
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSluggish Li2O2 dissolution – a key to unlock high-capacity lithium–oxygen batteries†
Lu
He‡
,
Shuo
Wang‡
,
Fengjiao
Yu
 and
Yuhui
Chen
*
and
Yuhui
Chen
*
State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: cheny@njtech.edu.cn
First published on 2nd December 2024
Abstract
While lithium–oxygen batteries have a high theoretical specific energy, the practical discharge capacity is much lower due to the passivation of the solid discharge product, Li2O2, on the electrode surface. Herein, we studied and quantified the deposition and dissolution kinetics of Li2O2 using an electrochemical quartz crystal microbalance (EQCM). It is found that the orientation of the electrode greatly influences the formation path and deposition amount of Li2O2. We identified two distinct dissolution modes: surface dissolution and bulk fragmentation, with the latter 100 times faster than the former. By revealing the underlying factors affecting dissolution, 80% of Li2O2 can dissolve within 3 minutes when a desorption potential of 2.9 V is applied. Consequently, we designed an intermittent-desorption discharge strategy, which increased the discharge capacity by an order of magnitude. This work shows that high practical specific energy of Li–O2 batteries can be achieved once problems of Li2O2 dissolution are addressed.
Introduction
Lithium–oxygen (Li–O2) batteries are strong competitors for the next generation of energy storage devices due to their extremely high theoretical energy density.1–3 Compared to lithium-ion batteries, they can provide a 10-fold higher discharge capacity.4 However, Li–O2 batteries still face many challenges in practical applications,5–11 particularly the practical discharge capacity being significantly lower than the theoretical discharge capacity,12–14 which severely undermines their competitiveness. The primary reason for the low discharge capacity is surface passivation, mainly caused by the accumulation of discharge products.15–17 During discharge, the solid product lithium peroxide (Li2O2) accumulates on the electrode surface, covering the active sites and thus leading to electrode passivation. This makes it difficult for further deposition of discharge products, reducing both discharge efficiency and capacity, and severely limiting the cycling performance.Therefore, addressing surface passivation is crucial for improving the performance of Li–O2 batteries.18 Researchers have used redox mediators to assist in the decomposition and formation of products.19–23 While redox mediators can significantly enhance battery capacity and cycle life, their own stability, compatibility, and shuttle effect must be considered.24–28 Alternatively, methods such as using molten salt electrolytes and additives can form soluble products (soluble intermediates).29–33 However, generating soluble products might also present challenges in terms of electrolyte stability and side reactions. Therefore, practical applications require balancing various factors.34 How can we maximize the discharge capacity of Li–O2 batteries under normal temperature and pressure conditions without additives? We recognize the importance of a dissolved product layer to maintain the catalytic activity of the electrode surface and catalyst sites. On the other hand, for the charging process, in many catalyst studies, Li2O2 must dissolve before it can be oxidized in the form of small molecules. For example, many calculations of Li2O2 oxidation decomposition are based on the oxidation decomposition of Li2O2 molecules rather than the solid-state Li2O2.35–37
Here, we studied the kinetics of Li2O2 deposition and dissolution processes using electrochemical quartz crystal microbalance (EQCM).38,39 We found that due to the presence of two different types of Li2O2 (strongly adsorbed Li2O2 and weakly adsorbed Li2O2), the orientation of the electrode has a great influence on the formation path and deposition amount of Li2O2, resulting in different degrees of passivation of the electrode surface. Additionally, we found that the Li2O2 on the electrode surface dissolves in two modes: bulk fragmentation mode and surface dissolution mode. The rate of the former is two orders of magnitude higher than the latter. The sluggish surface dissolution mode is the main reason for electrode passivation, with its kinetics influenced by both the electrode materials and the solvents. Based on the surface Li2O2 dissolution rate, we designed an intermittent discharge strategy to achieve a high discharge capacity. Furthermore, we estimated the limiting charging current during the charging process based on the Li2O2 dissolution–oxidation process.
Results and discussion
The deposition of Li2O2
To study the impact of electrode orientation on the deposition process of Li2O2, we deposited a layer of Li2O2 on the EQCM electrode at different angles in a tetraethylene glycol dimethyl ether (TEGDME) based electrolyte, specifically 0° (upward) and 90° (vertical) as shown in Fig. 1a. The detailed experimental procedure is described in the ESI.† As shown in Fig. 1b and c, linear sweep voltammetry (LSV) was carried out to reduce O2 to produce Li2O2 at 2.5 V (vs. Li+/Li). The characterization of the discharge product will be discussed later. The mass on the vertical electrode (denoted as Au-90°) increased with the reduction current, and the changes in charge and mass were synchronized (Fig. 1b). However, for the upward electrode (denoted as Au-0°), there was a distinct delay in the mass change between 2.25 V and 2.4 V. When a reduction current appeared on the upward electrode, Li2O2 began to form but did not immediately deposit onto the EQCM electrode, leading to no immediate mass response. Approximately 70 seconds later, the mass on the upward electrode began to increase rapidly when Li2O2 clusters or particles settled onto the surface (Fig. 1c). The rate of mass increase on the upward electrode was significantly higher than that on the vertical electrode.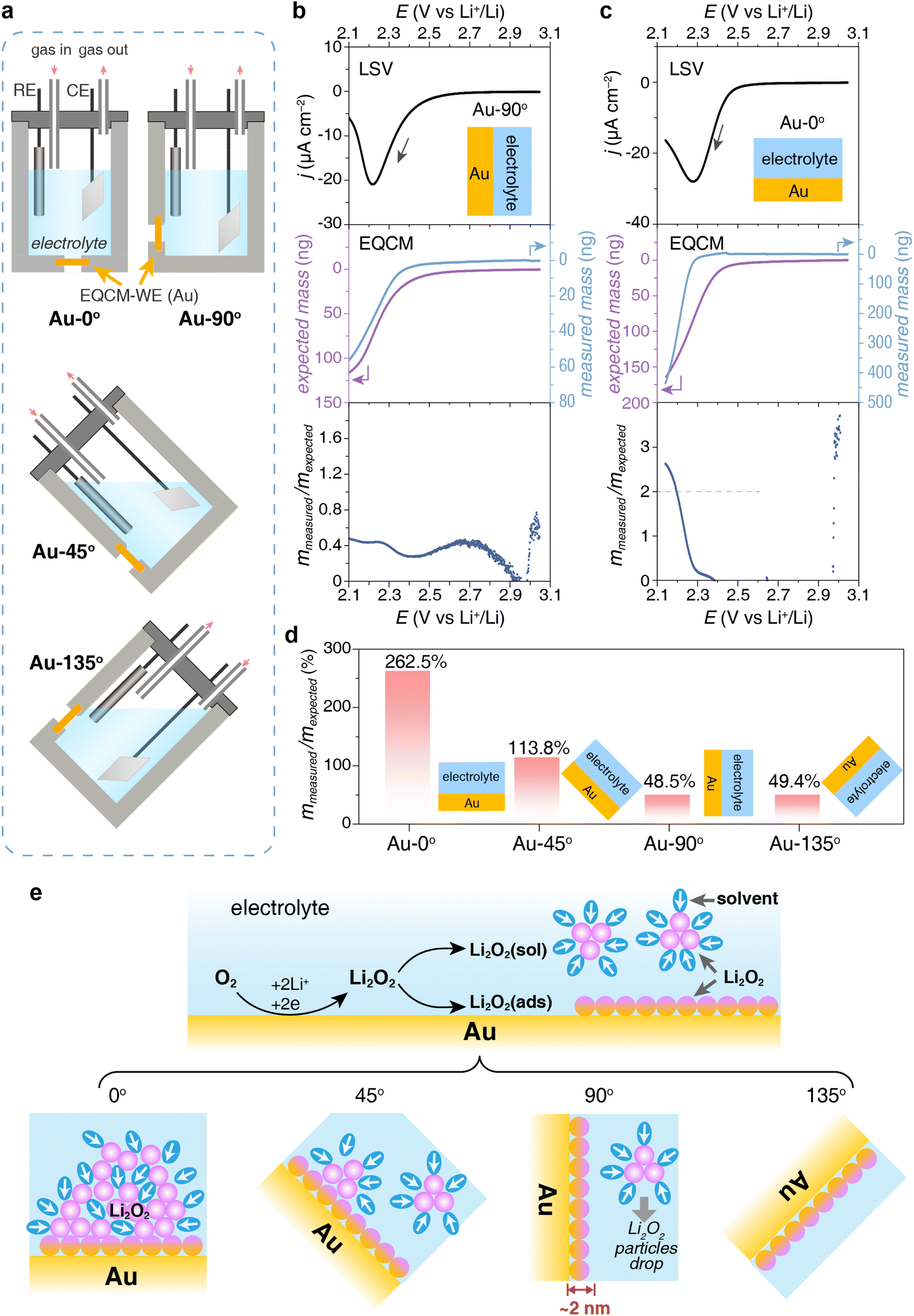 | ||
