
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow production of γ-valerolactone from methyl-levulinate promoted by MOF-derived Al2O3–ZrO2/C catalysts†
Marina
Ronda-Leal
a,
Alina M.
Balu
a,
Rafael
Luque
 bc,
Francesco
Mauriello
d,
Alberto
Ricchebuono
ef,
Christophe
Len
g,
Antonio A.
Romero
a and
Emilia
Paone
*df
bc,
Francesco
Mauriello
d,
Alberto
Ricchebuono
ef,
Christophe
Len
g,
Antonio A.
Romero
a and
Emilia
Paone
*df
aDepartamento de Quimica Organica, Universidad de Cordoba, Ctra Nnal IV-A, Km 396, E14014, Cordoba, Spain
bUniversidad ECOTEC, Km 13.5 Samborondón, Samborondón, EC092302, Ecuador
cNational University of Science and Technology Politehnica Bucharest, 1-7 Gh Polizu Street, Bucharest, Romania
dDipartimento DICEAM, Università degli Studi Mediterranea di Reggio Calabria Loc, Feo di Vito, I-89122 Reggio Calabria, Italy. E-mail: emilia.paone@unirc.it
eDipartimento di Chimica, INSTM and NIS Centre, Università di Torino, Via Quarello 15, I-10135 Torino, Italy
fConsorzio Interuniversitario per la Scienza e la Tecnologia dei Materiali (INSTM), 50121 Firenze, Italy
gInstitute of Chemistry for Life and Health Sciences, Chimie ParisTech, PSL Research University, Paris, France
First published on 12th March 2025
Abstract
This study investigates the catalytic transfer hydrogenation (CTH) of methyl levulinate (ML) into γ-valerolactone (GVL) using mixed metal oxides derived from metal–organic frameworks (MOFs) under continuous flow conditions. A series of MOF-derived Al2O3–ZrO2/C catalysts with different Al/Zr molar ratios were investigated, revealing a synergistic effect that significantly enhances catalytic efficiency. Physico-chemical characterization demonstrates that the incorporation of aluminum into zirconium dioxide increases the surface area as well as the presence of catalytically active acid and basic sites, which are essential for the efficient transfer hydrogenation of ML into GVL. Al2O3–ZrO2/C (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) exhibited the highest ML conversion rate (80%) and GVL yield (72%) at 200 °C within 30 minutes. The study also emphasizes the critical role of reaction parameters in maximizing GVL production. The stability and reusability of the Al2O3–ZrO2/C (1:1) catalyst, following appropriate thermal treatment, were also assessed.
:1) exhibited the highest ML conversion rate (80%) and GVL yield (72%) at 200 °C within 30 minutes. The study also emphasizes the critical role of reaction parameters in maximizing GVL production. The stability and reusability of the Al2O3–ZrO2/C (1:1) catalyst, following appropriate thermal treatment, were also assessed.
Sustainability spotlightThe production of γ-valerolactone (GVL), a renewable bio-based chemical and fuel additive, plays a crucial role in achieving Sustainable Development Goal 7 (SDG 7) by fostering affordable and clean energy solutions. This study highlights a novel approach using MOF-derived Al2O3–ZrO2/C catalysts to produce GVL through a scalable and highly efficient process. By replacing precious metals with cost-effective and sustainable catalysts, this method reduces dependence on non-renewable resources, directly supporting SDG 12 by promoting responsible consumption and production. Furthermore, the integration of renewable feedstocks and the optimization of catalyst reusability minimize environmental impacts, aligning with SDG 13 by contributing to climate action. These advancements underscore the potential of this process to drive greener industrial practices and accelerate the shift towards a circular, low-carbon economy. |
Introduction
The transition towards renewable energy sources, combined with the adoption of circular economy principles, serves as a critical strategy in promoting sustainable development. This approach emphasizes effective resource management by minimizing environmental impacts and maximizing economic benefits through the reuse, recycling, and regeneration of materials.1–3 The shift towards these practices necessitates innovative technological advancements, particularly in catalysis, which plays a vital role as a key technology that facilitates the efficient and selective conversion of renewable resources into valuable products.4,5 One promising direction in achieving circularity and sustainability is the use of lignocellulosic biomass as a renewable feedstock for producing fuels, chemicals, and materials.6–11 Biomass-derived levulinic acid and its alkyl esters can be utilized to synthesize value-added chemicals, including γ-valerolactone (GVL).12,13 GVL, recognized for its degradability, non-toxicity, and potential as a green solvent and fuel additive, highlights the necessity for sustainable and economically viable production processes.14An effective method to produce GVL from levulinates is the catalytic transfer hydrogenation (CTH) process, employing H-donor solvents as an indirect hydrogen source, thus addressing challenges linked with the direct use of molecular H2, such as purchase, transportation and safety risks, thereby enhancing the sustainability of industrial processes.15–20
To reduce the reliance on conventional noble metal-based catalysts for the production of γ-valerolactone (GVL) from alkyl levulinates,15–17 several non-precious metals have been investigated.21–25 In this context, ZrO2-based catalysts have emerged as promising non-precious metal alternatives in these reactions. These catalysts, often part of bifunctional catalytic systems that incorporate metals like zirconium (Zr) and niobium (Nb), along with Brønsted and Lewis acid sites, are effective in converting C5 carbohydrates and alcohols into alkyl levulinates.26–30
Furthermore, mixed metal oxides are favored in heterogeneous catalysis due to their cost-effectiveness, regenerative capabilities, and superior physical–chemical properties, which include enhanced surface area and thermal stability.31,32 Recent studies have demonstrated that the coexistence of acid and basic sites in catalysts like ZrO2 and Al2O3 can synergistically improve catalytic performance in MPV reactions.33,34
Accordingly, metal–organic frameworks (MOFs) have attracted significant attention as catalytic systems, due to their tunable properties and extensive surface area.
Among their advantages, the porous structure enables the encapsulation of active compounds that act as catalytic sites, thereby offering enhanced stability and selectivity in heterogeneous catalysis. These features facilitate efficient and selective conversion of lignocellulosic biomass.39–44 While MOFs have demonstrated proven efficacy under liquid batch conditions, their application in continuous flow systems poses challenges, primarily due to their structural instability and leaching during chemical reactions.45 Nevertheless, MOFs can be effectively utilized as templates to create MOF-derived catalysts.46–51 Their porous structure enables the encapsulation of active compounds that act as catalytic sites, thereby offering enhanced stability and selectivity in catalysis. During this templating process, calcination plays a crucial role in removing the organic ligands from the MOF structure.52 This leaves behind metal oxide nanoparticles or clusters, which further contributes to the stabilization of the catalyst structure. The addition of new properties to the material could address the challenges associated with MOF-flow systems that have been previously reported and may also enhance the batch catalytic performance of both MOFs and MOF-derived oxides. Several studies have already reported the use of MOF-derived catalysts in catalytic reactions for the production of GVL under batch conditions i.e. Ru–ZrO2@C,53 Ir@ZrO2@C,54 and Ru@C–Al2O3.55 However, these materials contain noble metals. On the other hand, flow-catalytic reactions from methyl levulinate to GVL have been studied (Table 1) in some research.
| Catalyst | Conditions | Conversion (%) | Selectivity (%) | Ref. |
|---|---|---|---|---|
| ZrO2 | ML:2-PrOH = 1:10 (molar ratio), τ = 1 s, % mol N2:ML:EtOH = 90.1:0.9:9, 250 °C |
>99 | 80 | 35 |
| UiO-66 (Zr) | ML:2-PrOH = 1:5.4 (molar ratio), time on stream: 1 h, 240 °C |
>99 | 74 | 36 |
| 5% Ru/C | ML:2-PrOH 1:50, 20 bar, 0.4 mL min−1, 150 °C |
>95 | 85 | 37 |
| Zr10/HBEA (75) | 0.3 M methyl levulinate and 0.01 M pyridine in 2-propanol, flow rate of 0.2 mL min−1, at 200 °C and 30 bar pressure | 45.5 | 96 | 38 |
| Al2O3–ZrO2/C | ML:2PrOH = 1:100, flow 0.5 mL min−1, 30 bar, 220 °C, 0.5 g cat |
>99 | 85 | This work |
Our study investigates the use of MOF-derived Al2O3–ZrO2/C catalysts for converting methyl levulinate into GVL under continuous flow conditions, focusing on the effect of varying Al/Zr molar ratios on catalytic efficiency and selectivity. By enhancing the selectivity for GVL production, a potential green fuel, this research advances the development of highly active non-noble metal catalysts and promotes sustainable catalytic processes.
Experimental section
Synthetic procedures
All chemicals were acquired as analytical grade compounds from commercial sources and used without any further purification: terephthalic acid (>99%, Acros), zirconium(IV) chloride (ZrCl4) (>99.9%, Sigma-Aldrich), aluminum nitrate nonahydrate (Al(NO3)3·9H2O) (>99%, Merck), methanol (MeOH) (>99.9%, Panreac), N,N-dimethylformamide (DMF) (>99.9%, Panreac), methyl levulinate (>98%, Sigma-Aldrich), and isopropanol (>99.9%, Panreac).Bimetallic MOFs, utilizing UiO-66 (Zr) and MIL-53 (Al) as sacrificial templates, were prepared for a series of Al2O3–ZrO2/C catalysts with varying Al/Zr ratios (0:1; 1:2; 1:1; 2:1). The preparation began with solution A, consisting of terephthalic acid and zirconium chloride in 50 mL of DMF, and solution B, consisting of terephthalic acid and aluminum nitrate in 50 mL of DMF. Initially, terephthalic acid was added to zirconium chloride with 50 mL of DMF (solution A) and mixed until it formed a clear solution over 30 minutes. Concurrently, terephthalic acid was mixed with aluminum nitrate in an equal volume of DMF (solution B) for another 30 minutes. Subsequently, solution A was added to solution B under stirring for 1 hour, during which the ratios of Al to Zr were adjusted. The combined solution was then transferred to a 125 mL stainless steel-lined Teflon autoclave and heated at 120 °C for 24 hours. The resultant precipitate was filtered and washed several times with DMF and MeOH. Finally, the precipitate was dried overnight at 80 °C and pyrolyzed in a muffle furnace under a mixed atmosphere (N2/air) at 600 °C for 2 hours.
Methodology
The crystal structure and phase composition of the synthesized Al2O3–ZrO2/C catalysts were characterized using a Bruker D8 DISCOVER with Cu Ka radiation (l = 1.5418 Å) and operated at a scanning speed of 1° min−1 over a range from 5° to 80° 2θ. The textural properties of the samples were examined using physisorption isotherms with N2, measured on a Micromeritics ASAP 2000 at 77 K. Samples were degassed under a vacuum of 0.1 Pa for 24 h at 130 °C prior to measurement.The crystal structure and phase composition of the synthesized Al2O3–ZrO2/C catalysts were characterized using a Bruker D8 DISCOVER with Cu Ka radiation (l = 1.5418 Å) and operated at a scanning speed of 1° min−1 over a range from 5° to 80° 2θ.
In situ IR spectroscopy of adsorbed CO2 was used to characterize the acidic and basic sites on the surface of the MOF-derived catalysts. Transmission IR spectra were collected using a Bruker Vertex 70 spectrophotometer equipped with a Mercury–Cadmium–Telluride (MCT) cryo-detector in the 4000–600 cm−1 range with a resolution of 2 cm−1. The samples, gently pelletized (applied pressure ca. 0.2 Ton per cm2) into self-supporting thin pellets (thickness ∼ 0.01 mg mm−2), were placed in a custom cell, equipped with KBr windows, designed for thermal treatments in a controlled atmosphere. Prior to analysis, the samples were activated on a vacuum line by degassing the pellets at 500 °C (ramp rate 5 °C min−1) until the pressure reached 5 × 10−4 mbar, then kept overnight. Hereafter, 100 mbar O2 was dosed into the cell and maintained for 15 min. This process was repeated three times, followed by a 2 h long outgassing step. Lastly, the cell was cooled down to room temperature and, without exposure to air, connected to a second vacuum line and placed inside the IR spectrometer. Experiments were carried out by sending an initial dose of 25 mbar CO2 on the catalysts at beam temperature. Spectra were continuously measured for 15 min, allowing the adsorbate–adsorbent system to equilibrate. Finally, the cell was opened to a dynamic vacuum to observe the behaviour of the species formed upon CO2 adsorption during evacuation at beam temperature. The evolution of the system under these conditions was monitored by collecting IR spectra for an additional 15 min. The collected spectra were a posteriori corrected for the sample thickness in order to allow the semi-quantitative considerations.
Surface activity was further studied using pulse chromatographic titration at 200 °C. Pyridine (PY) and 2,6-dimethylpyridine (DMPY) served as titrant bases in a cyclohexane solution using a Hewlett Packard 5890 Series II gas chromatograph. The catalysts were saturated by small injections (2 µL each) of these molecules. The probe molecules were then detected in the GC with an FID detector, assuming that pyridine titrates both Brønsted and Lewis sites, while 2,6-dimethylpyridine selectively titrates Brønsted acid sites.
X-ray photoelectron spectroscopy (XPS) was conducted to analyze the chemical composition of the surface materials. The internal structure of the catalysts was examined using a JEOL JEM-2100F transmission electron microscope (TEM). The same instrument was used for energy-dispersive X-ray (EDX) testing to verify the good dispersion of the MOFs and oxides. Morphological characteristics were assessed through scanning electron microscopy (JEOL JSM 6300), with an energy-dispersive X-ray detector (EDS) used for qualitative analysis of the sample's constituent elements.
Reactions were conducted using a ThalesNano Phoenix continuous flow reactor (see Scheme S1†). The reactor was loaded with 0.5 g of catalyst, packed into a 70 mm long ThalesNano CatCart, and inserted into the reactor. A solution of 0.2 M methyl levulinate in 2-propanol was passed through the CatCart at a flow rate of 0.1 to 0.5 mL min−1, under a nitrogen pressure of 30 bar and temperatures ranging from 160 to 220 °C. Samples were collected at specific times on stream; the analyzed samples correspond to the reactor's outlet stream collected over specified intervals. Therefore, the GC results represent an average of the reaction mixture's composition from the start of the reaction up to the respective sampling time.
Product identification was performed by comparing with commercially available samples using an Agilent 7820A GC/5977B equipped with an HP-5ms column (30 m × 0.25 mm × 0.25 µm). Products were quantified using an Agilent 6890A gas chromatograph equipped with an FID detector and a non-polar fused silica hydroxyl column, SUPELCO EQUITY TM-1 (60 m × 0.25 mm × 0.25 µm). The carrier gas used was N2, with a flow rate of 1.2 mL min−1. The heating program started with a 1 min isotherm at 60 °C, followed by an increase to 180 °C at a rate of 5 °C min−1, maintained for 5 min, then increased to 250 °C at a rate of 15 °C min−1 and maintained for another 5 min.
The methyl levulinate conversion, product selectivity and product yield were calculated as follow:
 | (1) |
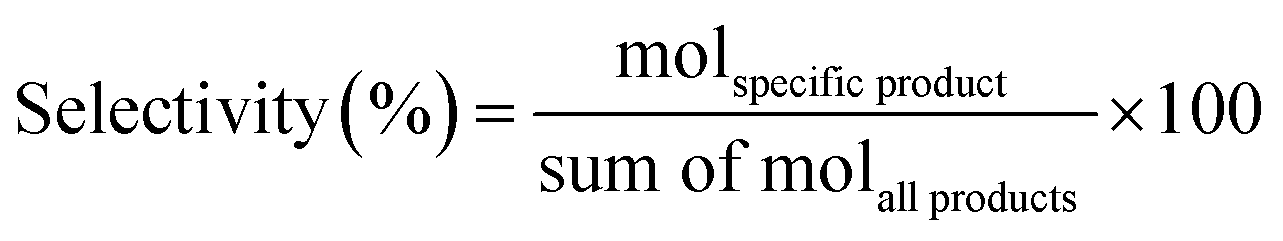 | (2) |
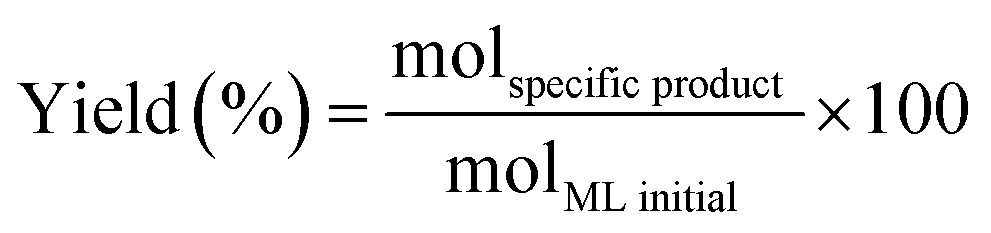 | (3) |
Carbon balance was assessed by GC-FID quantification, obtaining a loss-value in all cases lower than 2% of carbon loss.
