
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3DSMILES-GPT: 3D molecular pocket-based generation with token-only large language model†
Jike Wang‡
a,
Hao Luo‡a,
Rui Qin‡ a,
Mingyang Wanga,
Xiaozhe Wanb,
Meijing Fanga,
Odin Zhanga,
Qiaolin Goua,
Qun Sua,
Chao Shena,
Ziyi Youa,
Liwei Liu*b,
Chang-Yu Hsieh*a,
Tingjun Hou*a and
Yu Kang*a
a,
Mingyang Wanga,
Xiaozhe Wanb,
Meijing Fanga,
Odin Zhanga,
Qiaolin Goua,
Qun Sua,
Chao Shena,
Ziyi Youa,
Liwei Liu*b,
Chang-Yu Hsieh*a,
Tingjun Hou*a and
Yu Kang*a
aCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, Zhejiang, China. E-mail: yukang@zju.edu.cn; tingjunhou@zju.edu.cn; kimhsieh@zju.edu.cn
bAdvanced Computing and Storage Laboratory, Central Research Institute, 2012 Laboratories, Huawei Technologies Co., Ltd, Nanjing 210000, Jiangsu, China. E-mail: liuliwei5@huawei.com
First published on 4th December 2024
Abstract
The generation of three-dimensional (3D) molecules based on target structures represents a cutting-edge challenge in drug discovery. Many existing approaches often produce molecules with invalid configurations, unphysical conformations, suboptimal drug-like qualities, limited synthesizability, and require extensive generation times. To address these challenges, we present 3DSMILES-GPT, a fully language-model-driven framework for 3D molecular generation that utilizes tokens exclusively. We treat both two-dimensional (2D) and 3D molecular representations as linguistic expressions, combining them through full-dimensional representations and pre-training the model on a vast dataset encompassing tens of millions of drug-like molecules. This token-only approach enables the model to comprehensively understand the 2D and 3D characteristics of large-scale molecules. Subsequently, we fine-tune the model using pair-wise structural data of protein pockets and molecules, followed by reinforcement learning to further optimize the biophysical and chemical properties of the generated molecules. Experimental results demonstrate that 3DSMILES-GPT generates molecules that comprehensively outperform existing methods in terms of binding affinity, drug-likeness (QED), and synthetic accessibility score (SAS). Notably, it achieves a 33% enhancement in the quantitative estimation of QED, meanwhile the binding affinity estimated by Vina docking maintaining its state-of-the-art performance. The generation speed is remarkably fast, with the average time approximately 0.45 seconds per generation, representing a threefold increase over the fastest existing methods. This innovative 3DSMILES-GPT approach has the potential to positively impact the generation of 3D molecules in drug discovery.
Introduction
In recent years, deep generative models have attracted extensive attention, demonstrating remarkable advancements across diverse domains, ranging from natural language processing to video synthesis. These models exhibit remarkable proficiency in encoding and synthesizing data within continuous domains. However, as the focus shifts towards more intricate and discrete data types, notably chemical molecules, there is a growing emphasis on developing generative models capable of generating authentic and efficacious data within these realms. The progression of deep generative models has spurred the development of various methodologies aimed at tackling the challenge of molecular generation, offering a promising avenue for innovative drug molecule design.During earlier periods, ligand-based molecular generation (LBMG) gained significant popularity. These methodologies can be categorized into two primary types based on how generated molecules are represented: graph-based molecular generation and sequence-based molecular generation. The fundamental principle involves representing molecules as graphs or sequences, thus framing the generation task as either a graph structure generation or natural language generation problem. Techniques such as Bayesian optimization (BO) and reinforcement learning are employed to guide the model in generating the desired drug molecules.
Molecules inherently possess structures resembling graphs, rendering it intuitive to express their information graphically. Consequently, methods for molecular design grounded in graph representations and traditional heuristic algorithms have long been established. For example, Brown et al. devised a molecular optimization algorithm based on molecular graphs by employing genetic algorithms in 2004,1 while in 2013, Virshup et al. introduced the ACSESS algorithm.2 With the progression of graph neural networks (GNN) in recent years, these networks have exhibited remarkable adaptability across diverse challenges rooted in graph-structured data. De Cao et al. pioneered the integration of GNN for drug design with their 2018 proposal of MolGAN,3 thereby forging novel pathways in molecular design. As graph-based methodologies continue to advance rapidly, an escalating number of researchers are capitalizing on molecular graph representations for drug design.4–8
When compared to GNN-driven strategies for molecular synthesis, sequence-based methodologies offer a more succinct avenue. This stems from the fact that chemical compounds can be effectively represented through chemical languages such as the Simplified Molecular Input Line Entry System (SMILES)9 or SELFES,10 which mirror the structure of natural language. Consequently, a plethora of scholarly works on molecular design have proposed frameworks based on recurrent neural networks (RNNs) or transformer.11 In 2016, ChemVAE amalgamated variational autoencoders (VAEs) with BO to explore the latent state space in search of molecules with desired attributes.12 In 2017, Olivecrona et al. harnessed reinforcement learning to fine-tune the generation process of RNN-based molecules, yielding structures similar to specified ones or possessing predetermined activities.13 In 2021, Wang et al. shifted towards a transformer decoder instead of RNNs for generation, combining knowledge distillation and reinforcement learning to develop MCMG.14 Over time, a variety of sequence-based molecular generation methodologies have emerged.15–26
However, 2D molecular generation exhibits a significant limitation since these techniques neglect the crucial 3D structural complementarity between protein pockets and molecules. Given the pivotal role of ligand–protein conformational selection in drug design, evaluating such complementary features requires an understanding grounded in the intrinsic 3D structures of protein pockets and molecules. Consequently, there has been emerging interest in 3D structure-based molecular generation.
With the advent of deep geometric learning, numerous studies on autoregressive 3D molecular generation have surfaced. For instance, Gebauer introduced G-SchNet,27 an autoregressive deep neural network that generates diverse small organic molecules by sequentially positioning atoms in Euclidean space. Subsequently, models such as LiGAN,28 GraphBP,29 SBDD,30 and Pocket2Mol31 have been developed to directly generate molecules within pockets.32–35 However, autoregressive methodologies are susceptible to error accumulation, which has spurred the exploration of diffusion-based 3D molecular generation approaches.36–39 These methods facilitate the simultaneous generation of entire molecules, rather than sequentially producing atoms. So far, these strategies have yet to effectively capture the distribution of chemical bonds, leading to the creation of impractical molecular structures.
The 3D molecular generation methods discussed above primarily rely on GNN. While language models (LMs) effectively extract abundant 2D molecular insights from extensive drug-like datasets during 2D molecular design, their ability to represent continuous 3D molecular architectures remains limited. However, with the recent proliferation of large-scale language models (LLMs), numerous studies suggest that LLMs can adeptly acquire continuous numerical representations. Born et al. presented the Regression Transformer,40 which accomplishes unified regression and prediction tasks by encoding numerical values as tokens. The approach demonstrates the potential of Transformers for regression tasks by encoding numerical values as tokens, though it faces limitations in capturing complex 3D molecular structures and may be inefficient when scaled to larger datasets. Furthermore, Flam-Shepherd et al. utilized Cartesian coordinates xyz token to represent the 3D structures of molecules.41 However, this approach may struggle with generating physically plausible conformations due to the discrete nature of tokenization, and may encounter difficulties in maintaining chemical validity. Both methodologies have shown promising effectiveness in their respective drug design endeavors. Recently, Feng et al. proposed Lingo3DMol,42 a fragment-based LM-centric 3D molecular generation model, which exhibits promising performance surpassing that of graph network-based models during their benchmark assessments.
The aforementioned endeavors highlight the adeptness of LMs in discerning intricate details pertaining to the inherent 3D structural characteristics of molecules. Compared to the complex diffusion and GNN-based methods for molecular generation, autoregressive approaches grounded in LMs offer simpler and more efficient training processes. Moreover, token-only paradigms seamlessly integrate with existing universal LLMs. Consequently, we explore the feasibility of employing a simpler and more explicit method to delineate the structural features of molecules and protein pockets. Based on this understanding, we confirm that large language models (LLMs) can effectively capture and utilize positional information related to molecules and protein pockets, enabling them to generate new molecular structures within target pockets. Herein, we present 3DSMILES-GPT, an innovative token-only framework designed for explicit 3D molecular generation, firmly rooted in LLM. As shown in Fig. 1, the architectural blueprint of 3DSMILES-GPT centers on a transformer decoder. By framing the task of generating 2D and 3D structures as a natural language generation endeavor, 3DSMILES-GPT encodes atomic 3D coordinates as tokens, facilitating the acquisition of molecular 2D and 3D information. To maximize the intrinsic capabilities of LMs, our methodology begins with the pretraining phase of 3DSMILES-GPT using an extensive dataset with drug-like molecules. After fine-tuning on a specified protein–ligand dataset, we integrate surface atomic coordinates from pockets along with ligand molecules. Furthermore, to enhance the model's ability to extract information from protein pockets, we introduce a protein encoder as a detachable modular component. Additionally, the application of reinforcement learning methodologies enables the refinement of generated molecules across a diverse range of properties. The experimental results demonstrate that, compared to existing state-of-the-art (SOTA) methodologies, 3DSMILES-GPT achieves optimal performance across 8 out of 10 benchmark metrics including bioactivity, drug-likeness, and synthetic accessibility. Moreover, the targeted case studies on 5 distinct protein targets further elucidate its efficacy in generating drug-like molecules with robust binding strength in practical scenarios.
 | ||
| Fig. 1 The overview of 3DSMILES-GPT. | ||
Results
The competency of language models in generating 2D molecular configurations is undeniable, owing to their adeptness in processing discrete data. By utilizing chemical languages like SMILES as input, these models demonstrate proficiency in acquiring knowledge of the inherent 2D topological arrangements of molecules. However, the pivotal question remains whether language models can effectively capture the distribution of continuous data, including molecular conformations. Thus, in this segment, we begin by assessing the quality of conformations generated by 3DSMILES-GPT. Subsequently, we conduct an analysis of the properties and binding efficacy of the generated molecules. Finally, we evaluate the generalization capacity of 3DSMILES-GPT with respect to specific drug targets.Quality of generated conformation
A significant challenge in DL-based molecular generation is the frequent production of non-physically plausible structures, an issue further magnified in existing token-only LLM-based methodologies. This limitation stems from the inherent difficulty that token-only LLM-based approaches face in effectively processing continuous data, often leading to the generation of non-physical conformations. Addressing this critical concern, we aim to identify molecules that exhibit physical conformations. Building upon this premise, we delve into further exploration involving enhanced molecular affinity and desirable drug-likeness, taking into consideration the protein pocket as a constraint. In this aspect of assessment, distinct from mere generation of conformers, our focus lies predominantly on the physical plausibility of the generated molecules within the pocket.For molecules bound within protein pockets, each type of bond lengths exhibits a robust distribution due to the constrained degrees of freedom resulting from lower flexibility. These bond lengths do not vary significantly across different constrained pockets. To evaluate the performance, we compared the distributions of some common bond lengths between the generated molecules and training molecules across various types of chemical bonds, using the Jensen–Shannon divergence (JSD)43 as a quantification metric. We categorized the data based on the types of chemical bonds to visualize the distributions of bond lengths (Fig. 2a). It shows that our method maintains a most balanced overall performance with no significant weaknesses across all types of bonds, namely, no JSD values smaller than 0.4. In the first two groups shown in Fig. 2a, which include bonds with carbon atoms that form the basic skeleton of drug molecules, 3DSMILES-GPT generally achieved suboptimal results, slightly inferior to TargetDiff, a SOTA method based on diffusion models. Positively, our model achieved the best results for most chalcogen-related chemical bonds. Furthermore, when considering other types of chemical bonds, we found that the bond length quality, as measured by JSD, is consistently distributed between 0.4 and 0.6, indicating low deviation and controllable variation across different bond types. These bonds occur less frequently in drug molecules compared to other categories, but they remain crucial in key functional groups such as nitro and sulfonic groups, which are often found in antibacterial drugs. The success of 3DSMILES-GPT in these bonds is likely attributable to the GPT model's capability to retain and recall information from scarce samples, allowing it to accurately reproduce the relative positions of atoms in molecules containing these relatively rare groups.
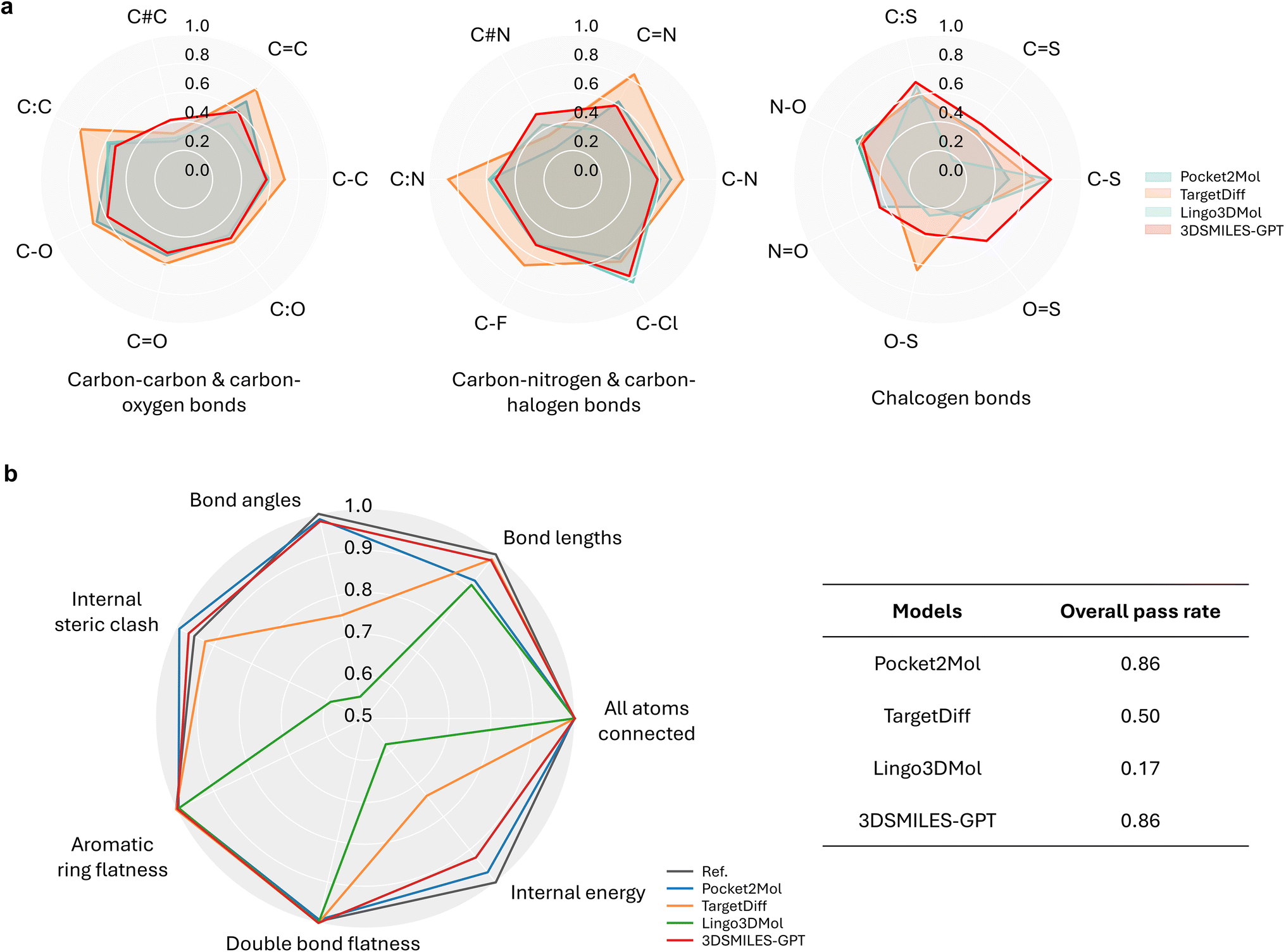 | ||
| Fig. 2 Quality of generated conformations. (a) The Jensen–Shannon Divergence (JSD) of common chemical bond lengths between 3DSMILES-GPT and other models, compared to reference molecules. For ease of visual comparison, the values are presented as 1-JSD, where values closer to 1 indicate better performance. (b) The performance on each metrics and the overall pass rate of each model tested with PoseBusters. | ||
As shown in Table S1† in more detail, our model achieves the best performance in over one-third of the bond length types and demonstrates comparably close to or superior performance in other bond length types compared to other models. On a global scale, our model's predictive capability for bond lengths in pocket generation tasks still lags behind TargetDiff, this discrepancy may be attributed to potential forgetting phenomena in transfer learning. The results indicate that, when compared to other methods specifically designed for pocket generation tasks utilizing GNNs or language models, the performance of our model is essentially equivalent and, in some instances, even superior. This finding underscores that using atomic coordinates as tokens for prediction can effectively reproduce the distributions of molecular bonds.
For a broader assessment, we employed PoseBusters,44 a suite designed to examine the physical and chemical inconsistencies in docking and molecular generation. It offers diverse metrics for inspecting potential errors in molecular conformations. Thus, we aim for our generated molecules to achieve high pass rates across those all metrics evaluating validity, sub-structure and stereochemistry plausibility, rather than excelling solely in specific ones. As shown in Fig. 2b and Table S2,† our model consistently achieves over an 85% pass rate across multiple metrics, indicating that the majority of generated molecules adhere to the physical and chemical plausibility as observed in natural states. In contrast, other models such as Lingo3DMol and TargetDiff, while achieving optimal performance in individual metrics, exhibit subpar performance in certain specific metrics like bond angles or steric clashes, with pass rates ranging from 50% to 70%. Compared to Pocket2Mol, our model performs almost equally well across multiple metrics, achieving a pass rate of over 90% in various independent metrics. Furthermore, the bond length analysis using PoseBusters supports the findings of the bond length analysis based on JSD. It is evident that both 3DSMILES-GPT and TargetDiff achieved nearly 100% pass rates in bond length metrics, while other models only exceeded 80%. This demonstrates that the majority of molecules generated by 3DSMILES-GPT have bond lengths within an acceptable range, consistent with the results presented in Fig. 2a, where the majority of chemical bonds relative to the reference molecule exhibited JSD values evenly distributed within the desirable range of 0.4–0.6.
In addition, while Pocket2Mol generated molecules with low molecular weights, molecules generated by 3DSMILES-GPT whose molecular weights were closer to the reference (reported real active molecules), thereby better aligning with the requirements of real-world drug discovery scenarios (Table 1). Consequently, our superior performance across various metrics and the overall higher pass rate with PoseBusters underscore the robustness and applicability of our approach. In conclusion, 3DSMILES-GPT demonstrates commendable performance in generating molecular conformations.
| Metrics | Ref. | Pocket2Mol | TargetDiff | Lingo3DMol | 3DSMILES-GPT |
|---|---|---|---|---|---|
| Mean Vina score (↓) | −7.45 | −7.15 | −7.11 | −7.68 | −7.72 |
| Mean QED (↑) | 0.48 | 0.57 | 0.57 | 0.26 | 0.76 |
| Mean SAS (↓) | 3.43 | 3.16 | 4.33 | 4.51 | 3.07 |
| Drug-like molecules % (↑) | 74% | 94% | 81% | 30% | 100% |
| Mol size | 22.75 | 17.74 | 22.65 | 40.68 | 23.71 |
| Mol weight | 332.35 | 241.25 | 298.41 | 480.50 | 329.10 |
| Validity (↑) | — | 1.00 | 0.97 | 0.99 | 0.99 |
| Diversity (↑) | — | 0.96 | 0.96 | 0.92 | 0.89 |
| BR % (↑) | — | 48% | 48% | 58% | 53% |
| BR-QED (↑) | — | 0.56 | 0.59 | 0.27 | 0.76 |
| BR-SAS (↓) | — | 3.52 | 4.78 | 4.51 | 3.10 |
| Time/s (↓) | — | 13.63 | 12.19 | 1.32 | 0.45 |
Molecular properties and binding mode
Initially, an assessment was conducted to evaluate the binding strength of the generated molecules by scoring them directly using QuickVina 2 with Vina score (kcal mol−1).45 As shown in Table 1, it was observed that 3DSMILES-GPT achieved notably higher average Vina scores compared to other baseline methods, even surpassing those of genuine molecules.Molecules with large size are more likely to occupy protein pockets, leading to higher Vina scores. This phenomenon emphasizes the importance of considering the physicochemical properties of generated molecules comprehensively. As demonstrated by Feng et al.,42 certain large, multi-ring structured molecules are unsuitable for many cases of drug development. Therefore, a thorough evaluation of the generated molecules is essential. An examination of Table 1 indicates that the molecules generated by our model align more closely with authentic molecules in terms of molecular weight compared to other baseline methods. Regarding molecular size, TargetDiff closely resembles authentic molecules, exhibiting similar characteristics with our model. Conversely, the molecules generated by Pocket2Mol and Lingo3Dmol display undersized and oversized dimensions, respectively, in both molecular size and weight.
Subsequent analyses involved a comparison of the SAS and QED of the generated molecules. As shown in Table 1, our model demonstrates significant advantages in both QED and SAS metrics compared to alternative pocket-aware molecular generation approaches. Notably, our model exhibits an approximate 33% improvement in QED performance over the top-performing baseline, Pocket2Mol. This highlights the superior drug-like characteristics of the molecules generated by 3DSMILES-GPT, thereby enhancing their potential pharmaceutical utility. Furthermore, when compared to other baseline models, the molecules generated by 3DSMILES-GPT also demonstrate higher SAS values, indicative of improved synthetic feasibility. Additionally, in comparison to other methodologies, our approach demonstrates a superior molecular generation speed, of only 0.45 s per generation, as evaluated using an NVIDIA Tesla V100 GPU.
To further explore the interplay among the QED, SAS, and various other molecular properties, we utilized heatmap visualization (Fig. 3). Fig. 3a illustrates the relationship between the quantity of molecules and their corresponding QED and SAS values. Notably, a substantial proportion of molecules generated by 3DSMILES-GPT cluster in the bottom-left quadrant, indicating a prevalence of molecules exhibiting heightened QED and diminished SAS compared to other models. Furthermore, we investigated the influence of molecular weight on this relationship, revealing that the molecules generated by Pocket2Mol, characterized by elevated QED and reduced SAS, tend to possess smaller molecular weights (Fig. 3b). Subsequently, we explored the correlation between the Vina scores and QED/SAS. As depicted in Fig. 3c, the notably lighter coloration in the bottom-left quadrant for 3DSMILES-GPT indicates lower Vina scores and, consequently, reduced binding energies.
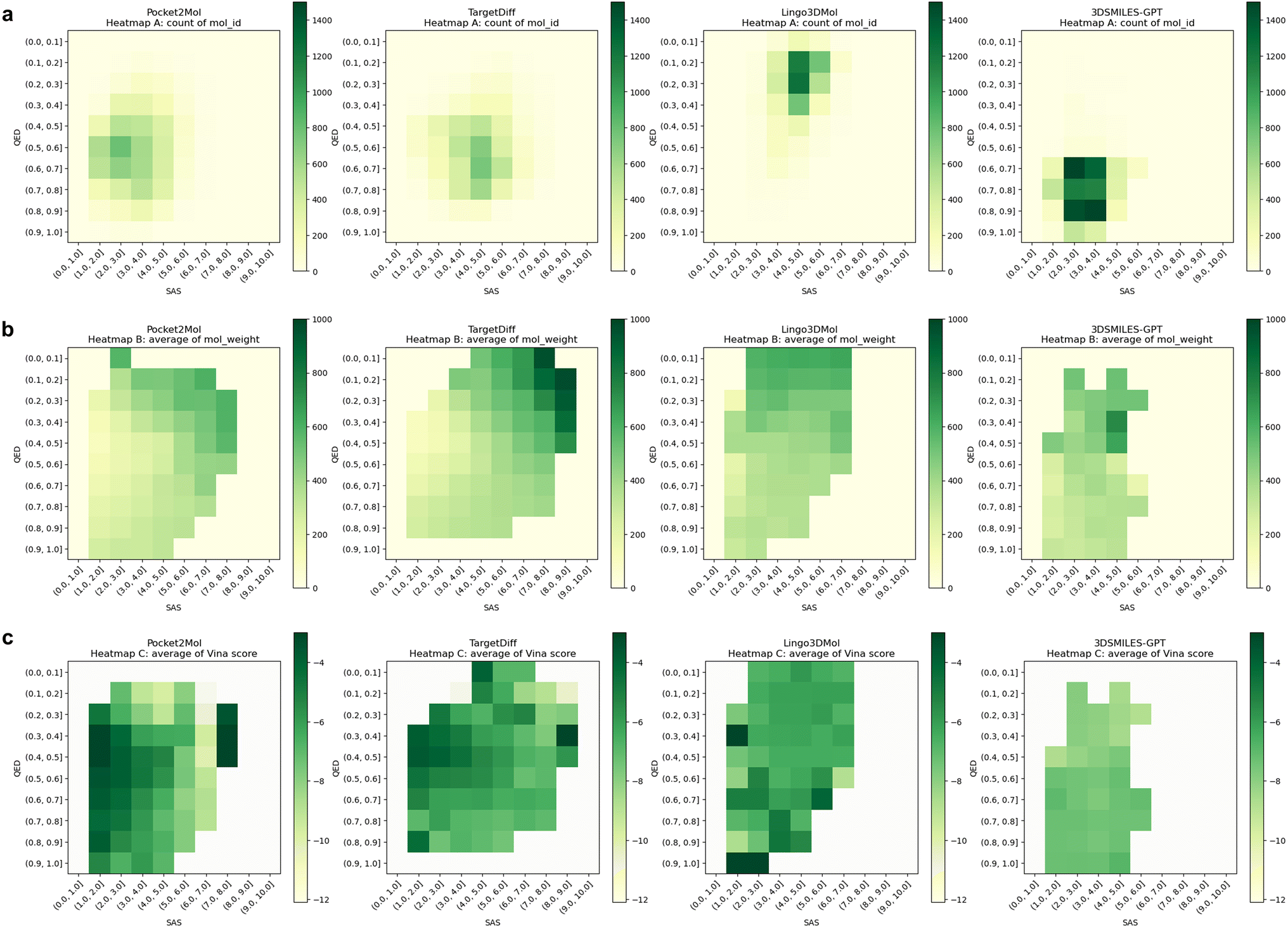 | ||
| Fig. 3 The distribution heatmaps of QED, SAS, and other properties for the molecules generated by each model. (a) QED, SAS and number of molecules, (b) QED, SAS and molecular weight, and (c) QED, SAS and Vina score. | ||
In our endeavor to create molecules, our objective is to generate compounds with properties that surpass those of currently available ones. To assess the model's effectiveness in achieving this objective, we thoroughly examined the molecules generated by each model, using the ground truth molecules from the test set as a reference point. We refer to results where the affinity is superior to the reference molecules as “Better than References” (BR). The analysis revealed that our model exhibits reduced diversity compared to others, a finding that aligns with our initial expectations. This decline in diversity can be attributed to the imposition of constraints related to physicochemical properties during the training phase of 3DSMILES-GPT, coupled with additional restrictions on QED and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P during the process of molecule generation, ultimately resulting in a lower diversity of generated molecules.
P during the process of molecule generation, ultimately resulting in a lower diversity of generated molecules.
We quantified the number of the molecules generated by 3DSMILES-GPT that achieved lower Vina scores compared to the reference molecules. Notably, 53% of the molecules generated by 3DSMILES-GPT exhibited lower Vina scores compared to the reference molecules, while Pocket2Mol and TargetDiff achieved 48%. However, although the molecules generated by Lingo3DMol often exhibit higher affinity than the reference molecules, they tend to cluster within a narrow range according to the Vina scores (Fig. 3). This might be suitable for certain drug discovery tasks, but if the reference molecules generally have high affinity, such as with kinase targets like ATK1 and CDK2, the proportion of BR molecules may decrease. On the other hand, 3DSMILES-GPT shows the ability to explore chemical space with higher affinity for the target, which is an advantage of our model. We also computed the average QED and SAS of molecules from the BR set (Table 1), consistently demonstrating that 3DSMILES-GPT keeps superior performance, particularly in QED, with an improvement of approximately 33%.
In summary, 3DSMILES-GPT demonstrates the capability to generate molecules with higher binding affinity and improved QED and SAS metrics. Notably, it can produce conformations comparable to those obtained through redocking without requiring the redocking process, an outstanding capability of direct generation with physical conformation, which is absent in other models.
Structure-based drug design for specific targets
Many reported pocket-based molecular generation methods lack testing on real targets outside the training set, raising doubt on their practical efficacy in real drug design tasks. To address this, we selected four protein targets independent from the training and testing sets: AKT1 (4gv1), SARS-COV2 3CL proteinase (7d3i), CDK2 (1h00) and DDR1 (5bvk).These targets have been utilized in virtual screening46,47 and molecular generation tasks,33,34,48 allowing us to simulate real drug discovery scenarios. As shown in Fig. 4a, the Vina scores of the molecules generated by 3DSMILES-GPT in the target pockets of these four different protein families predominantly fall within range of −10 to −5 kcal mol−1. This distribution is closer to the left side of the horizontal axis compared to both Pocket2Mol and TargetDiff, indicating higher binding energy and better affinity. Compared to Lingo3DMol, the affinity distribution of the molecules generated by Lingo3DMol results is concentrated between −10 and −7, with a greater focus on the high affinity range than 3DSMILES-GPT. However, our model generates more molecules in the high affinity range (Vina score ≤ −10) for targets other than DDR1. In practical drug discovery scenarios, high affinity is not the only pursuit, and molecules with better drug-likeness are also needed. Therefore, we filtered all molecules and re-examined the distribution of the Vina scores for the filtered molecules (Fig. 4b). It can be observed that the distribution of the Vina scores for the molecules generated by 3DSMILES-GPT does not change significantly compared to before filtering, indicating that the vast majority of the generated molecules meet our drug-likeness criteria. Additionally, most high-affinity molecules with Vina scores smaller or equal than −10 generated by other baseline models were filtered out, with the majority of molecules in this range being generated by 3DSMILES-GPT. In the virtual screening process, due to the limited number of molecules that can undergo activity validation, medicinal chemists typically select a few molecules with better docking scores for testing. 3DSMILES-GPT can generate molecules with both high affinity and better drug-likeness for specific targets, implying that the model has a promising potential to discover lead compounds that may achieve activity validation at the molecular or cellular level in practical drug discovery applications.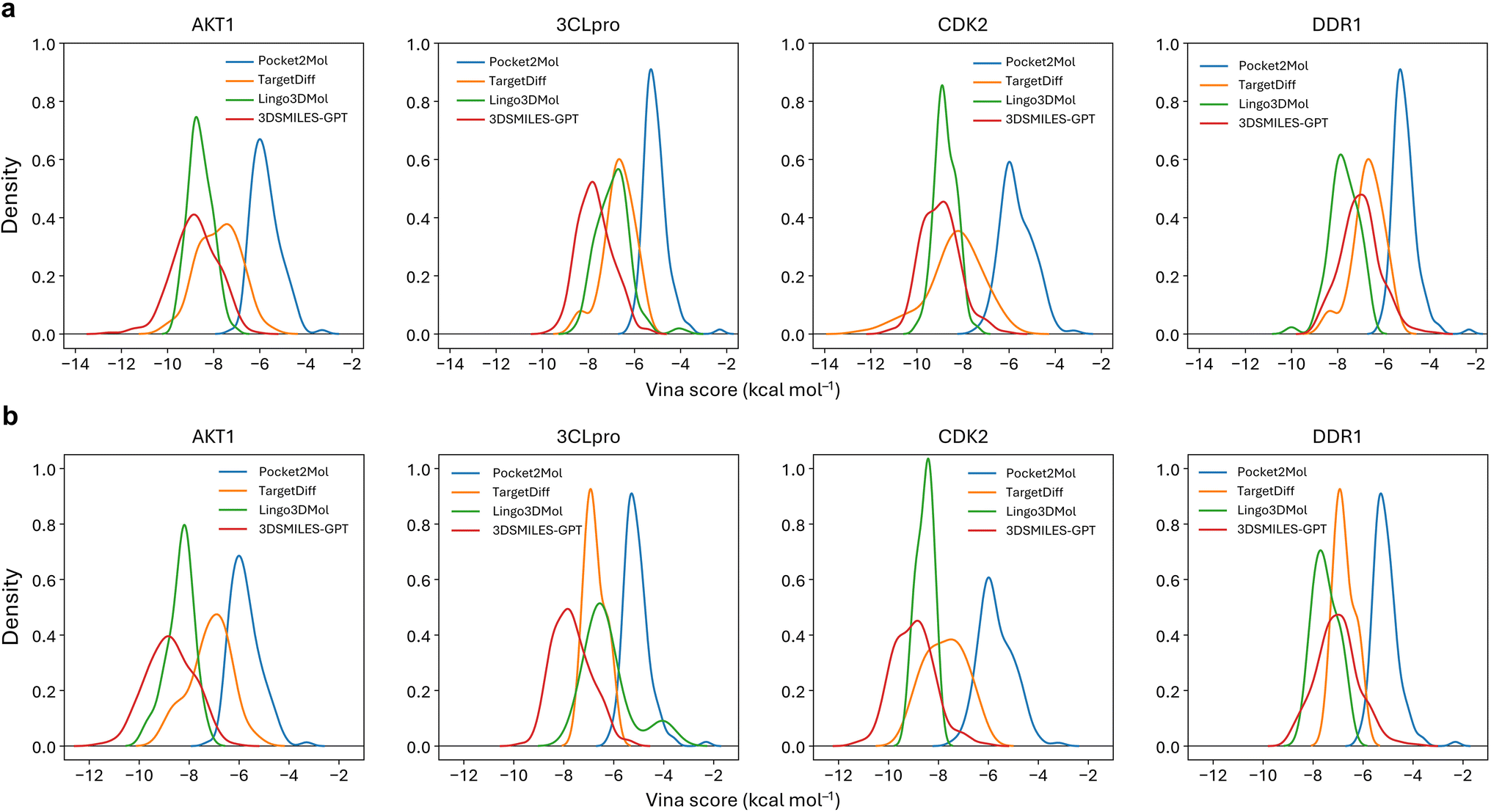 | ||
| Fig. 4 The distributions of Vina scores for the specific targets. (a) The distribution of all generated molecules for each model. (b) The distribution after drug-likeness screening (QED ≥ 0.3, SAS ≤ 5). The horizontal axis to the left represents better affinity. | ||
Beyond affinity, the purpose of generating molecules within specific target pockets also involves comparing the structural and binding mode similarity of the generated molecules to known ligands. As shown in Fig. 5, we selected the highest affinity molecules generated by each model to demonstrate their binding modes. Among the baseline models, Pocket2Mol and TargetDiff tend to generate either simple aromatic ring derivatives or complex macrocyclic compounds, lacking distinct target specificity. The molecules generated by Lingo3DMol are excessively large compared to the original ligands, consistent with the average molecular weight of 480 Daltons obtained in the test set results. Furthermore, in the case of molecule within the CDK2 pocket, there are issues of fragmentation and clash with the protein pocket, caused by large molecule size and unreasonable bond angles (Fig. S1†). Compared to other baseline models, the molecules generated by 3DSMILES-GPT exhibit more reasonable structures, and their binding modes within the pocket are relatively close to those of the original ligands. However, in the DDR1 pocket, the binding modes of the molecules are more exterior compared to that of the original ligand, which may explain why 3DSMILES-GPT does not show a significant advantage in affinity for the DDR1 target over other baseline models.
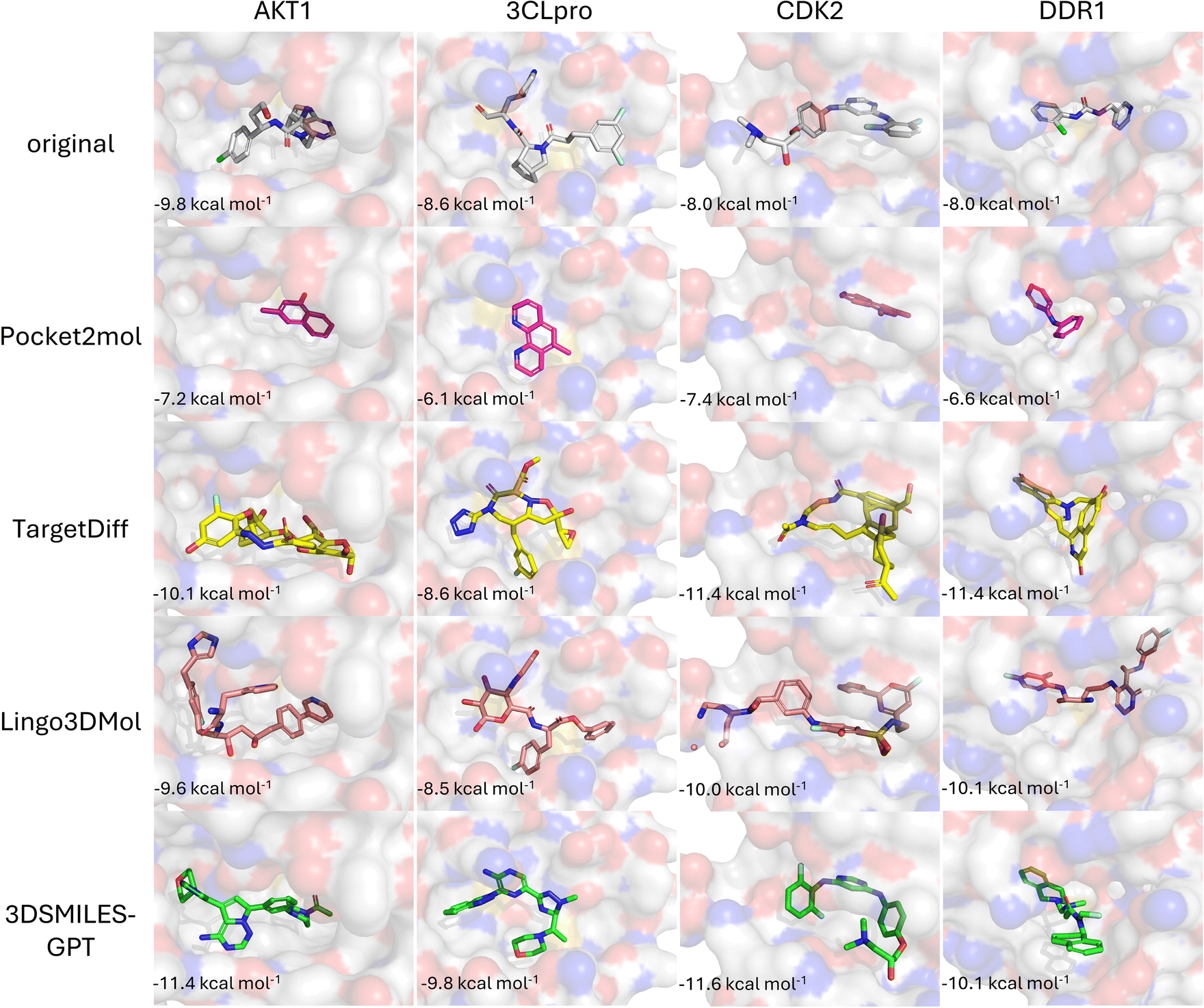 | ||
| Fig. 5 The binding modes of the molecules with optimal affinity generated by each model for specific targets, and the comparison with the binding modes of the original ligands. | ||
Overall, the tests on specific target pockets demonstrated that 3DSMILES-GPT, compared to previous baseline models, can generate more molecules that meet drug-likeness criteria with high affinity for the target, while also exhibiting more reasonable structures and higher structural specificity for different targets.
Conclusion
In this study, we present 3DSMILES-GPT, an innovative token-only pocket-aware molecular generation method for creating 3D molecular structures within protein pockets. This method leverages the robust capabilities of large language models to perceive and generate molecular structures that are not only chemically valid but also exhibit optimal biophysical and chemical properties. 3DSMILES-GPT exhibits exceptional performance across various benchmark metrics, outperforming existing methods in generating molecules with superior Vina docking scores and enhanced drug-likeness. Notably, the QED of the generated molecules improves by 33%, indicating that our model produces molecules more closely aligned with the pharmaceutical industry's criteria for drug candidates. Furthermore, 3DSMILES-GPT achieves a threefold increase in generation speed compared to the fastest existing methods, fulfilling the demand for rapid identification of drug candidates. Out-of-dataset evaluations validate the model's capability to generate drug-like molecules with strong binding affinities to specific targets, highlighting its potential in real-world drug discovery applications. Leveraging the foundation of 3DSMILES-GPT, future efforts will focus on developing a universal drug design language model by integrating advanced large model techniques with comprehensive training data. In summary, 3DSMILES-GPT signifies a paradigm shift in molecular generation for drug discovery, leveraging the capabilities of large language models to tackle complex biological challenges.Methods
Backbone
The architecture of 3DSMILES-GPT comprises an 8-layer transformer decoder with 12 attention heads, facilitating the autoregressive prediction of both 2D and 3D molecular structures while explicitly expressing them. The multi-head attention mechanism serves as a cornerstone of the Transformer model, allowing the model to attend to different subspaces of the input simultaneously, thus capturing richer information. Within the multi-head attention mechanism, each attention head learns a set of weights to compute attention weights for different positions in the input sequence, which are then used to weigh the input sequence representations. By performing parallel computation across multiple attention heads, the model gains the ability to interpret input sequences from various perspectives, thereby enhancing its representational capacity and generalization performance. The attention mechanism is shown in eqn (1):
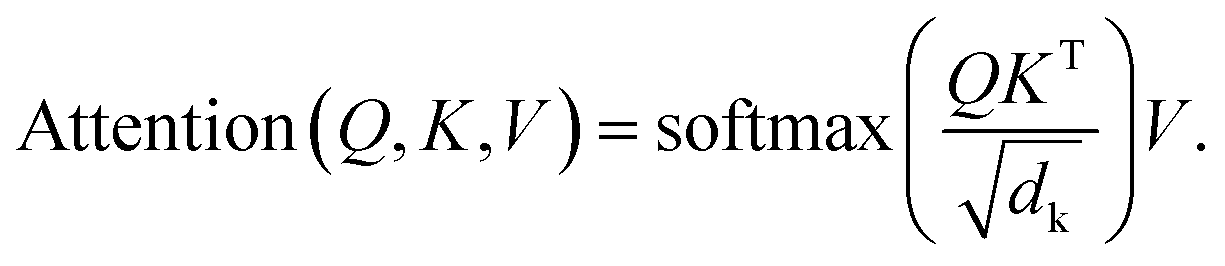 | (1) |
Detachable pocket encoder
To augment the extraction of protein pocket information, a detachable pocket encoder has been devised. We implemented a spatial positional encoding strategy proposed by Zhou et al.,49 which based Gaussian kernel to describe the atomic relative positions. The D-dimensional positional encoding between atom pairs can be expressed by the following equation:| pij = {G(A(dij, tij; u, v), μs, σs)|s ∈ [1, D]} | (2) |
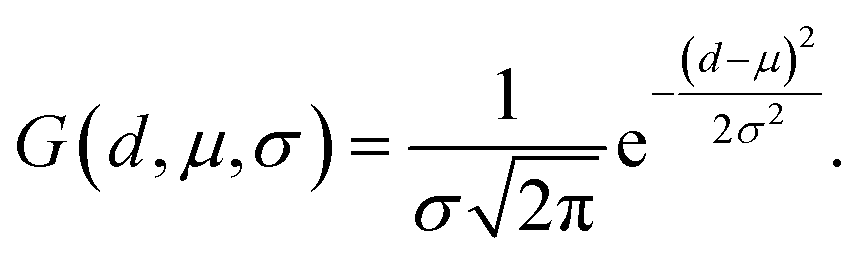 | (3) |
| A(d, t; u, v) = utd + vt | (4) |
Therefore, the information of i and j can be computed as:
| qij = W1GELU(W2pij) | (5) |
Reinforcement learning
The utilization of reinforcement learning to enhance sequence-based molecular generation models, such as REINVENT,13 is a well-established practice. However, its application to the specific task of generating 3D molecules in protein pockets remains relatively uncommon. By simplifying the intricacies of 3D molecular generation and representing molecular coordinates as tokens, we facilitate the direct adoption of methodologies akin to REINVENT for refining the generated molecules. Nevertheless, within this context, we opt for a more explicit strategy entailing multiple iterations employing policy gradient50 techniques to refine the model.In the present context, Pθ(M|C) denotes the initial policy governing our model, with C representing the protein pocket and M signifying the molecule. Simultaneously, D0 stands for the initial fine-tuning dataset. At each iteration, K molecules are sampled for every protein pocket, with those demonstrating favorable properties being incorporated into the fine-tuning dataset Dt at the t time step for subsequent iterations. Throughout each fine-tuning iteration, policy gradient methods are employed to iteratively refine the policy Pθ(M|C) of the model.
Dataset and data preprocessing
The pretraining phase encompassed a selection of 10 million molecules from the PubChem drug dataset,51 excluding those exceeding 48 atoms or containing elements beyond ‘C’, ‘O’, ‘N’, ‘S’, ‘P’, ‘F’, ‘Cl’, ‘Br’, and ‘I’. Each molecule underwent stereoisomer enumeration using RDKit, followed by the generation of two conformations per stereoisomer, subsequently minimized using the MMFF94 force field. Conformational centering involved the subtraction of the coordinate center from each conformation.For fine-tuning, the CrossDocked202052 dataset was employed following the Pocket2mol methodology, with poses featuring RMSD greater than 2 Å discarded. A 6 Å residue perimeter surrounding the ligand was isolated as pocket data, and the coordinates of the pocket surface were computed utilizing the MSMS.53 The resultant coordinates underwent sparsification via pymesh.
Further data processing was conducted to meet the model's input requirements. Initially, the QED and logP (partition coefficient) values were computed. Molecules with QED exceeding 0.5 or logP values falling between −1 and 3 were assigned a label of 1, while those outside these ranges were labeled as 0. During the fine-tuning phase, we employed a similar approach to label the training data and augmented it with Vina score labels. Molecules with a Vina score less than −0.75 were labeled as 1, while those with a score equal to or greater than −0.75 were labeled as 0.
For the 2D molecular structure representation, SMILES notation was utilized, with SMILES sequences encoded at the character byte level instead of byte-level tokenization.54 The initial vocabulary comprised 72 characters extracted from the SMILES alphabet. Following tokenization, it was segmented into 1000 most commonly encountered tokens.
To address 3D molecular structures, data augmentation techniques were employed to instill three-dimensional equivariance into the model. This entailed random translation and rotation of the 3D structures, with each coordinate represented by a distinct token. For the details of data augmentation, each molecule undergoes random rotations around the X, Y, and Z axes, with the rotation angles uniformly sampled from 0° to 360° independently for each axis. This approach ensures that the model is exposed to molecules in all possible orientations, thereby promoting rotational invariance. Moreover, random translations are applied to the molecular coordinates, using translation vectors uniformly sampled within a range of −10 Å to 10 Å along each axis. This specific range was selected to introduce subtle positional variations without significantly displacing the molecules from their original locations. To preserve the consistency of the augmented dataset, the same augmentation techniques are uniformly applied to all molecules, maintaining the original distribution of molecular properties.
Baseline
We have selected SOTA models that represent three distinct approaches for pocket generation: Pocket2Mol,31 an autoregressive model based on GNN. TargetDiff, which adopts diffusion-based methodologies for one-shot generation. Lingo3DMol,42 rooted in language models. Pocket2Mol and TargetDiff underwent training utilizing the CrossDocked2020 dataset, whereas Lingo3DMol was initially pretrained on a dataset consisting of 12 million drug-like molecules, followed by fine-tuning on the PDBbind202055 dataset. For the evaluation in this study, we directly employed the code and pretrained models provided by the respective works.
Training and generation protocol
In the training phase, we prefixed the molecular QED and logP labels to the SMILES string, and appended the corresponding atoms' coordinates to smiles tail. Coordinates between identical atoms were delineated by commas, while those between distinct atoms were enclosed within curly braces. In the fine-tuning stage, we converted the processed coordinates of the protein pocket surface into a prefix-form input string, allowing the model to comprehend the ligand coordinate boundaries. The sequence's start and end were indicated by ‘〈s〉’ and ‘〈/s〉’ tokens, respectively. Throughout the training regimen, we adopted a self-supervised methodology to acquaint the model with the SMILES and coordinates strings of molecules. The primary optimization objective entailed minimizing the negative log-likelihood, as described in eqn (6):
 | (6) |
This objective was accomplished by iteratively refining the loss function through gradient descent until convergence.
In the generation stage, we combined the processed protein pocket information with the specified molecular properties, forming the input for the model. The autoregressive process for generating smiles and coordinates strings follows eqn (7):
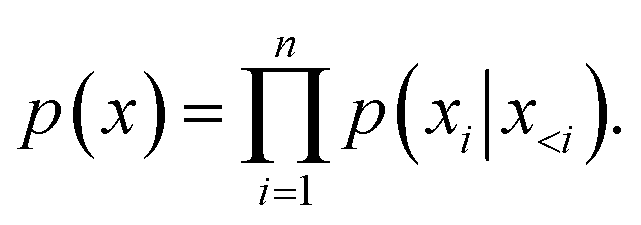 | (7) |
Data availability
The pocket-based molecular generation dataset is provided at https://drive.google.com/drive/folders/1CzwxmTpjbrt83z_wBzcQncq84OVDPurM. The code used in the study is publicly available from the GitHub repository: https://github.com/ashipiling/GPT_3DSMILES.Author contributions
T. J. H., Y. K. and C. Y. H. designed the research study. H. L., J. K. W. and R. Q. developed the method and wrote the code. J. K. W., H. L., R. Q., M. Y. W. performed the analysis. J. K. W., R. Q., M. Y. W., G. Q. L., Y. K., C. Y. H. and T. J. H. wrote the paper. All authors read and approved the manuscript. Zhejiang University supports in all GPU-related technologies and work content for this study.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (92370130, 22303083, 82373791), China Postdoctoral Science Foundation (2023M733128, 2023TQ0285), Postdoctoral Fellowship Program of CPSF (GZB20230657), and Scientific Research Fund of Zhejiang Provincial Education Department (Y202457041).References
- N. Brown, B. McKay, F. Gilardoni and J. Gasteiger, A graph-based genetic algorithm and its application to the multiobjective evolution of median molecules, J. Chem. Inf. Comput. Sci., 2004, 44, 1079–1087 Search PubMed.
- A. M. Virshup, J. Contreras-García, P. Wipf, W. Yang and D. N. Beratan, Stochastic voyages into uncharted chemical space produce a representative library of all possible drug-like compounds, J. Am. Chem. Soc., 2013, 135, 7296–7303 Search PubMed.
- N. De Cao and T. Kipf, MolGAN: an implicit generative model for small molecular graphs, arXiv, 2018, preprint, arXiv:1805.11973, DOI:10.48550/arXiv.1805.11973.
- W. Jin, R. Barzilay and T. Jaakkola, Junction tree variational autoencoder for molecular graph generation, arXiv, 2018, preprint, arXiv:1802.04364, DOI:10.48550/arXiv.1802.04364.
- Q. Liu, M. Allamanis, M. Brockschmidt and A. L. Gaunt, Constrained graph variational autoencoders for molecule design, arXiv, 2018, preprint, arXiv:1805.09076, DOI:10.48550/arXiv.1805.09076.
- B. Samanta, A. De, G. Jana, P. K. Chattaraj, N. Ganguly and M. G. Rodriguez, presented in part at the Proceedings of the AAAI Conference on Artificial Intelligence, 07/17, 2019 Search PubMed.
- V. Mnih, K. Kavukcuoglu, D. Silver, A. Graves, I. Antonoglou, D. Wierstra and M. Riedmiller, Playing atari with deep reinforcement learning, arXiv, 2013, preprint, arXiv:1312.5602, DOI:10.48550/arXiv.1312.5602.
- C. Zang and F. Wang, presented in part at the Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Virtual Event, CA, USA, 2020 Search PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CAS.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Self-referencing embedded strings (SELFIES): a 100% robust molecular string representation, Mach. Learn.: Sci. Technol., 2020, 1, 045024 Search PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser and I. Polosukhin, presented in part at the Advances in Neural Information Processing Systems, Long Beach, California, USA, 2017 Search PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, Molecular de-novo design through deep reinforcement learning, J. Cheminf., 2017, 9, 48 Search PubMed.
- J. Wang, C.-Y. Hsieh, M. Wang, X. Wang, Z. Wu, D. Jiang, B. Liao, X. Zhang, B. Yang, Q. He, D. Cao, X. Chen and T. Hou, Multi-constraint molecular generation based on conditional transformer, knowledge distillation and reinforcement learning, Nat. Mach. Intell., 2021, 3, 914–922 CrossRef.
- A. Gupta, A. T. Müller, B. J. H. Huisman, J. A. Fuchs, P. Schneider and G. Schneider, Generative recurrent networks for de novo drug design, Mol. Inf., 2018, 37, 1700111 CrossRef.
- E. Jannik Bjerrum and R. Threlfall, Molecular generation with recurrent neural networks (RNNs), arXiv, 2017, preprint, arXiv:1705.04612, DOI:10.48550/arXiv.1705.04612.
- P. Pogány, N. Arad, S. Genway and S. D. Pickett, De novo molecule design by translating from reduced graphs to SMILES, J. Chem. Inf. Model., 2019, 59, 1136–1146 CrossRef.
- X. Liu, K. Ye, H. W. T. van Vlijmen, A. P. Ijzerman and G. J. P. van Westen, An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: a case for the adenosine A2A receptor, J. Cheminf., 2019, 11, 35 Search PubMed.
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- X. Yang, J. Zhang, K. Yoshizoe, K. Terayama and K. Tsuda, ChemTS: an efficient python library for de novo molecular generation, Sci. Technol. Adv. Mater., 2017, 18, 972–976 CrossRef CAS PubMed.
- F. Grisoni, M. Moret, R. Lingwood and G. Schneider, Bidirectional molecule generation with recurrent neural networks, J. Chem. Inf. Model., 2020, 60, 1175–1183 CrossRef CAS.
- D. Merk, L. Friedrich, F. Grisoni and G. Schneider, De novo design of bioactive small molecules by artificial intelligence, Mol. Inf., 2018, 37, 1700153 CrossRef.
- M. Popova, O. Isayev and A. Tropsha, Deep reinforcement learning for de novo drug design, Sci. Adv., 2018, 4, eaap7885 CrossRef CAS PubMed.
- M. Wang, C.-Y. Hsieh, J. Wang, D. Wang, G. Weng, C. Shen, X. Yao, Z. Bing, H. Li, D. Cao and T. Hou, RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design, J. Med. Chem., 2022, 65, 9478–9492 CrossRef CAS PubMed.
- J. Wang, X. Wang, H. Sun, M. Wang, Y. Zeng, D. Jiang, Z. Wu, Z. Liu, B. Liao, X. Yao, C.-Y. Hsieh, D. Cao, X. Chen and T. Hou, ChemistGA: A Chemical Synthesizable Accessible Molecular Generation Algorithm for Real-World Drug Discovery, J. Med. Chem., 2022, 65, 12482–12496 CrossRef CAS PubMed.
- J. Wang, Y. Zeng, H. Sun, J. Wang, X. Wang, R. Jin, M. Wang, X. Zhang, D. Cao, X. Chen, C.-Y. Hsieh and T. Hou, Molecular Generation with Reduced Labeling through Constraint Architecture, J. Chem. Inf. Model., 2023, 63, 3319–3327 CrossRef CAS.
- N. W. A. Gebauer, M. Gastegger and K. T. Schütt, Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules, arXiv, 2019, preprint, arXiv:1906.00957, DOI:10.48550/arXiv.1906.00957.
- M. Ragoza, T. Masuda and D. R. Koes, Generating 3D molecules conditional on receptor binding sites with deep generative models, Chem. Sci., 2022, 13, 2701–2713 RSC.
- M. Liu, Y. Luo, K. Uchino, K. Maruhashi and S. Ji, Generating 3D molecules for target protein binding, arXiv, 2022, preprint, arXiv:2204.09410, DOI:10.48550/arXiv.2204.09410.
- S. Luo, J. Guan, J. Ma and J. Peng, A 3D Generative Model for Structure-Based Drug Design, arXiv, 2022, preprint, arXiv:2203.10446, DOI:10.48550/arXiv.2203.10446.
- X. Peng, S. Luo, J. Guan, Q. Xie, J. Peng and J. Ma, Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets, arXiv, 2022, preprint, arXiv:2205.07249, DOI:10.48550/arXiv.2205.07249.
- Y. Li, J. Pei and L. Lai, Structure-based de novo drug design using 3D deep generative models, Chem. Sci., 2021, 12, 13664–13675 RSC.
- O. Zhang, J. Zhang, J. Jin, X. Zhang, R. Hu, C. Shen, H. Cao, H. Du, Y. Kang, Y. Deng, F. Liu, G. Chen, C.-Y. Hsieh and T. Hou, ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling, Nat. Mach. Intell., 2023, 5, 1020–1030 CrossRef.
- O. Zhang, T. Wang, G. Weng, D. Jiang, N. Wang, X. Wang, H. Zhao, J. Wu, E. Wang, G. Chen, Y. Deng, P. Pan, Y. Kang, C.-Y. Hsieh and T. Hou, Learning on topological surface and geometric structure for 3D molecular generation, Nat. Comput. Sci., 2023, 3, 849–859 CrossRef PubMed.
- H. Du, D. Jiang, O. Zhang, Z. Wu, J. Gao, X. Zhang, X. Wang, Y. Deng, Y. Kang, D. Li, P. Pan, C.-Y. Hsieh and T. Hou, A flexible data-free framework for structure-based de novo drug design with reinforcement learning, Chem. Sci., 2023, 14, 12166–12181 RSC.
- J. Guan, W. W. Qian, X. Peng, Y. Su, J. Peng and J. Ma, 3D Equivariant Diffusion for Target-Aware Molecule Generation and Affinity Prediction, arXiv, 2023, preprint, arXiv:2303.03543, DOI:10.48550/arXiv.2303.03543.
- E. Hoogeboom, V. c. G. Satorras, C. Vignac and M. Welling, presented in part at the Proceedings of the 39th International Conference on Machine Learning, Proceedings of Machine Learning Research, 2022 Search PubMed.
- L. Huang, H. Zhang, T. Xu and K.-C. Wong, MDM: Molecular Diffusion Model for 3D Molecule Generation, arXiv, 2022, preprint, arXiv:2209.05710, DOI:10.48550/arXiv.2209.05710.
- M. Xu, A. S. Powers, R. O. Dror, S. Ermon and J. Leskovec, presented in part at the Proceedings of the 40th International Conference on Machine Learning, Proceedings of Machine Learning Research, 2023 Search PubMed.
- J. Born and M. Manica, Regression Transformer enables concurrent sequence regression and generation for molecular language modelling, Nat. Mach. Intell., 2023, 5, 432–444 CrossRef.
- D. Flam-Shepherd and A. Aspuru-Guzik, Language models can generate molecules, materials, and protein binding sites directly in three dimensions as XYZ, CIF, and PDB files, arXiv, 2023, arXiv:2305.05708, DOI:10.48550/arXiv.2305.05708.
- W. Feng, L. Wang, Z. Lin, Y. Zhu, H. Wang, J. Dong, R. Bai, H. Wang, J. Zhou, W. Peng, B. Huang and W. Zhou, Generation of 3D molecules in pockets via a language model, Nat. Mach. Intell., 2024, 6, 62–73 CrossRef.
- M. L. Menéndez, J. A. Pardo, L. Pardo and M. C. Pardo, The Jensen-Shannon divergence, J. Franklin Inst., 1997, 334, 307–318 CrossRef.
- M. Buttenschoen, G. M. Morris and C. M. Deane, PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences, Chem. Sci., 2024, 15, 3130–3139 RSC.
- A. Alhossary, S. D. Handoko, Y. Mu and C.-K. Kwoh, Fast, accurate, and reliable molecular docking with QuickVina 2, Bioinformatics, 2015, 31, 2214–2216 CrossRef CAS PubMed.
- J. Qiao, Y.-S. Li, R. Zeng, F.-L. Liu, R.-H. Luo, C. Huang, Y.-F. Wang, J. Zhang, B. Quan, C. Shen, X. Mao, X. Liu, W. Sun, W. Yang, X. Ni, K. Wang, L. Xu, Z.-L. Duan, Q.-C. Zou, H.-L. Zhang, W. Qu, Y.-H.-P. Long, M.-H. Li, R.-C. Yang, X. Liu, J. You, Y. Zhou, R. Yao, W.-P. Li, J.-M. Liu, P. Chen, Y. Liu, G.-F. Lin, X. Yang, J. Zou, L. Li, Y. Hu, G.-W. Lu, W.-M. Li, Y.-Q. Wei, Y.-T. Zheng, J. Lei and S. Yang, SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model, Science, 2021, 371, 1374–1378 CrossRef CAS PubMed.
- A. Clyde, S. Galanie, D. W. Kneller, H. Ma, Y. Babuji, B. Blaiszik, A. Brace, T. Brettin, K. Chard, R. Chard, L. Coates, I. Foster, D. Hauner, V. Kertesz, N. Kumar, H. Lee, Z. Li, A. Merzky, J. G. Schmidt, L. Tan, M. Titov, A. Trifan, M. Turilli, H. Van Dam, S. C. Chennubhotla, S. Jha, A. Kovalevsky, A. Ramanathan, M. S. Head and R. Stevens, High-Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Noncovalent Inhibitor, J. Chem. Inf. Model., 2022, 62, 116–128 CrossRef CAS.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov, A. Asadulaev, Y. Volkov, A. Zholus, R. R. Shayakhmetov, A. Zhebrak, L. I. Minaeva, B. A. Zagribelnyy, L. H. Lee, R. Soll, D. Madge, L. Xing, T. Guo and A. Aspuru-Guzik, Deep learning enables rapid identification of potent DDR1 kinase inhibitors, Nat. Biotechnol., 2019, 37, 1038–1040 CrossRef CAS PubMed.
- G. Zhou, Z. Gao, Q. Ding, H. Zheng, H. Xu, Z. Wei, L. Zhang and G. Ke, Uni-mol: A universal 3d molecular representation learning framework, ChemRxiv, 2023, preprint, chemrxiv-2022-jjm2020j, DOI:10.26434/chemrxiv-2022-jjm0j-v4.
- R. S. Sutton, D. McAllester, S. Singh and Y. Mansour, presented in part at the Proceedings of the 12th International Conference on Neural Information Processing Systems, Denver, CO, 1999 Search PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, PubChem Substance and Compound databases, Nucleic Acids Res., 2015, 44, D1202–D1213 CrossRef PubMed.
- P. G. Francoeur, T. Masuda, J. Sunseri, A. Jia, R. B. Iovanisci, I. Snyder and D. R. Koes, Three-Dimensional Convolutional Neural Networks and a Cross-Docked Data Set for Structure-Based Drug Design, J. Chem. Inf. Model., 2020, 60, 4200–4215 CrossRef CAS PubMed.
- M. F. Sanner, A. J. Olson and J.-C. Spehner, Reduced surface: An efficient way to compute molecular surfaces, Biopolymers, 1996, 38, 305–320 CrossRef CAS PubMed.
- R. Sennrich, B. Haddow and A. Birch, Neural Machine Translation of Rare Words with Subword Units, in Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers), Association for Computational Linguistics, Berlin, Germany, 2016, pp. 1715–1725 Search PubMed.
- R. Wang, X. Fang, Y. Lu and S. Wang, The PDBbind Database: Collection of Binding Affinities for Protein−Ligand Complexes with Known Three-Dimensional Structures, J. Med. Chem., 2004, 47, 2977–2980 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06864e |
| ‡ Equivalent authors. |
| This journal is © The Royal Society of Chemistry 2025 |