| Fig. 1 EQCM experiments with electrodes settling at different angles. (a) Schematic of a homemade EQCM cell with Au-coated quartz crystal electrodes at different angles. (b and c) Discharging current and mass deposition on (b) Au-90° and (c) Au-0° EQCM electrodes in 1 M LiTFSI–TEGDME using LSV. Sweep rate: 2 mV s−1. (d) The ratio of measured mass deposition to expected mass deposition at EQCM electrodes with various angles. (e) Schematics of the discharge process and Li2O2 deposition at electrodes with various angles. | ||
Their deposition behavior exhibited significant differences. As shown in Fig. 1b, when the Au crystal electrode was placed vertically in the electrolyte (Au-90°), the deposition mass on the electrode measured by EQCM was much smaller than the theoretically expected value. According to the charge passed, 116.3 ng of discharge product was expected to deposit on the electrode, while only 56.3 ng mass was detected, exhibiting a low mmea/mexp ratio of 48.4% and suggesting only part of the discharge product was deposited on the electrode. However, when the Au electrode was placed horizontally in the electrolyte (Au-0°, Fig. 1c), the expected deposition mass was 165.4 ng and the measured deposition mass was 434.3 ng, with the mmea/mexp ratio reaching 262.5%. This high mmea/mexp ratio will be explained in the later text. The measured deposition mass on Au-0° was approximately 7-fold greater than that on Au-90°, suggesting most discharge products did not deposit on the Au-90° electrode due to its vertical position.
To understand the impact of the placement angle of the electrode on the mass of deposition, we carried out a set of comparison experiments that depositing discharge product Li2O2 at various angles including upward, tilted upward (denoted as Au-45°, Fig. S1a†), vertical, and tilted downward (denoted as Au-135°, Fig. S1b†) using a homemade EQCM cell as shown in Fig. 1a. Fig. 1d shows the mmea/mexp ratio decreased from 262.5% to 48.5% when the electrode was rotated from 0° to 90°. The amount of discharge product deposited on Au-0° was 7 times greater than on Au-90°, and 2.5 times greater than on Au-45° (Fig. 1d). When the electrode was further turned facing down, the mmea/mexp ratio no longer changed, indicating the discharge products on Au-90° and Au-135° stuck tightly and nothing else could drop. Apparently, gravity and surface adsorption play important roles in the deposition process of discharge products, which will be discussed in detail below.
The difference in the mmea/mexp ratio originated from two competitive reaction pathways of the discharging process, i.e. solution pathway and surface pathway, which are two main pathways to form Li2O2 on discharge in lithium–oxygen batteries. We are not going to explain these two mechanisms in detail here because many reviews have discussed the discharging mechanism of lithium–oxygen batteries.40–42 Briefly, the solution pathway is based on the nucleation and growth of Li2O2 in the solution phase,43 with this type of Li2O2 depositing on the electrode surface as particles. In contrast, the surface pathway involves surface adsorption of the intermediate at the surface and Li2O2 product grows as a thin layer on the electrode surface. Both pathways take place during discharge and their competition relies on many factors such as potential, solvents, electrode surface, etc.
For Au-0° which faced upward, the discharge products from both pathways (i.e. Li2O2 film and Li2O2 particles) precipitated on the electrode due to gravity (Fig. 1e). Surprisingly, its mmea/mexp ratio reached 262.5%, indicating that the measured deposition mass of the discharge products was much higher than expected. However, the Raman spectrum and the chemical yield titration of the discharged electrode identify Li2O2 as the major discharge product (Fig. 2a). To maximize the Raman signal of the tiny amount of the discharge product, a roughened gold electrode was prepared and discharged for the surface enhanced Raman spectroscopy. The experimental details are stated in the ESI.† As shown in Fig. 2a, Li2O2 and LiTFSI-based electrolytes were identified as the depositing product on Au-0° electrode. Additionally, the Li2O2 yield was titrated and found to be 90%, which indicates that the primary discharge reaction was the formation of Li2O2 and is consistent with the ref. 44 and 45. Therefore, the additional mass depositing on the electrode surface in this case may result from the co-deposition of Li2O2 and electrolyte. Unlike the metal deposition (i.e. Cu plating and Ag plating) that forms a compact deposition product and real-time response, the Li2O2 deposition exhibits a delay in mass increase (Fig. 1c) and a relatively loose structure containing electrolyte (Fig. 1e, as determined by specific measurements, with Li2O2 and LiTFSI-4G electrolyte depositing in a ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio).
:1 molar ratio).
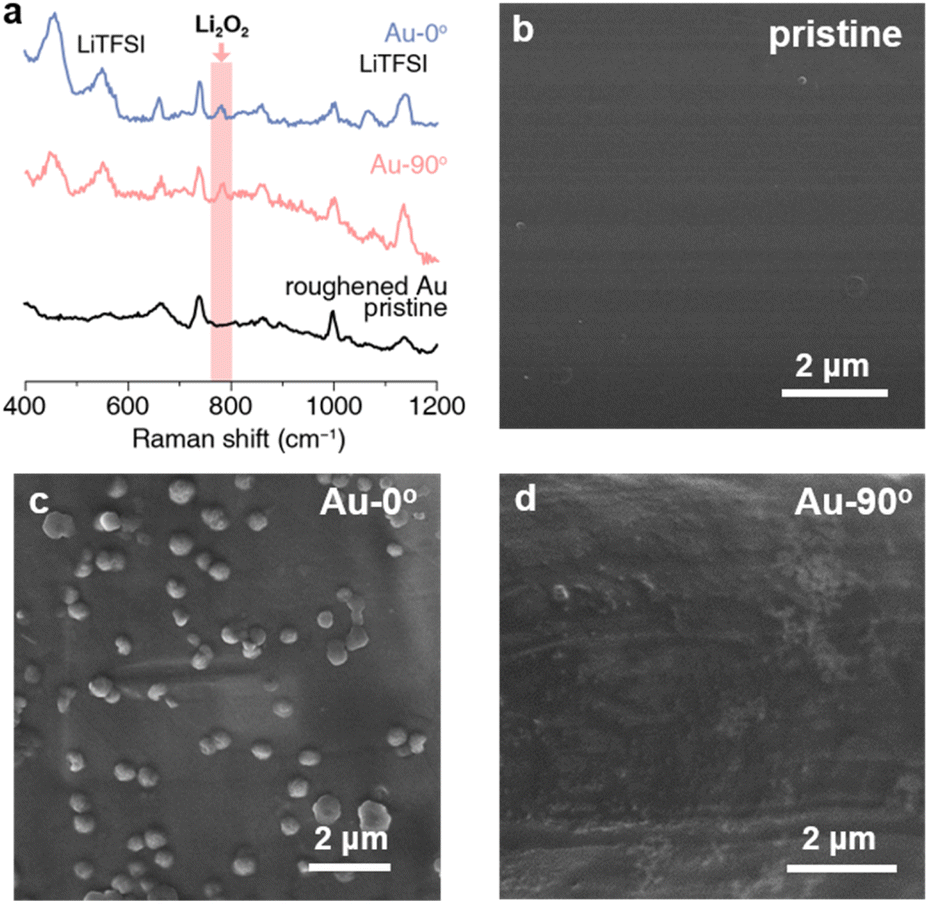 | ||
| Fig. 2 Characterization of discharge products on Au crystal electrodes at 0° and 90°. (a) Raman spectroscopy images display the types of discharge products on the Au (roughened) electrode. (b–d) SEM image of (b) the pristine Au electrode surface; (c) discharge products on Au-0° electrode and (d) Au-90° electrode surface. | ||
When the electrode was placed at 45°, the mmea/mexp ratio decreased by half to 113.8%, indicating that only some of the discharge products were deposited onto the electrode. When the electrode was placed at 90°, the mmea/mexp ratio further decreased to only 48.5% (Fig. 1e). On Au-90° electrode, some discharge products, especially Li2O2 particles from the solution pathway, did not adhere to the electrode thus leading to a loss of mmea and a low mmea/mexp ratio.
Therefore, we believe that the mass deposition on Au-90° can only be attributed to Li2O2 adsorbing on the electrode surface via the surface pathway without the contribution of Li2O2 particles via the solution pathway. The SEM image of Au-90° (Fig. 2d) showed a smooth surface of the vertical electrode, with no particulate products, which was distinctly different from Au-0° (Fig. 2c). Based on the deposited mass, the thickness of the Li2O2 layer on the electrode surface was estimated to be 2.1 nm, which is detailed in ESI.† This thickness matches the properties of Li2O2 thin films and is consistent with our previous results regarding the surface pathway using flow cell setup.46,47
Surface adsorption of Li2O2
To verify that this layer of Li2O2 product originates from surface adsorption, we conducted the same Li2O2 deposition but with the electrode tilted downward, namely Au-135° as shown in Fig. 1a and e. The Li2O2 particles are difficult to settle on the electrode facing downward. Therefore, the detected mass increase on the electrode can only be attributed to the Li2O2 product generated through strong surface adsorption. This is confirmed by the similar mmea/mexp ratio of Au-90° and Au-135° in Fig. 1d.To confirm the critical role of surface adsorption on Au-90° and Au-135°, carbon and platinum crystal electrodes were used to alter the strength of surface adsorption. The oxygen species and intermediates have weak adsorption on the carbon surface but strong adsorption on platinum surface. By keeping the electrodes vertical (Pt-90° and C-90°) and applying a reduction potential to deposit Li2O2, we observed significant differences in the amount of product deposition, Fig. 3a and b. For Pt-90°, the mass of expected and measured deposition was 79.0 ng and 50.7 ng, respectively, showing a mmea/mexp ratio of 64.1% (Fig. 3a). For C-90°, the mmea/mexp ratio is only 40.1%, based on the expected and measured deposition of 80.6 ng and 32.3 ng, respectively (Fig. 3b).
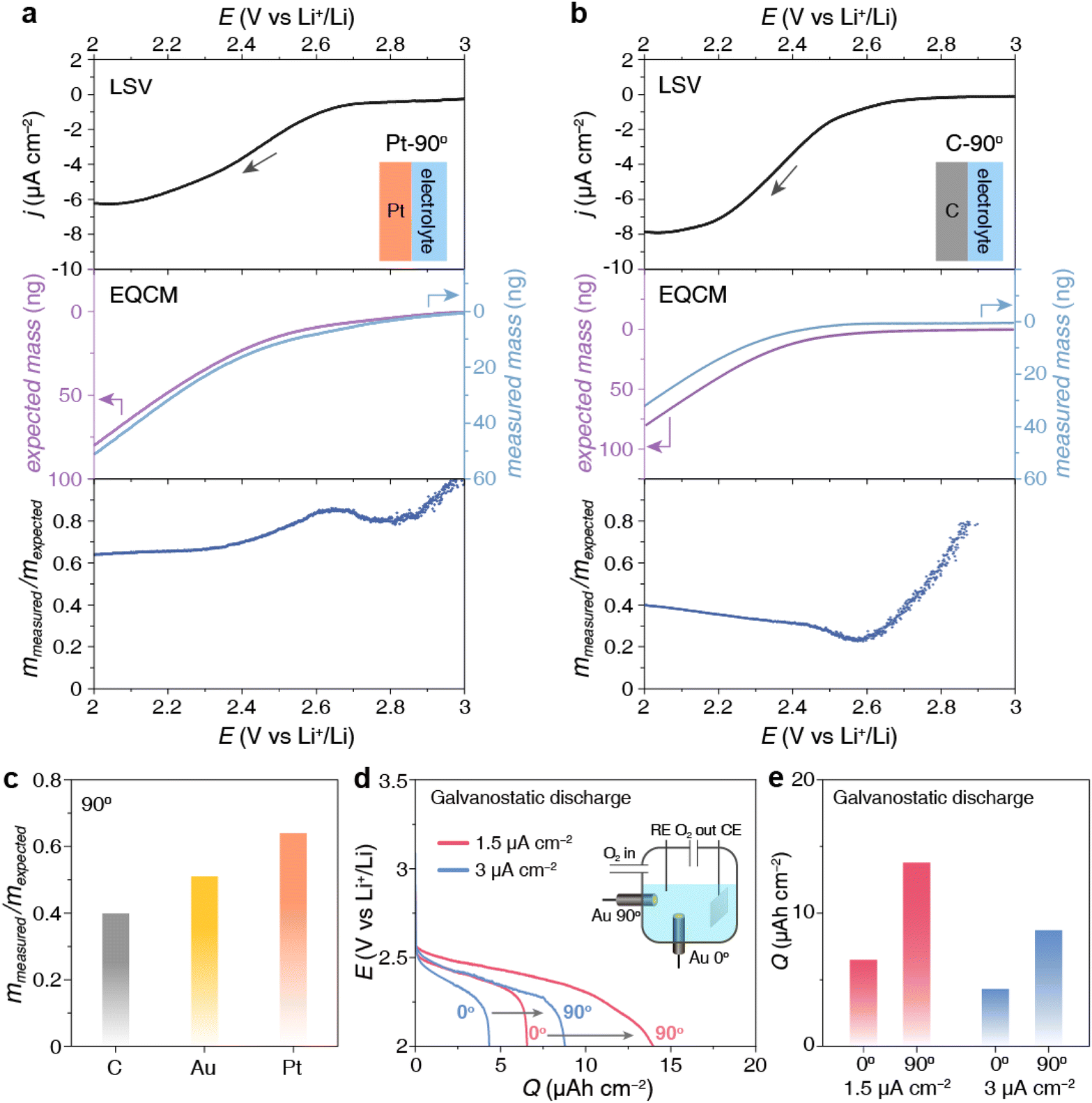 | ||
| Fig. 3 Impact of surface adsorption on the discharge process. (a and b) EQCM results at (a) a C electrode and (b) a Pt electrode placed at 90° using LSV, and the difference between the expected deposition mass and the measured deposition mass. Sweep rate: 2 mV s−1. (c) Comparison of the amount of product deposition at vertical Au/C/Pt electrodes. (d and e) Comparison of the load curves and capacities of the galvanostatic discharging using Au electrodes placed at 0° and 90° in O2-saturated 1 M LiTFSI–TEGDME electrolyte under a current density of 1.5 and 3 μA cm−2. | ||
As shown in Fig. 3c, the distinct comparison mmea/mexp ratios at carbon, Au, and Pt surface indicates that Li2O2 deposition at the vertical electrode surface was dominated by the surface adsorption pathway, which is substantially influenced by the electrode materials. Pt owns the strongest adsorption of oxygen species, resulting in the highest mmea/mexp ratio among three electrodes, whereas C owns the lowest adsorption, resulting in the lowest mmea/mexp ratio.
The above results demonstrate that the orientation of the electrodes determines the Li2O2 formation pathway. On a vertical electrode like Au-90°, apart from some adsorbed Li2O2 product, Li2O2 particles via the solution pathway will fall off to the remain the electrode surface clean (Fig. 1e), delaying discharge termination caused by surface passivation as much as possible. On an upward-facing electrode like Au-0°, products generated via both pathways will settle on the upward-facing surface. These deposited Li2O2 particles impede oxygen mass transport and passivate the electrode surface, thereby limiting the discharge capacity. As shown in Fig. 3d and e, the discharge capacity of Au-90° is twice that of Au-0° at both 1.5 and 3 μA cm−2. This comparison confirms that product accumulation on the upward-facing electrode more easily causes passivation of the electrode, while the vertical electrode can achieve a larger discharge capacity.
Therefore, within a practical porous electrode for lithium–oxygen batteries, surfaces oriented in different directions have distinct roles. Assuming a pore is a square room, the vertical surface provides reaction sites for the oxygen reduction to form Li2O2, and the upward-facing surface is responsible for supporting the Li2O2 products. This implies that the catalysts on upward-facing surfaces will quickly become passivated and fail. To approach the theoretical specific energy of lithium–oxygen batteries, it is essential to maximize the filling ratio of discharge products within the porous electrode, targeting a filling ratio of over 80%. How to leverage this characteristic to design the pore structure of electrodes and optimize the distribution of catalytic active sites is a crucial question. For primary lithium–oxygen batteries, what kind of electrode structure can separate the Li2O2 production zone from the Li2O2 storage zone and delay the passivation of catalytic sites in the Li2O2 production zone, thereby further increasing discharge capacity? For rechargeable lithium–oxygen batteries, if the discharge products fall off from vertical surfaces, they will not be oxidized and decomposed at their original sites but on other surfaces during subsequent charging. Therefore, ensuring the contact between discharge products and the charging catalyst on other surfaces to conduct the charging process is critical and must be considered.