Results and discussion
Catalyst synthesis and characterization
MOF-derived Al2O3–ZrO2/C catalysts were prepared using a solvothermal method as described by McKinstry and colleagues.56 Briefly, the synthesis involves combining solutions of terephthalic acid with zirconium chloride and/or aluminum nitrate in dimethylformamide (DMF). This mixture is then heated in an autoclave to form the MOF structure. After washing and drying, the material is subjected to pyrolysis in a nitrogen atmosphere to convert the precursors into the desired Al2O3–ZrO2/C catalysts. A schematic representation of the synthesis and preparation process of the samples is depicted in Scheme 1.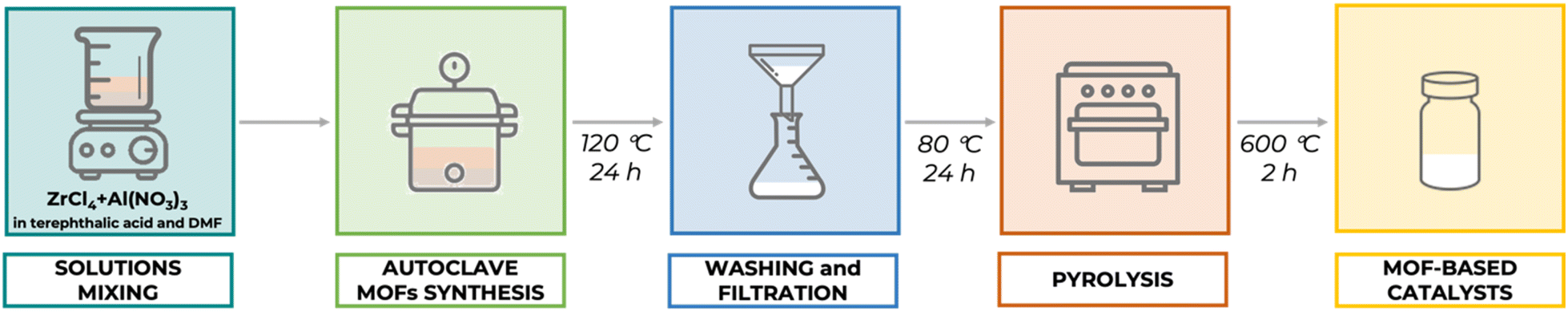 | ||
| Scheme 1 Schematic representation of the synthesis and preparation process of the bimetallic MOF-derived Al2O3–ZrO2/C catalysts. | ||
N2 physisorption was utilized to investigate the textural properties of the catalysts (Table 2). In contrast to the precursor MOFs, which typically exhibit type I isotherms, all catalysts displayed type IV isotherms indicative of mesoporous materials (Fig. S1†). This transformation can be attributed to the removal of organic linkers during MOF calcination, which originally contributed to microporosity. The highest surface area was recorded for Al2O3/C at 232 m2 g−1, while ZrO2/C exhibited the lowest at 31 m2 g−1. Materials containing Al2O3 in their structure (Al2O3–ZrO2/C mixtures) demonstrated enhanced ZrO2/C surface areas, mostly in ratios of 1:1 and 1:2. This trend extends to total pore volume and pore diameter, where an increased content of Al2O3/C correlates with large total pore volumes and diameters (Table 2).
| Catalyst | Al/Zr ratio | SA (m2 g−1) | V BJH (cm3 g−1) | V MESO (cm3 g−1) | D BJH (Å) | Total acidity (µmol m−2) | Total acidity (µmol g−1) | Brönsted acidity (µmol DMPY g−1) |
|---|---|---|---|---|---|---|---|---|
| ZrO2/C | — | 31 | 0.16 | 0.08 | 80 | 1 | 32 | <5 |
| Al2O3–ZrO2/C (1:2) |
1:2 |
165 | 0.75 | 0.54 | 100 | 0.18 | 30 | <5 |
| Al2O3–ZrO2/C (1:1) |
1:1 |
210 | 0.85 | 0.50 | 100 | 0.37 | 77 | <5 |
| Al2O3–ZrO2/C (2:1) |
2:1 |
96 | 0.42 | 0.21 | 70 | 0.26 | 25 | <5 |
| Al2O3/C | — | 232 | 1.44 | 0.94 | 300 | 0.18 | 43 | <5 |
Oxide crystal structure was investigated by XRD analysis (Fig. 1). ZrO2/C exhibits a mixed phase between monoclinic and tetragonal phases.35–38,56–60 Al2O3/C shows a characteristic amorphous structure.61 Peaks attributed to monoclinic ZrO2/C appear at 28 (111) and 62 (312) 2θ degrees while the tetragonal phase appears at 30 (101), 35 (110), 50 (112) and 59 (131) 2θ degrees. On the other hand, mixed oxide structures combine characteristics from both pure oxides, resulting in materials that exhibit unique properties, leading to a structure more amorphous than pure ZrO2/C due to the presence of Al2O3/C. Furthermore, it is noteworthy that these mixed oxide structures tend to maintain the tetragonal phase of ZrO2/C as the predominant phase. Other studies involving the synthesis of these mixed oxides have also shown similar trends.62
 | ||
| Fig. 1 XRD patterns of calcined MOF-derived Al2O3–ZrO2/C catalysts. | ||
The acidity of the catalysts was determined through pyridine adsorption followed by titration, with the acid site density results presented in Table 2, normalized for both surface area (m2) and mass (g). Notable differences were observed in the acid site densities between the pure oxides ZrO2/C and Al2O3/C. ZrO2/C, which has a relatively lower specific surface area of 31 m2 g−1, demonstrated a higher acid site density of 1 µmol m−2. This is attributed to a more pronounced aggregation of acid sites on the limited surface area available. In contrast, Al2O3/C, with a significantly higher specific surface area of 232 m2 g−1, showed the lowest acid site density of 0.18 µmol m−2, likely resulting from the broader dispersion of acid sites across the extensive surface. However, when quantified based on the total number of acid centers per gram, Al2O3/C, with 43 µmol g−1, is comparably effective to ZrO2/C, which has 32 µmol g−1. This indicates that the functional performance of Al2O3/C compensates for its lower density per unit area through a greater overall quantity of accessible acid sites.
The mixed oxides exhibit textural properties that represent a balance between the enhanced surface area provided by Al2O3 and the strong inherent acidity of ZrO2. A tendency was observed, where an excess of either component – Al2O3 or ZrO2 – results in acid site densities (µmol g−1) similar to those of the individual oxides. The optimal acid site density was observed in the Al2O3–ZrO2/C (1:1) catalyst, achieving the highest values both per gram (77 µmol g−1) and per unit area (0.37 µmol m−2).
Furthermore, it is important to note that all materials predominantly exhibit Lewis acid sites, with the absence of Brønsted acid sites. This characterization underscores the Lewis acid sites as the primary contributors to the acidity of these catalysts.
To further elucidate the acid–base properties of the materials, we added CO2 as a probe molecule in this study. This approach allows the simultaneous assessment of the materials' acidic and basic characteristics, contributing to a more complete understanding of their catalytic properties. Following this line, an in situ IR study exploiting CO2 as a probe molecule was performed to characterize the acid and basic sites on the surface of the catalysts. Two spectral regions were analyzed separately: (i) acid site region (within the 2400–2300 cm−1 range), where the adsorption bands are ascribed to the Σu+ mode of CO2 linearly adsorbed on cationic sites, responsible for Lewis acidity;63–66 and (ii) basic site region (within the 1800–1200 cm−1 range), where the adsorption bands are ascribed to carbonate and bicarbonate species (unidentate, bidentate, or bridged bidentate) formed after the adsorption of CO2 on electron-rich oxygen atoms on the surface.65,67,68
Fig. 2 reports the IR spectra in the 2400–2300 cm−1 region, relative to the linear adsorption of CO2 on Lewis acid sites. In agreement with the existing literature, the overall absorption of CO2 on ZrO2/C is shifted towards higher wavenumbers than on Al2O3/C, indicating that the acid sites on the surface of the former material are generally stronger than the ones on the latter. The spectra of CO2 on both ZrO2/C and Al2O3/C can be decomposed into 2–3 contributions, suggesting the presence of at least a few different families of acid sites on each material, as previously reported.67,69 Mixed oxides also show distinct contributions centered at intermediate wavenumbers between pure Al2O3/C and ZrO2/C due to which unambiguous assignation to CO2 adsorption on Al3+ or Zr4+ is not straightforward. The intensity of the overall band in the thickness-corrected spectra can be correlated with the abundance of the acid sites, pointing out substantial differences in the inspected materials. Pure oxides demonstrate lower acidity compared to some mixed oxides, such as Al2O3–ZrO2/C (1:1) and Al2O3–ZrO2/C (2:1). Results are in line with those in Table 2 obtained using pyridine, confirming the equimolar mixture of Al2O3–ZrO2/C (1:1) as the material with the highest abundance of Lewis acid sites.
 | ||
| Fig. 2 In situ IR spectra of CO2 adsorbed on all the pure and mixed oxides recorded at beam temperature. The spectra are corrected for the pellet thickness and focus on the 2400–2300 cm−1 range, relative to linear CO2 complexes. | ||
Fig. 3 presents the IR spectra in the 1800–1200 cm−1 region, recorded to follow the adsorption of CO2 on the basic sites of the pure ZrO2/C and Al2O3/C, which serve as references for the mixed materials. The measurements are divided into two sections. Spectra in Fig. 3A and B are meant to follow the evolution of the carbonate species formed upon CO2 adsorption under static conditions. The blue and orange spectra in Fig. 3A and B were collected immediately after CO2 dosing (25 mbar), while the black spectra correspond to the final situation, obtained after waiting 15 minutes maintaining the same CO2 pressure. Spectra in Fig. 3C and D are recorded degassing CO2 under dynamic vacuum. The blue and orange spectra in Fig. 3C and D were collected immediately after exposing the sample to dynamic vacuum, while the black spectra were collected after a degassing of 15 minutes.
