of dibenzolactones to fluorenes via Ni-or Pd-catalyzed decarboxylation, J. Org. Chem., 2023, 88, 10252–10256 CrossRef CAS PubMed.
- A. Albini and M. Alpegiani, The photochemistry of the N-oxide function, Chem. Rev., 1984, 84, 43–71 CrossRef CAS.
- G. G. Spence, E. C. Taylor and O. Buchardt, Photochemical reactions of azoxy compounds, nitrones, and aromatic amine N-oxides, Chem. Rev., 1970, 70, 231–265 CrossRef CAS.
- M. S. H. Salem, Y. M. A. Aziz, M. S. Elgawish, M. M. Said and K. A. Abouzid, Design, synthesis, biological evaluation and molecular modeling study of new thieno [2,3-d] pyrimidines with anti-proliferative activity on pancreatic cancer cell lines, Bioorg. Chem., 2020, 94, 103472 CrossRef CAS.
- A. R. Gardouh, A. S. Srag El-Din, M. S. H. Salem, Y. Moustafa and S. Gad, Starch nanoparticles for enhancement of oral bioavailability of a newly synthesized thienopyrimidine derivative with anti-proliferative activity against pancreatic cancer, Drug Des., Dev. Ther., 2021, 15, 3071–3093 CrossRef.
- H. Van der Plas and H. Jongejan, Ring transformations in reactions of heterocyclic compounds with nucleophiles (III): conversion of pyrimidine and some of its methyl derivatives by hydrazine and by methylhydrazine sulfate into pyrazoles and methylpyrazoles, Recl. Trav. Chim. Pays-Bas, 1968, 87, 1065–1072 CrossRef CAS.
- G. L. Bartholomew, F. Carpaneto and R. Sarpong, Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion, J. Am. Chem. Soc., 2022, 144, 22309–22315 CrossRef CAS PubMed.
- S. Huh, G. J. Saunders and A. K. Yudin, Single atom ring contraction of peptide macrocycles using Cornforth rearrangement, Angew. Chem., Int. Ed., 2023, 62, e202214729 CrossRef CAS PubMed.
- D. M. Lemal and T. W. Rave, Diazenes from Angeli's salt, J. Am. Chem. Soc., 1965, 87, 393–394 CrossRef CAS.
- W. D. Hinsberg III and P. B. Dervan, Synthesis and direct spectroscopic observation of a 1, 1-dialkyldiazene. Infrared and electronic spectrum of N,-(2,2,6,6-tetramethylpiperidyl) nitrene, J. Am. Chem. Soc., 1978, 100, 1608–1610 CrossRef.
- S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Skeletal editing through direct nitrogen deletion of secondary amines, Nature, 2021, 593, 223–227 CrossRef CAS PubMed.
- J. Masson-Makdissi, R. F. Lalisse, M. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, Evidence for dearomatizing spirocyclization and dynamic effects in the quasi-stereospecific nitrogen deletion of tetrahydroisoquinolines, J. Am. Chem. Soc., 2024, 146, 17719–17727 CrossRef CAS PubMed.
- B. A. Wright, A. Matviitsuk, M. J. Black, P. García-Reynaga, L. E. Hanna, A. T. Herrmann, M. K. Ameriks, R. Sarpong and T. P. Lebold, Skeletal editing approach to bridge-functionalized bicyclo [1.1.1] pentanes from azabicyclo [2.1.1] hexanes, J. Am. Chem. Soc., 2023, 145, 10960–10966 CrossRef CAS.
- X. Zou, J. Zou, L. Yang, G. Li and H. Lu, Thermal rearrangement of sulfamoyl azides: reactivity and mechanistic study, J. Org. Chem., 2017, 82, 4677–4688 CrossRef CAS.
- H. Qin, W. Cai, S. Wang, T. Guo, G. Li and H. Lu, N-Atom deletion in nitrogen heterocycles, Angew. Chem., Int. Ed., 2021, 60, 20678–20683 CrossRef CAS PubMed.
- C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, Stereoselective synthesis of cyclobutanes by contraction of pyrrolidines, J. Am. Chem. Soc., 2021, 143, 18864–18870 CrossRef CAS.
- S. F. Kim, H. Schwarz, J. Jurczyk, B. R. Nebgen, H. Hendricks, H. Park, A. Radosevich, M. W. Zuerch, K. Harper and M. C. Lux, Mechanistic investigation, wavelength-dependent reactivity, and expanded reactivity of N–aryl azacycle photomediated ring contractions, J. Am. Chem. Soc., 2024, 146, 5580–5596 CrossRef CAS PubMed.
- T. P. McFadden, C. I. Nwachukwu and A. G. Roberts, An amine template strategy to construct successive C–C bonds: synthesis of benzo[h]quinolines by a deaminative ring contraction cascade, Org. Biomol. Chem., 2022, 20, 1379–1385 RSC.
- E. K. Kirkeby, Z. T. Schwartz, M. A. Lovasz and A. G. Roberts, Deaminative ring contraction for the synthesis of polycyclic heteroaromatics: a concise total synthesis of toddaquinoline, Chem. Sci., 2023, 14, 10508–10514 RSC.
- X. Zhou, Q. Zhuo, T. Shima, X. Kang and Z. Hou, Denitrogenative ring-contraction of pyridines to a cyclopentadienyl skeleton at a dititanium hydride framework, J. Am. Chem. Soc., 2024, 146, 31348–31355 Search PubMed.
- R. Davenport, M. Silvi, A. Noble, Z. Hosni, N. Fey and V. K. Aggarwal, Visible-light-driven strain-increase ring contraction allows the synthesis of cyclobutyl boronic esters, Angew. Chem., Int. Ed., 2020, 59, 6525–6528 CrossRef CAS PubMed.
- L. D. Pennington and D. T. Moustakas, The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization, J. Med. Chem., 2017, 60, 3552–3579 CrossRef CAS PubMed.
- X. Li, J. Xu and Z.-G. Xu, Precision single-atom editing: new frontiers in nitrogen insertion and substitution for the generation of N-heterocycles, Org. Chem. Front., 2024, 11, 4041–4053 RSC.
- T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D.-I. Park and M. D. Levin, Aromatic nitrogen scanning by ipso-selective nitrene internalization, Science, 2023, 381, 1474–1479 CrossRef CAS.
- N. Kotwal and P. Chauhan, Accessing pyridines via a nitrene internalization process, Angew. Chem., Int. Ed., 2024, 63, e202317228 CrossRef CAS.
- R. J. Sundberg, S. R. Suter and M. Brenner, Photolysis of ortho-substituted aryl azides in diethylamine. Formation and autoxidation of 2-diethylamino-1H-azepine intermediates, J. Am. Chem. Soc., 1972, 94, 513–520 CrossRef CAS.
- S. C. Patel and N. Z. Burns, Conversion of aryl azides to aminopyridines, J. Am. Chem. Soc., 2022, 144, 17797–17802 CrossRef CAS PubMed.
- J. Woo, C. Stein, A. H. Christian and M. D. Levin, Carbon-to-nitrogen single-atom transmutation of azaarenes, Nature, 2023, 623, 77–82 CrossRef CAS.
- H. Lu, Y. Zhang, X.-H. Wang, R. Zhang, P.-F. Xu and H. Wei, Carbon–nitrogen transmutation in polycyclic arenol skeletons to access N-heteroarenes, Nat. Commun., 2024, 15, 3772 CrossRef CAS PubMed.
- J. Nociarová, A. Purkait, R. Gyepes and P. Hrobárik, Silver-catalyzed skeletal editing of benzothiazol-2(3H)-ones and 2-halogen-substituted benzothiazoles as a rapid single-step approach to benzo[1,2,3]thiadiazoles, Org. Lett., 2024, 26, 619–624 CrossRef.
- T. Zincke and G. Weisspfenning, Über dinitrophenylisochinoliniumchlorid und dessen umwandlungsprodukte, Justus Liebigs Ann. Chem., 1913, 396, 103–131 CrossRef CAS.
- T. Zincke, G. Heuser and W. Möller, I. Ueber dinitrophenylpyridiniumchlorid und dessen umwandlungsproducte, Justus Liebigs Ann. Chem., 1904, 333, 296–345 CrossRef.
- R. Sagitullin, S. Gromov and A. Kost, Alkylamino group exchange upon recyclization of pyridinium salts into anilines, Tetrahedron, 1978, 34, 2213–2216 CrossRef CAS.
- A. Kost, S. Gromov and R. Sagitullin, Pyridine ring nucleophilic recyclizations, Tetrahedron, 1981, 37, 3423–3454 CrossRef CAS.
- T. Morofuji, K. Inagawa and N. Kano, Sequential ring-opening and ring-closing reactions for converting para-substituted pyridines into meta-substituted anilines, Org. Lett., 2021, 23, 6126–6130 CrossRef CAS PubMed.
- T. Morofuji, S. Nagai, A. Watanabe, K. Inagawa and N. Kano, Streptocyanine as an activation mode of amine catalysis for the conversion of pyridine rings to benzene rings, Chem. Sci., 2023, 14, 485–490 RSC.
- L. Schmerling and W. Toekelt, Direct conversion of pyridine to benzoic acid, J. Am. Chem. Soc., 1964, 86, 1259–1259 CrossRef CAS.
- A. Conboy and M. F. Greaney, Synthesis of benzenes from pyridines via N to C switch, Chem, 2024, 10, 1940–1949 CAS.
- F.-P. Wu, M. Lenz, A. Suresh, A. R. Gogoi, J. L. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Nitrogen-to-functionalized carbon atom transmutation of pyridine, Chem. Sci., 2024, 15, 15205–15211 Search PubMed.
- B. J. Uhlenbruck, C. M. Josephitis, L. de Lescure, R. S. Paton and A. McNally, A deconstruction-reconstruction strategy for pyrimidine diversification, Nature, 2024, 631, 87–93 CrossRef CAS.
- Q. Cheng, D. Bhattacharya, M. Haring, H. Cao, C. Mück-Lichtenfeld and A. Studer, Skeletal editing of pyridines through atom-pair swap from CN to CC, Nat. Chem., 2024, 16, 741–748 CrossRef CAS PubMed.
- B. R. Boswell, Z. Zhao, R. L. Gonciarz and K. M. Pandya, Regioselective pyridine to benzene edit inspired by water-displacement, J. Am. Chem. Soc., 2024, 146, 19660–19666 CrossRef CAS PubMed.
- S. Lee, R. Jena and A. L. Odom, Substituted pyridines from isoxazoles: scope and mechanism, Org. Biomol. Chem., 2022, 20, 6630–6636 RSC.
- A. G. Schultz and M. Shen, Aromatic ring synthesis by N-aminopyrrole Diels-Alder reaction. Total synthesis of juncusol, Tetrahedron Lett., 1981, 22, 1775–1778 CrossRef CAS.
- S. Liu, A.-J. Wang, M. Li, J. Zhang, G.-D. Yin, W.-M. Shu and W.-C. Yu, Rh(III)-catalyzed tandem reaction access to (quinazolin-2-yl) methanone derivatives from 2,1-benzisoxazoles and α-azido ketones, J. Org. Chem., 2022, 87, 11253–11260 CrossRef CAS PubMed.
- W. Lv, X. Kong, Y. Qing, J. Zheng, Y. Yin, Y. Zhou and D. Wang, Skeletal editing of benzodithiol-3-ones for the assembly of benzo[d,][1,3]oxathiin-4-ones, Org. Chem. Front., 2024, 11, 4979–4985 Search PubMed.
- Y. Hamada and I. Takeuchi, Syntheses of nitrogen-containing heterocyclic compounds. 26. Reaction of benzo [f, or h] quinolines and their N-oxides with methylsulfinyl carbanion, J. Org. Chem., 1977, 42, 4209–4213 CrossRef CAS.
- T. Wang, C. Li, J. Mi, H. Wang, L. Chen, Q. Kong, N. Jiao and S. Song, Skeletal editing of pyridine and quinoline N-oxides through nitrogen to carbon single atom swap, CCS Chem., 2024, 1–23, DOI:10.31635/ccschem.024.202404133.
- N. A. Falcone, S. He, J. F. Hoskin, S. Mangat and E. J. Sorensen, N-Oxide-to-carbon transmutations of azaarene N-oxides, Org. Lett., 2024, 26, 4280–4285 CrossRef CAS PubMed.
- J. Nan, Q. Huang, X. Men, S. Yang, J. Wang and Y. Ma, Palladium-catalyzed denitrogenation/vinylation of benzotriazinones with vinylene carbonate, Chem. Commun., 2024, 60, 3571–3574 RSC.
- D. Kim, J. You, D. H. Lee, H. Hong, D. Kim and Y. Park, Photocatalytic furan-to-pyrrole conversion, Science, 2024, 386, 99–105 CrossRef CAS PubMed.
- X. Jiang, X. He, J. Wong, S. Scheeff, S. C.-K. Hau, T. H. Wong, Y. Qin, C. H. Fan, B. Ma, N. L. Chung, J. Huang, J. Zhao, Y. Yan, M. Xiao, X. Song, T. K. C. Hui, Z. Zuo, W. K.-K. Wu, H. Ko, K. H.-M. Chow and B. W.-L. Ng, Lactone-to-lactam editing alters the pharmacology of bilobalide, JACS Au, 2024, 4, 3537–3546 CrossRef CAS.
- F. H. Nelissen, M. Tessari, S. S. Wijmenga and H. A. Heus, Stable isotope labeling methods for DNA, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 96, 89–108 CrossRef CAS.
- K. Gevaert, F. Impens, B. Ghesquière, P. Van Damme, A. Lambrechts and J. Vandekerckhove, Stable isotopic labeling in proteomics, Proteomics, 2008, 8, 4873–4885 CrossRef CAS PubMed.
- D. R. Cole and S. Chakraborty, Rates and mechanisms of isotopic exchange, Rev. Mineral. Geochem., 2001, 43, 83–223 CrossRef CAS.
- W. von Philipsborn and R. Müller, 15N-NMR spectroscopy—new methods and applications [new analytical methods (28)], Angew. Chem., Int. Ed. Engl., 1986, 25, 383–413 CrossRef.
- M. Nowakowski, S. Saxena, J. Stanek, S. Żerko and W. Koźmiński, Applications of high dimensionality experiments to biomolecular NMR, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 90, 49–73 CrossRef PubMed.
- R. Marek and A. Lycka, 15N NMR spectroscopy in structural analysis, Curr. Org. Chem., 2002, 6, 35–66 CrossRef CAS.
- M. Kirihara, R. Nakamura, K. Nakakura, K. Tujimoto, M. S. H. Salem, T. Suzuki and S. Takizawa, DAST-mediated ring-opening of cyclopropyl silyl ethers in nitriles: facile synthesis of allylic amides via a Ritter-type process, Org. Biomol. Chem., 2022, 20, 6558–6561 RSC.
- M. Sako, K. Kanomata, M. S. H. Salem, T. Furukawa, H. Sasai and S. Takizawa, Metal-free C(aryl)–P bond cleavage: experimental and computational studies of the Michael addition/aryl migration of triarylphosphines to alkynyl esters, Org. Chem. Front., 2022, 9, 2187–2192 RSC.
- R. V. Shchepin, D. A. Barskiy, A. M. Coffey, T. Theis, F. Shi, W. S. Warren, B. M. Goodson and E. Y. Chekmenev, 15N hyperpolarization of imidazole-15N2 for magnetic resonance pH sensing via SABRE-SHEATH, ACS Sens., 2016, 1, 640–644 CrossRef CAS.
- G. L. Bartholomew, S. L. Kraus, L. J. Karas, F. Carpaneto, R. Bennett, M. S. Sigman, C. S. Yeung and R. Sarpong, 14N to 15N isotopic exchange of nitrogen heteroaromatics through skeletal editing, J. Am. Chem. Soc., 2024, 146, 2950–2958 CrossRef CAS PubMed.
- Z. A. Tolchin and J. M. Smith, 15NRORC: an azine labeling protocol, J. Am. Chem. Soc., 2024, 146, 2939–2943 CrossRef CAS.
- H. M. Nguyen, D. C. Thomas, M. A. Hart, K. R. Steenback, J. N. Levy and A. McNally, Synthesis of 15N-pyridines and higher mass isotopologs via Zincke imine intermediates, J. Am. Chem. Soc., 2024, 146, 2944–2949 CrossRef CAS PubMed.
- G. Tan and F. Glorius, Stereochemical editing, Angew. Chem., Int. Ed., 2023, 62, e202217840 CrossRef CAS PubMed.
- P.-Z. Wang, W.-J. Xiao and J.-R. Chen, Light-empowered contra-thermodynamic stereochemical editing, Nat. Rev. Chem., 2023, 7, 35–50 CrossRef.
- A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Catalytic deracemization of chiral allenes by sensitized excitation with visible light, Nature, 2018, 564, 240–243 CrossRef.
- M. Huang, L. Zhang, T. Pan and S. Luo, Deracemization through photochemical E/Z isomerization of enamines, Science, 2022, 375, 869–874 CrossRef CAS.
- Z. Zhang and X. Hu, Visible-light-driven catalytic deracemization of secondary alcohols, Angew. Chem., Int. Ed., 2021, 60, 22833–22838 CrossRef CAS PubMed.
- C. Zhang, A. Z. Gao, X. Nie, C.-X. Ye, S. I. Ivlev, S. Chen and E. Meggers, Catalytic α-deracemization of ketones enabled by photoredox deprotonation and enantioselective protonation, J. Am. Chem. Soc., 2021, 143, 13393–13400 CrossRef CAS PubMed.
- L. Wen, J. Ding, L. Duan, S. Wang, Q. An, H. Wang and Z. Zuo, Multiplicative enhancement of stereoenrichment by a single catalyst for deracemization of alcohols, Science, 2023, 382, 458–464 CrossRef CAS.
- Y. Wang, H. M. Carder and A. E. Wendlandt, Synthesis of rare sugar isomers through site-selective epimerization, Nature, 2020, 578, 403–408 CrossRef CAS.
- Y.-A. Zhang, X. Gu and A. E. Wendlandt, A change from kinetic to thermodynamic control enables trans-selective stereochemical editing of vicinal diols, J. Am. Chem. Soc., 2021, 144, 599–605 CrossRef.
- H. M. Carder, Y. Wang and A. E. Wendlandt, Selective axial-to-equatorial epimerization of carbohydrates, J. Am. Chem. Soc., 2022, 144, 11870–11877 CrossRef CAS.
- C. J. Oswood and D. W. MacMillan, Selective isomerization via transient thermodynamic control: dynamic epimerization of trans to cis diols, J. Am. Chem. Soc., 2021, 144, 93–98 CrossRef PubMed.
- Y.-A. Zhang, V. Palani, A. E. Seim, Y. Wang, K. J. Wang and A. E. Wendlandt, Stereochemical editing logic powered by the epimerization of unactivated tertiary stereocenters, Science, 2022, 378, 383–390 CrossRef CAS PubMed.
- M. S. H. Salem, M. I. Khalid, M. Sako, K. Higashida, C. Lacroix, M. Kondo, R. Takishima, T. Taniguchi, M. Miura, G. Vo-Thanh, H. Sasai and S. Takizawa, Electrochemical synthesis of hetero[7]helicenes containing pyrrole and furan rings via an oxidative heterocoupling and dehydrative cyclization sequence, Adv. Synth. Catal., 2023, 365, 373–380 CrossRef CAS.
- S. Yamahara, M. S. H. Salem, T. Kawai, M. Watanabe, Y. Sakamoto, T. Okada, Y. Kimura, S. Takizawa and M. Kirihara, Green and efficient protocols for the synthesis of sulfonyl fluorides using potassium fluoride as the sole fluorine source, ACS Sustainable Chem. Eng., 2024, 12, 12135–12142 CrossRef CAS.
| This journal is © the Partner Organisations 2025 |