The dissolution of Li2O2
In addition to the strong adsorption processes on the electrode surface, there is a competing dissolution process of Li2O2 product, otherwise, Li2O2 could not be efficiently oxidized by heterogeneous catalysts during charging. As shown in Fig. 4, after depositing a certain amount of Li2O2 on the EQCM Au electrode, we immediately ceased the applied potential, keeping the cell at an open circuit and monitoring the mass change of the EQCM electrode. Consequently, we found that the deposited Li2O2 would dissolve and detach from the electrode surface, leading to a decreasing mass of EQCM electrode. The dissolution of Li2O2 shows different kinetics under different conditions, including electrode angle, deposition pathway, and deposition amount, suggesting there might be several different dissolution modes.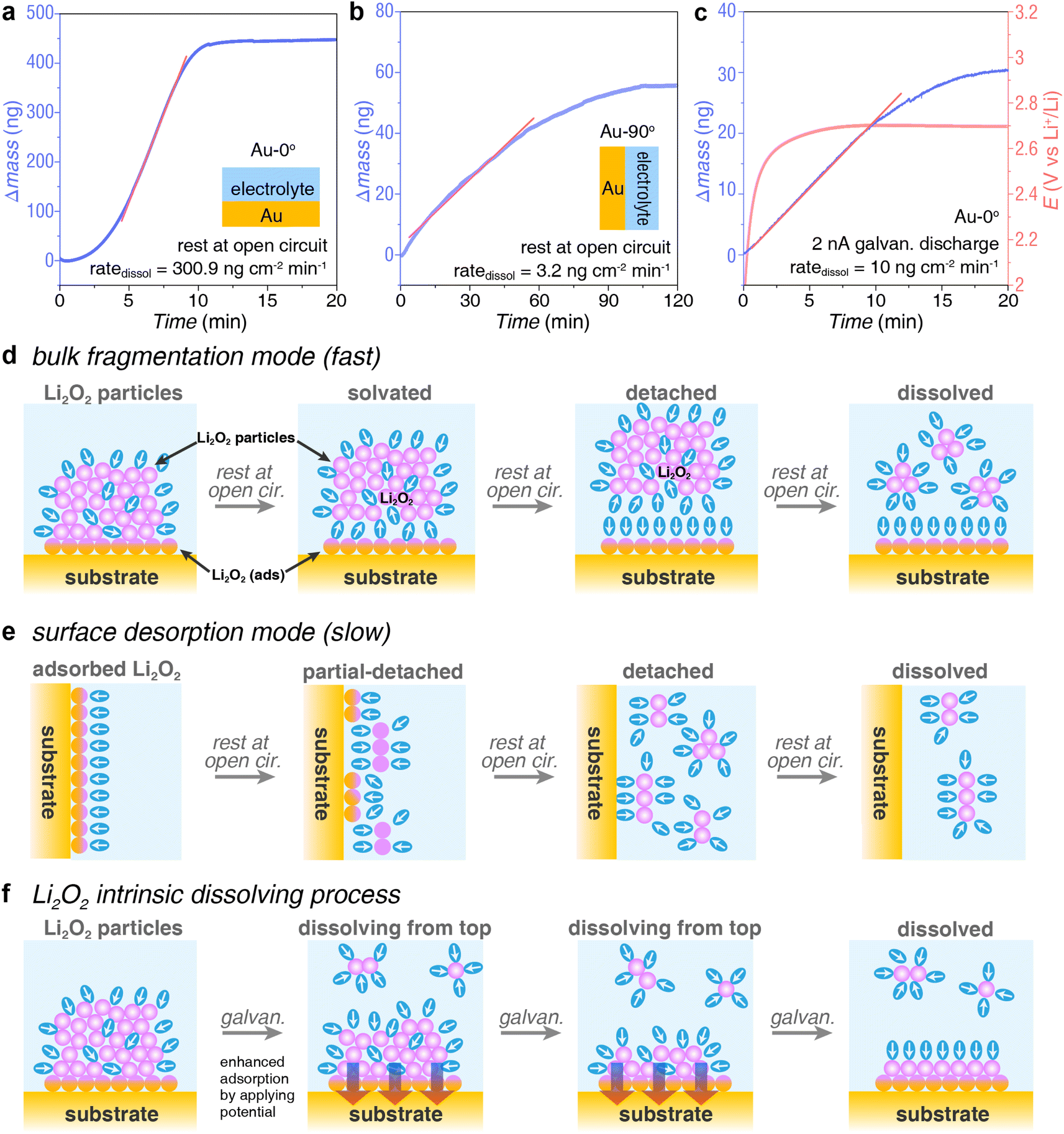 | ||
| Fig. 4 Kinetics of dissolution process of Li2O2. (a and b) Rate of natural dissolution of Li2O2 that was deposited on (a) Au-0° electrode and (b) Au-90° electrode at open circuit. (c) Rate of Li2O2 dissolution on Au-0° electrode at a reduction potential to maintain surface adsorption. A tiny discharging current of 2 nA (equivalent to 0.12 ngLi2O2 cm−2 min−1 deposition) was applied to keep the cell under a discharging status. (d–f) Schematics of the various dissolving processes corresponding to EQCM results in a–c. | ||
The electrode angle significantly influences the dissolution rate (ratedissol) of Li2O2 products. Li2O2 dissolved rapidly on Au-0° (Fig. 4a) but slowly on Au-90° (Fig. 4b). In Fig. 4a, at open circuit, the mass of Li2O2 on Au-0° electrode decreased rapidly within 10 minutes at a rate of 300.9 ng cm−2 min−1. This process dissolved approximately 415 ng of Li2O2, accounting for 95.5% of the total mass of Li2O2 deposition. In contrast, for Au-90° electrode at an open circuit, Li2O2 dissolved slowly at a rate of 3.2 ng cm−2 min−1 (Fig. 4b). This process dissolved approximately 54 ng of Li2O2, accounting for 95.9% of the total mass of deposition.
The different rate of dissolution between Au-0° and Au-90° arises from the differences in the dissolution mode, i.e. bulk fragmentation mode and surface desorption mode. When the electrode was upward-facing (Fig. 4d), the solution pathway dominated during discharging, leading to Li2O2 particles deposition onto the crystal electrode. This resulted in a loosely bound structure of the discharge product that was easily penetrated and solvated by the solvent. Therefore, the high ratedissol, in this case, was due to large chunks of Li2O2 detaching from the electrode surface (specifically at the interface of Li2O2(ads) and sub-layer of Li2O2) with the assistance from the bulk electrolyte and the electrolyte contained within the particles, as shown in Fig. 4d. Once the large chunks of Li2O2 were solvated and detached from the surface, a large mass loss was detected by EQCM in Fig. 4a. In contrast, when the electrode was vertical (Fig. 4e), the Li2O2 product came from the surface pathway during discharging. Despite the small amount of Li2O2 deposition, it was tightly bound to the electrode and less prone to detachment. Therefore, the dissolution process was slower in this case. The key to this dissolution process lies in the competition between solvation of the products and surface adsorption. In summary, the entire dissolution process can be divided into two concurrent parts: dissolution of the surface adsorption layer at electrode|Li2O2(ads) interface (a slow process) and dissolution of bulk product at Li2O2(ads)|Li2O2(bulk) interface, leading to bulk fragmentation (a fast process). The ratedissol of the slow process is two orders of magnitude lower than the fast process.
Solvation could start at both electrode|Li2O2(ads) interface and Li2O2(ads)|Li2O2(bulk) interface, leading to the dissolution and detachment of the products. Unfortunately, the ratedissol measured in these two modes does not represent the intrinsic ratedissol of Li2O2. Solvation at the former interface is affected by the competition of solvation energy and adsorption energy. Due to surface adsorption, the ratedissol of Li2O2(ads) tends to be slower. Solvation at the latter interface is affected by the thickness and density of the product layer, which influences the speed of solvent penetration and thus the dissolution time. As discussed above, the discharge products deposited on the electrode are loose and contain electrolytes interspersed within, which accelerates dissolution and bulk fragmentation. The measured ratedissol tends to be faster.
Interestingly, when we apply a slight reduction potential to the electrode while measuring ratedissol, a very small amount of discharging reaction occurs on the electrode surface (reduction of O2 to form Li2O2). This approach prevents the desorption of Li2O2(ads) and allows the dissolved oxygen in the electrolyte to quickly diffuse to the electrode surface and be reduced to Li2O2 as soon as gaps form during the solvation of Li2O2(bulk). The newly formed Li2O2 acts like glue, preventing the adsorption layer from detaching, meanwhile holding the cracked Li2O2 together and preventing its further fragmentation. Consequently, both dissolution pathways are inhibited, resulting in only the Li2O2 at the Li2O2(bulk)|electrolyte interface undergoing dissolution (Fig. 4f). Given the great error in estimating the amount of Li2O2 produced during potentiostatic measurement due to switching current ranges, we employed the galvanostatic measurement. As shown in Fig. 4c, after applying a discharging current of 2 nA (corresponding to a potential around 2.7 V), we observed the ratedissol of 10 ng cm−2 min−1. The deposition rate of Li2O2 at the 2 nA is 0.12 ng cm−2 min−1 (Fig. S2†), which is two orders of magnitude lower than the dissolution rate and can be neglected. Fig. S3† shows that the ratedissol is same even if the initial amount of Li2O2 at the EQCM electrode is several times higher, indicating that this rate appears to be the intrinsic dissolution rate of Li2O2 taking place at the Li2O2(bulk)|electrolyte interface.
The composition of the electrolyte also affects the dissolution rate of the products. To study the effect of different solvent molecules, the solvent in the electrolyte was replaced with dimethyl sulfoxide (DMSO) with a high donor number. In Fig. S4,† the dissolution process of products in DMSO-based electrolyte is faster, with a dissolution rate of up to 22.7 ng cm−2 min−1. Firstly, in high donor number electrolytes, the formation of solution-phase products is facilitated, making the dissolution process easier. Secondly, DMSO molecules are smaller and more polar, allowing the solvent to insert more rapidly between the electrode and the product. Therefore, compared to TEGDME-based electrolyte, DMSO-based electrolyte can provide more discharge capacity.
Implications of sluggish dissolution
The sluggish dissolution of discharge products results in two distinct consequences for the discharge and charge process of lithium–oxygen batteries, respectively. For the discharge process, to avoid electrode passivation by discharge products, cells can discharge at a low current, allowing the rate of product formation and dissolution to reach equilibrium, thus achieving high capacity and specific energy. This is why many studies on lithium–oxygen batteries employ low current on discharge to achieve higher capacities.48 However, due to the tiny discharge current, this approach does not meet the demands of applications.Intermittent-desorption discharge can provide time for Li2O2 dissolution to prevent electrode blockage as much as possible, thereby increasing discharge capacity. Electrode blockage refers to the difficulty of oxygen passing through the discharge product layer, leading to high overpotential and low discharge capacity. Our previous study has demonstrated that more than 70% of discharge products are formed at the electrode|Li2O2 interface.49 This means that oxygen needs to pass through the product layer to reach the electrode and catalyst surface for reduction, making this mass transfer process the bottleneck of the discharge reaction. Intermittent discharge leaves time for Li2O2 partial dissolution, exposing fresh active sites, and increasing discharge capacity. However, during intermittent discharge, the natural rest to equilibrium potential is slow and takes a long time due to the sluggish dissolution as discussed above.
According to the kinetics of dissolution, if the electrode was applied 2.9 V (lower than  of 2.96 V) after Li2O2 deposition to encourage the Li2O2 desorption, 80% of deposited Li2O2 detached from the surface within 3 min (Fig. 5a). This is 3 times faster than the dissolution under simple open circuit (Fig. 4a). We constructed lithium–oxygen cells and carried out the intermittent-desorption discharge. As shown in Fig. S5,† after every 10 minutes of galvanostatic discharge, the cell is held at 2.9 V for 3 minutes to allow the product to desorb. If a potential above 3 V is applied, it becomes the subsequent charging process. To ensure that the discharge process remains uninterrupted, we hold the potential at 2.9 V, although it is not an optimized potential. This cycle is repeated continuously until reaching the cutoff voltage. Compared to the continuous galvanostatic discharge, the cell achieved a 10-fold capacity at the same current density (Fig. 5b and c).
of 2.96 V) after Li2O2 deposition to encourage the Li2O2 desorption, 80% of deposited Li2O2 detached from the surface within 3 min (Fig. 5a). This is 3 times faster than the dissolution under simple open circuit (Fig. 4a). We constructed lithium–oxygen cells and carried out the intermittent-desorption discharge. As shown in Fig. S5,† after every 10 minutes of galvanostatic discharge, the cell is held at 2.9 V for 3 minutes to allow the product to desorb. If a potential above 3 V is applied, it becomes the subsequent charging process. To ensure that the discharge process remains uninterrupted, we hold the potential at 2.9 V, although it is not an optimized potential. This cycle is repeated continuously until reaching the cutoff voltage. Compared to the continuous galvanostatic discharge, the cell achieved a 10-fold capacity at the same current density (Fig. 5b and c).
 | ||
| Fig. 5 The consequence of slow dissolution process on discharging and charging process. (a) The dissolution of the Li2O2 deposited on the electrode (Au-0°) surface when applying a potential of 2.9 V. (b) Load curve for discharging a Li–O2 cell with intermittent-desorption discharge mode. After every 10 minutes of galvanostatic discharge, the cell is held at 2.9 V for 3 minutes. Current density: 0.22 mA cm−2. (c) Comparison between intermittent-desorption discharge and continuous galvanostatic discharge at the same current density of 0.22 mA cm−2. (d) The estimate of the maximum charging current under the given discharge capacity and product particle size. | ||
For the charging process, Li2O2 may decompose in two pathways: one is direct oxidative decomposition at the catalyst|Li2O2 interface, which is a solid–solid interface. In this case, only products in strong contact with the surface can be decomposed, and it is challenging to maintain solid–solid contact, similar to the challenges in solid-state batteries.50,51 The other way is that the Li2O2 dissolves and diffuses to the electrode surface in the form of molecules or clusters, where they are oxidized and decomposed. This is the common model used in calculating the decomposition of Li2O2 on different catalyst surfaces.
If the catalyst can only oxidize dissolved Li2O2 molecules or clusters, the charging current is so low that achieving a reasonable charging current is challenging. In this model, the decomposition current is significantly related to the discharge capacity and particle size. Fig. 5d shows the maximum charging current that could be achieved if the dissolved molecules can be quickly decomposed. The details of the estimate have been stated in the ESI.† Achieving a current density of 0.4 mA cmareal−2 requires small particle size (i.e. <3 nm) and high capacity (i.e. >5 mA h cm−2), which is almost impractical. Therefore, new catalytic pathways need to be designed for the charging process.
Conclusions
Herein, we studied the deposition and dissolution processes of solid Li2O2 products in lithium–oxygen batteries, revealing that the electrode orientation significantly impacts product deposition. When the electrode is vertical in the electrolyte, only the adsorbed Li2O2 layer can adhere to the vertical electrode, Li2O2 particles from the solution pathway are difficult to adhere to, resulting in a small amount of product deposition. The Li2O2 particles fall onto the upward electrode surface, continuously accumulating and forming a loose product layer. This finding can guide the rational design of electrode pore structures by decoupling the reaction zone (providing catalytic active sites) and the product storage zone (storing discharge product), thereby achieving their respective functions, suppressing active site passivation, and maximizing the filling ratio of the porous electrode, pursuing the theoretical specific energy of lithium–oxygen batteries.The dissolution of Li2O2 deposition has two modes: the surface desorption mode and the bulk fragmentation mode. The latter is 2–3 orders of magnitude faster than the former. In the surface desorption mode, solvent molecules are inserted between the electrode and the Li2O2 adsorbed layer, resulting in a relatively slow process (3.2 ng cm−2 min−1). In the bulk fragmentation mode, solvent molecules insert into the Li2O2(ads)|Li2O2(bulk) interface, making chunk Li2O2 rapidly detach from the electrode surface in clusters. Because Li2O2 falls off in chunks, the dissolution rate is very fast (>300 ng cm−2 min−1) and the dissolution rate is related to the amount of deposited product. The solvent plays an important role in the dissolution process. Since the discharge product is not dense and contains many electrolyte molecules, these molecules accelerate Li2O2 dissolution. It is worth noting that this rate is only the rate at which Li2O2 particles fall off the electrode surface, not the intrinsic dissolution rate of Li2O2 particles. The intrinsic dissolution rate of Li2O2 at the Li2O2|electrolyte interface is between the rates of these two modes (10 ng cm−2 min−1). If the charging process is based on the mode of Li2O2 dissolution-diffusion-molecular oxidation process, it is difficult to achieve a satisfactory charging current.
Based on these dissolution kinetics, we designed an intermittent-desorption discharge lithium–oxygen battery by applying a low potential lower than 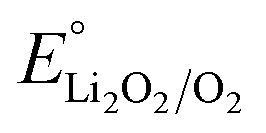 during a period of discharging to encourage the rapid dissolving of the innermost adsorbed Li2O2, resetting the active sites on the electrode surface, thereby increasing the discharge capacity by an order of magnitude. Furthermore, by adjusting the chemical composition of the electrolyte and the physical properties of the electrode surface, the dissolution of Li2O2 can be promoted as well. These findings provide a new approach to addressing the issue of electrode surface passivation in lithium–oxygen batteries in the pursuit of its ultrahigh theoretical specific energy.
during a period of discharging to encourage the rapid dissolving of the innermost adsorbed Li2O2, resetting the active sites on the electrode surface, thereby increasing the discharge capacity by an order of magnitude. Furthermore, by adjusting the chemical composition of the electrolyte and the physical properties of the electrode surface, the dissolution of Li2O2 can be promoted as well. These findings provide a new approach to addressing the issue of electrode surface passivation in lithium–oxygen batteries in the pursuit of its ultrahigh theoretical specific energy.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. H. and S. W. performed experiments. Y. C. conceived the project and wrote the manuscript. All authors discussed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by Jiangsu Province Carbon Peak and Neutrality Innovation Program (Industry tackling on prospect and key technology) (BE2022031-4, BE2022002-3), National Natural Science Foundation of China (52173173), Natural Science Foundation of Jiangsu Province (BK20220051). We are grateful to the High Performance Computing Center of Nanjing Tech University for supporting the computational resources.Notes and references
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS.
- Y. Chen, J. Xu, P. He, Y. Qiao, S. Guo, H. Yang and H. Zhou, Sci. Bull., 2022, 67, 2449–2486 CrossRef CAS.
- J. Zhang, Y. Zhao, B. Sun, Y. Xie, A. Tkacheva, F. Qiu, P. He, H. Zhou, K. Yan, X. Guo, S. Wang, A. M. McDonagh, Z. Peng, J. Lu and G. Wang, Sci. Adv., 2022, 8, eabm1899 CrossRef.
- Z. Sun, X. Lin, C. Wang, Y. Tan, W. Dou, A. Hu, J. Cui, J. Fan, R. Yuan, M. Zheng and Q. Dong, Adv. Mater., 2024, 36, 2404319 CrossRef CAS PubMed.
- P. Zhang, M. Ding, X. Li, C. Li, Z. Li and L. Yin, Adv. Energy Mater., 2020, 10, 2001789 CrossRef CAS.
- L. A. Archer, P. G. Bruce, E. J. Calvo, D. Dewar, J. H. J. Ellison, S. A. Freunberger, X. Gao, L. J. Hardwick, G. Horwitz, J. Janek, L. R. Johnson, J. W. Jordan, S. Matsuda, S. Menkin, S. Mondal, Q. Qiu, T. Samarakoon, I. Temprano, K. Uosaki, G. Vailaya, E. D. Wachsman, Y. Wu and S. Ye, Faraday Discuss., 2024, 248, 392–411 RSC.
- Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J.-G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2013, 23, 987–1004 CrossRef CAS.
- J.-H. Kang, J. Lee, J.-W. Jung, J. Park, T. Jang, H.-S. Kim, J.-S. Nam, H. Lim, K. R. Yoon, W.-H. Ryu, I.-D. Kim and H. R. Byon, ACS Nano, 2020, 14, 14549–14578 CrossRef CAS.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- M. Asadi, B. Sayahpour, P. Abbasi, A. T. Ngo, K. Karis, J. R. Jokisaari, C. Liu, B. Narayanan, M. Gerard, P. Yasaei, X. Hu, A. Mukherjee, K. C. Lau, R. S. Assary, F. Khalili-Araghi, R. F. Klie, L. A. Curtiss and A. Salehi-Khojin, Nature, 2018, 555, 502–506 CrossRef CAS.
- Y. Zhou and S. Guo, eScience, 2023, 3, 100123 CrossRef.
- C. Prehal and S. A. Freunberger, Joule, 2019, 3, 321–323 CrossRef.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed.
- P. Albertus, G. Girishkumar, B. McCloskey, R. S. Sánchez-Carrera, B. Kozinsky, J. Christensen and A. C. Luntz, J. Electrochem. Soc., 2011, 158, A343 CrossRef CAS.
- V. Viswanathan, K. S. Thygesen, J. S. Hummelshøj, J. K. Nørskov, G. Girishkumar, B. D. McCloskey and A. C. Luntz, J. Chem. Phys., 2011, 135, 214704 CrossRef CAS.
- H. Yan, W.-W. Wang, T.-R. Wu, Y. Gu, K.-X. Li, D.-Y. Wu, M. Zheng, Q. Dong, J. Yan and B.-W. Mao, J. Am. Chem. Soc., 2023, 145, 11959–11968 CrossRef CAS PubMed.
- J. Wang, L. Ma, J. Xu, Y. Xu, K. Sun and Z. Peng, SusMat, 2021, 1, 345–358 CrossRef CAS.
- Z. Zhao, X. Zhang, Z. Zhou, E. Wang and Z. Peng, Nano Lett., 2022, 22, 501–507 CrossRef CAS PubMed.
- X. Gao, Y. Chen, L. R. Johnson, Z. P. Jovanov and P. G. Bruce, Nat. Energy, 2017, 2, 17118 CrossRef CAS.
- Y. Ren, J.-s. Fan and Y.-z. Fu, Energy Mater., 2023, 3, 300015 CAS.
- W.-J. Kwak, A. Mahammed, H. Kim, T. T. Nguyen, Z. Gross, D. Aurbach and Y.-K. Sun, Mater. Horiz., 2020, 7, 214–222 RSC.
- Y. Dou, Z. Xie, Y. Wei, Z. Peng and Z. Zhou, Natl. Sci. Rev., 2022, 9, nwac040 CrossRef PubMed.
- L. Liu, C. Zhou, W. Fang, Y. Hou and Y. Wu, Energy Mater., 2023, 3, 300011 CAS.
- D. J. Lee, H. Lee, Y.-J. Kim, J.-K. Park and H.-T. Kim, Adv. Mater., 2016, 28, 857–863 CrossRef CAS.
- D. Zhou, J. Zhang, T. Bian, Y. Tao, X. Liu, Q. Han, Z. Liu, S. Chen, J. Wang, P. Zhang and Y. Zhao, Adv. Energy Mater., 2024, 14, 2303192 CrossRef CAS.
- W.-J. Kwak, S.-J. Park, H.-G. Jung and Y.-K. Sun, Adv. Energy Mater., 2018, 8, 1702258 CrossRef.
- T. Liu, J. T. Frith, G. Kim, R. N. Kerber, N. Dubouis, Y. Shao, Z. Liu, P. C. M. M. Magusin, M. T. L. Casford, N. Garcia-Araez and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 1428–1437 CrossRef CAS.
- G. Sun, Y. Wang, D. Yang, Z. Zhang, W. Lu and M. Feng, Chin. Chem. Lett., 2024, 35, 108469 CrossRef CAS.
- Q. Zhao, N. Katyal, I. D. Seymour, G. Henkelman and T. Ma, Angew. Chem., Int. Ed., 2019, 58, 12553–12557 CrossRef CAS.
- J. Lu, Y. Jung Lee, X. Luo, K. Chun Lau, M. Asadi, H.-H. Wang, S. Brombosz, J. Wen, D. Zhai, Z. Chen, D. J. Miller, Y. Sub Jeong, J.-B. Park, Z. Zak Fang, B. Kumar, A. Salehi-Khojin, Y.-K. Sun, L. A. Curtiss and K. Amine, Nature, 2016, 529, 377–382 CrossRef CAS PubMed.
- L.-N. Song, L.-J. Zheng, X.-X. Wang, D.-C. Kong, Y.-F. Wang, Y. Wang, J.-Y. Wu, Y. Sun and J.-J. Xu, J. Am. Chem. Soc., 2024, 146, 1305–1317 CrossRef CAS PubMed.
- M. Li, J. Wu, Z. You, Z. Dai, Y. Gu, L. Shi, M. Wu and Z. Wen, Angew. Chem., Int. Ed., 2024, 63, e202403521 CrossRef CAS.
- Z. Jiang, B. Wen, Y. Huang, Y. Guo, Y. Wang and F. Li, Angew. Chem., Int. Ed., 2024, 63, e202315314 CrossRef CAS.
- K. Nishioka, M. Tanaka, H. Fujimoto, T. Amaya, S. Ogoshi, M. Tobisu and S. Nakanishi, Angew. Chem., Int. Ed., 2022, 61, e202112769 CrossRef CAS.
- M. D. Radin and D. J. Siegel, Energy Environ. Sci., 2013, 6, 2370–2379 RSC.
- J. B. Varley, V. Viswanathan, J. K. Nørskov and A. C. Luntz, Energy Environ. Sci., 2014, 7, 720–727 RSC.
- Q. Cui, Y. Zhang, S. Ma and Z. Peng, Sci. Bull., 2015, 60, 1227–1234 CrossRef CAS.
- G. Vanhoutte, S. Schaltin, M. Wu, F. Bardé and J. Fransaer, ECS Meeting Abstracts, 2015, vol. MA2015-03, p. 454 Search PubMed.
- Y. Ji, Z.-W. Yin, Z. Yang, Y.-P. Deng, H. Chen, C. Lin, L. Yang, K. Yang, M. Zhang, Q. Xiao, J.-T. Li, Z. Chen, S.-G. Sun and F. Pan, Chem. Soc. Rev., 2021, 50, 10743–10763 RSC.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- D. G. Kwabi, M. Tułodziecki, N. Pour, D. M. Itkis, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2016, 7, 1204–1212 CrossRef CAS PubMed.
- C. Shen, P. Andrei and J. P. Zheng, ECS Meeting Abstracts, 2018, vol. MA2018-02, p. 351 Search PubMed.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- Y. J. Lee, W.-J. Kwak, Y.-K. Sun and Y. J. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 526–533 CrossRef CAS.
- W. Wang, C. Tan, L. He, F. Yu, X. Gao and Y. Chen, J. Phys. Chem. Lett., 2024, 15, 583–589 CrossRef CAS PubMed.
- C. Tan, W. Wang, Y. Wu and Y. Chen, Faraday Discuss., 2024, 248, 160–174 RSC.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1778 RSC.
- C. Tan, D. Cao, L. Zheng, Y. Shen, L. Chen and Y. Chen, J. Am. Chem. Soc., 2022, 144, 807–815 CrossRef CAS PubMed.
- N. Sun, Q. Liu, Y. Cao, S. Lou, M. Ge, X. Xiao, W.-K. Lee, Y. Gao, G. Yin, J. Wang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 18647–18653 CrossRef CAS PubMed.
- L. Xu, S. Tang, Y. Cheng, K. Wang, J. Liang, C. Liu, Y.-C. Cao, F. Wei and L. Mai, Joule, 2018, 2, 1991–2015 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05911e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |