
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulation of STING G-quadruplex for rescuing cellular senescence and Aβ phagocytic capacity of microglia†
Heying
Yuan‡
ab,
Jie
Yang‡
ab,
Geng
Qin
ab,
Yue
Sun
ab,
Chuanqi
Zhao
 ab,
Chunyu
Wang
c,
Jinsong
Ren
ab and
Xiaogang
Qu
*ab
ab,
Chunyu
Wang
c,
Jinsong
Ren
ab and
Xiaogang
Qu
*ab
aLaboratory of Chemical Biology and State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: xqu@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, Anhui 230029, China
cState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun 130012, China
First published on 27th November 2024
Abstract
Alzheimer's disease (AD), the most common form of dementia, affects millions of people worldwide and its cause is very complicated. Besides the classical amyloid cascade hypothesis, oxidative stress, metal ion imbalance, cellular senescence and neuroinflammation are also considered crucial triggers of AD. Therefore, therapeutic strategies other than inhibiting Aβ deposition are very promising. As a crucial innate immune pathway, the abnormal activation of the cGAS-STING pathway in AD has attracted much attention and become a promising target for AD treatment. Here, we identify a highly conserved and stable G-quadruplex (G4) in the STING promoter region, and further verify its function in transcriptional inhibition of STING by using CRISPR technology to precisely target STING G4. Intriguingly, down-regulation of STING expression can alleviate cellular senescence and restore the Aβ phagocytic capacity of microglia. Our results highlight the compelling therapeutic potential of STING promoter G4 for regulation of the abnormal activation of the cGAS-STING pathway in AD. Different from the existing therapeutic strategies for AD, this work provides an alternative way of targeting the functional gene secondary structure, such as the STING promoter region, which may promote the design and synthesis of drug candidates for AD.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that commonly occurs in the elderly population.1 It is characterized by the accumulation of amyloid-β (Aβ) in plaques, the aggregation of hyperphosphorylated tau protein in neurofibrillary tangles and neuroinflammation, which ultimately lead to neurodegeneration and cognitive decline.2,3 Neuroinflammation, mitochondrial dysfunction and aging are suggested as the main risk factors of AD.4–9 Extensive efforts have been devoted to slowing down disease progression.10,11 Nevertheless, clinical drug treatments still suffer from unsatisfactory and limited effects for AD patients and are even accompanied by severe side effects.3,10,11 Therefore, it is important and urgent to further study the pathogenic mechanism of AD and explore new therapeutic strategies for AD treatment.The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, as the principal effector of cell's response to abnormal cytoplasmic double-stranded DNA (dsDNA), establishes an effective innate immune response by activating the transcription of interferon-stimulated genes (ISGs) and proinflammatory cytokines, which play a significant role in the organism's response to tissue damage and pathogen invasion.12 Moreover, it is also involved in diverse cellular processes, such as DNA damage repair, autophagy, protein synthesis, cellular condensation, senescence and cell death.13,14 Many recent studies have demonstrated that inhibiting the abnormal activation of the cGAS-STING pathway can prevent the progression of AD and cellular senescence.9,15,16 However, current regulation of the cGAS-STING pathway has mainly focused on its activation for tumor immunotherapy.17–19 It is imperative to explore new methods for suppressing the cGAS-STING pathway for the treatment of AD.
G-quadruplexes (G4s) are nonclassical secondary structures formed in the G-rich regions of DNA and RNA sequences and are widely found at telomere ends, genomic promoter regions and splice recombination sites.20 They have attracted extensive attention due to their unique conformation and crucial cellular functions. Among them, the regulation of G4s has been identified as a method of regulating numerous signal pathways and plays a significant role in the treatment of diseases. For example, naphthalenediimide derivatives restrained the RAS/MEK/ERK and PI3K/AKT pathways by inducing the conformational transition of the Epidermal Growth Factor Receptor (EGFR) promoter towards a G4 structure, broadening the treatment methods for metastatic castration-resistant prostate cancer (mCRPC).21 The G4 structure stabilizer developed by Jing-Jer Lin's group effectively repressed the migration and invasion of tumor by suppressing the Wnt1 signal pathway.22 In addition, the G4 ligand 20A balances apoptosis and senescence in cancer cells through the modulation of the ATM/autophagy pathway,23 while the regulation of the G4 structure in the Hypoxia Inducible Factor 1 Alpha (HIF1α) promoter region by the benzo[c]phenanthridine derivative M3 can effectively reduce the expression of VEGF and GluT1, which are downstream genes of the HIF1α hypoxia signal pathway, shedding light on the treatment of tumor.24 Thus, targeting G4 is an attractive and promising strategy for the regulation of cell signal pathways.
In this work, we found a highly conserved and stable G4 in the STING promoter region, and identified its function in transcriptional inhibition of STING by using Clustered Regularly Interspersed Short Palindromic Repeats (CRISPR) technology to precisely target STING G4. The down-regulation of STING expression rescued cellular senescence and restored the Aβ phagocytic capacity of microglia. This work provides a new approach for suppression of the abnormally activated cGAS-STING pathway in AD by targeting STING G4 to rescue microglia senescence and restore Aβ phagocytic capacity Scheme 1.
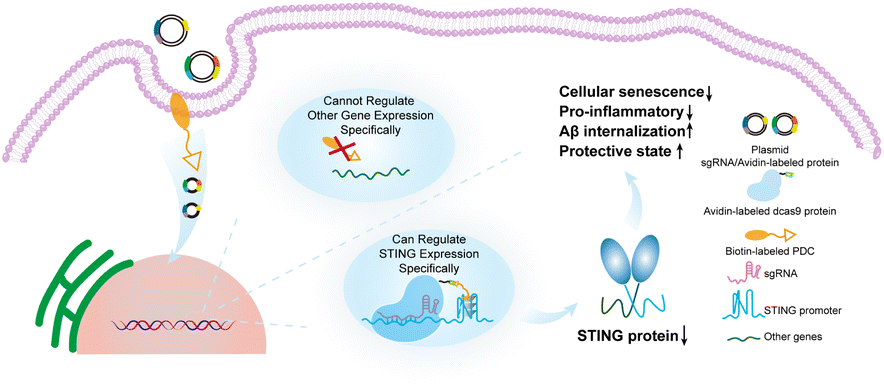 | ||
| Scheme 1 Precise regulation of the highly conserved G4 sequence in the STING promoter through CRISPR technology for the treatment of Alzheimer's disease. | ||
Results and discussion
Bioinformatics sequence analysis reveals that guanine (G)-rich sequences capable of forming G-quadruplexes (G4s) are widespread in the human genome, particularly within 1 kb upstream of gene transcription start sites. These potential G4-forming sequences (PQSs) within promoter regions directly participate in regulating transcription levels.25 Considering the significant role played by G4s in gene regulation within promoter regions, we conducted an analysis of the PQSs within the promoter region of the STING gene. First, we screened all G4s in the STING promoter region using three independent G4s prediction tools, QGRS26 and Pqsfinder,27 and G4Hunter (G4H).28 As shown in Fig. 1A, fifteen PQSs were identified in the STING promoter region. By comprehensively evaluating the parameters of G4 formation, including the QGRS, Pqsfinder and G4H scores, we selected one putative PQS (PQS-1735) in the STING promoter region for further investigation. Given that cumulative mutations of STING may confer resistance to various drugs, we subsequently analyzed the conservation of PQS-1735 among different species (Fig. 1B and C).29 The conservation of a particular nucleotide is reflected by the height of the letter, which indicates the relative frequency that nucleotide at that position. The result suggested that PQS-1735 is highly conserved among different species, indicating its potential for further study.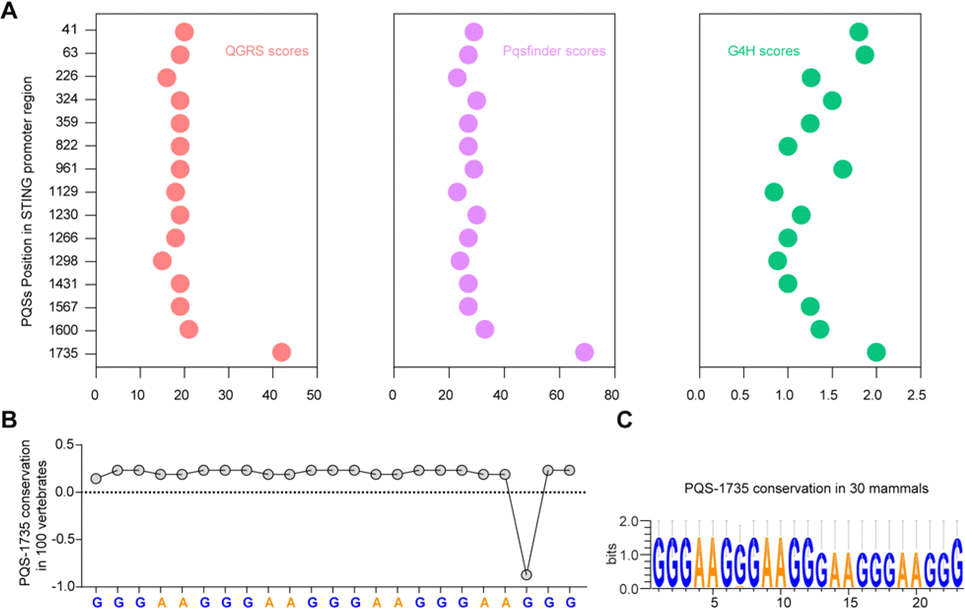 | ||
| Fig. 1 Identification and annotation of G4s in the STING promoter. (A) The scores of putative G4s in the STING promoter, which are calculated by using QGRS, Pqsfinder and G4H, respectively. (B and C) The analysis of the conservation of the selected PQS across different species. | ||
To prove the formation of G4, we first synthesized the wild-type PQS-1735 (STING-WT) and its mutant (STING-Mut, with G/A mutations) (Fig. 2A), and then adopted several methods to verify whether STING-WT could form a G4 structure. First, by conducting native polyacrylamide gel electrophoresis (PAGE) experiments, we discovered that STING-WT displayed a faster migrating band, indicating that STING-WT forms a compact secondary structure (Fig. 2B). Meanwhile, the fluorescence resonance energy transfer (FRET) assays further confirmed the folding of STING-WT (Fig. 2C).30 Then, the capability of STING-WT to fold into G4 was demonstrated by fluorescence turn-on assays using the classical G4 fluorescent ligand, N-methyl mesophorphyrin IX (NMM), as evidenced by the enhanced fluorescence level of STING-WT, but not its mutant, in K+ buffer (Fig. 2D). Subsequently, we investigated the G4 formation of STING-WT through circular dichroism (CD) spectroscopy. CD spectra showed a negative peak at around 240 nm and positive peaks at both 260 nm and 290 nm (Fig. 2E), which is characteristic of the hybrid G4 structure, suggesting that STING-WT folded into a hybrid G4 topology.31,32 Meanwhile, 1H nuclear magnetic resonance (NMR) was performed to confirm STING-WT G4 formation. Chemical shifts at 10.5–12.0 ppm are characteristic of the Hoogsteen hydrogen interactions between the guanines in G-tetrads and are considered indicative of the G4 structure.33 Obviously, the 1H- NMR spectroscopy of STING-WT showed distinct imino proton peaks in the G4 characteristic region, further confirming the formation of STING-WT G4 (Fig. 2F). According to the above results, we can conclude that STING-WT forms the hybrid G4 structure with three G-tetrad layers. In addition, we performed stopped-flow assays to explore the kinetic folding process of STING-WT G4. It was shown that STING-WT G4 formed within 25 seconds, indicating moderate folding kinetics (Fig. 2G). Besides, we conducted G4 chromatin immunoprecipitation (G4-ChIP) with BG4 to detect the formation of STING G4 in cells. We found that compared with the negative control groups TMCC2 and NFASC, STING promoter fragments with STING-WT sequences were significantly pulled-down by BG4 (Fig. 2H), indicating that STING G4 can form in cells. In conclusion, the results confirmed that the STING-WT sequence can fold into stable G4s both in vitro and in cells.
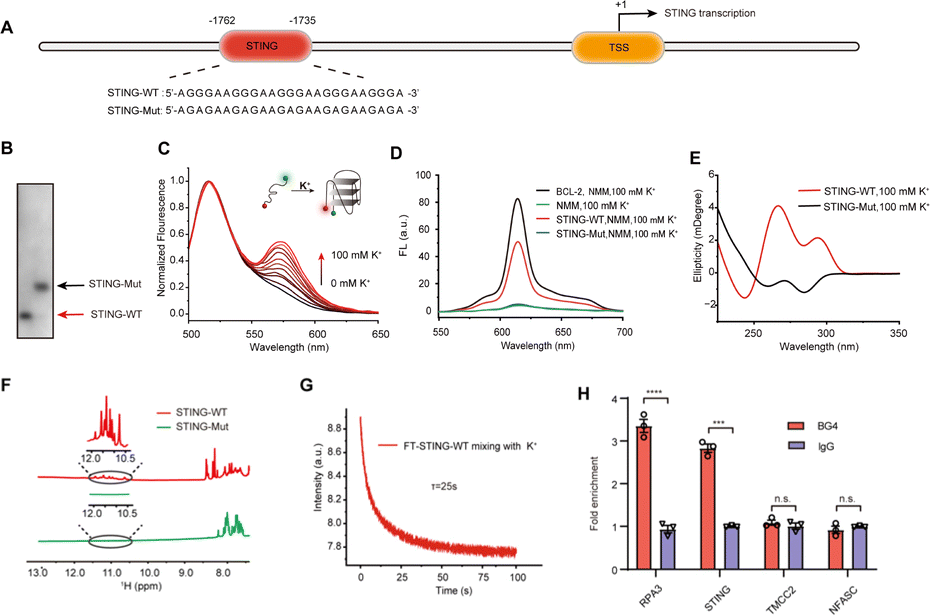 | ||
| Fig. 2 Characterization of STING G4 formation in vitro. (A) The location of G4 in STING promoter regions. (B) Native polyacrylamide gel electrophoresis analysis of STING-WT and its mutant. Lane 1, STING-WT (2 μM); Lane 2, STING-Mut (2 μM). (C) FRET spectra of fluorescence-labeled STING-WT(FT-STING-WT) under different K+ conditions. (D) Fluorescence turn-on assays of STING-WT and its mutant. DNA samples (1 μM) were mixed with NMM (2 μM) under 100 mM K+. BCL-2 G4 was used as the positive control. (E) CD spectra of STING-WT (15 μM) and STING-Mut (15 μM) under 100 mM K+. (F) 1H-NMR spectra of STING-WT (1 mM) and STING-Mut (1 mM) under 100 mM K+. (G) Typical stopped-flow trace of the FT-STING-WT sample mixed with 200 mM K+ buffer. (H) G4 chromatin immunoprecipitation (G4-ChIP) for detecting the formation of the STING promoter in HMC3 cells. Error bars represent SEM, the standard error of the mean. ***P < 0.001, ****P < 0.0001. | ||
Recently, we proposed to combine CRISPR technology with G4 regulation strategies to target specific DNA G4, which only requires changing the target sequence in the sgRNAs to recruit G4-stabilizing ligands to selectively target the G4 of interest.34 Inspired by this, we designed a regulatory system capable of specifically targeting STING G4 to investigate the effect of STING G4 formation on gene expression. The CRISPR-dCas9 and biotin-labeled pyridodicarboxamide (Bio-PDC) system (CRISPR–PDC system) is presented in Fig. 3A. The system contains the following two parts: the identifier module to target the sequence selectively using CRISPR-dCas9 and Bio-PDC as the G4 regulator. The modification of biotin enabled a convenient and accessible conjugation process with avidin-labeled dCas9 protein. First, we evaluated the interaction of Bio-PDC with STING G4. As shown in Fig. 3B, the thermal stability of STING G4 was significantly enhanced by Bio-PDC, indicating its strong interaction with G4. Then, we designed two guide RNAs (sg1 and sg2, Fig. S1†) to examine the possible effect of G4 formation on STING expression with high selectivity. Before performing cell experiments, we first evaluated the cytotoxicity of Bio-PDC using CCK-8 assay. As shown in Fig. S2,† the cytotoxicity of Bio-PDC on HEK293T cells was negligible in the range of 0.1–4.0 μM. Then, using RT-qPCR and western blot assays, we found that both the mRNA and protein levels of STING decreased, indicating the inhibition of STING expression by the CRISPR–PDC system (Fig. 3C and D). In addition, to further verify the suppressive effect of promoter G4 formation on gene expression, we inserted the STING promoter in the 5′UTR of the luciferase gene sequence to construct a luciferase reporter system (pGL3-STI vector, Fig. S5†). Using the luciferase reporter system, our results suggested that the formation of STING G4 led to the downregulation of gene expression in cells and the presence of the CRISPR-dCas9 system allowed PDC to specifically target STING G4 (Fig. S6†), consistent with the above RT-qPCR assays. All these results demonstrated that the CRISPR–PDC system could effectively stabilize STING G4 and impede STING expression in living cells.
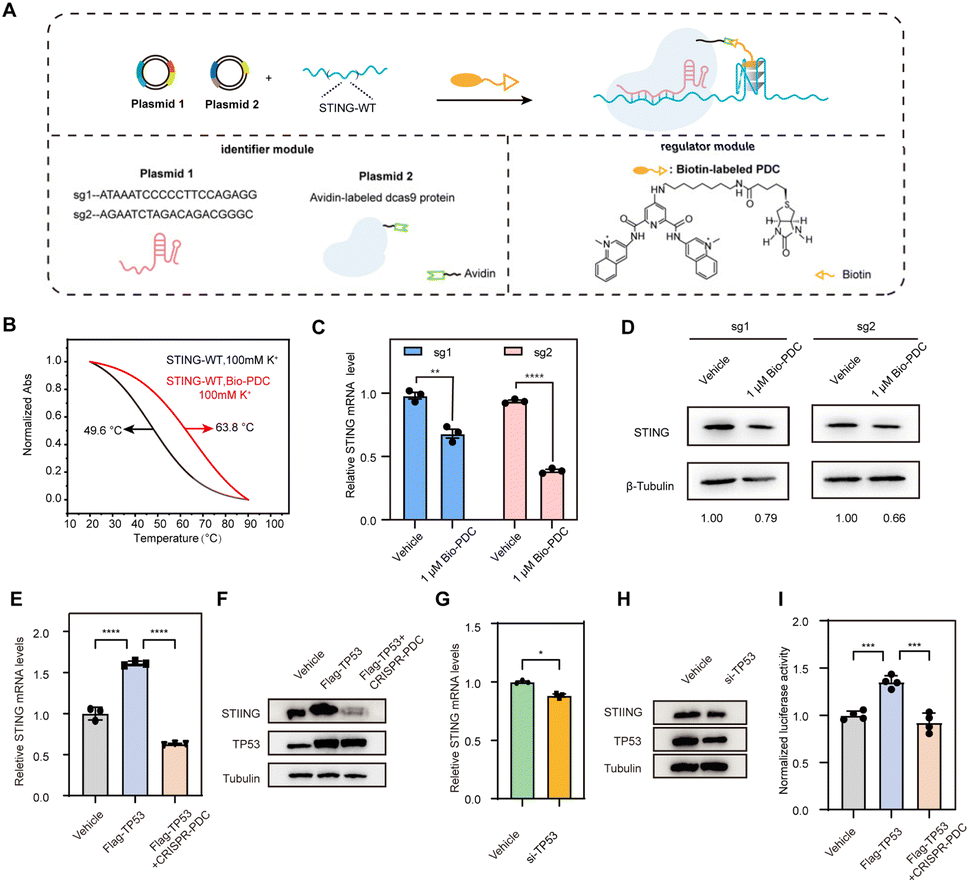 | ||
| Fig. 3 Effects of the G4-specific ligand on the stabilization of STING-WT G4 and gene expression. (A) Schematic diagram of the CRISPR–PDC system. PDC accurately regulates STING G4 through biotin–avidin interaction between PDC and dCas9 protein. (B) Normalized UV melting curves of STING-WT (2 μM) and STING-Mut (2 μM) treated with Bio-PDC (3 μM) under 100 mM K+. (C and D) The inhibition of STING expression in HEK293T cells with CRISPR–PDC (1 μM) treatment for 48 h, detected using RT-qPCR assays (C) and western blot assays (D). (E and F) CRISPR–PDC (1 μM) treatment inhibited TP53 mediated STING transcriptional activation after 48 h, as detected using qRT-PCR assays (E) and western blot assays (F). (G and H) TP53 siRNA reduced the transcription level of STING, as detected using qRT-PCR assays (G) and western blot assays (H) after 48 h of transfection. (I) Luciferase activity was reduced following treatment with the CRISPR–PDC system for 48 h. Data are shown as mean ± SEM of three independent experiments, two-tailed Student's t-test. *P < 0.05, **P < 0.01,***P < 0.001, and ****P < 0.0001. | ||
To provide more convincing evidence for the transcriptional repression caused by STING G4 formation, we explored the potential mechanism through which the formation of G4 reduces STING expression. After predicting the transcription factors of STING through bioinformatics methods, we discovered that there was a statistically positive correlation between the expression of TP53 and STING in the brain (Fig. S7 and S8†), indicating that TP53 may bind to the STING promoter region to activate STING transcription. To demonstrate the positive relationship between TP53 and STING in HMC3 cells, RT-qPCR assays and western blot assays were conducted. The results indicated that the overexpression of TP53 activated STING transcription (Fig. 3E and F) while the loss of TP53 inhibited STING transcriptional activation (Fig. 3G and H). Furthermore, treatment with CRISPR–PDC could significantly restrain the transcription activation of STING mediated by TP53 (Fig. 3E and F). Next, we used the constructed luciferase reporter system to further confirm the transcriptional repression mediated by STING G4 formation. As shown in Fig. 3I, CRISPR–PDC treatment significantly reduced the up-regulation of luciferase expression induced by TP53 overexpression. Collectively, our results suggest that the STING G4 formation mediated by CRISPR–PDC treatment repressed gene expression by interfering with TP53 binding to the promoter region.
Given the sequence specificity, the CRISPR–PDC system should suppress STING expression rather than other genes containing G4s (Fig. 4A). To confirm this, we conducted RT-qPCR assays to quantify the STING mRNA levels with or without CRISPR-mediated specificity. HEK293T cells steadily transfected with an empty plasmid served as the control. As shown in Fig. 4B, CRISPR–PDC showed a stronger inhibitory effect on STING expression, indicating the satisfactory sequence specificity of the designed system. Since many G4s in the human genome have been reported, such as cKIT,35 HRAS,36 HIF1α37and KRAS,38 we studied the mRNA levels of these typical G4s treated with the designed CRISPR–PDC system using RT-qPCR assays (Fig. 4C–F and S10–S13†). Obviously, the application of sg2 had little effect on the suppression of gene expression for other genes. Collectively, compared with other genes, the STING mRNA showed much more down-regulation due to the sequence specificity of the CRISPR–PDC system. These results indicate that the designed CRISPR–PDC system displayed the selective regulation of STING G4.
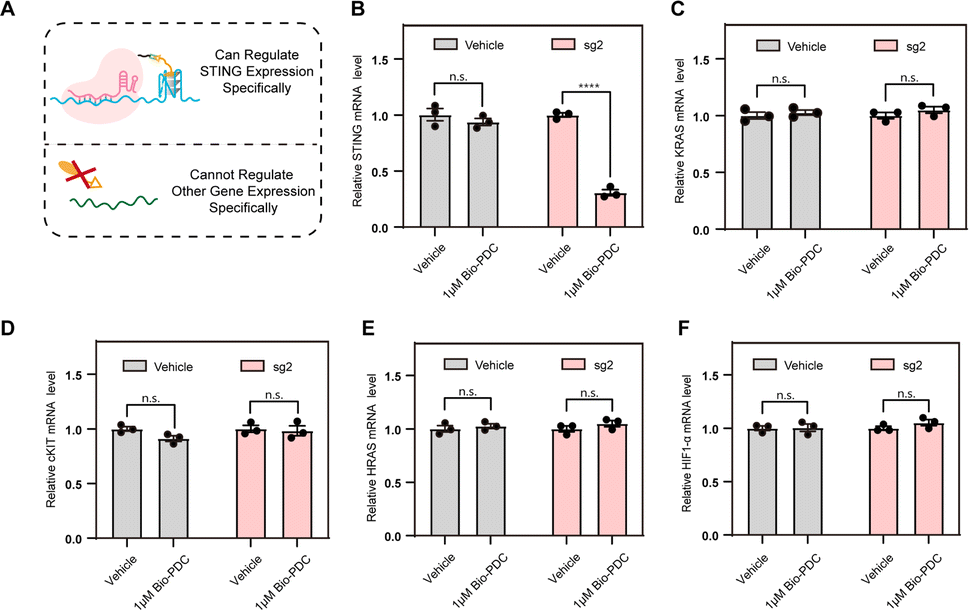 | ||
| Fig. 4 Evaluation of the CRISPR–PDC system as a gene-specific G4-targeting strategy in cells. (A) Schematic diagram of the specific regulation by the CRISPR–PDC system. (B) Comparison of Bio-PDC and CRISPR–PDC systems in inhibiting STING expression, as detected using RT-qPCR assays. The cells were collected after 48 h of treatment. (C–F) The expression levels of cKIT, HRAS, HIF1α and KRAS treated with or without the CRISPR–PDC system measured using RT-qPCR assays, respectively. Data are shown as mean ± SEM of three independent experiments, two-tailed Student's t-test. n.s.: not significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
Aging, characterized by cellular senescence, is a primary risk factor for multiple neurodegenerative disorders, such as AD.8 Increased senescent microglia, the senescence-associated secretory phenotype (SASP) and senescence-associated β-galactosidase activity (SA-β-gal) have been observed in the brain tissue of AD patients.39 Therefore, senolytic therapy based on the removal of senescent cells is expected to show beneficial effects in the treatment of AD. Zhang et al. reported that the selective removal of senescent cells from AD mice helps reduce neuroinflammation and Aβ accumulation and improves cognitive deficits, confirming the efficacy of senolytic therapy in AD treatment.40 Recent studies have reported that the cGAS-STING pathway might be a potential target for the regulation of cellular senescence in addition to a key innate immune pathway of organisms.9,41 Thus, we speculate that the STING G4 formation mediated by the CRISPR–PDC system may influence cellular senescence in AD. Microglial cells were used for further studies because of their function of maintaining central nervous system homeostasis and their close relationship with neurodegeneration.42 Etoposide (Eto) has been often used to induce DNA damage and cellular senescence, serving as a paradigm model for studying age-related disease.43 So, in our studies, human microglial cells (HMC3) were treated with Eto (3 μM) with or without the CRISPR–PDC system for 48 h. Obviously, up-regulation of STING gene expression induced by Eto was observed, which was effectively mitigated by CRISPR–PDC treatment at both mRNA and protein levels in senescent cells (Fig. 5A and B).9 Subsequently, to investigate whether CRISPR–PDC treatment could alleviate cellular senescence, we conducted RT-qPCR assays and found that the mRNA levels of senescence markers, CDKN2B, p16INK4a and p21, all decreased compared to cells treated with Eto alone (Fig. 5C). Furthermore, the SASP, including proinflammatory cytokines and chemokines such as IL1β, IL6, TNF-α and CXCL10, as one of the main characteristics of senescence, was down-regulated obviously after CRISPR–PDC treatment (Fig. 5D), further supporting that CRISPR–PDC could reduce cellular senescence. The result of the SA-β-gal staining assay further supported the above conclusion (Fig. S14†). To further confirm that CRISPR–PDC reduces cellular senescence by decreasing STING levels, we used siRNA to inhibit STING expression (Fig. S15†) and the results displayed that the mRNA levels of senescence markers and SASP could not be further reduced by CRISPR–PDC in STING-deficient cells (Fig. 5C and D), thus indicating that the suppression of cellular senescence is achieved through STING inhibition mediated by the CRISPR–PDC system.
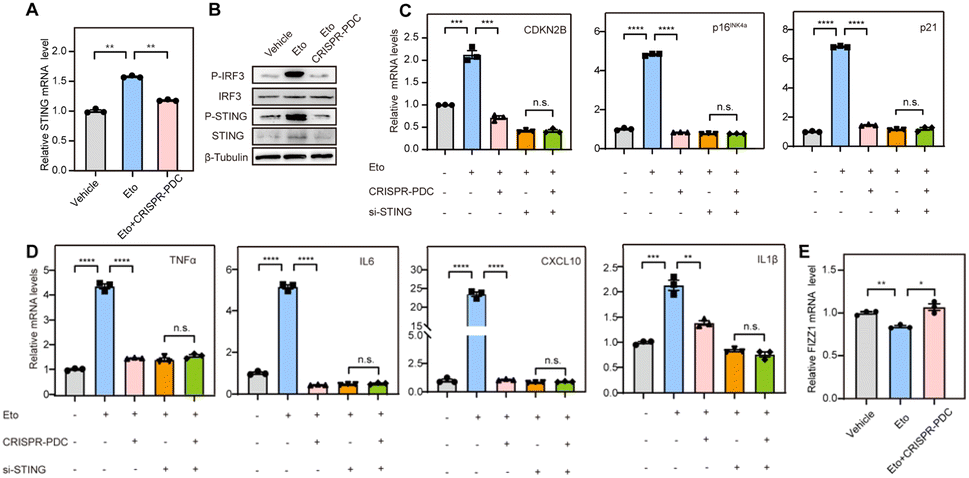 | ||
| Fig. 5 STING G4 formation induced by the CRISPR–PDC system attenuates cellular senescence in AD. Cells were treated with Vehicle, Eto (3 μM for 48 h) or/and the CRISPR–PDC system (1 μM) for 48 h in the following experiments. (A and B) The expression of STING under different conditions in HMC3 cells, detected using RT-qPCR assays (A) and western blot assays (B). (C–E) The mRNA levels of senescence markers(C), SASP (D) and protective microglia markers (E) in HMC3 cells detected using RT-qPCR assays. Data are shown as mean ± SEM of three independent experiments, two-tailed Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
As resident phagocytes in the central nervous system, microglia cells can phagocytose cell debris and Aβ aggregates to improve neuronal activity and maintain cerebral homeostasis under normal conditions.44 However, recent studies have demonstrated that the anti-inflammatory and phagocytic functions of microglial cells are impaired in AD.45–47 Thereby, restoring anti-inflammatory and phagocytosis functions of microglia cells is of great significance for delaying neurodegenerative progression. Microglia cells exhibit anti-inflammatory activity by up-regulating the expression of Found in inflammatory zone 1 (FIZZ1), which implies that FIZZ1 can be used as a marker for the neuroprotective state of microglia.48 Therefore, we assessed the expression level of FIZZ1 mRNA after CRISPR–PDC-treatment. As shown in Fig. 5E, the CRISPR–PDC-treatment clearly enhanced the level of FIZZ1 mRNA in HMC3 cells compared to the group where senescence was induced by ETO, indicating the recovery of the anti-inflammatory function of microglia cells. Lastly, we visualized Aβ internalization in HMC3 cells using a confocal laser scanning microscope (CLSM) (Fig. S16†). As shown in Fig. S16,† microglia basically lost the ability to phagocytose Aβ after Eto treatment, while CRISPR–PDC treatment restored the phagocytosis of Aβ in contrast to the ETO-only treatment group, indicating that the proposed CRISPR–PDC strategy could rescue the dysfunction of aging microglia.49 Collectively, the results suggested that the CRISPR–PDC system, with the ability to down-regulate STING expression, can attenuate microglial dysfunction by rescuing cellular senescence.
It is worth mentioning that a number of small-molecule inhibitors or degraders targeting STING have gradually emerged after continued efforts. Several of these molecules are currently in different stages of preclinical development and demonstrate varying efficacy against distinct STING mutants. However, adaptive resistance and reactivation of STING signaling after their treatment may occur as these molecules are occupancy driven. We demonstrated that CRISPR-guided PDC effectively stabilizes STING G4 to inhibit STING transcription, suggesting its promising potential for targeted regulation of STING at transcriptional levels. Thus, targeting STING G4 is an alternative strategy, which can not only complement existing inhibitors but also facilitate the realization of the full potential of modulating the STING pathway. Furthermore, in addition to STING G4, G4s also exist in the promoter regions of other key genes in the cGAS-STING pathway, such as TANK Binding Kinase 1(TBK1) and Interferon Regulatory Factor 3 (IRF3) (Fig. S17†), which implies more possibilities for the regulation of G4 and cGAS-STING pathways, providing a new insight for the development of cGAS-STING pathway inhibitors.
Conclusion
In summary, we identified and characterized G4 formation in the STING promoter region both in vitro and in living cells, and comprehensively investigated the inhibition effects of STING expression by using the precisely targeting CRISPR system. More importantly, we found that the downregulation of STING expression can alleviate cellular senescence and restore Aβ phagocytic capacity of microglia, providing a new approach for regulation of the abnormal activation of the cGAS-STING pathway in AD. This is the first example of suppressing the cGAS-STING pathway by targeting the STING G4 structure. Our work may provide new insights into the design and synthesis of drug candidates for AD by alternatively targeting the functional gene secondary structure.Data availability
The data are available upon request from the authors.Author contributions
J. R. and X. Q. conceived the project. H. Y. and J. Y. carried out the experiments, analyzed the data and drafted the manuscript. Y. S., C. W. helped collect some of the experimental data. G. Q., C. Z., J. R., and X. Q. supervised the study and revised the manuscript. All authors have given approval to the final version of the manuscript. H. Y. and J. Y. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Key R&D Program of China (2019YFA0709202 and 2021YFF1200700) and the National Nature Science Foundation of China (22437006 and 22237006).References
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS.
- D. S. Knopman, H. Amieva, R. C. Petersen, G. Chételat, D. M. Holtzman, B. T. Hyman, R. A. Nixon and D. T. Jones, Nat. Rev. Dis. Primers, 2021, 7, 33 CrossRef PubMed.
- W. K. Self and D. M. Holtzman, Nat. Med., 2023, 29, 2187–2199 CrossRef CAS PubMed.
- F. Leng and P. Edison, Nat. Rev. Neurol., 2021, 17, 157–172 CrossRef PubMed.
- C. Ising, C. Venegas, S. Zhang, H. Scheiblich, S. V. Schmidt, A. Vieira-Saecker, S. Schwartz, S. Albasset, R. M. McManus, D. Tejera, A. Griep, F. Santarelli, F. Brosseron, S. Opitz, J. Stunden, M. Merten, R. Kayed, D. T. Golenbock, D. Blum, E. Latz, L. Buée and M. T. Heneka, Nature, 2019, 575, 669–673 CrossRef CAS PubMed.
- T. Ashleigh, R. H. Swerdlow and M. F. Beal, Alzheimer's Dementia, 2023, 19, 333–342 CrossRef CAS.
- Q. Zhang, Q. Song, R. Yu, A. Wang, G. Jiang, Y. Huang, J. Chen, J. Xu, D. Wang, H. Chen and X. Gao, Adv. Sci., 2023, 10, 2204596 CrossRef CAS PubMed.
- Y. Hou, X. Dan, M. Babbar, Y. Wei, S. G. Hasselbalch, D. L. Croteau and V. A. Bohr, Nat. Rev. Neurol., 2019, 15, 565–581 CrossRef.
- Y. Hou, Y. Wei, S. Lautrup, B. Yang, Y. Wang, S. Cordonnier, M. P. Mattson, D. L. Croteau and V. A. Bohr, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2011226118 CrossRef CAS.
- E. Solopova, W. Romero-Fernandez, H. Harmsen, L. Ventura-Antunes, E. Wang, A. Shostak, J. Maldonado, M. J. Donahue, D. Schultz, T. M. Coyne, A. Charidimou and M. Schrag, Nat. Commun., 2023, 14, 8220 CrossRef CAS.
- S. Salloway, M. Farlow, E. McDade, D. B. Clifford, G. Wang, J. J. Llibre-Guerra, J. M. Hitchcock, S. L. Mills, A. M. Santacruz, A. J. Aschenbrenner, J. Hassenstab, T. L. S. Benzinger, B. A. Gordon, A. M. Fagan, K. A. Coalier, C. Cruchaga, A. A. Goate, R. J. Perrin, C. Xiong, Y. Li, J. C. Morris, B. J. Snider, C. Mummery, G. M. Surti, D. Hannequin, D. Wallon, S. B. Berman, J. J. Lah, I. Z. Jimenez-Velazquez, E. D. Roberson, C. H. Van Dyck, L. S. Honig, R. Sánchez-Valle, W. S. Brooks, S. Gauthier, D. R. Galasko, C. L. Masters, J. R. Brosch, G.-Y. R. Hsiung, S. Jayadev, M. Formaglio, M. Masellis, R. Clarnette, J. Pariente, B. Dubois, F. Pasquier, C. R. Jack, R. Koeppe, P. J. Snyder, P. S. Aisen, R. G. Thomas, S. M. Berry, B. A. Wendelberger, S. W. Andersen, K. C. Holdridge, M. A. Mintun, R. Yaari, J. R. Sims, M. Baudler, P. Delmar, R. S. Doody, P. Fontoura, C. Giacobino, G. A. Kerchner, R. J. Bateman, M. Formaglio, S. L. Mills, J. Pariente and C. H. Van Dyck, The Dominantly Inherited Alzheimer Network–Trials Unit, Nat. Med., 2021, 27, 1187–1196 CrossRef CAS.
- Q. Chen, L. Sun and Z. J. Chen, Nat. Immunol., 2016, 17, 1142–1149 CrossRef CAS.
- C. Chen and P. Xu, Trends Cell Biol., 2023, 33, 630–648 CrossRef CAS PubMed.
- Z. Zhang, H. Zhou, X. Ouyang, Y. Dong, A. Sarapultsev, S. Luo and D. Hu, Signal Transduction Targeted Ther., 2022, 7, 394 CrossRef CAS.
- X. Xie, G. Ma, X. Li, J. Zhao, Z. Zhao and J. Zeng, Nat. Aging, 2023, 3, 202–212 CrossRef CAS.
- M. F. Gulen, N. Samson, A. Keller, M. Schwabenland, C. Liu, S. Glück, V. V. Thacker, L. Favre, B. Mangeat, L. J. Kroese, P. Krimpenfort, M. Prinz and A. Ablasser, Nature, 2023, 620, 374–380 CrossRef CAS.
- A. Decout, J. D. Katz, S. Venkatraman and A. Ablasser, Nat. Rev. Immunol., 2021, 21, 548–569 CrossRef CAS PubMed.
- X. Tian, F. Xu, Q. Zhu, Z. Feng, W. Dai, Y. Zhou, Q.-D. You and X. Xu, Eur. J. Med. Chem., 2022, 244, 114791 CrossRef CAS PubMed.
- J. Liu, L. Yuan, Y. Ruan, B. Deng, Z. Yang, Y. Ren, L. Li, T. Liu, H. Zhao, R. Mai and J. Chen, J. Med. Chem., 2022, 65, 6593–6611 CrossRef CAS.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS.
- M. Recagni, M. L. Greco, A. Milelli, A. Minarini, N. Zaffaroni, M. Folini and C. Sissi, Eur. J. Med. Chem., 2019, 177, 401–413 CrossRef CAS PubMed.
- J.-M. Wang, F.-C. Huang, M. H.-J. Kuo, Z.-F. Wang, T.-Y. Tseng, L.-C. Chang, S.-J. Yen, T.-C. Chang and J.-J. Lin, J. Biol. Chem., 2014, 289, 14612–14623 CrossRef CAS.
- J. Beauvarlet, P. Bensadoun, E. Darbo, G. Labrunie, B. Rousseau, E. Richard, I. Draskovic, A. Londono-Vallejo, J.-W. Dupuy, R. Nath Das, A. Guédin, G. Robert, F. Orange, S. Croce, V. Valesco, P. Soubeyran, K. M. Ryan, J.-L. Mergny and M. Djavaheri-Mergny, Nucleic Acids Res., 2019, 47, 2739–2756 CrossRef CAS PubMed.
- H. Chen, H. Long, X. Cui, J. Zhou, M. Xu and G. Yuan, J. Am. Chem. Soc., 2014, 136, 2583–2591 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- O. Kikin, L. D'Antonio and P. S. Bagga, Nucleic Acids Res., 2006, 34, W676–W682 CrossRef CAS.
- J. Hon, T. Martínek, J. Zendulka and M. Lexa, Bioinformatics, 2017, 33, 3373–3379 CrossRef CAS PubMed.
- A. Bedrat, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2016, 44, 1746–1759 CrossRef.
- G. E. Crooks, G. Hon, J.-M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef CAS PubMed.
- J.-L. Mergny and J.-C. Maurizot, ChemBioChem, 2001, 2, 124–132 CrossRef CAS.
- A. I. Karsisiotis, N. M. Hessari, E. Novellino, G. P. Spada, A. Randazzo and M. Webba da Silva, Angew. Chem., Int. Ed., 2011, 50, 10645–10648 CrossRef CAS.
- R. del Villar-Guerra, J. O. Trent and J. B. Chaires, Angew. Chem., Int. Ed., 2018, 57, 7171–7175 CrossRef CAS.
- M. Adrian, B. Heddi and A. T. Phan, Methods, 2012, 57, 11–24 CrossRef CAS.
- G. Qin, Z. Liu, J. Yang, X. Liao, C. Zhao, J. Ren and X. Qu, Nat. Cell Biol., 2024, 26, 1212–1224 CrossRef CAS.
- M. Bejugam, S. Sewitz, P. S. Shirude, R. Rodriguez, R. Shahid and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 12926–12927 CrossRef CAS PubMed.
- A. Membrino, S. Cogoi, E. B. Pedersen and L. E. Xodo, PLoS One, 2011, 6, e24421 CrossRef CAS.
- R. De Armond, S. Wood, D. Sun, L. H. Hurley and S. W. Ebbinghaus, Biochemistry, 2005, 44, 16341–16350 CrossRef CAS PubMed.
- S. Cogoi and L. E. Xodo, Nucleic Acids Res., 2006, 34, 2536–2549 CrossRef CAS PubMed.
- N. He, W.-L. Jin, K.-H. Lok, Y. Wang, M. Yin and Z.-J. Wang, Cell Death Dis., 2013, 4, e924 CrossRef CAS PubMed.
- P. Zhang, Y. Kishimoto, I. Grammatikakis, K. Gottimukkala, R. G. Cutler, S. Zhang, K. Abdelmohsen, V. A. Bohr, J. Misra Sen, M. Gorospe and M. P. Mattson, Nat. Neurosci., 2019, 22, 719–728 CrossRef CAS.
- S. Glück, B. Guey, M. F. Gulen, K. Wolter, T.-W. Kang, N. A. Schmacke, A. Bridgeman, J. Rehwinkel, L. Zender and A. Ablasser, Nat. Cell Biol., 2017, 19, 1061–1070 CrossRef.
- T. Bartels, S. De Schepper and S. Hong, Science, 2020, 370, 66–69 CrossRef CAS.
- H. Yang, H. Wang, J. Ren, Q. Chen and Z. J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4612–E4620 CAS.
- M. Prinz, S. Jung and J. Priller, Cell, 2019, 179, 292–311 CrossRef CAS.
- B. L. Heckmann, B. J. W. Teubner, B. Tummers, E. Boada-Romero, L. Harris, M. Yang, C. S. Guy, S. S. Zakharenko and D. R. Green, Cell, 2019, 178, 536–551 CrossRef CAS.
- J. V. Pluvinage, M. S. Haney, B. A. H. Smith, J. Sun, T. Iram, L. Bonanno, L. Li, D. P. Lee, D. W. Morgens, A. C. Yang, S. R. Shuken, D. Gate, M. Scott, P. Khatri, J. Luo, C. R. Bertozzi, M. C. Bassik and T. Wyss-Coray, Nature, 2019, 568, 187–192 CrossRef CAS PubMed.
- H. Scheiblich, C. Dansokho, D. Mercan, S. V. Schmidt, L. Bousset, L. Wischhof, F. Eikens, A. Odainic, J. Spitzer, A. Griep, S. Schwartz, D. Bano, E. Latz, R. Melki and M. T. Heneka, Cell, 2021, 184, 5089–5106 CrossRef CAS PubMed.
- Z. Li, J. Xiao, X. Xu, W. Li, R. Zhong, L. Qi, J. Chen, G. Cui, S. Wang, Y. Zheng, Y. Qiu, S. Li, X. Zhou, Y. Lu, J. Lyu, B. Zhou, J. Zhou, N. Jing, B. Wei, J. Hu and H. Wang, Sci. Adv., 2021, 7, eabb6260 CrossRef CAS PubMed.
- H. Yang, X. Li, L. Zhu, X. Wu, S. Zhang, F. Huang, X. Feng and L. Shi, Adv. Sci., 2019, 6, 1901844 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04453c |
| ‡ Yuan Heying and Yang Jie contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |