
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn elastic siderophore synthetase and rubbery substrates assemble multimeric linear and macrocyclic hydroxamic acid metal chelators†
Kate P.
Nolan
,
Callum A.
Rosser
,
James L.
Wood
 ,
Josep
Font
,
Athavan
Sresutharsan
,
Joseph
Wang
,
Todd E.
Markham
,
Renae M.
Ryan
and
Rachel
Codd
*
,
Josep
Font
,
Athavan
Sresutharsan
,
Joseph
Wang
,
Todd E.
Markham
,
Renae M.
Ryan
and
Rachel
Codd
*
The University of Sydney, School of Medical Sciences, New South Wales 2006, Australia. E-mail: rachel.codd@sydney.edu.au
First published on 25th November 2024
Abstract
The trihydroxamic acid bacterial siderophore desferrioxamine B (DFOB, 1) produced by the DesABCD biosynthetic cluster coordinates metals beyond Fe(III), which identifies potential to modify this chelator type to broaden metal sequestration and/or delivery applications. Rather than producing discrete chelators by total chemical synthesis from native monomers including N-hydroxy-N-succinyl-cadaverine (HSC, 2), the recombinant siderophore synthetase from Salinispora tropica CNB-440 (StDesD) was used with different substrate combinations to produce biocombinatorial mixtures of hydroxamic acid chelators. The mixtures were screened with Ga(III) or Zr(IV) as surrogates of immunological positron emission tomography (PET) imaging radiometals 68Ga(III) or 89Zr(IV) to inform known or new coordination chemistry. The last-in-line enzyme DesD forms amide bonds between two equivalents of 2 and N-hydroxy-N-acetyl-cadaverine to produce trimeric 1. Although hexadentate 1 is the terminal product evolved for Fe(III) complexation, it was conceived amine-containing 1 might remain a viable DesD substrate for further iteration with 2 to generate higher-order hydroxamic acid multimers. Incubation of StDesD, cofactors ATP and Mg(II), and 1 and 2, generated the octadentate hydroxamic acid DFOB-HSC (3) (previously characterised and named DFO*), decadentate DFOB-(HSC)2 (4), dodecadentate DFOB-(HSC)3 (5) and tetradecadentate DFOB-(HSC)4 (6). The system with StDesD and 2 alone generated a set of linear multimers containing flanking amine and carboxylic acid groups (HSC)x-L (x = 2 (7), x = 3 (8), x = 4 (9), x = 5 (10)) and a subset of the cognate ring-closed macrocycles (HSC)x-MC (x = 3 (12), x = 4 (13), x = 5 (14), with x = 2 (11) not detected). Liquid chromatography-mass spectrometry metal screening experiments detected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes of Ga(III) or Zr(IV) and 1, 3–5, 8–10, and 12–14. Complexes of 2:1 stoichiometry were formed between Ga(III) and the high-denticity, high-cavity-volume chelators 4–6, and 14. A processive intra-cavity assembly mechanism has been posited for this flexible siderophore synthetase in delivering a large set of multimeric chelators.
:1 complexes of Ga(III) or Zr(IV) and 1, 3–5, 8–10, and 12–14. Complexes of 2:1 stoichiometry were formed between Ga(III) and the high-denticity, high-cavity-volume chelators 4–6, and 14. A processive intra-cavity assembly mechanism has been posited for this flexible siderophore synthetase in delivering a large set of multimeric chelators.
Introduction
The set of synthetic chelators used for metal binding applications in biomedicine and the environment has narrow structural diversity. Widening chemical space in any class of molecule, including metal chelators, has the potential to broaden function. The structural diversity inherent to natural product metal chelators provides a useful platform to bioengineer new analogues to expand chemical space. The natural product hydroxamic acid chelator desferrioxamine B (DFOB, 1) biosynthesized by the DesABCD enzyme cluster, is produced by many soil actinomycetes for Fe(III) acquisition.1–3 The three hard base O′,O′-bidentate hydroxamic functional groups in the 1 backbone have evolved to match the coordination demands of the hard acid Fe(III) to form a stable 1:1 octahedral complex (logK = 30.5).4,5 Early studies of the biosynthetic pathway of 16,7 identified the possibility in the current work of using recombinant forms of DesABCD to machine pools of known and new hydroxamic acid chelators. This could generate metal chelators with a range of denticities and cavity sizes to expand applications.
The biosynthesis of 1 begins with the decarboxylation of L-lysine (DesA) to produce cadaverine (1,5-diaminopentane), which is mono-N-hydroxylated (DesB) to produce N-hydroxy-cadaverine (HC). The HC intermediate is processed (DesC) with succinyl-coenzyme A (Suc-CoA) or acetyl-coenzyme A (Ac-CoA) to produce N-succinyl-HC (HSC, 2) or N-acetyl-HC (HAC, 2a), respectively.6–14 In one proposed onward pathway, the terminal siderophore synthetase DesD condenses 2a with 2 to produce the HAC-HSC heterodimer 2b, which in a second DesD cycle is condensed with a second equivalent of 2 to produce 1 (Scheme 1, orange box).7 DesD requires cofactors ATP and Mg(II) to adenylate 2 primed for nucleophilic attack from the amine group in 2a (Cycle 1) or 2b (Cycle 2).
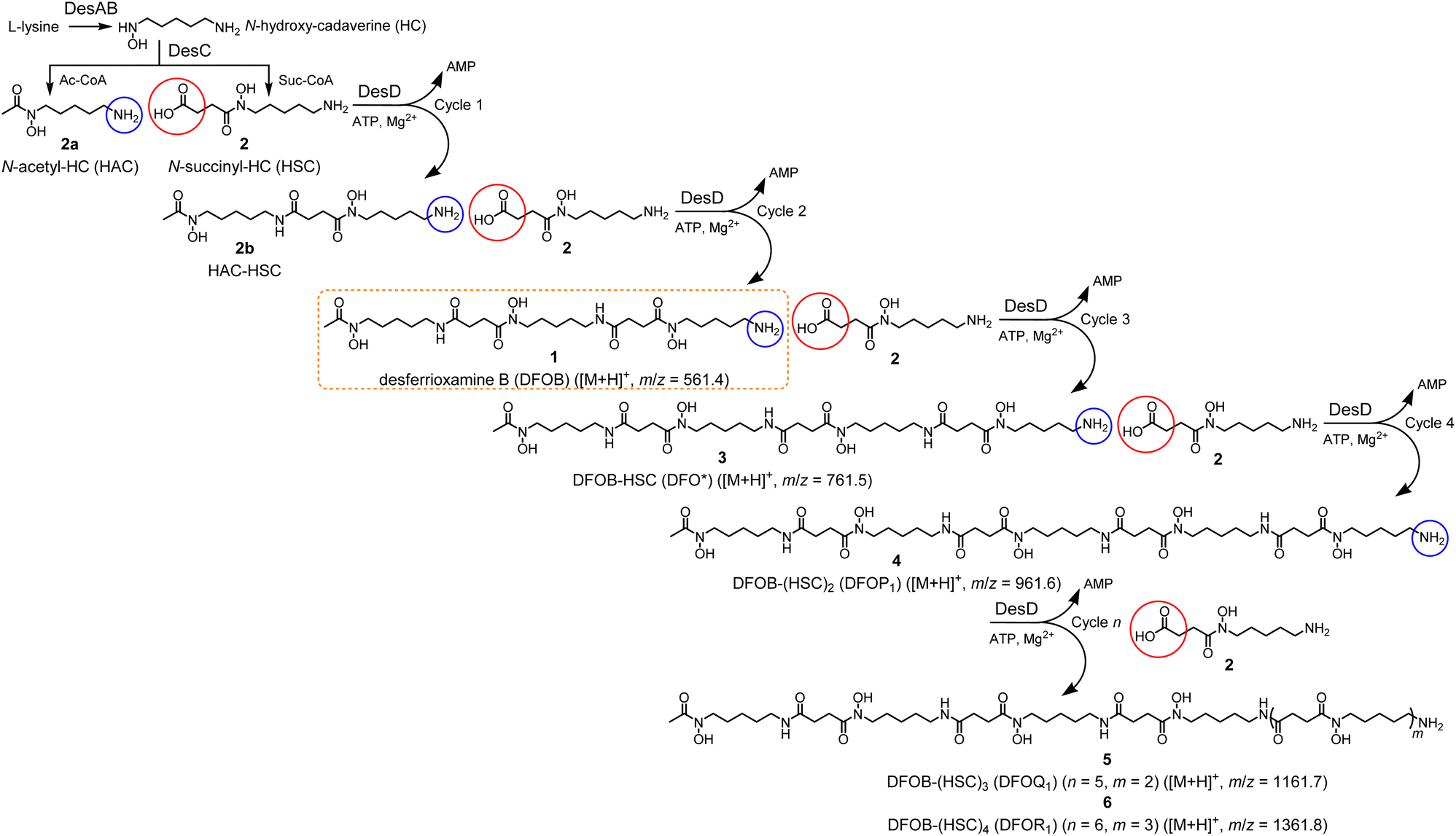 | ||
| Scheme 1 One pathway for the native biosynthesis of desferrioxamine B (DFOB, 1, orange box) involving two iterative DesD-mediated condensation steps (Cycle 1, 2) with two equivalents of the substrate HSC (2); and higher-order linear multimers (3–6) from further DesD-mediated iterations with 2. | ||
This work sought to examine the potential of using recombinant DesD from Salinispora tropica CNB-440 (StDesD) in a chemoenzymatic synthesis approach to generate biocombinatorial pools of hydroxamic acid compounds, and to probe these pools with metal ions to assess coordination chemistry. This organism was selected based on the availability of the S. tropica CNB-440 genome15 and the verified production of a wide range of linear and macrocyclic hydroxamic acid siderophores.16,17 The work showed that StDesD could use as substrates a combination of 1 and 2, or 2 alone, in multiple iterative cycles to generate known and new linear and macrocyclic hydroxamic acid compounds with higher-order multiplicities and octa-/deca-/dodeca- and tetradeca-dentate denticities, which displayed different coordination chemistry towards Ga(III) and Zr(IV). These data support StDesD as an elastic machine able to accommodate rubbery substrates to provide a facile pathway to widening the chemical space and potential function of this class of metal chelator.
Results and discussion
Biosynthetic capacity of the siderophore synthetase StDesD
This work began by interrogating whether 1, which is produced by the DesABCD biosynthetic cluster of many actinomycetes as the terminal product optimised for Fe(III) chelation,1 could remain as a nucleophile substrate in onward StDesD-mediated condensation reactions with carboxylate-bearing substrates to produce higher-order multimers. Experiments involved incubating mixtures of recombinant StDesD18 and co-factors ATP and Mg(II), together with 1 in the presence of the native substrate 2 (two-substrate system), or with 2 alone (single-substrate system), and analysing product mixtures by liquid chromatography-mass spectrometry (LC-MS) and LC-MS/MS. With StDesD in-hand and optimised in-house capacity to synthesize this substrate class including 2,19–21 the study was well positioned to establish the feasibility of using this facile method to expand the chemical space of hydroxamic acid chelators. The work sought to explore the limits of multimer assembly, expand insight into assembly mechanisms, and examine metal complexation.DFOB (1) is a substrate of StDesD
The LC-MS trace detected by total ion current (TIC) (positive ion mode‡)7,18,20 from the reaction solution containing StDesD, 1 and 2 showed multiple signals between 9–13.5 min attributable to enzyme-mediated compound production (Fig. 1a), as compared with the enzyme-free matched control which contained unreacted 1 and 2 (Fig. 1b). Signals from the StDesD system were evident in the extracted ion chromatograms (EICs) set to report the chain-extended analogues 3 (Fig. 1e) and 4 (Fig. 1g), with 3 confirmed from re-acquisition of the solution containing exogenous synthetic 3 (Fig. 1e, gray). Signals for 3 (Fig. 1f) and 4 (Fig. 1h) were absent in the enzyme-free system. These data showed 1 was not a terminal product but remained a viable substrate for StDesD-mediated iterations with 2 to assemble higher-order multimers. The iterative capacity of StDesD using 1 beyond a terminal product was evident in DesD from other actinomycete producers. The LC-MS siderophore profile of commercial 1 supplied at 95% purity from Streptomyces pilosus fermentation showed the presence of 3 and 4, and higher-order multimers consistent with multi-cycle SpDesD iterations. In this work, commercial 1 was pre-purified (Fig. S1†) to reduce the potential for confounding factors posed by the presence of multimers in the 1 substrate solution.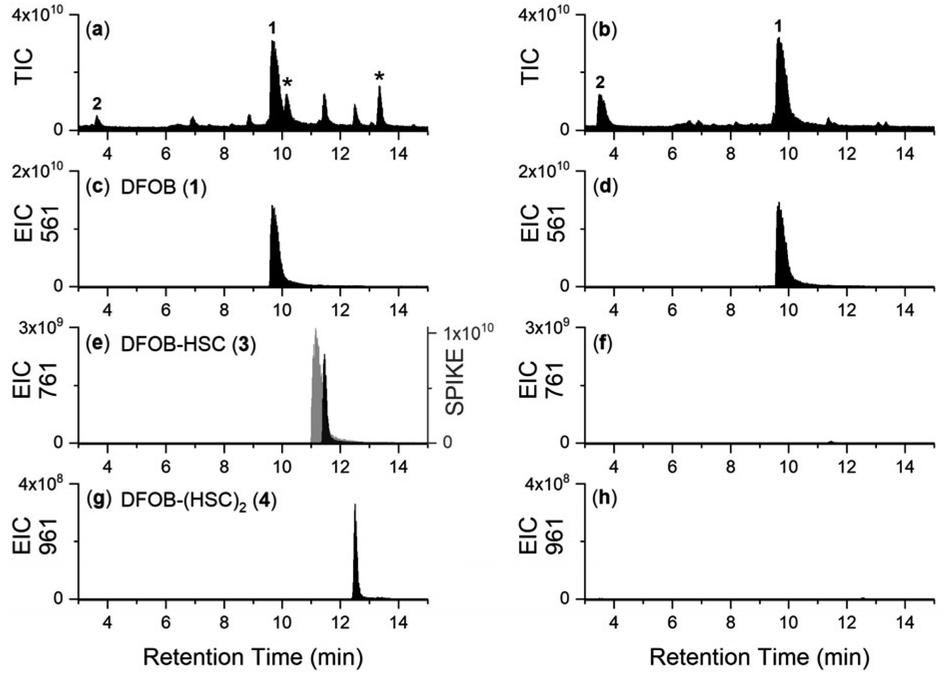 | ||
| Fig. 1 LC-MS traces from solutions of 1 and 2 with MgCl2 and ATP, incubated (37 °C, 2 h, pH 8) in the presence (a, c, e and g) or absence (b, d, f and h) of StDesD, as detected by TIC (a and b), or shown as an EIC with values set to report the [M + H]+ adducts of (c and d) 1, (e and f) 3, or (g and h) 4. The trace in gray (e) was acquired from the solution following the addition of authentic 3. Asterisked signals in (a) are due to species assembled from 2 alone. | ||
Although there is debate about the biosynthetic sequence for 1 and its multimers, only one mechanism can be invoked for producing 3 from 1 and 2, namely the condensation between the primary amine group of 1 and the activated adenylated 2 monomer. This mechanism parallels that for the condensation of 2b with 2 to form 1. Ambiguity in the assembly mechanism arises in the case of 4, which could be formed from a reaction between 3 and 2, and/or between 1 and the homodimer of 2 (equivalent to 7). Both sequences towards 4 are reasonable and are not mutually exclusive, with further insights into assembly mechanisms provided in a later section.
The upper boundary for the StDesD-mediated production of 1-based multimers was examined using a higher-sensitivity LC-MS system (Fig. 2), which showed EIC signals and MS/MS fragmentation patterns matching theoretical patterns (Fig. S2†) characteristic of 3, 4, 5 and 6, as a set of high-denticity (octa-, deca-, dodeca-, tetradeca-) siderophores. Assuming similar ionisation properties among these structural analogues, and using peak areas normalised to 3 (100%), the relative concentrations of 4, 5 and 6 were estimated as 29%, 2% and 0.1%, respectively. There was a positive correlation between the number of HC units present in 1, 3, 4, 5 and 6 and the increase in the reverse-phase LC retention time, as a correlate of the increase in compound hydrophobicity.
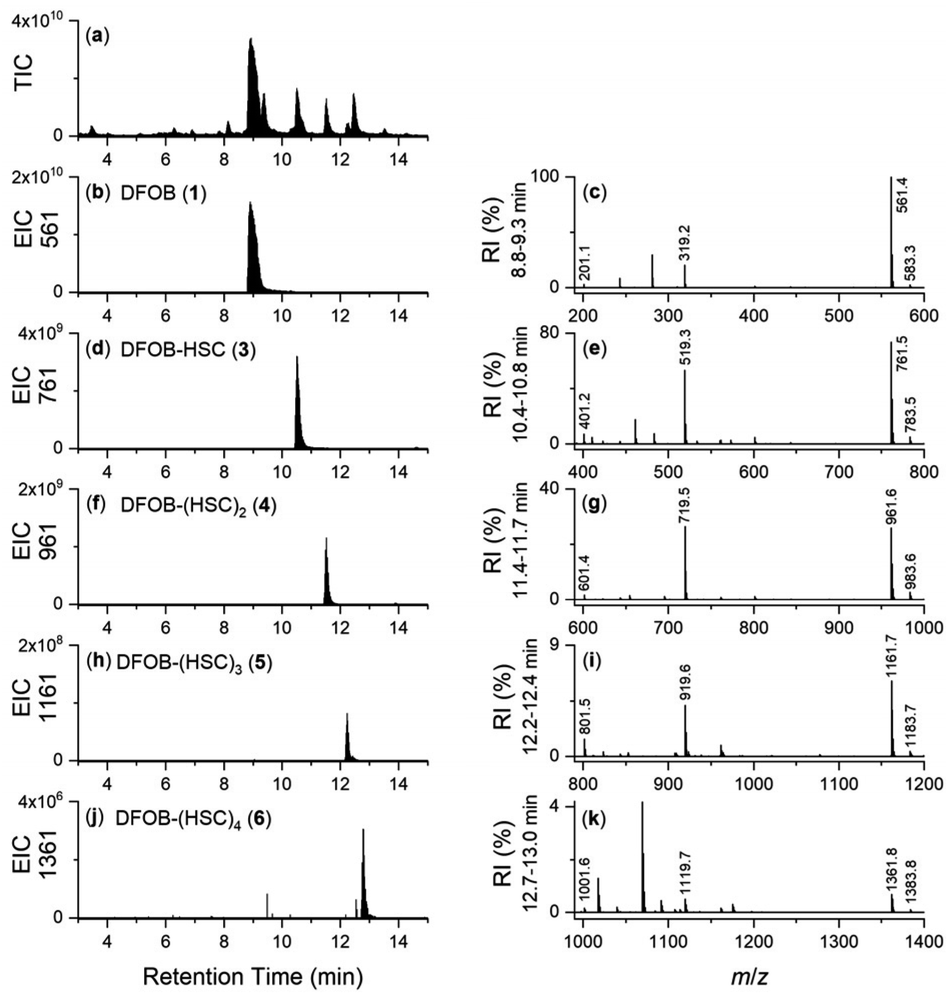 | ||
| Fig. 2 LC-MS traces from solutions of 1 and 2 with MgCl2 and ATP, incubated (37 °C, 2 h, pH 8) in the presence of StDesD, as detected by TIC (a), or shown as an EIC with values set to report the [M + H]+ adducts of (b) 1, (d) 3, (f) 4, (h) 5, or (j) 6, with signature regions of the MS fragmentation pattern aligned at right (c, e, g, i and k). | ||
HSC (2) as a single substrate of StDesD
Together with the DFOB-(HSC)x (x = 1–4) multimers, the reaction solution from StDesD, 1 and 2, generated products assembled solely from 2 (Fig. 1a, asterisked). This product profile was subsequently analysed from a reaction solution of StDesD and 2 alone (Fig. 3). Based on previous in vitro studies and knowledge of these natural products, expected compounds included linear multimers containing amine and carboxylic acid termini and the cognate ring-closed macrocycles, although the multiplicity boundary was uncertain (Scheme 2).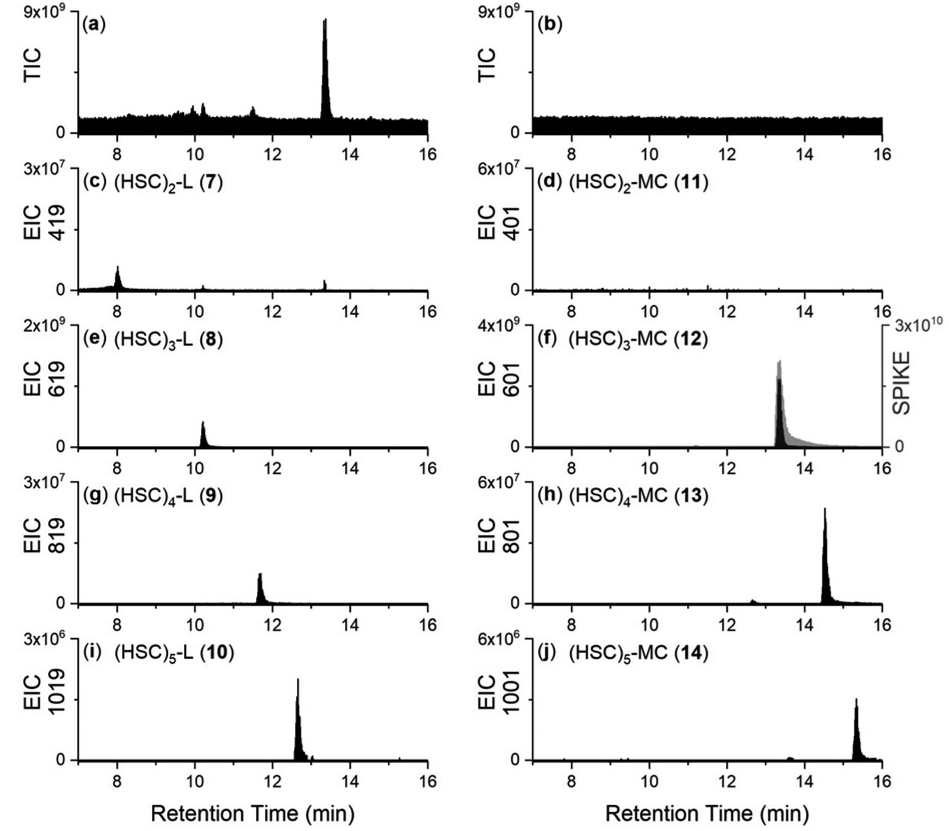 | ||
| Fig. 3 LC-MS traces from solutions of 2 with MgCl2 and ATP, incubated (37 °C, 2 h, pH 8) in the presence (a) or absence (b) of StDesD, as detected by TIC (a and b), or shown as an EIC with values set to report the [M + H]+ adducts of (c) 7, (d) 11, (e) 8, (f) 12, (g) 9, (h) 13, (i) 10, or (j) 14. The trace in gray (f) was acquired from the solution following the addition of authentic 12. | ||
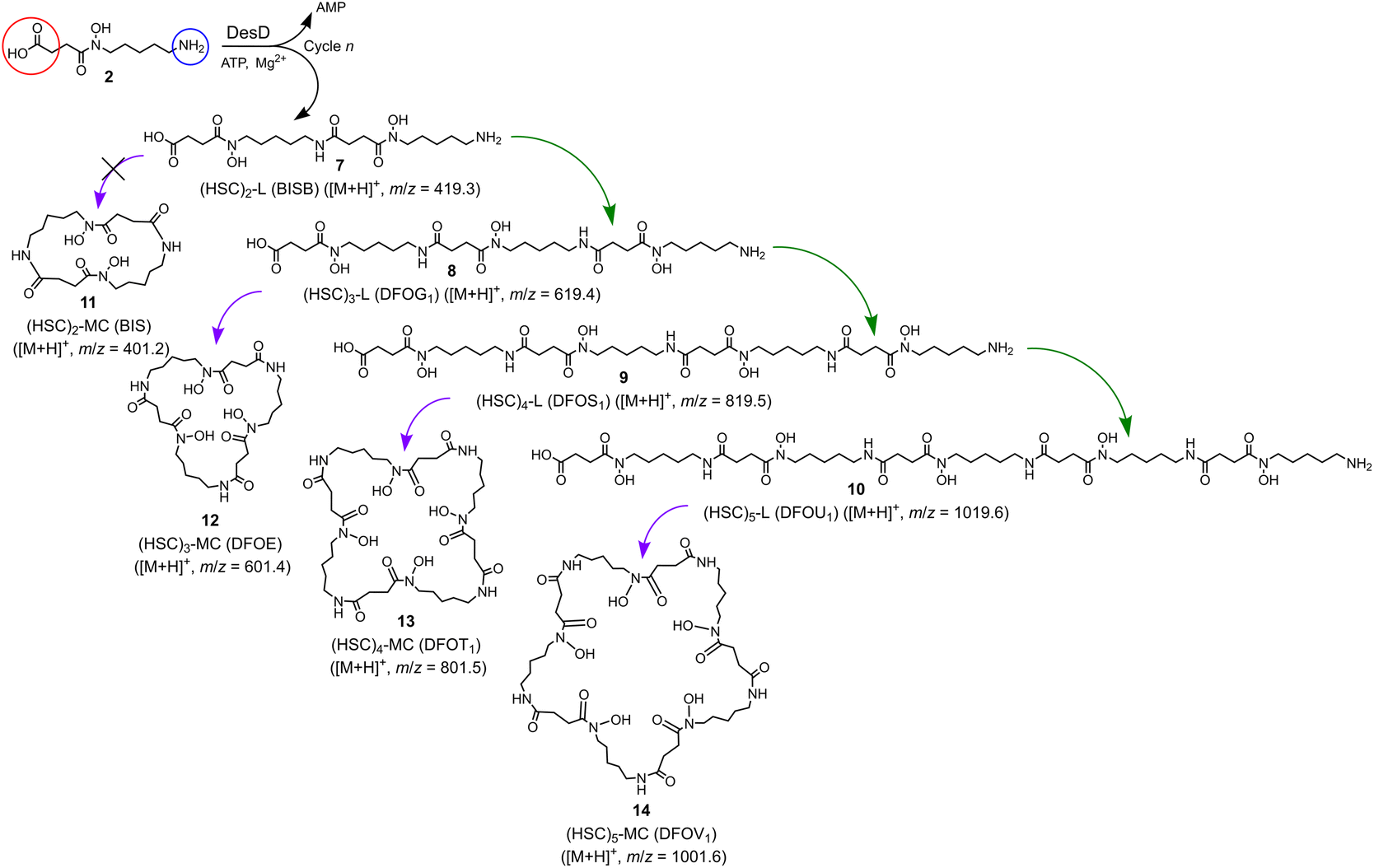 | ||
| Scheme 2 Linear (7, 8, 9, 10) and cognate macrocyclic hydroxamic acid multimers (11 (not observed), 12, 13, 14) produced from StDesD-mediated condensation reactions with 2 as sole substrate. | ||
The LC-MS trace from the StDesD and 2 system showed a major signal at tR 13.4 min attributed to the macrocycle (HSC)3-MC (desferrioxamine E (DFOE)) (12) (Fig. 3f, black; with exogenous addition of authentic 12 to the sample shown in gray), which is the macrocyclic product of linear (HSC)3-L (desferrioxamine G1 (DFOG1)) (8) which eluted at tR 10.2 min (Fig. 3e). The asterisked signals in the LC trace from the StDesD, 1 and 2 system (Fig. 1a) at 10.2 min and 13.4 min corresponded with 8 and 12, respectively. The minimal linear compound (HSC)2-L (7), known as bisucaberin B,23 was detected in the StDesD and 2 system (Fig. 3c), although the corresponding natural product bisucaberin macrocycle (HSC)2-MC (11)24,25 was not (Fig. 3d), which might indicate some strain in the macrocyclic pre-complex that prevented macrocyclisation.
The StDesD synthetase has evolved to produce siderophores optimised for Fe(III) binding,16,17 with hexadentate, macrocyclic 12 (logK = 32.5)26 ideal for this function, in accord with 12 as the major product. The system gave lower intensity signals that corresponded with (HSC)4-L (9) (Fig. 3g) and (HSC)4-MC (13) (Fig. 3h), with this latter macrocycle (known as DFOT1) previously characterised in nature,27 and in both in vitro14 and synthetic studies.28 Signals and MS/MS fragmentation patterns consistent with the linear hydroxamic acid amino-carboxylic acid pentamer (HSC)5-L (10) (Fig. 3i) and the cognate pentameric macrocycle (HSC)5-MC (14) (Fig. 3j) were detected. In the system with 2 as sole substrate under these analytical conditions, linear 10 and macrocyclic 14 appeared to mark the upper boundary of the multimeric assembly capacity of StDesD (Table 1).
| No. | Species | Alternative name | RT (min) | EIC [M + H]+ | Ref. |
|---|---|---|---|---|---|
| a The first report of DFOB-HSC named the compound DFO*. The name DFOB-HSC is used here to maintain consistency among the set of DFOB-(HSC)x multimers. b Reported in ref. 14; named in this work. c N/D not detected. | |||||
| 1 | DFOB | DFO | 8.9 | 561.4 | 29 |
| 3 | DFOB-HSCa | DFO* | 10.5 | 761.5 | 30 |
| 4 | DFOB-(HSC)2 | DFOP1 | 11.5 | 961.6 | This work |
| 5 | DFOB-(HSC)3 | DFOQ1 | 12.2 | 1161.7 | This work |
| 6 | DFOB-(HSC)4 | DFOR1 | 12.8 | 1361.8 | This work |
| 7 | (HSC)2-L | Bisucaberin B | 8.0 | 419.3 | 23 |
| 8 | (HSC)3-L | DFOG1 | 10.2 | 619.4 | 31 |
| 9 | (HSC)4-L | DFOS1b | 11.7 | 819.5 | 14 |
| 10 | (HSC)5-L | DFOU1 | 12.7 | 1019.6 | This work |
| 11 | (HSC)2-MC | Bisucaberin | N/Dc | 401.2 | 24 |
| 12 | (HSC)3-MC | DFOE | 13.4 | 601.4 | 32 |
| 13 | (HSC)4-MC | DFOT1 | 14.5 | 801.5 | 14 and 27 |
| 14 | (HSC)5-MC | DFOV1 | 15.3 | 1001.6 | This work |
Screening coordination chemistry
The biocombinatorial pools of multimeric chelators generated from StDesD-mediated synthesis using co-substrates 1 and 2 (products: 3–6)§ or 2 as sole substrate (products: 7–10, 12–14) were incubated with an excess of either Ga(III) or Zr(IV) and analysed using LC-MS to screen in situ complex formation. Both Ga(III) (logβ > 30) and Zr(IV) (logβ > 40) have been established by experiment and calculation to form 1:1 complexes with hydroxamic acid-based chelators with high stability constants.33–35
Complex formation was evident at a macroscopic level based on the change in the TIC signal profiles between the free chelator mixtures and those containing Ga(III) or Zr(IV) (Fig. 4). EIC traces were assessed for individual metal-chelator species (Chart 1) with a match between experimental and theoretical36m/z values and isotope patterns supporting the formation of known and new complexes. The structures (Chart 1) depict a single representative isomer, noting the possibility of coordination isomers depending on the combination of participant hydroxamic acid units. Species were detected as intrinsically charged adducts, as single-protonated adducts or for low-concentration species, as double-protonated adducts (Chart 1, Table 2). The coordination spheres of the native complexes would be expected to contain additional ancillary ligands (aqua, hydroxyl) which as labile species would be displaced under the LC-MS acquisition conditions, as shown in previous work on metal-hydroxamic acid speciation.37
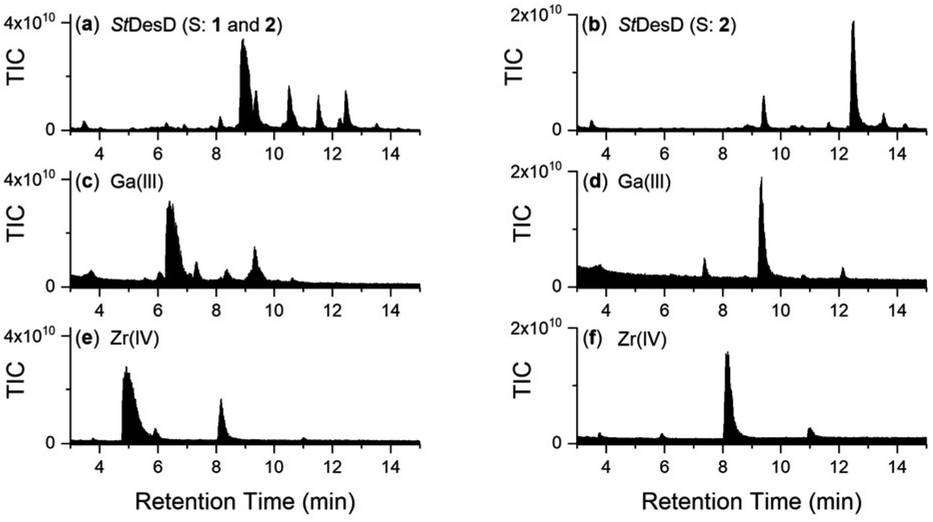 | ||
| Fig. 4 LC-MS traces detected by TIC from the reaction solution of StDesD with MgCl2 and ATP with substrate(s): (a) 1 and 2, and following the addition to (a) of (c) Ga(III) or (e) Zr(IV); or (b) 2 alone, and following the addition to (b) of (d) Ga(III) or (f) Zr(IV). | ||
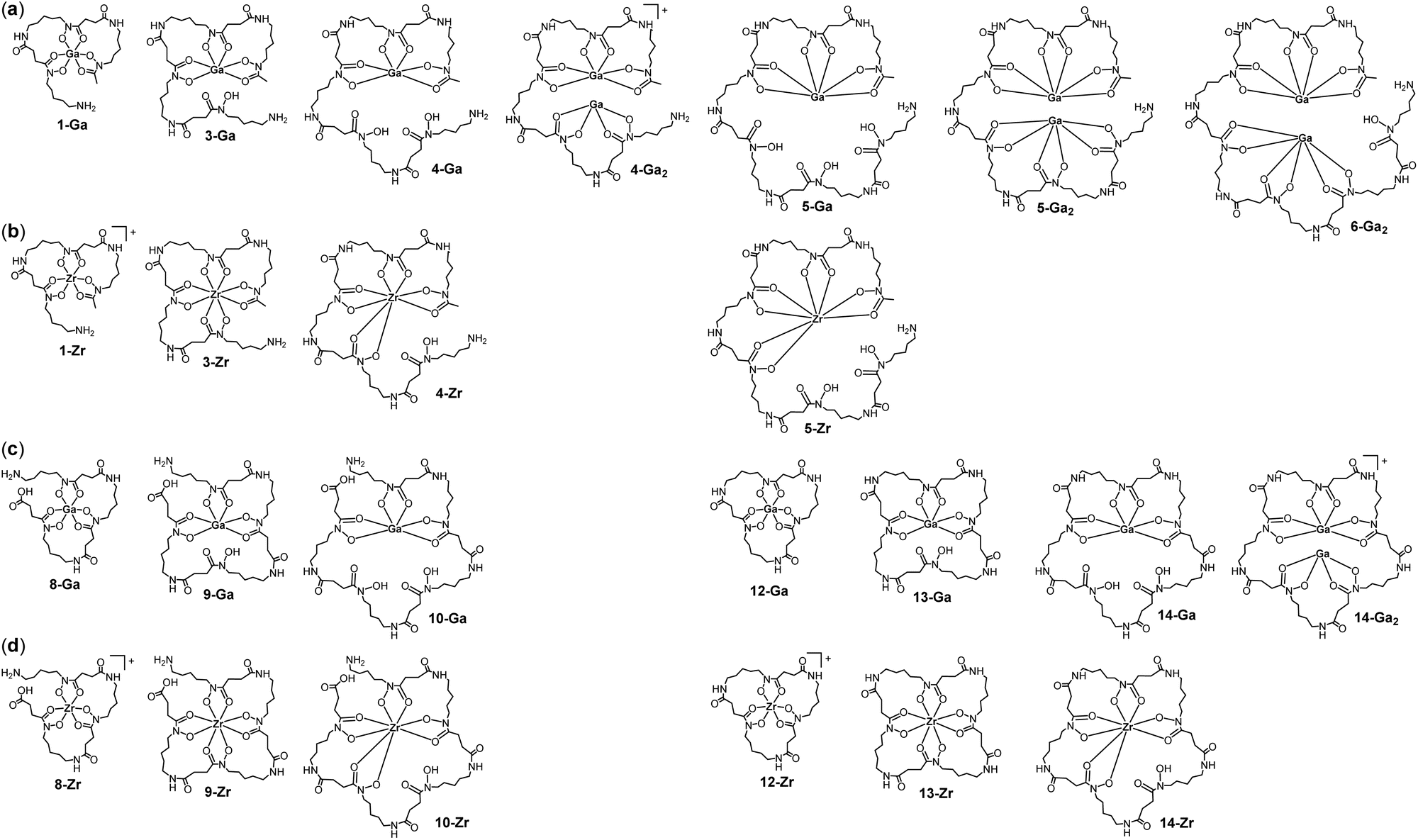 | ||
| Chart 1 Representative metal–ligand complexes (noting the possibility of coordination isomers) detected in solution by LC-MS measurements from (a) Ga(III) or (b) Zr(IV) and a mixture of 1–6; and (c) Ga(III) or (d) Zr(IV) and a mixture of 7–14. | ||
| No. | Species | Adduct | RT (min) | EIC calc. | EIC obs | No. | Species | Adduct | RT (min) | EIC calc. | EIC obs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1-Ga | [Ga(1(3−))] | [M − 3H + Ga + H]+ | 6.1 | 627.3 | 627.3 | 1-Zr | [Zr(1(3−))]+ | [M − 3H + Zr]+ | 4.9 | 647.2 | 647.2 |
| 3-Ga | [Ga(3(3−))] | [M − 3H + Ga + H]+ | 9.0 | 827.4 | 827.4 | 3-Zr | [Zr(3(4−))] | [M − 4H + Zr + H]+ | 7.9 | 847.4 | 847.4 |
| 4-Ga | [Ga(4(3−))] | [M − 3H + Ga + H]+ | 10.5 | 1027.5 | 1027.5 | 4-Zr | [Zr(4(4−))] | [M − 4H + Zr + H]+ | 9.6 | 1047.5 | 1047.5 |
| 5-Ga | [Ga(5(3−))] | [M − 3H + Ga + H]+ | 11.2 | 1227.6 | 1227.6 | 5-Zr | [Zr(5(4−))] | [M − 4H + Zr + 2H]2+ | 10.8 | 624.3 | 624.3 |
| 8-Ga | [Ga(8(3−))] | [M − 3H + Ga + H]+ | 6.7 | 685.3 | 685.3 | 8-Zr | [Zr(8(3−))]+ | [M − 3H + Zr]+ | 5.8 | 705.2 | 705.2 |
| 9-Ga | [Ga(9(3−))] | [M − 3H + Ga + H]+ | 9.1 | 885.4 | 885.4 | 9-Zr | [Zr(9(4−))] | [M − 4H + Zr + H]+ | 8.2 | 905.4 | 905.4 |
| 10-Ga | [Ga(10(3−))] | [M − 3H + Ga + H]+ | 10.4 | 1085.5 | 1085.5 | 10-Zr | [Zr(10(4−))] | [M − 4H + Zr + H]+ | 9.8 | 1105.5 | 1105.5 |
| 12-Ga | [Ga(12(3−))] | [M − 3H + Ga + H]+ | 9.0 | 667.3 | 667.3 | 12-Zr | [Zr(12(3 −))]+ | [M − 3H + Zr]+ | 8.2 | 687.2 | 687.2 |
| 13-Ga | [Ga(13(3−))] | [M − 3H + Ga + H]+ | 11.5 | 867.4 | 867.4 | 13-Zr | [Zr(13(4−))] | [M − 4H + Zr + H]+ | 10.8 | 887.3 | 887.3 |
| 14-Ga | [Ga(14(3−))] | [M − 3H + Ga + H]+ | 12.8 | 1067.5 | 1067.5 | 14-Zr | [Zr(14(4−))] | [M − 4H + Zr + H]+ | 12.4 | 1087.5 | 1087.5 |
| 4-Ga2 | [Ga2(4(5−))]+ | [M − 5H + 2Ga]+ | 8.1 | 1093.4 | 1093.4 | ||||||
| 5-Ga2 | [Ga2(5(6−))] | [M − 6H + 2Ga + H]+ | 10.3 | 1293.5 | 1293.5 | ||||||
| 6-Ga2 | [Ga2(6(6−))] | [M − 6H + 2Ga + 2H]2+ | 11.2 | 747.3 | 747.3 | ||||||
| 14-Ga2 | [Ga2(14(5−))]+ | [M − 5H + 2Ga]+ | 10.4 | 1133.4 | 1133.4 |
As would be expected, the addition of Ga(III) or Zr(IV) to the co-substrate system generated high intensity signals for 1-Ga or 1-Zr, respectively, due to the presence of unreacted 1 (Fig. S3†). Signals in the co-substrate system correlating with 1:1 complexes 3-Ga, 4-Ga, 5-Ga (Fig. 5a–c) and 3-Zr, 4-Zr, 5-Zr (Fig. 5g–i) were detected, with trends in relative concentration reflecting free ligand concentrations. Complexes with 6 were not detected likely due to its presence in low concentration. In the single-substrate system, 1:1 complexes 8-Ga, 9-Ga, 10-Ga, and 8-Zr, 9-Zr, 10-Zr were detected (Fig. S3†), together with signals for the cognate macrocycles 12-Ga, 13-Ga, 14-Ga (Fig. 5d–f) and 12-Zr, 13-Zr, 14-Zr (Fig. 5j–l). For each set of related chelators (blunt-end linear multimers (1, 3, 4, 5), open-chain linear multimers (8, 9, 10), or macrocycles (12, 13, 14)), the retention time of Ga(III) and Zr(IV) complexes increased as a function of the chelator multiplicity, in accord with the increased number of methylene units in the free chelators. The product profiles of the apo-multimers and metal complexes was robust, with repeat experiments giving reproducible results.
 | ||
| Fig. 5 EIC traces from biocombinatorial mixtures of chelators acquired following the addition of excess Ga(III) or Zr(IV) with EIC values set to report complexes between Ga(III) (a–f) or Zr(IV) (g–l) and 3–5, or 12–14, with isotope patterns (upper: experiment; lower: calculated) from the major or asterisked peak in the inset, and the relevant species as a cartoon. | ||
New coordination chemistry was identified with Ga(III) and the high-multiplicity chelators 4, 5, 6, and 14, with denticities and larger cavity sizes that could conceivably enable the formation of 2:1 Ga(III):chelator complexes. Signals were observed that correlated with EIC traces set to report 4-Ga2, 5-Ga2, 6-Ga2, and 14-Ga2, with each giving an isotope pattern that matched the calculated pattern distinct for a complex with two Ga(III) ions, and a compressed isotope pattern for 6-Ga2, which was detected as the double-protonated adduct (Fig. 6). Signals correlating with 2:1 metal:chelator complexes formed between Zr(IV) and this set of chelators were not detectable. This likely reflects one or more factors including analytical detection limits and the different coordination chemistry demands of Ga(III) (hexadentate) and Zr(IV) (octadentate), which could predict a requirement for even higher denticity chelators to form 2:1 Zr(IV):chelator complexes. Decadentate HOPO–O10 has been reported to form 2:1 metal:ligand complexes with La(III) and Tb(III).38
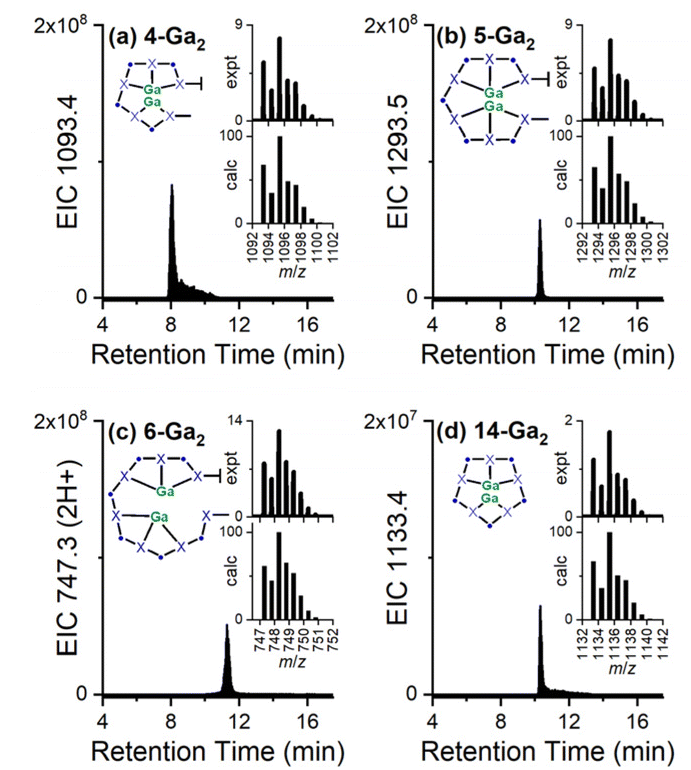 | ||
| Fig. 6 EIC traces from biocombinatorial mixtures of chelators acquired following the addition of excess Ga(III) with EIC values set to report complexes between Ga(III) and 4–6 (a–c) or 14 (d), with isotope patterns (upper: experiment; lower: calculated) in the inset, and the relevant species as a cartoon. | ||
Together, this part of the study shows the use of a chemo-enzymatic approach to generate biocombinatorial pools of metal chelators amenable for screening with a given metal ion to inform known and new coordination chemistry.
Posing a processive assembly mechanism
The StDesD-mediated production of higher-order multimers from the single-substrate (2) and two-substrate (1, 2) systems prompted the consideration of two assembly mechanisms (Scheme 3, M1, M2) with each applied to the single-substrate (M1-1, M2-1) or two-substrate system (M1-2, M2-2).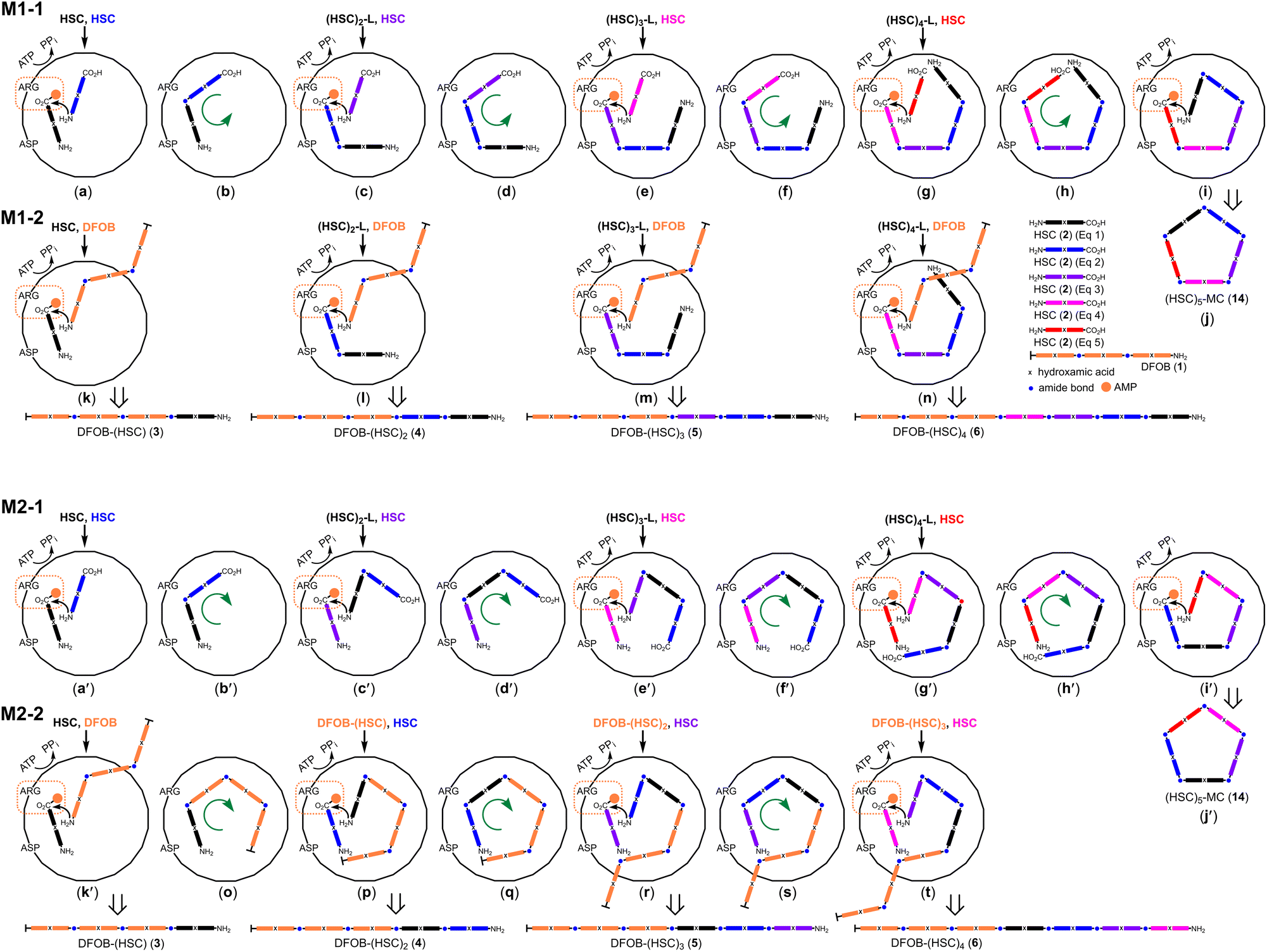 | ||
| Scheme 3 Putative StDesD-mediated assembly of macrocyclic (HSC)5-MC (14) by a processive mechanism with 2 predominating as the nucleophilic (M1-1: (a–j)) or carboxylate-activated (M2-1: (a′–j′)) substrate. Putative assembly of DFOB-(HSC)x (x = 1–4) (3–6, respectively) with 1 conserved as the nucleophilic substrate for condensation with pre-assembled carboxylate-activated 2 or 2 oligomers (M1-2: (k–n)), or 2 conserved as the carboxylate-activated substrate for condensation with 1 or 3–5 as pre-assembled nucleophilic substrates (M2-2: (k′–t)). Intra-cavity procession of intermediate substrates (green arrow) would enable the position of the carboxylic acid group of 2 or 2 oligomers is conserved for activation (broken orange box). | ||
In the single-substrate system with 2, the highest-order macrocycle detected was pentameric (HSC)5-MC (14), which is used to open the following discussion and draws upon this class of siderophore synthetase containing an activation site (carboxylic acid group positioned for adenylation) proximal to a condensation site (amine group as the nucleophile for amide bond formation),13 and the X-ray crystal structure of a complex between DesD from S. griseoflavus DSM 40698 (SgDesD) and an adenylated substrate mimic, which supports the presence of an activation site.12
It was considered reasonable that the stepwise assembly of 14 in both assembly mechanisms would begin following the entry of two equivalents of 2 into the active site (Scheme 3a and a′), with one equivalent positioned for adenylate-based carboxylic acid activation and the amine group of the other substrate positioned for condensation to form 7 (Scheme 3b and b′). The circle depicting the active site shows an aspartic acid and arginine residue lining the cavity predicted to stabilise 2 in the activation site, as identified (D497, R303) in the X-ray crystal structure of SgDesD bound to an adenylated 2 mimic.12 Both of these residues are preserved in the StDesD sequence (Fig. S4†).
At the point of the production of 7 (Scheme 3b and b′), the assembly mechanism can diverge. In one sequence (M1-1), the intermediate substrate 7 could re-orient within the active site cavity (Scheme 3b and c) to position its carboxylic acid group ready for activation, with the third equivalent of 2 entering as the nucleophile to generate 8, which would similarly re-orientate (Scheme 3d and e) to continue the stepwise assembly of 9 and 10 (Scheme 3f–i) for the final intramolecular condensation reaction to produce 14 (Scheme 3j). The intramolecular condensation of the discrete linear multimers 8 (Scheme 3e) or 9 (Scheme 3g) would produce 12 and 13, respectively.
This describes a processive assembly mechanism whereby the growing polymer chain is shunted around the active site cavity to position itself for carboxylic acid activation, with equivalents of 2 entering as the nucleophile (Scheme 3a–j). This first mechanism (M1) as applied to the two-substrate system with 1 and 2 (M1-2) produced the blunt-end multimers 3, 4, 5, and 6, which could be generated from the respective carboxylate-adenylated substrates 2 (Scheme 3k), 7 (Scheme 3l), 8 (Scheme 3m) or 9 (Scheme 3n) undergoing condensation with 1 as the nucleophile.
Returning to the production of 7 in the single-substrate system (Scheme 3b,b′), a different sequence (M2-1) could instead ascribe 7 (rather than 2) as the nucleophile. This would require 7 be re-positioned to the condensation site (Scheme 3c′) for reaction with adenylated 2 preserved in the activation site. This alternative logic would generate 8–10 (Scheme 3d′–h′) with 10 ultimately positioned (Scheme 3i′) as in the first sequence (M1-1) for intramolecular condensation to generate 14 (Scheme 3j′). This alternative sequence (M2-1) might imply the requirement for further substrate reorganisation to position the carboxylic acid group of 8 (Scheme 3d′) or 9 (Scheme 3f′) in the activation site for intramolecular condensation to generate 12 and 13, respectively.
In the two-substrate system in both sequences (M1-2, M2-2), blunt-end 3 as generated from 1 and 2, involves a unique condensation reaction (described earlier) between adenyl-activated 2 and 1 as the nucleophile (Scheme 3k and k′). Maintaining 3 as the nucleophile would require its re-organisation to enable condensation with the incoming equivalent of adenylated 2 (Scheme 3o and p), with this continued logic generating 4, 5 and 6 (Scheme 3q–t).
There was a difference in the multiplicity limit of the single- and two-substrate systems. The pentameric macrocycle 14 was the highest-order multimer detected in the single-substrate system with 2, with heptameric linear 6 detected in the system using 1 and 2. This might suggest capacity for longer-chain linear substrates to flex beyond the active site cavity at the point of entry and/or at the exit point for the chain-extended products.
These two sequences are united in proposing the processive movement of the growing polymer chain within the active-site cavity but differ in the positional assignment of co-substrates for activation or condensation. M1-1 assigns units of 2 as the diffusible nucleophile and the growing 2 multimer chain undergoing re-positioning for carboxylic acid activation, while M2-1 assigns the growing 2 multimer chain as the nucleophile and units of 2 entering to undergo carboxylic acid activation. Elements of these pathways could coalesce, with one example 9 being formed from the condensation between 7 positioned for carboxylate activation (Scheme 3c) with 7 positioned as the nucleophile (Scheme 3c′). It may be that the overall sequences or parts thereof are not mutually exclusive and operate to variable extents in parallel. The overarching proposition is of a processive mechanism allowing for the energetically preferred diffusion of relatively low-molecular-weight compounds (ATP, AMP, PPi, 2) and the growing multimer chains maintained within or extending beyond the active site (when exceeding a volume capacity limit) with products expelled along the way.
The wide product profile observed experimentally (3–10, 12–14) suggests the StDesD active site has sufficient elasticity to build and accommodate flexible (rubbery) multimers of variable length as intermediates or final products.
Conclusions
A recombinant siderophore synthetase from Salinispora tropica CNB-400 StDesD has been used to assemble biocombinatorial pools of hydroxamic acid multimeric chelators using either 1 and 2 as co-substrates or 2 as a sole substrate. This chemo-enzymatic approach is attractive, since this offers a more facile route to access sets of structurally diverse metal chelators compared to preparing each individual compound by total synthesis. Each biocombinatorial pool was well populated with series of blunt-end linear multimers as StDesD-mediated condensation products of 1 and 2 (3–6), or of linear multimers built from 2 (7–10) which, aside from 7, were viable pre-cursors of the cognate multimeric macrocycles (12–14). The upper assembly/detection limit of the blunt-end linear multimer class was tetradecadentate 6, and the decadentate macrocycle, 14.The integrity of chelator function was established by incubating each biocombinatorial pool with excess Ga(III) or Zr(IV) which formed known/expected 1:1 complexes, and in the case of high-denticity chelators, some new 2:1 Ga(III):chelator complexes (Scheme 4). Applications of 2:1 68Ga(III):chelator complexes could be useful as positron emission tomography (PET) imaging partners of targeted radiopharmaceutical agents, where the molar activity of the radiolabelled compound could be increased enabling a reduction in mass dose, which would reduce the toxicity of the agent itself and the risk of receptor blocking effects.
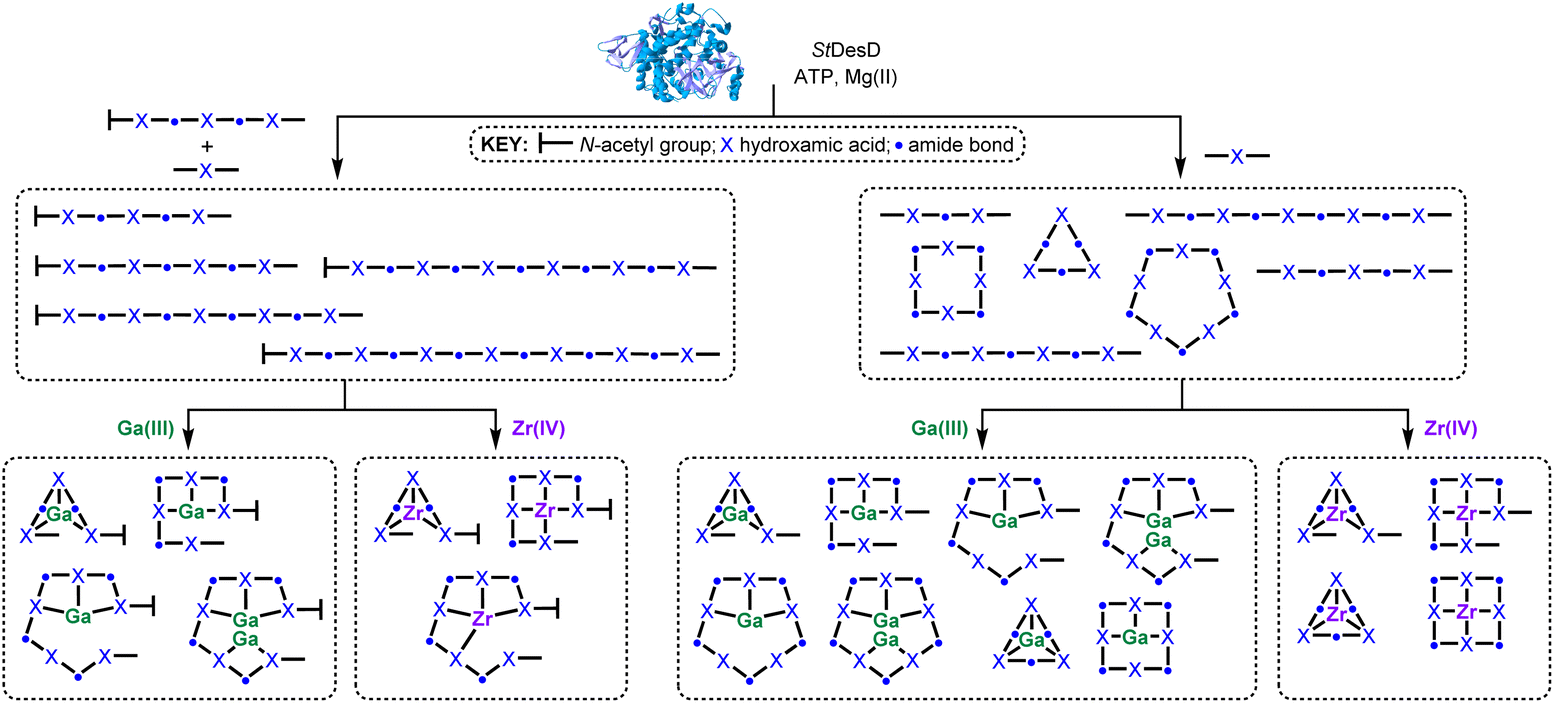 | ||
| Scheme 4 Biocombinatorial pools of hydroxamic acid chelators (3–14) in cartoon form produced by StDesD-mediated synthesis using as substrates 1 and 2 (left path; compounds contain a terminal amine group) or 2 (right path; linear compounds contain flanking amine and carboxylic acid groups) and coordination complexes characterised in situ upon incubation of each pool with Ga(III) or Zr(IV). The structure of StDesD was produced by AlphaFold.39 | ||
The array of multimeric chelators generated from simple substrates prompted consideration of assembly mechanisms which led to the posit of a processive intra-cavity assembly mechanism, with the growing multimer chain being shunted around the enzyme cavity to conserve the structural and functional integrity of the activation and condensation sites. Ongoing work using structural and predictive biology, site-directed mutagenesis, and molecular dynamics calculations is underway to further interrogate this mechanism. It could be that acidic and basic amino acid residues are systematically patterned around the active site cavity to accommodate the intra-cavity repositioning of 2 multimers.
The work highlights the general scope in using recombinant biosynthetic enzymes of natural products together with synthetically tractable substrates as a facile chemoenzymatic approach to generate biocombinatorial pools of structurally diverse analogues to screen for function.
Experimental
Materials and methods
Acetonitrile (ACN) (99.8%), adenosine 5′-triphosphate disodium salt hydrate (Grade II, 98.5% (HPLC) crystalline), desferrioxamine B mesylate salt (DFOB, ≥95%), formic acid ≥95%, gallium(III) nitrate (≥99%), magnesium chloride hexahydrate (≥99%), TRIS hydrochloride (TRIS/HCl) (≥99%) and zirconium(IV) chloride (≥99%) were from Merck. MilliQ water was used in all experiments requiring water.Production of StDesD and substrates and standards
The recombinant siderophore synthetase DesD from S. tropica CNB440 (StDesD) was expressed and purified18 and incubated (1 mg mL−1) with cofactors ATP (4 mM), MgCl2 (15 mM), and hydroxamic acid substrate(s) (2.4 mM) in a solution of Tris/HCl (25 mM, pH 8) in a final volume (200 μL) for 2 h at 37 °C. Control reaction solutions omitted DesD. Reactions were terminated by the addition of formic acid (2 μL of 10% v/v) and following centrifugation (15000 rpm, 15 min), the supernatant was filtered using a PTFE syringe filter (0.45 μm, 17 mm) before analysis by liquid chromatography-mass spectrometry, similar with protocols in previous work.6,13,18 Commercial 1 mesylate (93%) was obtained from Sigma-Aldrich and was purified using HPLC to about 99% purity (Fig. S1†). The macrocyclic siderophore desferrioxamine E (DFOE, 12) was obtained from EMC microcollections, and 2 and 3 were prepared using published methods.19,20,30
Solution chemistry with Ga(III) and Zr(IV)
An aliquot of the terminated reaction solution of StDesD with the two-substrate system with 1 and 2 (4 mM total substrate) was incubated with either Zr(IV)Cl4 (20 mM) or Ga(III)NO3 (12 mM) at 37 °C for 2 h. An aliquot of the terminated reaction solution of StDesD with the single-substrate system with 2 (2 mM substrate) was incubated with either Zr(IV)Cl4 (10 mM) or Ga(III)NO3 (6 mM) at 37 °C for 2 h. Samples were then centrifuged at 15000 rpm for 15 min, and the supernatant was filtered using a PTFE syringe filter (0.45 μm, 5 mm) prior to analysis by LC/MS.
Analytical procedures: LC-HRMS/MS
Samples were separated with a Thermo Fisher Vanquish Horizon UHPLC coupled to an Agilent Zorbax Eclipse XDB-C18 column (3.5 μm particle size, 150 mm length, 2.1 mm i. d., column oven set to 30 °C). Mobile phase A: aqueous formic acid (0.1%). Mobile phase B: formic acid (0.1%) in aqueous ACN (80% ACN). A gradient of 2–62% mobile phase B from 0–25 min was applied at the 0.2 mL min−1 flowrate. Tandem mass spectrometry fragmentation was performed with a Thermo Fisher Q Exactive HF-X Hybrid Quadrupole-Orbitrap Spectrometer. Collision energy voltages were set on the fly by the mass spectrometer for individual precursor ions (steps at 20, 25, and 30 V). The capillary voltage and the temperature of the heated transfer capillary were set to 4000 V and 300 °C, respectively. Positive ions were generated by electrospray, and the Orbitrap was operated in data-dependent acquisition mode. A survey scan of 100–1500 m/z was acquired (resolution = 60000, with an accumulation target value of 3000000 ions). Up to 10 of the most abundant ions (>1.7 × 105 ions), with charge state +1 were sequentially isolated and fragmented. An AGC setting of 100000 was used for MS/MS mode. Ions selected for MS/MS were dynamically excluded for 4 s. Xcalibur (version B.07.01) software was used for data acquisition and processing. Experimentally determined m/z values were used with allowable δ = 5 ppm to generate EICs.
Abbreviations
| Ac-CoA | Acetyl-coenzyme A |
| AMP | Adenosine monophosphate |
| ATP | Adenosine triphosphate |
| BIS | Bisucaberin |
| DFOB | Desferrioxamine B |
| EIC | Extracted ion chromatogram |
| HAC | N-Hydroxy-N-acetylcadaverine |
| HC | N-Hydroxycadaverine |
| HSC | N-Hydroxy-N-succinylcadaverine |
| L | Linear |
| LC-MS | Liquid chromatography-mass spectrometry |
| MC | Macrocycle |
| PET | Positron emission tomography |
| PPi | Pyrophosphate |
| SpDesD | Streptomyces pilosus DesD |
| StDesD | Salinispora tropica CNB-440 DesD |
| Suc-CoA | Succinyl-coenzyme A |
| TIC | Total ion current |
| t R | Retention time |
Data availability
All relevant data are presented in the main text and ESI† (general information, LC-MS data)Author contributions
The study was conceptualized by RC, with all authors contributing to different methodological aspects and elements of the study design. Initial recombinant protein production was undertaken by KPN, under the supervision of JF and RMR, and subsequently by CAR and JW. Experimental data including optimised syntheses of substrates was generated by KPN, CAR, JLW, AS, JW, and TEM, with these authors and RC contributing to the analysis and interpretation of results. The manuscript including data presentation was written and prepared by RC and KPN, with all authors contributing to manuscript review, editing, and approving the final submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Australian Research Council is acknowledged for research support to RC (DP220100101) and a Future Fellowship to RMR (FT220100717). The Sydney Mass Spectrometry (SydneyMS) Core Research Facility is acknowledged for technical support. The University of Sydney is acknowledged for providing each of KPN, CAR, JLW, AS, and JW with an Australian Government Research Training Program (RTP) Scholarship.Notes and references
- F. Barona-Gómez, U. Wong, A. E. Giannakopulos, P. J. Derrick and G. L. Challis, J. Am. Chem. Soc., 2004, 126, 16282–16283 CrossRef.
- P. Cruz-Morales, H. E. Ramos-Aboites, C. Licona-Cassani, N. Selem-Mójica, P. M. Mejía-Ponce, V. Souza-Saldívar and F. Barona-Gómez, FEMS Microbiol. Ecol., 2017, 93, 1–12 CrossRef CAS.
- R. Codd, T. Richardson-Sanchez, T. J. Telfer and M. P. Gotsbacher, ACS Chem. Biol., 2018, 13, 11–25 CrossRef CAS.
- S. Dhungana, P. S. White and A. L. Crumbliss, J. Biol. Inorg. Chem., 2001, 6, 810–818 CrossRef CAS PubMed.
- A. Evers, R. D. Hancock, A. E. Martell and R. J. Motekaitis, Adv. Inorg. Chem., 1989, 28, 2189–2195 CrossRef CAS.
- N. Kadi, D. Oves-Costales, F. Barona-Gómez and G. L. Challis, Nat. Chem. Biol., 2007, 3, 652–656 CrossRef CAS.
- T. J. Telfer, M. P. Gotsbacher, C. Z. Soe and R. Codd, ACS Chem. Biol., 2016, 11, 1452–1462 CrossRef CAS PubMed.
- S. Schmelz and J. H. Naismith, Curr. Opin. Struct. Biol., 2009, 19, 666–671 CrossRef CAS PubMed.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 CrossRef CAS.
- K. M. Hoffmann, E. S. Goncuian, K. L. Karimi, C. R. Amendola, Y. Mojab, K. M. Wood, G. A. Prussia, J. Nix, M. Yamamoto, K. Lathan and I. W. Orion, Biochemistry, 2020, 59, 3427–3437 CrossRef CAS PubMed.
- K. M. Hoffmann, J. S. Kingsbury, N. L. March, Y. Jang, J. H. Nguyen and M. M. Hutt, Molecules, 2022, 27, 6144 CrossRef CAS.
- J. Yang, V. S. Banas, K. D. Patel, G. S. M. Rivera, L. S. Mydy, A. M. Gulick and T. A. Wencewicz, J. Biol. Chem., 2022, 298, 102166 CrossRef CAS.
- S. Rütschlin and T. Böttcher, Chem.–Eur. J., 2018, 24, 16044–16051 CrossRef PubMed.
- J. Yang, V. S. Banas, G. S. M. Rivera and T. A. Wencewicz, ACS Chem. Biol., 2023, 18, 1266–1270 CrossRef CAS PubMed.
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci., 2007, 104, 10376–10381 CrossRef CAS PubMed.
- A. A. Roberts, A. W. Schultz, R. D. Kersten, P. C. Dorrestein and B. S. Moore, FEMS Microbiol. Lett., 2012, 335, 95–103 CrossRef CAS PubMed.
- N. Ejje, C. Z. Soe, J. Gu and R. Codd, Metallomics, 2013, 5, 1519–1528 CrossRef CAS.
- K. P. Nolan, J. Font, A. Sresutharsan, M. P. Gotsbacher, C. J. M. Brown, R. M. Ryan and R. Codd, ACS Chem. Biol., 2022, 17, 426–437 CrossRef CAS PubMed.
- C. J. M. Brown, M. P. Gotsbacher, J. P. Holland and R. Codd, Inorg. Chem., 2019, 58, 13591–13603 CrossRef CAS PubMed.
- C. J. M. Brown, M. P. Gotsbacher and R. Codd, Aust. J. Chem., 2020, 73, 969–978 CAS.
- T. E. Markham and R. Codd, J. Org. Chem., 2024, 89, 5118–5125 CrossRef CAS.
- A. A. H. Pakchung, T. Lifa and R. Codd, RSC Adv., 2013, 3, 16051–16059 RSC.
- M. J. Fujita, K. Nakano and R. Sakai, Molecules, 2013, 18, 3917–3926 CrossRef CAS PubMed.
- A. Takahashi, H. Nakamura, T. Kameyama, S. Kurasawa, H. Naganawa, Y. Okami, T. Takeuchi and H. Umezawa, J. Antibiot., 1987, 40, 1671–1676 CrossRef CAS PubMed.
- S. Rütschlin, S. Gunesch and T. Böttcher, Cell Chem. Biol., 2017, 24, 598–604 CrossRef PubMed.
- G. Anderegg, F. L'Eplattenier and G. Schwarzenbach, Helv. Chim. Acta, 1963, 46, 1400–1408 CrossRef CAS.
- G. J. Feistner, D. C. Stahl and A. H. Gabrik, Org. Mass Spectrom., 1993, 28, 163–175 CrossRef CAS.
- W. Tieu, T. Lifa, A. Katsifis and R. Codd, Inorg. Chem., 2017, 56, 3719–3728 CrossRef CAS PubMed.
- H. Bickel, R. Bosshardt, E. Gäumann, P. Reusser, E. Vischer, W. Voser, A. Wettstein and H. Zähner, Helv. Chim. Acta, 1960, 43, 2118–2128 CrossRef CAS.
- M. Patra, A. Bauman, C. Mari, C. A. Fischer, O. Blacque, D. Haussinger, G. Gasser and T. L. Mindt, Chem. Commun., 2014, 50, 11523–11525 RSC.
- W. Keller-Schierlein and V. Prelog, Helv. Chim. Acta, 1962, 45, 590–595 CrossRef.
- W. Keller-Schierlein and V. Prelog, Helv. Chim. Acta, 1961, 44, 1981–1985 CrossRef CAS.
- Y. Toporivska and E. Gumienna-Kontecka, J. Inorg. Biochem., 2019, 198, 110753 CrossRef CAS PubMed.
- Y. Toporivska, A. Mular, K. Piasta, M. Ostrowska, D. Illuminati, A. Baldi, V. Albanese, S. Pacifico, I. O. Fritsky, M. Remelli, R. Guerrini and E. Gumienna-Kontecka, Inorg. Chem., 2021, 60, 13332–13347 CrossRef CAS.
- J. P. Holland, Inorg. Chem., 2020, 59, 2070–2082 CrossRef CAS PubMed.
- L. Patiny and A. Borel, J. Chem. Inf. Model., 2013, 53, 1223–1228 CrossRef CAS PubMed.
- C. Z. Soe, T. J. Telfer, A. Levina, P. A. Lay and R. Codd, J. Inorg. Biochem., 2016, 162, 207–215 CrossRef CAS PubMed.
- I. Carbo-Bague, C. Li, B. L. McNeil, Y. Gao, A. W. McDonagh, M. Van de Voorde, M. Ooms, P. Kunz, H. Yang, V. Radchenko, G. Schreckenbach and C. F. Ramogida, Inorg. Chem., 2023, 62, 20549–20566 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04888a |
| ‡ Hydroxamic acid-based chelators and metal complexes under the acidic conditions of the LC-MS system are routinely detected as positively charged adducts. Studies on similar systems using LC-MS in positive and negative ion detection modes22 showed signals were significantly weaker in negative ion mode than positive ion mode and did not reveal core species beyond those detected by positive ion mode. |
| § The co-substrate system generated product set 3–6 (formed from 1 and 2) and product set 7–10, 12–14 (formed from 2), with the latter set also formed in the single-substrate system. For simplicity, the major discussion of the co-substrate system focussed on the system-exclusive products 3–6. |
| This journal is © The Royal Society of Chemistry 2025 |