
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllosteric release of cucurbit[6]uril from a rotaxane using a molecular signal†
Aneta
Závodná
 a,
Petr
Janovský
a,
Václav
Kolařík
a,
Jas S.
Ward
b,
Zdeňka
Prucková
a,
Michal
Rouchal
a,
Kari
Rissanen
b and
Robert
Vícha
*a
a,
Petr
Janovský
a,
Václav
Kolařík
a,
Jas S.
Ward
b,
Zdeňka
Prucková
a,
Michal
Rouchal
a,
Kari
Rissanen
b and
Robert
Vícha
*a
aDepartment of Chemistry, Faculty of Technology, Tomas Bata University in Zlín, Vavrečkova 5669, 760 01 Zlín, Czech Republic. E-mail: rvicha@utb.cz
bDepartment of Chemistry, University of Jyväskylä, P.O. Box 35, Survontie 9 B, 40014 Jyväskylä, Finland
First published on 5th November 2024
Abstract
Rotaxanes can be regarded as storage systems for their wheel components, which broadens their application potential as a complement to the supramolecular systems that retain a mechanically interlocked structure. However, utilising rotaxanes in this way requires a method to release the wheel while preserving the integrity of all molecular constituents. Herein, we present simple rotaxanes based on cucurbit[6]uril (CB6), with an axis equipped with an additional binding motif that enables the binding of another macrocycle, cucurbit[7]uril (CB7). We demonstrate that the driving force behind the wheel dethreading originates from the binding of the signalling macrocycle to the allosteric site, leading to an increase in the system's strain. Consequently, the CB6 wheel leaves the rotaxane station overcoming the mechanical barrier. Portal–portal repulsive interactions between the two cucurbituril units play a crucial role in this process. Thus, the repulsive strength and the related rate of slipping off can be finely tuned by the length of the allosteric binding motif. Finally, we show that the CB6 wheel can be utilised within complexes with other guests in the mixture once released from the rotaxane.
Introduction
Rotaxanes represent a classical family of mechanically interlocked supramolecular systems,1 alongside catenanes,2 molecular knots,3 Borromean rings,4 and suitanes.5 Since their very first synthesis almost sixty years ago,6 rotaxanes have been employed as key components of molecular machines,7 catalytic devices,8 responsive materials,9 drug delivery systems,10 and molecular memories.7a,11 Hand in hand with an increasing number of applications, various methods for rotaxane synthesis have been developed.12 In terms of the way the building blocks are transformed to more complex architectures, there are three distinguishable approaches: threading the axle into the wheel component (slipping),13 creating one of the components within the rotaxane (capping,14 clipping15), and manipulating the size of the components (shrinking the wheel,16 swelling the axle termini17). Syntheses can be performed under statistical control, using molecular recognition (including a passive metal template) or the state-of-the-art active metal template methodologies, which employ a mechanistically distinct approach in this context.18Within mechanically interlocked structures, two classes can be distinguished based on whether it is topologically necessary to break a covalent bond for the system to disassemble (e.g. catenanes) or not (e.g. rotaxanes). Rotaxanes and pseudorotaxanes are topologically identical systems, with the distinction that the latter can be disassembled under suitable conditions without breaking or significantly distorting the covalent bonds. The somewhat unclear boundary between them19 is represented by kinetically highly stable pseudorotaxanes, which, under certain conditions, are functionally equivalent to rotaxanes. Since our compounds 6a–6e exhibit high kinetic stability at temperatures relevant for their potential applications, we will refer to them as rotaxanes in this article. Regardless, rotaxanes can be considered storage devices for their wheel components, which can be released on demand. Indeed, methods for overcoming the mechanical barrier are required for this purpose. The most common approaches to releasing a wheel from the rotaxane are to destroy an axle or remove the stopping moiety.20 Unlike these destructive methods, the disassembly of rotaxanes into their intact molecular components is less frequently reported. In addition to dissociation by mechanical force,21 the recent procedures follow reverse synthetic approaches. The simple inversion of the slipping process requires a high temperature and a suitable competitor to prevent the wheel from remaining at the rotaxane station. As high temperatures are incompatible with many application areas (e.g. biosystems either in vitro or in vivo), some systems operating at ambient temperatures have been developed (Fig. 1). Leigh and co-workers reported the removal of the crown-ether wheel via pH changes in conjunction with the untying of the molecular knot, reducing the bulkiness of the stopper.22 Similarly, Li and co-workers modulated the speed of wheel slippage via conformational changes of H-bonded arylamide foldamers.23 Akine and co-workers utilised the dynamic nature of palladium complexes to accelerate 27-crown-9 removal from the metallorotaxane via the addition of bromide as an accelerator and caesium as a competitor for 27C9 (reverse capping).24 Finally, Heitz and co-workers inverted the shrinking process to remove a bis-porphyrin cage from its semirotaxane by the allosteric action of Ag+ ions.25
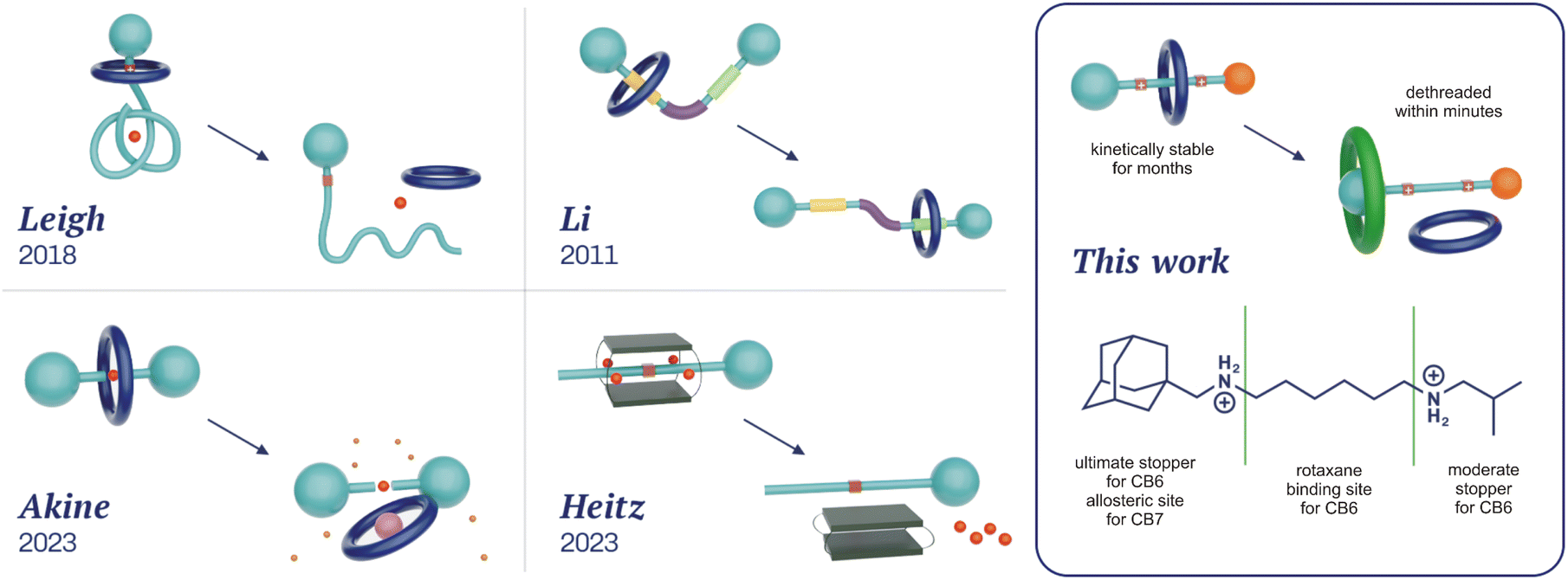 | ||
| Fig. 1 Previously published approaches for wheel dethreading and the design of our new system. | ||
Herein, we report a very simple approach for the release of the CB6 wheel by a stimulus that is orthogonal to the preparation of the rotaxane. We employed the extraordinary selectivity of cucurbit[n]urils (CBns)26 to arrange a system in which the originally favoured rotaxane site is no longer attractive for CB6 after the addition of CB7. CB7 strongly prefers the allosteric binding site and increases the strain of the system via portal–portal electrostatic repulsion, producing a metastable structure that relaxes by releasing the CB6 unit. This process inherently does not require structural changes of any component, nor any other additives (e.g. competitors).
Results and discussion
Structure justification and preparation of rotaxanes
We combined three previously published pieces of information in designing the axle component for our systems. First, Mock reported that the hexamethylene diammonium motif is an ideal binding site for CB6 (K = 2.9 × 108 M−1; 50 mM NaCl; 25 °C).27 Hereinafter, we will abbreviate hexamethylene diamine/diammonium as HMDA and related binding sites as HM. Second, Masson recognised an isobutyl (IB) stopper as a kinetic barrier of 121 kJ mol−1, allowing for CB6 to slip on at 120 °C while maintaining the system in a rotaxane manner at 30 °C.13a Third, it has been previously reported that CBn units repel each other via portal–portal electrostatic interactions.28 This results in the unlikely binding of two CBn portals to a single cationic moiety.Further, we used adamantane-derived motifs, which provide two functionalities. As the adamantane cage is too bulky to pass through the CB6 cavity, it serves as an ultimate stopper for this macrocycle. In contrast, the adamantane cage perfectly fits the interior of CB7 to provide an allosteric binding site with an exceptionally high affinity towards CB7. The binding strength of CB7 at adamantane-derived ammonium motifs can be modulated by the distance between the adamantane cage and the cationic moiety: the greater the distance, the weaker the complex.29 Indeed, the greater distance decreases the repulsion between the two CBn units, which is crucial for expelling CB6 from its rotaxane position. Doubt arose as to whether it is possible to balance these two effects so that CB6 is released at room temperature. Therefore, we prepared a series of five rotaxanes, 6a–6e, with variable allosteric binding motifs, as shown in Scheme 1.
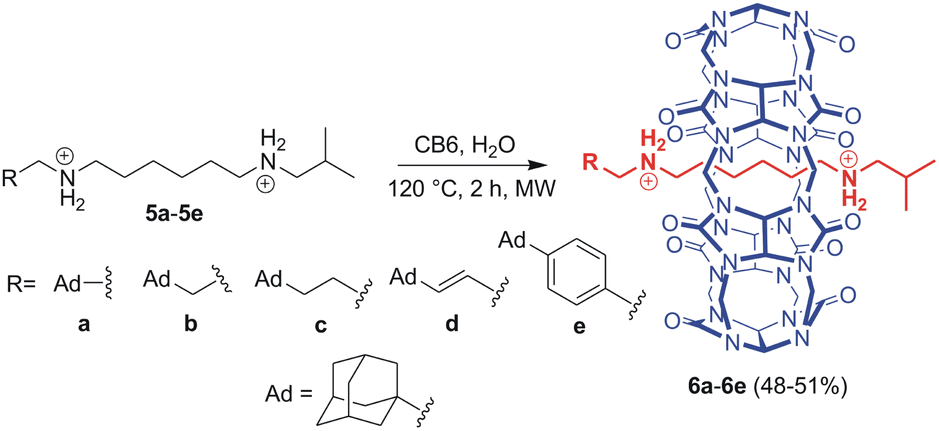 | ||
| Scheme 1 Synthesis of rotaxanes 6a–6e. Counterion chlorides are omitted for clarity. | ||
The axles were synthesised from hexamethylenediamine (HMDA, 1) via a sequence of Boc-protection, condensation with isobutyric aldehyde 7f, deprotection, and second condensation with adamantane-derived aldehydes 7a–7e. The final rotaxanes were prepared by slipping CB6 in water at 120 °C under microwave irradiation, with an overall yield of 7–14% with respect to the HMDA. Synthetic protocols and spectral characterisation for all intermediates and final rotaxanes 6a–6e are detailed in the SI (for 1H and 13C NMR spectra, see Fig. S1–S20;† for ESI-MS spectra, see Fig. S21–S30†). We succeeded in growing single crystals of 6a–6d to confirm the positioning of the CB6 unit at the HM binding site by X-ray diffraction analysis.30 It is worth noting that the CB6 unit, although sitting on the central HM site, is slightly shifted towards the Ad terminus in all four analysed structures. The magnitude of the shifting is given in Fig. 2 as the distance Δ between the respective CB6 and HM centres of gravity. Molecular models and further details are provided in Fig. 2, S60–S63 and Table S2.†
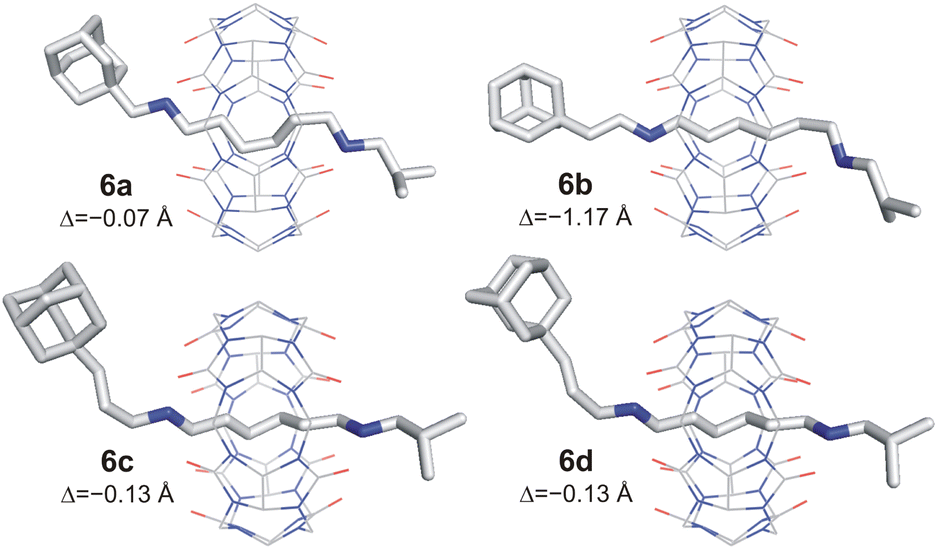 | ||
| Fig. 2 X-ray structures of 6a–6d. Minor disordered atoms, solvents, PF6− anions, and H-atoms are omitted for clarity. Δ = distance between centres of gravity of HM site and CB6 unit. | ||
As the rate of the slipping-on/off process depends on kinetic barriers, we initially thoroughly tested whether the complexes 6a–6e can be considered as rotaxanes, i.e. determining whether the dethreading is negligible at room temperature. We prepared a mixture of 6a (0.97 mM) with 1 equivalent of spermine·4HCl (hereinafter referred to as SP; KCB6 = 3.3 × 109 M−1, 50 mM NaCl, 25 °C)27b and monitored it using 1H NMR spectroscopy stored at room temperature. The binding strength of CB6 at the HM site within the rotaxanes could be assumed to be similar to that of HMDA itself. Thus, considering the affinities of CB6 towards HMDA and SP, approximately 90% of CB6 should be bound at SP in the thermodynamically equilibrated mixture of 5a, SP, and CB6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). However, we did not observe any changes in the NMR spectrum within ten months (Fig. S59†). Additionally, the complex 5a@CB6HM (i.e.6a) was not observed in the mixture of 6a (or 5a and CB6) and SP at 120 °C, implying that a kinetic barrier indeed prevents the dethreading of CB6 at 25 °C. Therefore, we consider the structures 6a–6e as kinetically interlocked rotaxanes at room temperature. Subsequently, we tested whether CB7 was able to unlock the rotaxane barrier.
:1:1). However, we did not observe any changes in the NMR spectrum within ten months (Fig. S59†). Additionally, the complex 5a@CB6HM (i.e.6a) was not observed in the mixture of 6a (or 5a and CB6) and SP at 120 °C, implying that a kinetic barrier indeed prevents the dethreading of CB6 at 25 °C. Therefore, we consider the structures 6a–6e as kinetically interlocked rotaxanes at room temperature. Subsequently, we tested whether CB7 was able to unlock the rotaxane barrier.
Allosteric triggering of the CB6 slipping off
The representative NMR spectra demonstrating the action of CB7 on rotaxane 6a are shown in Fig. 3. As can be seen, two new sets of signals appeared after the addition of 1 equivalent of CB7 into a 0.85 mM solution of 6a in D2O at 30 °C (line ii). In the first set (marked in red), H atoms at the isobutyl terminus (Ha and Hc) are deshielded, while the H atoms of the methylene group (Hj) at the adamantane terminus are shielded. In the second set (marked in blue), all these H atoms are significantly shielded. It is worth noting that all H atoms of the adamantane cage are substantially shielded in both new sets of signals. Whereas the first set disappeared within approximately 4 hours, the second set predominated, representing 90% of the axle 5a. It is well known that H atoms positioned inside the CBn cavity are strongly shielded, while there is a deshielding area outside near the portals.26b,28a Thus, the abovementioned data indicate that CB7 was instantly bound at the adamantane site to form a strained ternary complex28c5a@CB6HM,CB7Ad with the CB6 unit pushed somewhat towards the IB terminus (GFN2 molecular models31 indicate a shift of about 1.3 Å). This arrangement is not thermodynamically stable, and CB6 relatively rapidly (t½ = 36 min) surmounts the IB stopper to form the pseudorotaxane complex 5a@CB6IB,CB7Ad. The mutual orientations of axle 5a, CB7, and CB6 are represented by minimised molecular models in Fig. 3.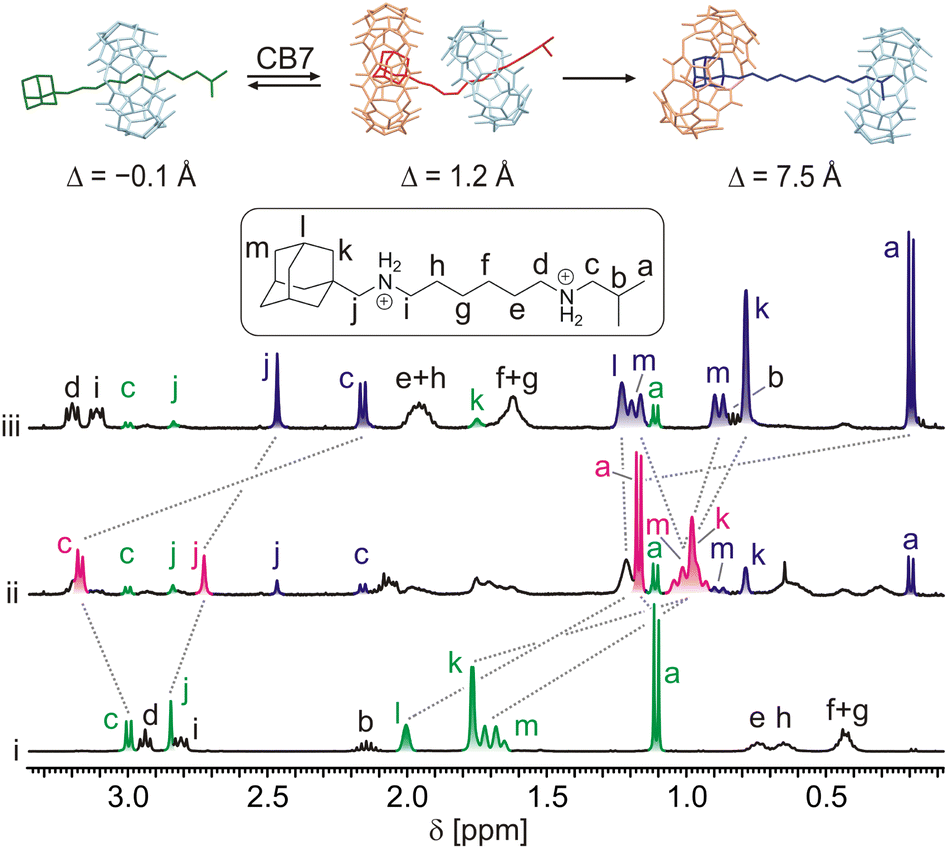 | ||
| Fig. 3 Portions of 1H NMR spectra (D2O, 30 °C, 400 MHz) and molecular models (GFN2-xTB) representing observed complexes. (i) 6a (0.85 mM); (ii) 6a + CB7 (1 equiv.) in t = 8 min; (iii) 6a + CB7 (1 equiv.) in t = 1608 min. Δ = distance between centres of gravity of HM site and CB6 units. Spectra are not to scale. | ||
Continuing our study, we examined other rotaxanes 6b–6e with CB7 to reveal how the length of the allosteric binding site influences the CB6 slipping off. The 1H NMR spectra showed that CB7 was bound to Ad sites (significant shielding of Ad H atoms) and CB6 slightly moved towards the IB termini (deshielding of IB H atoms) after the addition of 1 equivalent of CB7 into solutions of rotaxanes 6b–6e at 30 °C. However, as no other changes were observed in spectra for two months, the complexes 6b–6e@CB7Ad were considered stable at room temperature. Thus, we treated the mixtures at elevated temperatures. The rotaxanes 6b–6d released the CB6 unit in response to the CB7 signal at 60 °C displaying respective half-lives of 1.5, 17.8, and 9.6 days (kinetic curves for all rotaxanes are given in Fig. 4A). The activation parameters obtained from the Eyring plots (Fig. 4C), along with the half-lives, are given in Table 1. It is evident that the longer the spacer between the Ad cage and the adjacent ammonium cation, the slower the CB6 dethreading, in concert with the weaker repulsions between CBn portals (Fig. 4D). The inconsistency in this trend for 6c and 6d may be due to the fact that the ability of CB7 to occupy a specific position relative to the ammonium cation is actually more important than the mere length of the linker. Thus, even a slightly longer but sterically less bulky linker in substance 6d may allow CB7 to approach the ammonium cation more effectively. In the case of 6e@CB7, essentially no dethreading (<2%) was observed even at 120 °C (the temperature of rotaxane formation). This indicates that CB7 does not significantly influence the rotaxane binding site in the case of the phenylmethyl spacer, and positioning of the CB6 unit at the HM site of 5e is thermodynamically preferred even in the presence of CB7 at the Ad terminus.
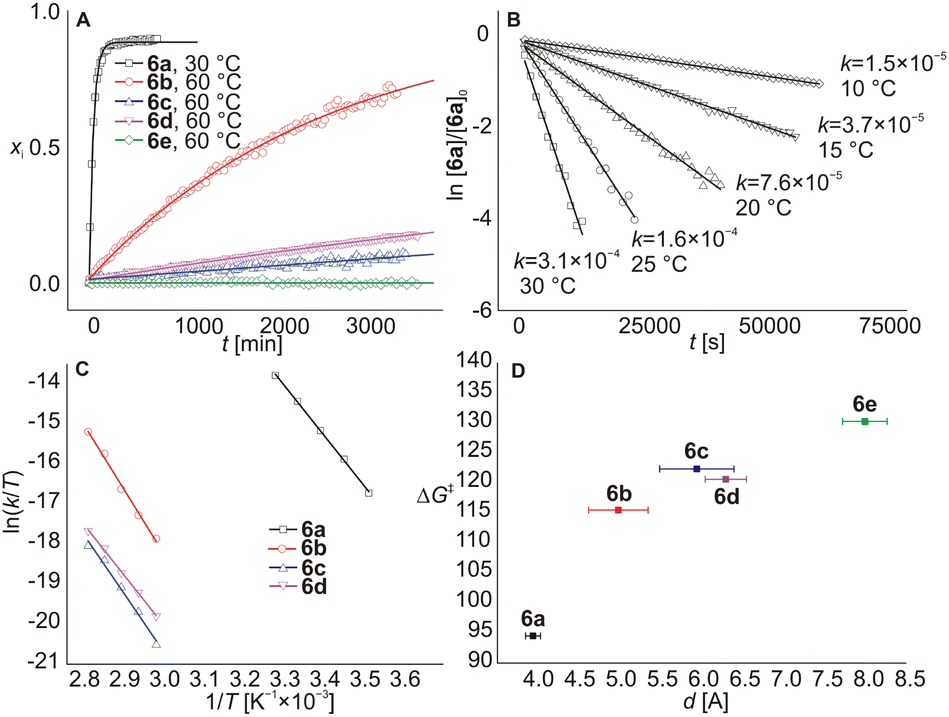 | ||
| Fig. 4 The kinetic curves for decomposition of 6a–6e@CB7 complexes (A), the first-order linearisation plot for 6a@CB7 (B), Eyring plots for 6a–6d (C) and the correlation plot of the allosteric binding site length d (obtained from molecular dynamics simulations) against the slipping-off activation energy (D). | ||
| Guest | ΔH‡a [kJ mol−1] | ΔS‡a [J mol−1 K−1] | ΔG‡a (30 °C) [kJ mol−1] | t ½ (30 °C) |
|---|---|---|---|---|
| a Calculated from Eyring plots. b Extrapolated from Eyring plots. c Estimated as detailed in text. na = not applicable. | ||||
| 6a | 104.9 ± 1.7 | 34.2 ± 0.9 | 94.5 ± 1.7 | 36.0 ± 1.4 min |
| 6b | 135 ± 6 | 58 ± 4 | 117 ± 6 | 200 ± 30b d |
| 6c | 122 ± 8 | −1.6 ± 0.2 | 122 ± 8 | 1500 ± 200b d |
| 6d | 105 ± 3 | −46 ± 2 | 119 ± 3 | 450 ± 30b d |
| 6e | na | na | ≥130c | ∞ |
Further, we made some effort to shed light on the mechanism of the CB7 action. First, we tested the interaction of 6a (0.83 mM) with CB7 in different ratios. Data in Fig. S55† demonstrate that the rate of the CB6 dethreading depends on the concentration of the 6a@CB7 complex and is not affected by an excess of CB7. In addition, the kinetic data matches the first-order model, as shown for 6a@CB7 disassembly in Fig. 4B. These experiments support our previous assumptions: the CB6 slipping off is the rate-determining step, while binding of CB7 at the allosteric Ad site is significantly faster.
Subsequently, kinetic data, along with the below-mentioned assumptions, allowed us to construct energetic profiles describing the involved supramolecular complexes and their transformations, as shown in Fig. 5. To obtain the energy gain of the first step, we determined the binding constants for the axles 5a–5e with CB6. The K values given in Table S1† demonstrate that the binding strength of CB6 at the IB terminus does not depend on the nature of the allosteric Ad binding site and yields approximately 37 kJ mol−1. In the second step, CB6 leaves the IB terminus and binds at the central HM station. It was found that the slipping on produced more than 95% CB6 bound at the HM site according to 1H NMR spectroscopy. The kinetic barrier for this process is 121 kJ mol−1, as previously determined by Masson.13a Thus, the barrier for the inverse slipping off must be at least about 130 kJ mol−1, i.e. the transformation of 5@CB6IB to 6 yields about 9 kJ mol−1. In the third step, the addition of CB7 and its binding at the Ad site produces 6@CB7Ad complexes. The relative energies for 6a–6d can be determined from the activation energies of the final dethreading step, as given in Table 1. We assume that the inherent slipping-off barrier is the same for all five rotaxanes, as the nature of the allosteric Ad site does not affect the opposite terminus. The experimentally determined differences in activation energy are related to the repulsions between two CBn units. The affinity of the CB6 unit for the HMDA site is also weakened by the fact that it shares one ammonium cation with CB7 (the binding of two CBn portals to a single cationic moiety was not observed in solution).28a,b,32 We further suppose that the slipping-on/off barriers for CB6 at 6e@CB7Ad are the same as in the case without CB7. The final step results in complexes 5@CB6IB,CB7Ad, whose relative energy gains can be estimated as the superposition of binding events at both termini (respective K values are in Table S1†).
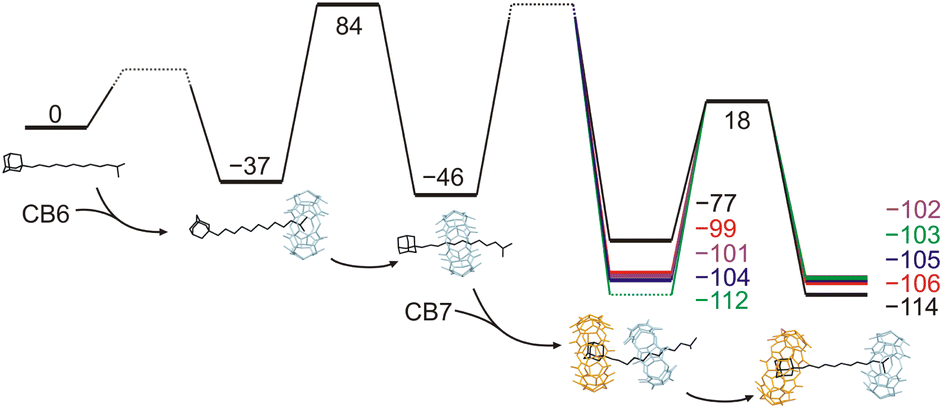 | ||
| Fig. 5 A free energy profile of the transformation of the axles 5 to 5@ CB6IB,CB7Ad through the rotaxanes 6. Optimised models (GFN2-xTB) related to 5a are given for illustration. 6a (black), 6b (red), 6c (blue), 6d (magenta), 6e (green). | ||
Releasing of CB6 to form other complexes
In contrast to pseudorotaxane systems that were published previously,28c our axles contain the terminal IB site, which binds CB6 once released from the rotaxane station. Therefore, we tested whether CB6 can be available for complexation with additional guests in the solution.First, we prepared an equimolar mixture of rotaxane 6a (0.68 mM) and SP, as described above, testing the stability of rotaxane. Subsequently, we added 1 equivalent of CB7 and monitored the progress by 1H NMR spectroscopy (Fig. 6). Immediately after the CB7 addition, the complex SP@CB7 (K = 4.8 × 108 M−1, H2O, 25 °C)33 was formed almost quantitatively (with respect to CB7). The value of the association constant of CB7 at the Ad site of 6a is not experimentally available; however, we infer that it is similar to or slightly higher than that for SP. Thus, a small portion of the strained complex 6a@CB7Ad was also observed after the CB7 addition.
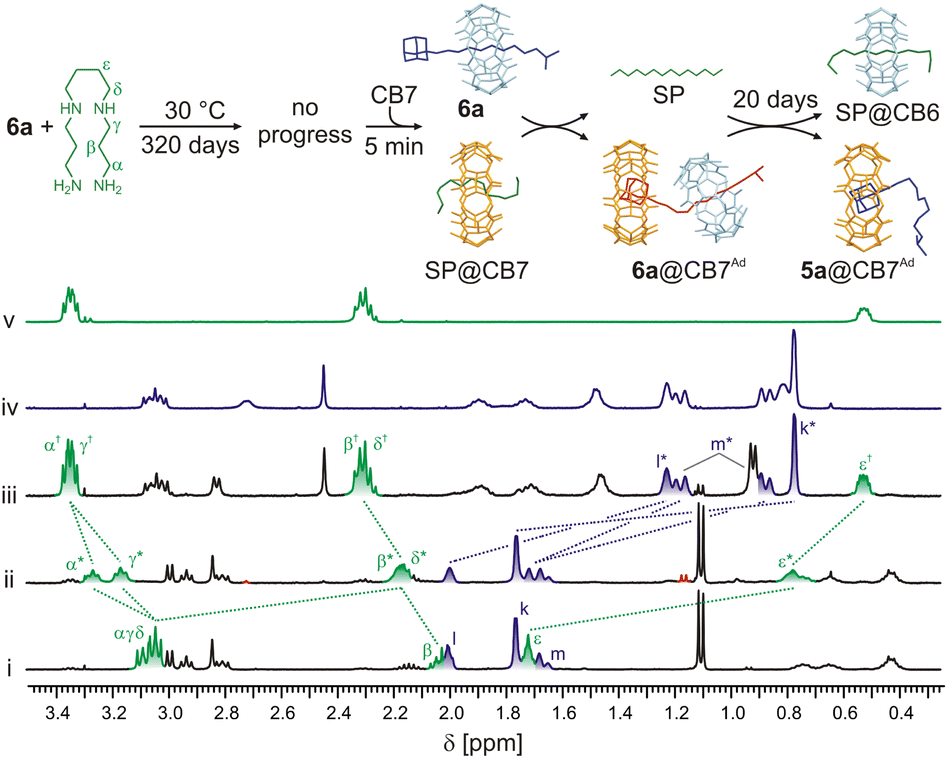 | ||
| Fig. 6 Portions of 1H NMR spectra (D2O, 30 °C, 400 MHz) and molecular models (GFN2-xTB) representing observed complexes and their transformations. (i) 6a (0.68 mM) + SP (1 equiv.); (ii) 6a + SP + CB7 (1 equiv.) in t = 5 min; (iii) the same sample as previous in 20 days; (iv) 5a (1.0 mM) + CB7 (1 equiv.); (v) SP (1.0 mM) + CB6 (1 equiv.). For signal assignment of the 6a, see Fig. 3. The signals of the component complexed with CB6 and CB7 are labelled with † and *, respectively. Spectra are not to scale. | ||
As described above, this strained complex released CB6 that is subsequently captured by SP in concert with its SP/IB selectivity of approximately 6200:1. Even SP within the complex SP@CB7 can be taken by CB6 since the SP selectivity towards CB6/CB7 is about 3300-fold in favour of CB6. The mixture reached thermodynamic equilibrium, containing 50% of SP@CB6, 46% of 5a@CB7, and 4% of the remaining 6a, in 20 days. Similarly, more than 80% of CB6 was released from the original rotaxane 6a by CB7 when HMDA was used as the final acceptor for CB6. Indeed, as the HMDA/IB selectivity of CB6 is lower than that of SP/IB, 4% of released CB6 remained bound at the IB terminus to form the 5a@CB6IB,CB7Ad complex (for 1H NMR spectra, see Fig. S57†). The rate of the equilibration negatively correlates with the affinity of CB7 towards the competitor. Fig. S58† shows the faster equilibration for the system with SP as the thermodynamic selectivity of CB7 towards HMDA/SP is 22:1.
Conclusions
We demonstrated that steric and/or electrostatic repulsion between two cucurbit[n]uril portals arranged around a single cationic moiety is strong enough to bring a stable kinetically locked system to a dynamic state, resulting in the dethreading of the wheel. We constructed rotaxanes where the stopper moiety at one end of the axle has an additional function: it serves as an allosteric binding site with high selectivity for the trigger macrocycle. The two macrocycles, i.e. CB6 as the rotaxane wheel and CB7 as the trigger molecule, are known for strong electrostatic repulsion between their carbonyl-lined portals. We proved that the rotaxane with a 1-adamantylmethyl allosteric site (6a) is strained enough by the addition of CB7 to release CB6 at 30 °C with a half-life of 36 min. In addition, we demonstrated that the length of the allosteric binding site can be used for tuning the rate of release. Thus, ethan-1,2-diyl (6b), propen-1,3-diyl (6d) and propan-1,3-diyl (6c) linkers between the adamantane cage and the adjacent ammonium cation provided half-lives of 204, 451 and 1524 days at 30 °C, respectively. The CB7 unit occupying the longest 4-(1-adamantyl)benzyl moiety (6e) has an insignificant impact on CB6 within the rotaxane, and the complex 6e@CB7Ad is the thermodynamically preferred arrangement even at 120 °C. As the isobutyl termini of the axles represent binding site with relatively high affinity towards CB6 (K ≈ 2 × 106, 50 mM NaCl, 30 °C), CB6, once released from the rotaxane, remains bound here to form 5@CB6IB,CB7Ad. However, we demonstrated that CB6 is still available for complexation with other guests, e.g. HMDA or SP, added into the mixture.In addition to the previously published time-dependent self-sorting systems based on cucurbiturils,34 we present here the first example of the expulsion of the cucurbituril wheel from a rotaxane by a molecular signal. These results are particularly significant considering the high selectivity and large range of association constants of CBns, which makes them promising components of molecular devices (for example, molecular memories, catalysts or drug delivery systems).
Presently, we are working on the incorporation and utilisation of the CB6-releasing trigger within more complex supramolecular systems, either in multicomponent mixtures or in an intramolecular manner.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds 6a, 6b, 6c, and 6d has been deposited at the Cambridge Crystallographic Data Centre under respective numbers 2312915–2312918 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Author contributions
AZ and RV conceived and designed the experiments. All authors performed experiments and data analysis. RV wrote the draft with contributions from all co-authors. AZ, PJ, VK, JSW, ZP and MR prepared the initial version of ESI.† All authors reviewed and edited the draft and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Professor Vladimír Šindelář from Masaryk University for valuable discussion over the manuscript. The financial support of this work by the Internal Funding Agency of Tomas Bata University in Zlín, project IGA/FT/2024/001, and the University of Jyväskylä is gratefully acknowledged.Notes and references
- (a) J.-X. Liu, K. Chen and C. Redshaw, Chem. Soc. Rev., 2023, 52, 1428–1455 RSC; (b) J. Yu, M. Gaedke and F. Schaufelberger, Eur. J. Org Chem., 2023, 26, e202201130 CrossRef CAS; (c) M. Nandi, S. Bej, T. Jana and P. Ghosh, Chem. Commun., 2023, 59, 14776–14790 RSC; (d) N. Pearce, M. Tarnowska, N. J. Andersen, A. Wahrhafting-Lewis, B. S. Pilgrim and N. R. Champness, Chem. Sci., 2022, 13, 3915–3941 RSC; (e) A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed; (f) D. Sluysmans and J. F. Stoddart, Trends Chem., 2019, 1, 185–197 CrossRef CAS.
- (a) H. Y. Au-Yeung and Y. Deng, Chem. Sci., 2022, 13, 3315–3334 RSC; (b) J. Webb, J. Am. Chem. Soc., 2020, 142, 18859–18865 CrossRef PubMed; (c) J. J. Danon, D. A. Leigh, S. Pisano, A. Valero and I. J. Vitorica-Yrezabal, Angew. Chem., Int. Ed., 2018, 57, 13833–13837 CrossRef CAS PubMed.
- (a) Z. Ashbridge, S. D. P. Fielden, D. A. Leigh, L. Pirvu, F. Schaufelberger and L. Zhang, Chem. Soc. Rev., 2022, 51, 7779–7809 RSC; (b) D. H. Kim, N. Singh, J. Oh, E.-H. Kim, J. Juang, H. Kim and K.-W. Chi, Angew. Chem., Int. Ed., 2018, 57, 5669–5673 CrossRef CAS; (c) D. A. Leigh, J. J. Danon, S. D. P. Fielden, J.-F. Lemonnier, G. F. S. Whitehead and S. L. Woltering, Nat. Chem., 2021, 13, 117–122 CrossRef CAS PubMed; (d) D. A. Leigh, F. Schaufelberger, L. Pirvu, J. H. Stenlid, D. P. August and J. Segard, Nature, 2020, 584, 562–568 CrossRef CAS.
- (a) Y. Lu, Y.-X. Deng, Y.-J. Lin, Y.-F. Han, L.-H. Weng, Z.-H. Li and G.-X. Jin, Chem, 2017, 3, 110–121 CrossRef CAS; (b) Y. Lu, P. D. Dutsche, J. Kinas, A. Hepp, G.-X. Jin and F. E. Hahn, Angew. Chem., Int. Ed., 2023, 62, e202217681 CrossRef CAS PubMed.
- (a) H. Xu, M.-D. Lin, J. Yuan, B. Zhou, Y. Mu, Y. Huo and K. Zhu, Chem. Commun., 2021, 57, 3239–3242 RSC; (b) X.-Y. Chen, D. Shen, K. Cai, Y. Jiao, H. Wu, B. Song, L. Zhang, Y. Tan, Y. Wang, Y. Feng, C. L. Stern and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 20152–20160 CrossRef CAS PubMed; (c) W. Liu, C. L. Stern and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 10273–10278 CrossRef CAS PubMed; (d) T.-H. Chiang, C.-Y. Tsou, Y.-H. Chang, C.-C. Lai, R. P. Cheng and S.-H. Chiu, Org. Lett., 2021, 23, 5787–5792 CrossRef CAS PubMed.
- I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89, 5723–5724 CrossRef CAS.
- (a) P. Wu, B. Dharmadhikari, P. Patra and X. Xiong, Nanoscale Adv., 2022, 4, 3418–3461 RSC; (b) L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 9397–9404 CrossRef CAS PubMed; (c) L. Binks, S. Borsley, T. R. Gingrich, D. A. Leigh, E. Penocchio and B. M. W. Roberts, Chem, 2023, 9, 2902–2917 CrossRef; (d) E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, Adv. Mater., 2019, 32, 1906036 CrossRef.
- (a) D. A. Leigh, V. Marcos and M. R. Wilson, ACS Catal., 2014, 4, 4490–4497 CrossRef CAS; (b) C. Kwamen and J. Niemeyer, Chem.–Eur. J., 2021, 27, 175–186 CrossRef CAS PubMed; (c) A. Martinez-Cuezva, A. Saura-Sanmartin, M. Alajarin and J. Berna, ACS Catal., 2020, 10, 7719–7733 CrossRef CAS.
- A. Saura-Sanmartin, Beilstein J. Org. Chem., 2023, 19, 873–880 CrossRef CAS.
- (a) F. d'Orchymont and J. P. Holland, Chem. Sci., 2022, 13, 12713–12725 RSC; (b) A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS PubMed; (c) R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS.
- (a) X. Ling, E. L. Samuel, D. L. Patchell and E. Masson, Org. Lett., 2010, 12, 2730–2733 CrossRef CAS; (b) A. C. Catalán and J. Tiburcio, Chem. Commun., 2016, 52, 9526–9529 RSC; (c) A. J. McConnell and P. D. Beer, Chem.–Eur. J., 2011, 17, 2724–2733 CrossRef CAS PubMed; (d) H. Masai, J. Terao, T. Fujihara and Y. Tsuji, Chem.–Eur. J., 2016, 22, 6624–6630 CrossRef CAS PubMed.
- (a) A. H. G. David, P. García-Cerezo, A. G. Campaña, F. Santoyo-González and V. Blanco, Chem.–Eur. J., 2019, 25, 6170–6179 CrossRef CAS PubMed; (b) M. A. Soto and M. J. MacLachlan, Org. Lett., 2019, 21, 1744–1748 CrossRef CAS; (c) M. D. Cornelissen, S. Pilon, L. Steemers, M. J. Wanner, S. Frölke, E. Zuidinga, S. I. Jørgensen, J. I. van der Vlugt and J. H. van Maarseveen, J. Org. Chem., 2020, 85, 3146–3159 CrossRef CAS PubMed; (d) P. Branná, M. Rouchal, Z. Prucková, L. Dastychová, R. Lenobel, T. Pospíšil, K. Maláč and R. Vícha, Chem.–Eur. J., 2015, 21, 11712–11718 CrossRef PubMed; (e) H.-L. Sun, H.-Y. Zhang, Z. Dai, X. Han and Y. Liu, Chem.–Asian J., 2017, 12, 265–270 CrossRef CAS PubMed.
- (a) M. Horn, J. Ihringer, P. T. Glink and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 4046–4054 CrossRef CAS PubMed; (b) J. Yin, S. Dasgupta and J. Wu, Org. Lett., 2010, 12, 1712–1715 CrossRef CAS PubMed.
- (a) I. Yoon, M. Narita, T. Shimizu and M. Asakawa, J. Am. Chem. Soc., 2004, 126, 16740–16741 CrossRef CAS PubMed; (b) S.-Y. Hsueh, J.-L. Ko, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2011, 48, 6643–6646 CrossRef.
- (a) C.-W. Chiu, C.-C. Lai and S.-H. Chiu, J. Am. Chem. Soc., 2007, 129, 3500–3501 CrossRef CAS PubMed; (b) K. Fujimura, Y. Ueda, Y. Yamaoka, K. Takasu and T. Kawabata, Angew. Chem., Int. Ed., 2023, 62, e202303078 CrossRef CAS PubMed.
- (a) M. Denis and S. M. Goldup, Nat. Rev. Chem, 2017, 1, 0061 CrossRef CAS; (b) R. Jamagne, M. J. Power, Z.-H. Zhang, G. Zango, B. Gibber and D. A. Leigh, Chem. Soc. Rev., 2024, 53, 10216–10252 RSC.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons, Hoboken, NJ, 2016 Search PubMed.
- (a) D. Tomas, D. J. Tetlow, Y. Ren, S. Kassem, U. Karaca and D. A. Leigh, Nat. Nanotechnol., 2022, 17, 701–707 CrossRef; (b) L. Feng, Y. Qiu, Q.-H. Guo, Z. Chen, J. S. W. Seale, K. He, H. Wu, Y. Feng, O. Farha, R. D. Astumian and J. F. Stoddart, Science, 2021, 374, 1215–1221 CrossRef CAS PubMed; (c) Y. Ren, R. Jamagne, D. J. Tetlow and D. A. Leigh, Nature, 2022, 612, 78–82 CrossRef CAS PubMed; (d) Q.-H. Guo, Y. Qiu, X. Kuang, J. Liang, Y. Feng, L. Zhang, Y. Jiao, D. Shen, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 14443–14449 CrossRef CAS PubMed; (e) T. Kench, P. A. Summers, M. K. Kuimova, J. E. M. Lewis and R. Vilar, Angew. Chem., Int. Ed., 2021, 60, 10928–10934 CrossRef CAS PubMed; (f) C. C. Slack, J. A. Finbloom, K. Jeong, C. J. Bruns, D. E. Wemmer, A. Pines and M. B. Francis, Chem. Commun., 2017, 53, 1076–1079 RSC; (g) A. Tamura and N. Yui, Polym. J., 2017, 49, 527–534 CrossRef CAS; (h) A. H. G. David, P. García-Cerezo, A. G. Campaña, F. Santoyo-González and V. Blanco, Org. Chem. Front., 2022, 9, 633–642 RSC; (i) S. C. Rajappan, D. R. McCarthy, J. P. Campbell, J. B. Ferrell, M. Sharafi, O. Ambrozaite, J. Li and S. T. Schneebeli, Angew. Chem., Int. Ed., 2020, 59, 16668–16674 CrossRef CAS; (j) M. Zhang and G. De Bo, J. Am. Chem. Soc., 2019, 141, 15879–15883 CrossRef CAS PubMed.
- T. Muramatsu, Y. Okado, H. Traeger, S. Schrettl, N. Tamaoki, C. Weder and Y. Sagara, J. Am. Chem. Soc., 2021, 143, 9884–9892 CrossRef CAS PubMed.
- D. A. Leigh, L. Pirvu, F. Schaufelberger, D. J. Tetlow and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 10484–10488 CrossRef CAS PubMed.
- K.-D. Zhang, X. Zhao, G.-T. Wang, Y. Liu, Y. Zhang, H.-J. Lu, X.-K. Jiang and Z.-T. Li, Angew. Chem., Int. Ed., 2011, 50, 9866–9870 CrossRef CAS PubMed.
- Y. Sakata, R. Nakamura, T. Hibi and S. Akine, Angew. Chem., Int. Ed., 2023, 62, e202217048 CrossRef CAS PubMed.
- R. Djemili, S. Adrouche, S. Durot and V. Heitz, J. Org. Chem., 2023, 88, 14760–14766 CrossRef CAS PubMed.
- (a) A. Kermagoret and D. Bardelang, Chem.–Eur. J., 2023, 29, e202302114 CrossRef CAS PubMed; (b) K. A. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC; (c) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- (a) W. L. Mock and N. Y. Shih, J. Org. Chem., 1983, 48, 3618–3619 CrossRef CAS; (b) M. V. Rekharsky, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Supramol. Chem., 2007, 19, 39–46 CrossRef CAS.
- (a) P. Branná, J. Černochová, M. Rouchal, P. Kulhánek, M. Babinský, R. Marek, M. Nečas, I. Kuřitka and R. Vícha, J. Org. Chem., 2016, 81, 9595–9604 CrossRef PubMed; (b) M. K. Sinha, O. Reany, M. Yefet, M. Botoshansky and E. Keinan, Chem.–Eur. J., 2012, 18, 5589–5605 CrossRef CAS PubMed; (c) Y.-C. Liu, W. M. Nau and A. Hennig, Chem. Commun., 2019, 55, 14123–14126 RSC.
- J. Tomeček, A. Čablová, A. Hromádková, J. Novotný, R. Marek, I. Durník, P. Kulhánek, Z. Prucková, M. Rouchal, L. Dastychová and R. Vícha, J. Org. Chem., 2021, 86, 4483–4496 CrossRef PubMed.
- Deposition numbers 2312915 (for 6a), 2312916 (for 6b), 2312917 (for 6c), and 2312918 (for 6d) contain the supplementary crystallographic data for this paper.
- (a) C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e01493 Search PubMed; (b) C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- (a) L. Yuan, R. Wang and D. H. Macartney, J. Org. Chem., 2007, 72, 4539–4542 CrossRef CAS PubMed; (b) I. W. Wyman and D. H. Macartney, J. Org. Chem., 2009, 74, 8031–8038 CrossRef CAS PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 20737–20742 CrossRef CAS PubMed.
- (a) I. Neira, M. D. García, C. Peinador and A. E. Kaifer, J. Org. Chem., 2019, 84, 2325–2329 CrossRef CAS PubMed; (b) S. Senler, B. Cheng and A. E. Kaifer, Org. Lett., 2014, 16, 5834–5837 CrossRef CAS PubMed; (c) M. H. Tootoonchi, S. Yi and A. E. Kaifer, J. Am. Chem. Soc., 2013, 135, 10804–10809 CrossRef CAS PubMed; (d) X. Ling and E. Masson, Org. Lett., 2012, 14, 4866–4869 CrossRef CAS PubMed; (e) E. Masson, X. Lu, X. Ling and D. L. Patchell, Org. Lett., 2009, 11, 3798–3801 CrossRef CAS PubMed; (f) G. Celtek, M. Artar, O. A. Scherman and D. Tuncel, Chem.–Eur. J., 2009, 15, 10360–10363 CrossRef CAS PubMed; (g) P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Single-crystal XRD data (CIF), experimental procedures, characterisation data, K values and their determination via ITC and 1H NMR spectroscopy. CCDC 2312915–2312918. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03970j |
| This journal is © The Royal Society of Chemistry 2025 |