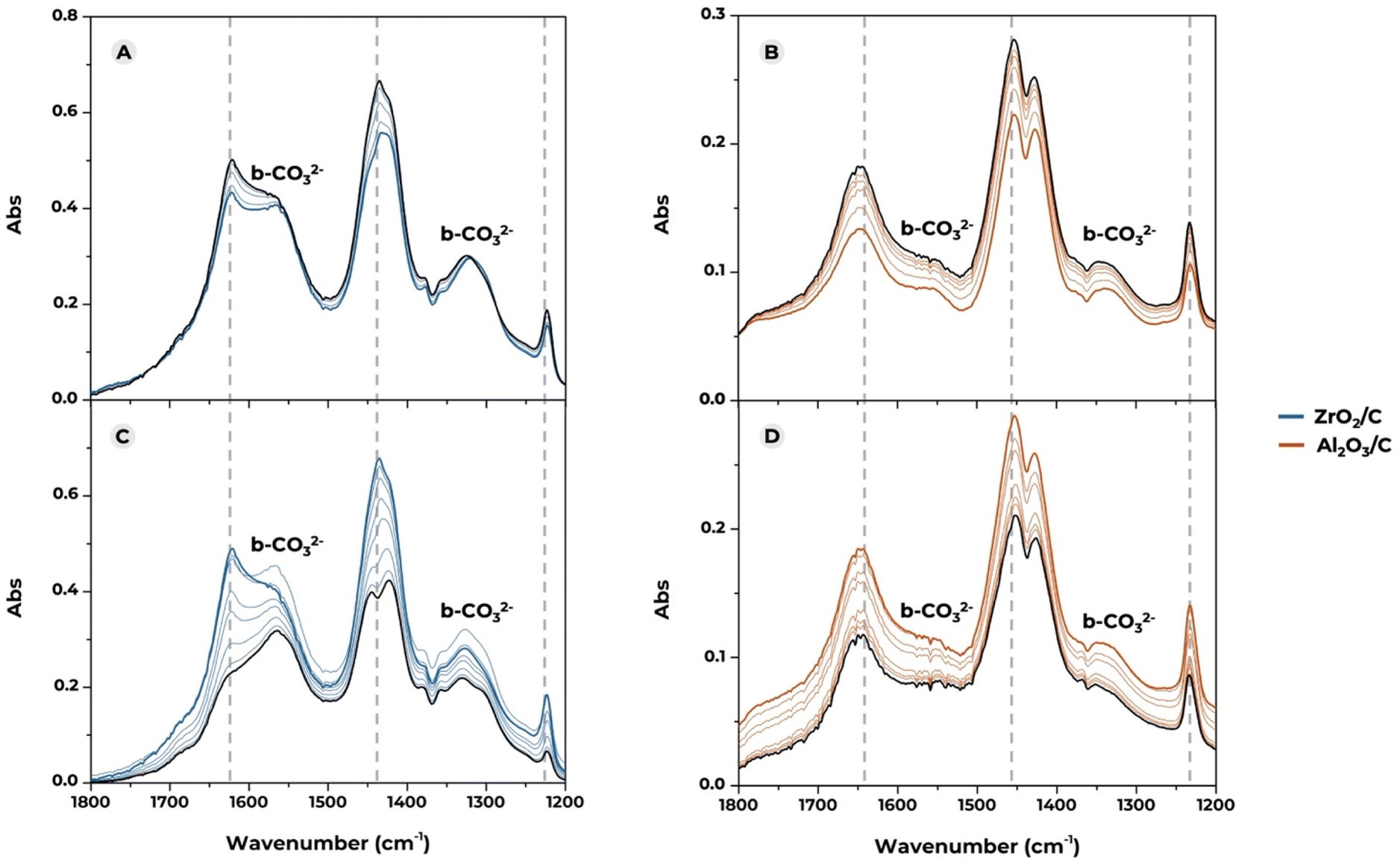 | ||
| Fig. 3 The in situ IR spectra of CO2 adsorption in the 1800–1200 cm−1 range are divided into two sections. Sections (A) and (B) correspond to the stabilization of the carbonate species on the surface of ZrO2 (A) and Al2O3 (B) in contact with 25 mbar CO2 (0 min corresponding to the blue and orange bold spectra and 15 min to the black bold spectra). Sections (C) and (D) follow the CO2 degassing step on the same materials (0 min corresponding to the blue and orange bold spectra and 15 min to the black bold spectra). | ||
Dotted vertical lines mark the positions of bicarbonate species (HCO32−)
• Symmetric C–O stretching (νsym C–O) at 1435–1480 cm−1. The band features contributions from monodentate (1435 cm−1) and bidentate bicarbonates (1455 cm−1), complicating the analysis.• Asymmetric C–O stretching (νasym C–O) at 1660–1680 cm−1
• Bending of the OH group (dOH) on bicarbonate species at 1230 cm−1.
Conversely, the bands centered at ∼1550 cm−1 and ∼1330 cm−1 indicate the presence of bidentate carbonates, as confirmed by the significant spectral separation (Dν = 200 cm−1).60,65,66
By comparing the spectra of the carbonate species formed on the surface of the two pure oxides, it emerges that ZrO2/C exhibits a strong affinity for CO2, forming both carbonates and bicarbonates. On the other hand, the spectra recorded on Al2O3/C present a much smaller contribution of carbonate bands, suggesting that the basicity profile of Al2O3/C is primarily provided by OH groups, which is in line with previous studies.70
Fig. 4 presents the thickness-corrected spectra of the five catalysts, with pure materials shown in Fig. 4A and mixed materials in Fig. 4B. All spectra are normalized and displayed on a consistent scale to simplify comparison. Two spectra, one recorded after contact with CO2 (solid line) and one upon degassing at room temperature (dotted line), are reported for each catalyst. In both cases, the reported spectrum corresponds to the situation after 15 minutes of equilibration and degassing, respectively. The overall higher intensity of the bands in the ZrO2/C spectrum compared with Al2O3/C indicates the higher basicity of the former pure catalyst (Fig. 4A).71 Regarding the mixed samples, it is evident that Al2O3–ZrO2/C (1:1) exhibits the highest abundance of basic sites (even compared with the pure ZrO2/C). The other mixed oxides display similar basicity to one another, although the formed species are different due to the varying Al2O3–ZrO2 molar ratios.72–75
 | ||
| Fig. 4 IR spectra of CO2 adsorption of pure oxides (A) and mixed oxides (B) with the solid lines corresponding to stabilization of 25 mbar CO2 and dotted lines to CO2 outgassing (5 × 10−5 mbar). | ||
Lastly, the dotted spectra in Fig. 4 allow the stability of the carbonate species to be assessed depending on the quantity that is retained after the degassing treatment at room temperature. The IR bands of the carbonate species decrease in intensity in all cases upon degassing, with the Al2O3–ZrO2/C (1:1) catalyst being able to retain the highest quantity of carbonates compared with the rest of the materials for the same degassing time.
X-ray Photoelectron Spectroscopy (XPS) was employed to characterize the surface chemical composition of the catalysts under investigation. Detailed binding energies for the O 1s, Al 2p, and Zr 3d orbitals are reported in Table 3 across all samples. For the Al2O3/C and ZrO2/C samples, the binding energies observed for O 1s, Al 2p, and Zr 3d are consistent with established signatures for Al2O3 and ZrO2 oxides, respectively. In mixed Al2O3–ZrO2/C samples, the O 1s spectrum exhibits a broad peak at 529.7 eV, reflecting contributions from oxygen in both Al2O3 and ZrO2 matrices, and at 530.8 eV, indicative of oxide defects. Additional contributions are noted at 531.5 eV and 533 eV, corresponding to carbonyl groups and ether linkages or possibly to water adsorbed on the surface. The Al 2p core-level spectra of these catalysts display peaks at approximately 73.7 and 75.2 eV, indicative of Al3+ in distinct chemical environments. The peak at 73.7 eV is attributed solely to Al2O3, while the peak at 75.2 eV is likely associated with hydrated aluminum oxides or hydroxide species. Similarly, Zr 3d core-level spectra show peaks with a separation of 2.4 eV, typical for ZrO2, at binding energies of 184.1 and 181.8 eV. Additional peaks at 185.4 eV and 183.2 eV are identified as originating from Zr(OH)2. These observations align with the known characteristics of ZrO2 derived from UiO-66 (Zr), underscoring the intricate composition of these catalytic materials.75
| Sample | Binding energy (eV) | |||||
|---|---|---|---|---|---|---|
| O 1s | Al 2p | Zr 3d | ||||
| Al2O3 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–O O/C–O |
ZrO2 | Al2O3 | ZrO2 | Zr(OH)2 | |
| ZrO2/C | — | — | 529.9 | — | 181.8–184.1 | 183.2–185.4 |
| Al2O3–ZrO2/C (1:2) |
533 | 531.9–530.8 | 530.7 | 75.2–73.7 | 181.8–184.1 | 183.2–185.4 |
| Al2O3–ZrO2/C (1:1) |
533 | 531.9–530.8 | 530.7 | 75.2–73.7 | 181.8–184.1 | 183.2–185.4 |
| Al2O3–ZrO2/C (2:1) |
533 | 531.9–530.8 | 530.7 | 75.2–73.7 | 181.8–184.1 | 183.2–185.4 |
| Al2O3/C | 533 | — | — | 75.2–73.7 | — | — |
An increase in the Zr binding energy was noted, suggesting that Zr and Al species are effectively dispersed within the mixed metal oxides corroborated by Energy Dispersive X-ray Spectroscopy (XPS) analysis (Fig. S2†). The positive charge on Zr atoms, attributable to the higher electronegativity of Al in Zr–O–Al bonds, likely enhances the number and efficacy of Lewis acid sites. This phenomenon is in agreement with previously reported literature findings.75
Transmission electron microscopy (TEM) analysis was carried out to elucidate the size and morphology of the catalyst structures. The Al2O3–ZrO2/C (1:1) sample displays aggregates of small particles, creating interparticle voids that contribute to a disordered structure (Fig. 5). This observation is consistent with the X-ray diffraction (XRD) results presented in Fig. 1.
 | ||
| Fig. 5 STEM image and EDX analysis of the Al2O3–ZrO2/C (1:1) catalyst. | ||
TEM analysis shows a homogeneous distribution of Zr and Al and distinctly captures the formation of γ-Al2O3/C, with darker areas indicating particle presence (Fig. S3†), visually confirming its structural composition, and with lighter areas indicating the formation of ZrO2. Scanning electron microscopy (SEM) was employed to further delineate the morphology of the materials. It reveals that the catalysts possess predominantly irregular, amorphous structures, either as blocks or planes. Notably, the Al2O3–ZrO2/C (1:1) sample exhibits a more porous structure compared to other materials, aligning with porosimetry results (Fig. S4†). Moreover, the EDS results presented in Fig. S5† confirm that the elemental ratios in Al2O3–ZrO2/C catalysts align with their respective nominal molar ratios, such as 2:1, 1:1 and 1:2.
Catalytic test
To expedite catalyst screening and gain preliminary insights into the ability of MOF-derived Al2O3–ZrO2/C catalysts, all prepared systems were tested in the transfer hydrogenation of methyl levulinate by using 2-propanol as an indirect H-source at 200 °C at 30 min (Fig. 6).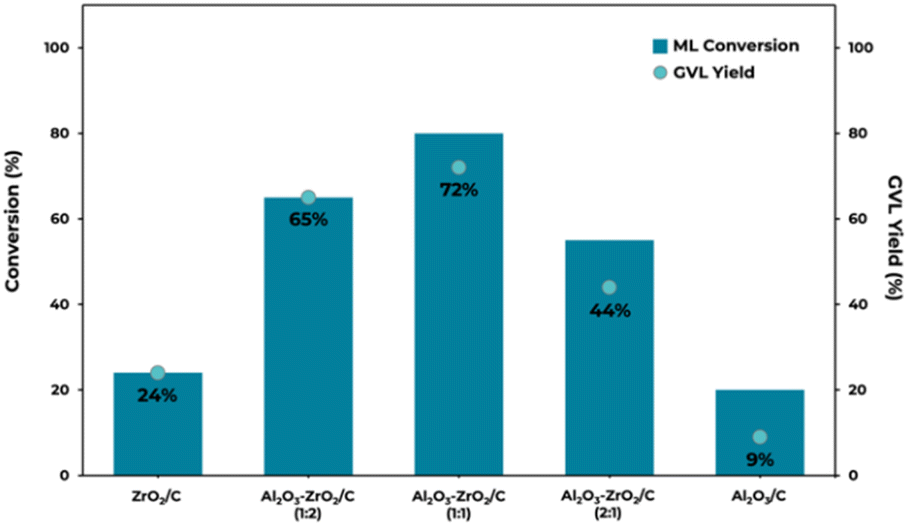 | ||
| Fig. 6 Catalyst screening in the CTH of methyl levulinate into g-valerolactone promoted by MOF-derived Al2O3–ZrO2/C catalysts by using 2-propanol as an H-source (reaction conditions: 0.5 mL min−1 flow rate; ML 0.2 M in 2-propanol; 0.500 g of catalyst; 30 bar pressure; time on stream: 30 min; reaction temperature: 200 °C). | ||
The data presented in Fig. 6 point to the significant role of Al2O3 content in modulating the catalytic efficiency of the examined systems.
Employing ZrO2/C at 200 °C for 30 minutes resulted in an ML conversion rate of 24%, with a 100% selectivity towards GVL. The use of a pure Al2O3 catalyst yielded a similar ML conversion rate of 20%, but with a significantly reduced GVL yield of 9%, where isopropyl levulinate (IPL) emerged as the predominant reaction product. When Al2O3 was integrated in a 1:2 molar ratio with ZrO2, a notable enhancement in both ML conversion and GVL yield, peaking at 65%, was observed. This marked improvement suggests a synergistic interaction within the mixed metal oxide matrix, potentially augmenting the number and efficacy of catalytic sites available for the hydrogenation reaction.
The optimum catalytic performance was recorded with an equimolar ratio of Al2O3 to ZrO2 in the Al2O3–ZrO2/C (1:1) catalyst. This configuration not only enhanced ML conversion to an impressive 80% but also pushed GVL yield (72%). The GC-MS chromatogram of this reaction at 30 min time on stream is depicted in Fig. S6.†
To assess the catalytic efficiency, we quantified the productivity of γ-valerolactone (GVL) under specific conditions: employing 0.5 g of the Al2O3–ZrO2/C (1:1) catalyst at a flow rate of 0.5 mL min−1, with a reaction temperature of 200 °C and a N2 pressure of 30 bar. The catalyst achieved a productivity rate of 9 mmol GVL per gram of catalyst per hour. This performance is noteworthy when compared with analogous systems such as ZrO2/SBA-15 and Zr-containing zeolites, which report productivities of 5.2 and 0.81 mmol GVL per gram of catalyst per hour, respectively.35–38,76
The performance data depict a tendency-curve where excessive addition of Al2O3 beyond the optimum ratio led to a diminished conversion rate (55%) and a comparably reduced GVL yield (44%) when using Al2O3–ZrO2/C.
This trend can be attributed to the substantial surface area and the dense distribution of acidic and basic sites associated with the Al2O3–ZrO2 ratio, where Al2O3–ZrO2/C (1:1) composition exhibits a higher density of acid (Table 2 and Fig. 2) and basic sites (Fig. 4). Moreover, it has been previously reported how basic sites contribute more importantly to the reaction than acid sites.77 In other words, while Al2O3 contributes beneficial properties to some extent, it may also induce over-adsorption and complex transformations of intermediates on the catalyst surface due to changes in the catalytic acid and basic sites. Additionally, an increase in the Al2O3 content was correlated with a decreased GVL selectivity, favoring the production of isopropyl levulinate (IPL), a notable by-product.
Having identified Al2O3–ZrO2/C (1:1) as an optimum performing catalyst, subsequent experiments were focused to identify the best reaction conditions to maximize GVL production and to assess its practical application potential.
Temperature tests in the range from 160 °C to 220 °C were subsequently carried out to establish the optimum thermal conditions that favor high GVL yield while minimizing the formation of by-products such as isopropyl levulinate (IPL). Results revealed that temperatures above 200 °C resulted in a higher rate of ML conversion with decreased selectivity towards GVL, indicating a shift in reaction dynamics towards the IPL pathway (Fig. 7). These results already improve not only those obtained in continuous flows (Table 1), but also those obtained under batch conditions with similar materials subjected to high temperatures, pressures and times (Table S1†).
 | ||
| Fig. 7 Effect of reaction temperature on ML conversion and product selectivity for Al2O3–ZrO2/C (1:1) under transfer hydrogenolysis conditions (reaction conditions: 0.5 mL min−1 flow rate; ML 0.2 M in 2-propanol; 0.500 g of catalyst; 30 bar pressure; time on stream: 30 min). | ||
The time effect of Al2O3–ZrO2/C (1:1) in the transfer hydrogenation of methyl levulinate (ML) at 200 °C was meticulously monitored to assess changes in catalytic activity over prolonged time on stream (Fig. 8). By using the Al2O3–ZrO2/C (1:1) catalyst, a consistent decline in performance was observed over 6 hours of continuous reaction. Notably, a significant drop in activity commenced after the first 60 minutes with the investigated catalyst achieving a 75% ML conversion rate and a 67.5% GVL yield. As the reaction progressed, ML conversion decreased to 35% and GVL yield to 31.5% by the end of 360 minutes. This decline in catalytic activity can primarily be attributed to the accumulation of humin deposits on the Al2O3–ZrO2/C (1:1) catalyst surface, which block active sites and progressively deactivate the catalyst.
 | ||
| Fig. 8 CTH of methyl levulinate into γ-valerolactone promoted by the Al2O3–ZrO2/C (1:1) catalyst against time (reaction conditions: 0.5 mL min−1 flow rate; ML 0.2 M in 2-propanol; 0.500 g of catalyst; 30 bar pressure; reaction temperature: 200 °C). The regeneration of the catalyst was performed for three consecutive cycles after 360 min of time-on-stream (dotted gray line). | ||
Other synthesized mixed oxide catalysts, Al2O3–ZrO2 (1:2) and Al2O3–ZrO2 (2:1), demonstrated similar deactivation patterns (Fig. S7†). This uniformity across different material compositions suggests that the presence of aluminum within the catalyst does not significantly impact catalytic stability. However, aluminum remains essential for optimizing both conversion and selectivity.
To address this challenge, a regeneration protocol was implemented. This involves halting the flow of the reagent mixture and removing the catalyst from the flow system. The catalyst is then subjected to a thermal treatment, where it is maintained at 400 °C in an air flow of 30 mL min−1 for two hours. This procedure aimed to promote the combustion of deposited carbonaceous residues, effectively restoring its activity. It is important to emphasize that the regeneration treatment is not expected to alter the chemical nature or properties of the Al2O3–ZrO2/C (1:1) catalyst. The catalyst underwent initial synthesis through a thermal treatment at 600 °C, a process far more intense than the subsequent regeneration at 400 °C. Consequently, the approximately 5% of carbon integrated into the material remains unchanged and is not oxidized into other species. Even in the unlikely event of oxidation, the minimal amount of carbon present would have negligible impact on the overall physico-chemical properties of the Al2O3–ZrO2/C (1:1) catalyst.
Analysis of the catalyst performance after regeneration reveals a noticeable decline in ML conversion efficiency, yet the selectivity towards GVL remains consistently high at 90%. This demonstrates the catalyst's capability to maintain its effectiveness in producing the desired product despite a reduction in overall activity. In comparison, when the catalyst is simply discharged and dried under normal conditions, it continues to function, yielding similar results to those observed during its last use. These results suggest that the catalyst's deactivation is primarily due to the blocking of active sites. Extensive literature indicates that in biomass processing, the deposition of humins (high-molecular-weight compounds formed by the polymerization of intermediates) on the catalytic surface is a major cause of catalyst deactivation.78–80 Similarly, in this study, humins are primarily suspected as the main cause of deactivation. Although these compounds are not detectable by gas chromatography (GC) due to their high molecular weight, the carbon balance analysis, which shows a maximum weight loss of only 2%, suggests that a small portion of ML is converted into humins through a competing polymerization reaction.
The continuous flow setup intensifies this effect, as the catalyst sites are constantly exposed to the reaction mixture. Attempts to detect humins on the exhausted catalyst using ATR spectroscopy were unsuccessful due to the instrument's insufficient sensitivity to detect significant spectral differences. Additionally, the analysis of changes in acid-base sites was unreliable due to the temperature activation required prior to measurement, as detailed in the Experimental section. In any case, the correlation between the loss of active sites and humin deposition is supported not only by the observed catalytic behavior (where conversion decreases while selectivity remains constant, indicating that the remaining active sites are still functional) but also by the catalyst's color change to a slightly darker hue. Moreover, these observations align with corroborative findings from previous studies within our group on the reductive upgrading of biomass derived molecules under continuous flow conditions with oxide materials derived from MOFs, further validating our conclusions.47,81
Different flow rates were subsequently assessed employing Al2O3–ZrO2/C (1:1) to investigate their effect on ML conversion and GVL yield at 200 °C. Results, including the residence time for each condition, are depicted in Fig. 9.
 | ||
| Fig. 9 CTH of methyl levulinate into g-valerolactone promoted by Al2O3–ZrO2/C (1:1) under a flow rate effect at different times on stream (reaction conditions: ML 0.2 M in 2-propanol; 0.500 g of catalyst; 30 bar pressure; reaction temperature: 200 °C). | ||
Initially, at 30 minutes, reducing the flow rates from 0.5 mL min−1 to 0.2 mL min−1 and 0.1 mL min−1 did not significantly influence ML conversion rates (80%, 81% and 83%, respectively at 0.5, 0.2 and 0.1 mL min−1) or GVL selectivity (90% at 0.5, 88.9% at 0.2 mL min−1 and 88% at 0.1 mL min−1). However, by using a lower flow rate, catalyst deactivation was markedly accelerated, evidenced by the significant drop in ML conversion to 4% at a lower flow rate of 0.1 mL min−1 after 360 minutes, likely due to prolonged contact between the reactants and the catalyst, as demonstrated by the increased residence time. This prolonged contact time also shifted product selectivity, primarily increasing the tendency for ML to undergo transesterification and likely leading to the deposition of humins on the catalyst surface at lower residence times. As a result, GVL selectivity decreased progressively over time, from 88.9%, 88.5%, and 86%, to 85% at 0.2 mL min−1 and from 88%, 86.7%, and 84.4%, to 82.8% at 0.1 mL min−1 for 30-, 60-, 180-, and 360 minutes of time on stream, respectively. Conversely, at a higher flow rate of 1 mL min−1, the system encountered complications such as blockages. This outcome emphasizes the importance of balancing contact time to maintain both the effectiveness and durability of the catalytic system as well as to prevent mechanical issues.
Understanding of GVL production pathways in the presence of 2-propanol
The conversion of methyl levulinate (ML) into g-valerolactone (GVL) via a transfer hydrogenation process is thoroughly documented and remains a focus of significant research interest.12,13,82–84 Utilizing 2-propanol both as a solvent and a hydrogen donor, ML is subjected to a variety of chemical transformations that may lead to GVL production. These transformations are primarily categorized into three potential pathways (Scheme 2): | ||
| Scheme 2 Reaction pathways in the CTH of methyl levulinate (ML) with 2-propanol to GVL under continuous flow conditions. | ||
(1) Direct (transfer) hydrogenation: the carbonyl group in ML is first CTH hydrogenated through catalytic transfer hydrogenation (CTH), leading to the formation of methyl 4-hydroxypentanoate that is then rapidly cyclized into GVL.
(2) Cyclization and subsequent (transfer) hydrogenation: ML might initially cyclize into various lactone intermediates, which are subsequently hydrogenated to produce GVL.
(3) ML undergoes transesterification with 2-propanol, resulting in the formation of isopropyl levulinate (IPL) that can be further converted into GVL via lactone cyclization.
Catalytic tests and physicochemical characterizations facilitate the interpretation of reported results and support the hypothesized mechanisms underlying GVL production. Results related to the effects of temperature and alumina content distinctly show that introducing alumina into the catalyst system significantly influences the course of these reactions. The presence of alumina notably enhances ML conversion, while its varying content critically impacts substrate conversion and liquid product selectivity. Notably, an increased Al/Zr ratio tends to increase ML conversion and alters the reaction pathway, especially under elevated temperatures (220 °C).
Experimental outcomes reveal that lower reaction temperatures (up to 180 °C) and a minimum presence of alumina (up to a 1:1 Al/Zr ratio) favor the activation of 2-propanol, thereby promoting transfer hydrogenation reactions over transesterification, which in turn directs product selectivity towards enhanced GVL production. Conversely, higher alumina content and increased reaction temperatures tend to favor intramolecular cyclization, predominantly leading to the formation of IPL. In any case, the absence of detectable methyl 4-hydroxypentanoate and lactone intermediates in experimental analyses restricts a definitive conclusion about whether GVL is primarily produced from the direct hydrogenation (pathway 1) or via cyclization and subsequent hydrogenation (pathway 2).
Conclusions
The effectiveness of MOF-derived Al2O3–ZrO2/C catalysts in the catalytic transfer hydrogenation of methyl levulinate to γ-valerolactone under continuous flow conditions was successfully demonstrated. By incorporating Al2O3 into ZrO2 using MOFs as sacrificial templates, the catalytic performance was significantly enhanced due to the synergistic interaction between these two oxides. The combination of alumina and zirconia in catalysts not only enhances the number of acid and base sites but also increases the specific surface areas of the resulting catalysts. Notably, all mixed Al–Zr oxides demonstrated superior catalytic activity compared to the individual ZrO2 and Al2O3 catalysts, with the Al2O3–ZrO2/C catalyst with a 1:1 ratio demonstrating superior performance, achieving more than 99% conversion of ML and 85% yield of GVL at 220 °C within 30 minutes.
Optimum reaction conditions were identified to maximize GVL production, with a reaction temperature of 200 °C and a duration of 30 minutes proving most effective. These conditions effectively balanced the catalytic activity, leading to high conversion rates and desired product yields. Extended time on stream up to 6 hours showed a decrease in ML conversion and GVL yield, primarily due to catalyst deactivation from carbonaceous deposits. However, thermal regeneration at 400 °C successfully restored the catalyst's activity, demonstrating the feasibility of catalyst reuse.
The study also highlights the importance of maintaining balanced Al/Zr ratios to optimize selectivity and avoid excessive production of by-products such as isopropyl levulinate (IPL) through transesterification reactions. Different flow rates were tested, and the optimal flow rate was determined to be 0.5 mL min−1, which maintained high conversion and selectivity while preventing catalyst deactivation.
The successful implementation of a thermal regeneration strategy additionally underscores the potential practical feasibility of this catalytic system for industrial applications. To further enhance the promise of this work and align with sustainable development goals, we have already begun to focus on two crucial advancements: (i) the synthesis of MOF-derived materials utilizing greener aprotic solvents, and (ii) the refinement of catalyst synthesis conditions to not only preserve but also optimize the high selectivity observed while improving stability and thereby increasing GVL production. Since γ-valerolactone (GVL) is one of the most promising chemicals in modern biorefineries, these enhancements in its catalytic production from methyl-levulinate are anticipated to significantly advance the development of a circular economy, optimizing resource efficiency and minimizing waste.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Marina Ronda-Leal: writing – review & editing, writing – original draft, methodology, investigation, formal analysis, data curation. Alina M. Balu: visualization, investigation. Rafael Luque: visualization, investigation, resources, project administration, funding acquisition. Francesco Mauriello: writing – review & editing, visualization, methodology, investigation. Alberto Ricchebuono: writing – review & editing, methodology, investigation. Christophe Len: visualization, investigation. Antonio A. Romero: writing – review & editing, validation, supervision, resources, project administration. Emilia Paone: writing – review & editing, validation, supervision, conceptualization.Conflicts of interest
The authors declare that they have no known conflicts of interest that could have appeared to influence the work reported in this paper.Acknowledgements
This publication was prepared with support and funding from the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant “Ecosistema TECH4YOU” (CUP: C33C22000290006, Spoke 3 – Goal 3.5). Emilia Paone gratefully acknowledges the Italian Ministry for University and Research (MUR) and the Università degli Studi Mediterranea di Reggio Calabria for funding under D.M. 1062/2021 and D.M. 737/2022 programs. A.A. Romero gratefully acknowledges MINECO for funding under project PID2019-109953GB-100, co-financed with FEDER funds. M. Ronda Leal also expresses gratitude to the MINECO project for providing an FPU contract (REF: FPU20/03875) associated with the PID2019-109953GB-100 project, carried out within the FQM-383 Group. The authors express their gratitude to the anonymous reviewers for their insightful comments and feedback, which have significantly enhanced the quality of the paper.References
- M. Ishaq, G. Ghouse, R. Fernandez-Gonzalez, F. Puime-Guillen, N. Tandir and H. M. S. de Oliveira, Front. Environ. Sci. Eng., 2022, 10, 941791 Search PubMed.
- United Nations, COP28 Ends with Call to ‘Transition Away’ from Fossil Fuels; UN Chief Says Phaseout Inevitable, https://unsdg.un.org/latest/stories/cop28-ends-call-%E2%80%98transition-away%E2%80%99-fossil-fuels-un-chief-says-phaseout-inevitable, accessed 2024-06-12 Search PubMed.
- L. Hens, C. Block, J. J. Cabello-Eras, A. Sagastume-Gutierez, D. Garcia-Lorenzo, C. Chamorro, K. Herrera Mendoza, D. Haeseldonckx and C. Vandecasteele, J. Cleaner Prod., 2018, 172, 3323–3333 Search PubMed.
- S. Abate, G. Centi and S. Perathoner, Natl. Sci. Rev., 2015, 2, 143–145 Search PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 Search PubMed.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Renewable Sustainable Energy Rev., 2020, 127, 109852 Search PubMed.
- C. Espro, E. Paone, F. Mauriello, R. Gotti, E. Uliassi, M. L. Bolognesi, D. Rodríguez-Padrón and R. Luque, Chem. Soc. Rev., 2021, 50, 11191–11207 RSC.
- A. Satira, E. Paone, V. Bressi, D. Iannazzo, F. Marra, P. S. Calabrò, F. Mauriello and C. Espro, Appl. Sci., 2021, 11, 10983 CrossRef CAS.
- E. Paone, M. Miceli, A. Malara, G. Ye, E. Mousa, E. Bontempi, R. Pietropaolo and F. Mauriello, ACS Sustainable Chem. Eng., 2022, 10, 2275–2281 Search PubMed.
- E. Paone, F. Fazzino, D. M. Pizzone, A. Scurria, M. Pagliaro, R. Ciriminna and P. S. Calabrò, Sustainability, 2021, 13, 2428 Search PubMed.
- T. Tabanelli, E. Paone, P. B. Vásquez, R. Pietropaolo, F. Cavani and F. Mauriello, ACS Sustainable Chem. Eng., 2019, 7, 9937–9947 Search PubMed.
- P. B. Vásquez, T. Tabanelli, E. Monti, S. Albonetti, D. Bonincontro, N. Dimitratos and F. Cavani, ACS Sustainable Chem. Eng., 2019, 7, 8317–8330 Search PubMed.
- L. e, Y. Han, J. Feng and X. Lu, Ind. Crops Prod., 2020, 144, 112031 CrossRef.
- M. G. Al-Shaal, M. Calin, I. Delidovich and R. Palkovits, Catal. Commun., 2016, 75, 65–68 Search PubMed.
- A. S. Amarasekara and M. A. Hasan, Catal. Commun., 2015, 60, 5–7 Search PubMed.
- L. Biancalana, S. Fulignati, C. Antonetti, S. Zacchini, G. Provinciali, G. Pampaloni, A. M. Raspolli Galletti and F. Marchetti, New J. Chem., 2018, 42, 17574–17586 Search PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 Search PubMed.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 Search PubMed.
- C. Espro, B. Gumina, T. Szumelda, E. Paone and F. Mauriello, Catalysts, 2018, 8, 313 Search PubMed.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338–1352 Search PubMed.
- T. Komanoya, K. Nakajima, M. Kitano and M. Hara, J. Phys. Chem. C, 2015, 119, 26540–26546 CrossRef CAS.
- Y. Kuwahara, W. Kaburagi, Y. Osada, T. Fujitani and H. Yamashita, Catal. Today, 2017, 281, 418–428 Search PubMed.
- S. Ostovar, H. Saravani and D. Rodríguez-Padrón, Renewable Energy, 2021, 178, 1070–1083 CrossRef.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233–12235 RSC.
- M. Boronat, A. Corma and M. Renz, J. Phys. Chem. B, 2006, 110, 21168–21174 CrossRef CAS PubMed.
- A. Osatiashtiani, A. F. Lee and K. Wilson, J. Chem. Technol. Biotechnol., 2017, 92, 1125–1135 Search PubMed.
- X. Chen, T. Zhao, X. Zhang, Y. Zhang, H. Yu, Q. Lyu, X. Jia, L. Han and W. Xiao, J. Energy Chem., 2021, 63, 430–441 Search PubMed.
- R. Q. Raguindin and J. G. Seo, Chem. Eng. J., 2024, 152141 Search PubMed.
- N. Karanwal, D. Verma, P. Butolia, S. M. Kim and J. Kim, Green Chem., 2020, 22, 766–787 RSC.
- M. B. Gawande, R. K. Pandey and R. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113–1125 RSC.
- J. C. Védrine, Catalysts, 2017, 7, 341 Search PubMed.
- C. García-Sancho, C. P. Jiménez-Gómez, N. Viar-Antuñano, J. A. Cecilia, R. Moreno-Tost, J. M. Mérida-Robles and P. Maireles-Torres, Appl. Catal., A, 2021, 609, 117905 CrossRef.
- J. He, H. Li, Y. M. Lu, Y. X. Liu, Z. B. Wu, D. Y. Hu and S. Yang, Appl. Catal., A, 2016, 510, 11–19 CrossRef CAS.
- T. Tabanelli, P. B. Vásquez, E. Paone, R. Pietropaolo, N. Dimitratos, F. Cavani and F. Mauriello, Chem. Proc., 2020, 2, 28 Search PubMed.
- W. Ouyang, D. Zhao, Y. Wang, A. M. Balu, C. Len and R. Luque, ACS Sustain. Chem. Eng., 2018, 6, 6742–6756 Search PubMed.
- D. Zhao, Y. Wang, F. Delbecq and C. Len, Mol. Catal., 2019, 475, 110456 CrossRef CAS.
- M. Cabanillas, A. Franco, N. Lázaroa, A. M. Balua, R. Luque and A. Pineda, Mol. Catal., 2019, 477, 110522 CrossRef.
- D.-P. Phan, M. H. Tran and E. Y. Lee, Mater. Today Chem., 2023, 33, 101691 CrossRef CAS.
- A. Kumari, S. Kaushal and P. P. Singh, Mater. Today Energy, 2021, 20, 100667 CrossRef CAS.
- X. Liu, Z. Liu and R. Wang, Adv. Polym. Technol., 2020, 2020, 1201923 Search PubMed.
- J. Gascon, A. Corma and F. Kapteijn, ACS Catal., 2014, 4, 361–378 Search PubMed.
- R. Fang, A. Dhakshinamoorthy and Y. Li, Chem. Soc. Rev., 2020, 49, 3638–3687 Search PubMed.
- Q. Fu, H. Jiang, Y. Wang, H. Wang and X. Zhao, Mater. Chem. Front., 2023, 7, 628–642 RSC.
- W. Ouyang, D. Zhao, Y. Wang, A. M. Balu, C. Len and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 6746–6752 Search PubMed.
- J. Li, S. Zhao, Z. Li, D. Liu, Y. Chi and C. Hu, Inorg. Chem., 2021, 60, 7785–7793 Search PubMed.
- M. Ronda-Leal, S. M. Osman, H. W. Jang, M. Shokouhimehr, A. A. Romero and R. Luque, Fuel, 2023, 333, 126221 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. Dhakshinamoorthy, S. Navalon, A. M. Asiri and H. Garcia, Chem. Commun., 2020, 56, 26–45 RSC.
- S. L. Griffiths, P. Boyall, J. P. Müller, A. M. Harrington, W. Sobolewska, W. R. Reynolds, A. B. Richard, K. Wu, S. M. Collins, M. Muldowney and T. W. Chamberlain, Nanoscale, 2023, 15, 17910–17921 RSC.
- M.-L. Hu, M. Y. Masoomi and A. Morsali, Coord. Chem. Rev., 2019, 387, 415–435 CrossRef CAS.
- K.-Y. Zou and Z.-X. Li, Chem. - Eur. J., 2018, 24, 6506–6518 Search PubMed.
- W. Cao, W. Luo, H. Ge, Y. Su, A. Wang and T. Zhanga, Green Chem., 2017, 19, 2201–2211 CAS.
- W. Cao, L. Lin, H. Qi, Q. He, Z. Wu, A. Wang, W. Luo and T. Zhang, J. Catal., 2019, 373, 161–172 CrossRef CAS.
- C. Van Nguyen, B. M. Matsagar, J.-Y. Yeh, W.-H. Chiang and K. C.-W. Wu, J. Mol. Catal., 2019, 475, 110478 CAS.
- R. J. McKinstry, E. J. Cathcart Cussen, A. J. Fletcher, S. V. Patwardhan and J. Sefcik, Chem. Eng. J., 2016, 285, 718–725 Search PubMed.
- N. J. A. Rahman, A. Ramli, K. Jumbri and Y. Uemura, Sci. Rep., 2019, 16223 Search PubMed.
- J. Sakfali, S. B. Chaabene, R. Akkari, F. Dappozze, G. Berhault, C. Guillard and M. S. Zina, J. Photochem. Photobiol., A, 2022, 430, 113970 Search PubMed.
- C. Guo, P. Wang, S. Liao, H. Si, S. Chen, G. Fan and Z. Wang, Appl. Sci., 2019, 9(19), 4145 CAS.
- M. Farrag, Sci. Rep., 2023, 13, 10123 CAS.
- S. Shi, S. Qian, X. Hou, J. Mu, J. He and X. Chou, Adv. Condens. Matter Phys., 2018, 1, 7598978 Search PubMed.
- N. Tao, J. Liu, Y. Xu, Y. Feng, Y. Wang, W. Liu and J. Lv, Chem. Pap., 2020, 74, 3503–3515 Search PubMed.
- C. Liu, J. Sun, H. M. Brown, O. G. Marin-Flores, J. T. Bays, A. M. Karim and Y. Wang, Catal. Today, 2016, 269, 103–109 CrossRef CAS.
- J. He, H. Li, Y.-M. Lu, Y.-X. Liu, Z.-B. Wu, D.-Y. Hu and S. Yang, Appl. Catal., A, 2016, 510, 11–19 CrossRef CAS.
- H. Bekhti, Y. Boucheffa, A. H. A. Blal and A. Travert, Vib. Spectrosc., 2021, 117, 103313 Search PubMed.
- J. Goscianska, M. Ziolek, E. Gibson and M. Daturi, Catal. Today, 2010, 152(1–4), 33–41 CrossRef CAS.
- V. Bolis, G. Magnacca, G. Cerrato and C. Morterra, Thermochim. Acta, 2001, 379(1–2), 147–161 CrossRef CAS.
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17(14), 4297–4303 CAS.
- C. Morterra, A. Zecchina, S. Coluccia and A. Chiorino, J. Chem. Soc., Faraday Trans., 1977, 1(73), 1544–1560 Search PubMed.
- H. Y. T. Chen, S. Tosoni and G. Pacchioni, Surf. Sci., 2016, 652, 163–171 CAS.
- J. Podobiński and J. Datka, Molecules, 2024, 29(8), 1726 Search PubMed.
- S. Mukhopadhyay, R. Shimoni, I. Liberman, R. Ifraemov, I. Rozenberg and I. Hod, Angew. Chem., 2021, 133, 13535–13541 CrossRef.
- X. Chen, X. Liu, L. Zhu, X. Tao and X. Wang, Chemosphere, 2022, 291, 133032 CrossRef CAS PubMed.
- Z. Dai, D. T. Lee, K. Shi, S. Wang, H. F. Barton, J. Zhu, J. Yan, Q. Ke and G. N. Parsons, J. Mater. Chem. A, 2020, 8, 3803–3813 RSC.
- M. A. Gondal, T. A. Fasasi, U. Baig and A. Mekki, J. Nanosci. Nanotechnol., 2018, 18, 4030–4039 CrossRef CAS PubMed.
- A. Osatiashtiani, S. A. Orr, L. J. Durndell, I. Collado García, A. Merenda, A. F. Lee and K. Wilson, Catal. Sci. Technol., 2022, 12, 5611 Search PubMed.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Sustainable Chem. Eng., 2017, 5(1), 1141–1152 CrossRef CAS.
- G. Caeiro, J. M. Lopes, P. Magnoux, P. Ayrault and F. R. Ribeiro, J. Catal., 2007, 249, 234–243 CrossRef CAS.
- D. G. Blackmond, J. G. Goodwin and J. E. Lester, J. Catal., 1982, 78, 34–43 CrossRef CAS.
- J. V Ibarra, C. Royo, A. Monzón and J. Santamaría, Vib. Spectrosc., 1995, 9(2), 191–196 Search PubMed.
- M. Ronda-Leal, N. Lázaro, A. Pineda, A. A. Romero and R. Luque, Sustainable Chem. Pharm., 2024, 38, 101467 Search PubMed.
- M. Cabanillas, A. Franco, N. Lazaro, A. M. Balu, R. Luque and A. Pineda, Mol. Catal., 2019, 477, 110522 Search PubMed.
- M. Douthwaite, B. Zhang, S. Iqbal, P. J. Miedziak, J. K. Bartley, D. J. Willock and G. J. Hutchings, Catal. Commun., 2022, 106430 CAS.
- M. Ma, X. Yan, P. Hou, J. Cao, H. Liu, X. Xu, H. Yue, G. Tian, S. Jing and S. Feng, ChemistrySelect, 2020, 5, 924–930 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00797b |
| This journal is © The Royal Society of Chemistry 2025 |