
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA non-hydrolysable peptidomimetic for mitochondrial targeting†
Yeray
Folgar-Cameán
 a,
Daniel
Torralba-Maldonado
b,
Patricia
Fulias-Guzmán
a,
Marta
Pazo
a,
Irene
Máximo-Moreno
a,
Miriam
Royo
c,
Ona
Illa
*b and
Javier
Montenegro
*a
a,
Daniel
Torralba-Maldonado
b,
Patricia
Fulias-Guzmán
a,
Marta
Pazo
a,
Irene
Máximo-Moreno
a,
Miriam
Royo
c,
Ona
Illa
*b and
Javier
Montenegro
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15705, Santiago de Compostela, Spain. E-mail: javier.montenegro@usc.es
bDepartament de Química, Universitat Autònoma de Barcelona, 08193, Cerdanyola del Vallès, Spain. E-mail: Ona.Illa@uab.cat
cInstituto de Química Avanzada de Cataluña-Consejo Superior de Investigaciones Científicas (IQAC-CSIC) and Centro de Investigación Biomédica en Red-Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), 08034, Barcelona, Spain
First published on 22nd January 2025
Abstract
Peptidomimetics, molecules that mimic the activity of natural peptides with improved stability or bioavailability, have emerged as interesting materials with applications in biomedicine. In this study, we describe a hybrid γ,γ-peptidomimetic that efficiently aims at mitochondria, a key therapeutic target associated with several disorders, in living cells. Peptide backbones with a component of cationic and hydrophobic amino acids have been shown to preferentially target mitochondria due to their high negative membrane potential and hydrophobic character of the membranous invaginations of these key organelles. We here exploit the advantageous bioorthogonal properties of a peptidomimetic scaffold that consists of an alternation of (1S,3R)-3-amino-2,2-dimethylcyclobutane-1-carboxylic acid and an Nα-functionalised cis-γ-amino-L-proline derivative. This peptidomimetic exhibited excellent membrane translocation efficiency, mitochondrial targeting ability, and biocompatibility. Mitochondrial targeting was confirmed to be dependent on the electrochemical potential generated by the electron transport chain. The presence of non-natural amino acids rendered the compound exceptionally stable in the presence of proteases, maintaining its integrity and functionality for targeting the organelle even after 1 week of incubation in serum. This stability, coupled with its targeting abilities and the low cytosolic/endosomal residual signal, facilitated the tracking of relevant mitochondrial dynamics, including fission events and intracellular movement. Additionally, this peptidomimetic scaffold allowed the sustained and precise mitochondrial targeting of a pH sensitive ratiometric probe, 5(6)-carboxy-SNARF-1, which enabled mitochondrial pH monitoring. In summary, our study introduces a biomimetic peptide with exceptional mitochondria-targeting properties, ensuring stability in biological media and offering insights into crucial mitochondrial processes.
Introduction
Peptides and peptide-derived materials constitute a powerful chemical tool for the development of biocompatible drugs or selective carriers, which have found numerous applications in biomedicine.1–3 Their simple and modular synthesis allows for the straightforward modification of the sequence and the attachment of other bioactive compounds.2,4,5 In addition, some peptides possess membrane transport properties, making them very useful for the delivery of impermeable probes or bioactive cargoes into the cell.6,7 Their sequence and physicochemical properties endow them with selectivity towards specific organisms8,9 or even cell types,2,4,5 either by specific interaction with receptors,2,10 differential affinity to membrane compositions,9 or an activatable mechanism.5 Furthermore, there are some cell penetrating peptides that show affinity for subcellular structures,11 providing an additional layer of specificity.However, despite all these advantages, peptide-based compounds present limitations that may hinder their widespread application. For instance, they are prone to enzymatic hydrolysis and typically have short half-lives (from minutes to hours) in biological media.12–14 In addition, the flexibility of the backbone can also lead to unwanted interactions due to changes in the orientation of the functional groups.15 In the last few decades, there has been interest in addressing these limitations through the use of peptidomimetics, peptide analogues that present structural modifications to improve stability, bioavailability or selectivity.15–18 For instance, peptoids retain a peptide backbone but feature side chains attached to the amide nitrogen rather than to the α-carbon,12 while azapeptides replace the α-carbon by nitrogen.19 Alternative scaffolds include further modifications of the backbone, such as the inclusion of triazole rings,12,20 the insertion of D-amino acids,21 or even the complete substitution of the peptide backbone by another molecular scaffold.22 Despite key structural differences, peptidomimetic scaffolds are emerging as valuable tools in biomedical research, with applications in cancer treatment,23 antimicrobial drug development,24,25 protein–protein interaction modulation,26 and tissue engineering.27,28
Efficient targeting of intracellular structures or organelles constitutes a key goal for synthetic carriers, with peptidomimetics offering potential advantages in this regard. Mitochondria not only are important for energy production in the cell but also play a central role in a wide range of metabolic and signalling cellular pathways.29–32 Thus, they are highly related to numerous human pathologies, including cancer,33 mitochondrial diabetes,34 cardiovascular diseases,35 and neurodegenerative disorders like Alzheimer's and Parkinson's diseases.29,36 Recently, the mitochondrion has become a highly sought-after target for the treatment of these conditions, either by the targeted delivery of therapeutics12 or by exploiting the potential of gene editing technology to specifically modify the mitochondrial genome.37,38 Although reaching this organelle can be challenging due to its complex structure and particular physiology,12 mitochondrion-targeted peptides and other related scaffolds have been developed and validated using different techniques.12,20,21,39–43
Recently, a group of hybrid γ,γ-peptidomimetic amphiphiles has been introduced, which are easily prepared via solid phase peptide synthesis with suitable building blocks.44–46 The combination of a rigid hydrophobic cyclobutane residue and a functionalised γ-amino-L-proline47 introduces conformational constraints in the peptide backbone that increase their stability and promote the formation of stable secondary structures, an aspect that has been associated with improved uptake in many cell penetrating peptides.13,44,48–51 In addition, the derivatisation of the Nα position of the proline with a guanidinium group provides a cationic charge optimal for cell internalisation.44,46,48,52,53 Intriguingly, some of these peptidomimetics have recently shown excellent biocompatibility with mammalian cells and also interesting delivery capacity of cancer drugs (e.g. doxorubicin) into the protozoan parasite Leishmania sp.44
Here, we report the synthesis and characterisation of a hybrid tetradecameric γ,γ-peptidomimetic incorporating a cyclobutane-containing amino acid coupled in alternation with a guanidinium-functionalised γ-amino-L-proline residue and equipped with a ratiometric probe, γ-SCC (Fig. 1(A)). This peptide surrogate is used as an effective membrane-penetrating probe with excellent mitochondria targeting capabilities. Our findings reveal that this peptidomimetic effectively crosses the plasma membrane by a combination of direct translocation and endocytic mechanisms, followed by selective mitochondria targeting guided by the characteristic membrane potential of this organelle. Remarkable for its high stability, this bioorthogonal peptide resists enzymatic hydrolysis in the presence of proteases, enabling the visualisation of mitochondrial dynamic processes and the monitoring of the organelle pH. This study underscores the potential of peptidomimetic designs as new tools for membrane transport and mitochondrial tracking as well as their broad applicability in biological studies.
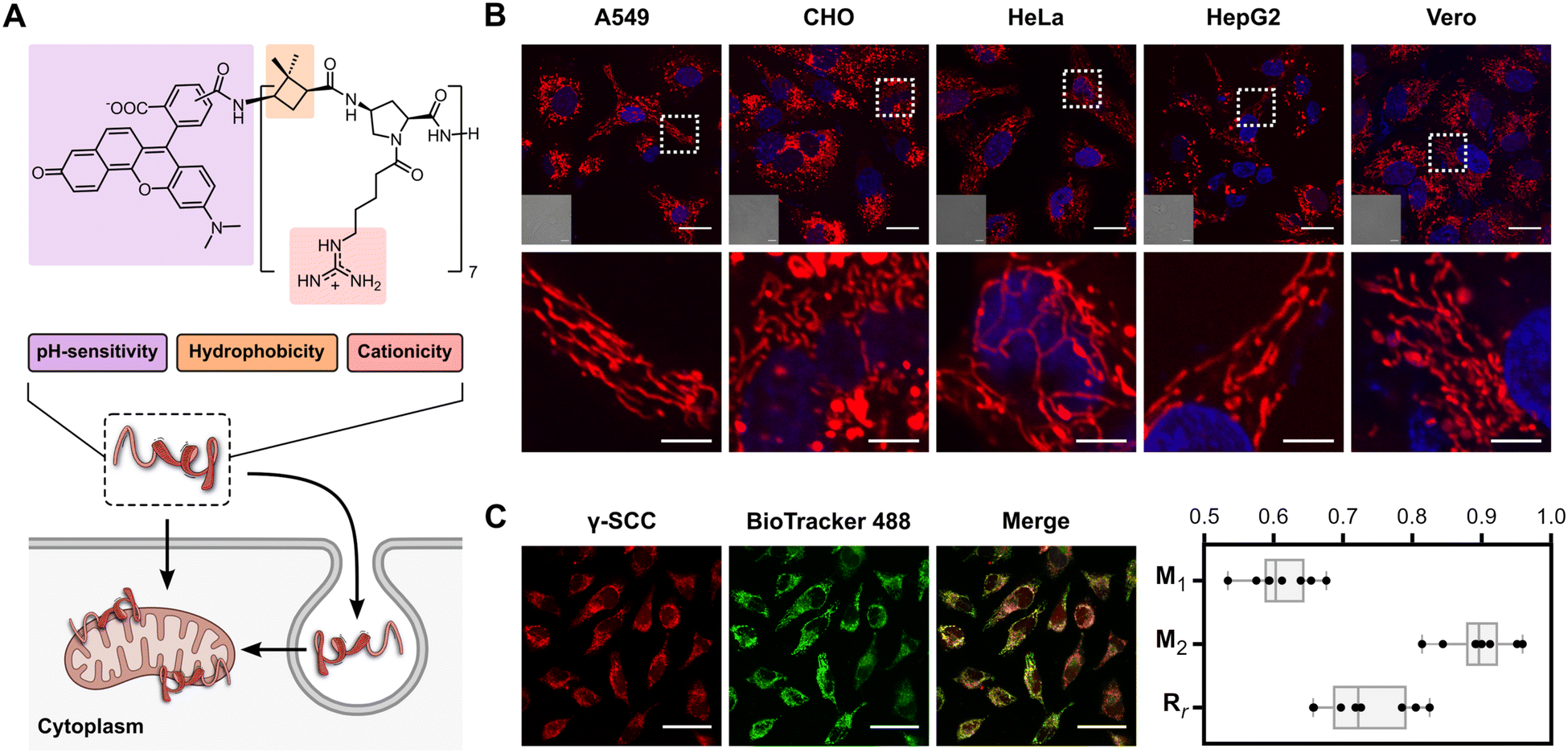 | ||
| Fig. 1 Peptidomimetic structure and mitochondria targeting. (A) Structure of γ-SCC and schematic representation of cellular internalisation and mitochondrial targeting. (B) Confocal microscopy images of γ-SCC intracellular distribution (in red) in various cell lines. Cells were incubated with the 75 μM peptide for 1 h, then samples were replaced, and cells were incubated in complete DMEM for 6 h. A representative cellular region is magnified in the bottom row. Nuclei were stained with Hoechst (in blue), and differential interference contrast (DIC) images are provided in insets. The scale bar is 20 μm (5 μm for the magnified images). See also Fig. S13 (ESI†). (C) Analysis of the peptide (25 μM, in red) colocalization with mitochondria using the BioTracker 488 Green Mitochondria Dye (in green). The scale bar is 50 μm. Manders’ overlap coefficients (M1 and M2) and Pearson's coefficient (Rr) were calculated from a set of 8 independent images (see Fig. S14, ESI†), with the obtained values represented as box plots. Whiskers indicate the total range of values within each sample. M1 refers to the fraction of γ-SCC overlapping BioTracker 488 Green, and M2 refers to the fraction of BioTracker 488 Green overlapping γ-SCC. | ||
Results and discussion
Peptide synthesis and intracellular distribution
The γ,γ-peptidomimetic tetradecameric hybrid oligomer, named γ-CC, is constructed by alternating non-natural hydrophobic units, specifically (1S,3R)-3-amino-2,2-dimethylcyclobutane-1-carboxylic acid ((1S,3R)-γ-CBAA) and cationic domains, featuring a cis-γ-amino-L-proline unit functionalised with a guanidinium group connected to the Nα position through a five-carbon spacer (Fig. 1(A)). The synthesis of such a peptide scaffold was conducted via solid phase peptide synthesis using a standard Fmoc/Boc strategy (see the ESI†). We fluorescently labelled the peptidomimetic at the N-terminus with the ratiometric 5(6)-carboxy-SNARF-1 probe (γ-SCC). The final γ-CC and γ-SCC peptides were purified by reverse phase HPLC and characterised by mass spectrometry (Fig. S11, ESI†).We next examined the potential plasma membrane translocation and intracellular distribution of the fluorescently labelled peptide using confocal microscopy in a broad range of different cell lines including A549 (human lung carcinoma), CHO (Chinese hamster ovary cells), HeLa (human cervical carcinoma), HepG2 (human hepatoblastoma), and Vero (African green monkey kidney cells). Remarkably, in all these different cell lines, the peptide was efficiently internalised and successfully stained an elongated network throughout the cytoplasm, resembling mitochondria (Fig. 1(B) and Fig. S13, ESI†). To confirm its localization within this organelle, colocalization assays were conducted with BioTracker 488 Green Mitochondria Dye (Fig. 1(C) and Fig. S14, ESI†). Manders' (M1, M2) and Pearson's (Rr) correlation coefficients showed a very strong association with mitochondria (M1 = 0.61 ± 0.05, M2 = 0.90 ± 0.05, Rr = 0.73 ± 0.07, and n = 8 in all cases), comparable to the best known mitochondrial targeting peptides.21,40,43,54–59 Colocalization with other organelles was lower than in the case of the mitochondria (e.g. lysosome, endoplasmic reticulum and nucleus, Fig. S14–S17, ESI†). In addition, the accumulation of γ-SCC in the mitochondria could be already observed after just 1.5 h of incubation, with a diffuse staining of cytosol, nucleus and nucleolus decreasing over time (Fig. S18, ESI†).
Control experiments by incubating cells with only the SNARF probe, at peptide tested concentrations, confirmed low internalisation and a punctate cytoplasmic pattern compatible with endosomal entrapment (Fig. S19, ESI†), which supports that the peptide is responsible for mitochondrial accumulation. Additionally, MTT assays confirmed excellent cell viability for the peptide carrier at the employed concentration regime (Fig. S20, ESI†).
Intracellular uptake studies in the presence of endocytic inhibitors were conducted in HeLa cells to investigate the γ-SCC peptidomimetic internalisation mechanism (Fig. 2(A)). Inhibitors targeting dynamin-dependent (dynasore, Dyn) and clathrin-mediated endocytosis (chlorpromazine, CPZ) reduced the total peptide uptake, indicating a partial role of endocytic internalisation in this process. In contrast, the inhibition of macropinocytosis (5-(N-ethyl-N-isopropyl)-amiloride, EIPA) had no impact on uptake levels. The partial contribution of energy-dependent pathways is also shown by the reduction in the internalisation observed when cells were treated with metabolic inhibitors that deplete intracellular ATP levels (sodium azide and 2-deoxy-D-glucose, NaN3/DOG).60 However, the persistence of around half of the intracellular peptide signal, even in the complete absence of energy-dependent pathways, suggests that the γ-SCC peptidomimetic can directly translocate across the plasma membrane into the cytosol of cells. The reduction of uptake after membrane depolarisation induced by the use of a high potassium buffer (K+PBS) indicates that, as for most cationic carriers,61 the membrane potential across the plasma membrane drives peptide translocation into the cell. Altogether, these findings suggest that the peptide enters into living cells through the combination of an energy- and endocytic-dependent process and a direct cytosol translocation mechanism driven by the negative plasma membrane potential. Importantly, as reflected by the mitochondria labelling and the lower punctate fluorescent patterns observed in confocal micrographs (Fig. 1(B)), the γ-SCC peptidomimetic also showed a suitable capacity to escape from potential endosomal compartments that could have been formed after the energy-dependent endocytic uptake.
 | ||
| Fig. 2 Internalisation and mitochondria-targeting mechanisms. (A) Uptake mechanism. HeLa cells were incubated with the peptide (50 μM) in the presence of different cellular internalisation inhibitors (10 mM NaN3/10 mM 2-DOG, 80 μM Dyn, 30 μM CPZ, 50 μM EIPA, or K+PBS), and the uptake was quantified by flow cytometry. Data were presented as mean and standard deviation. (B) Effect of mitochondrial depolarisation. HeLa cells were incubated with either 50 nM MitoTracker™ Red CMXRos, 200 nM TMRE or the 50 μM peptide, after a 5 min preincubation in the absence (top row) or presence of 50 μM of the protonophore CCCP (bottom row). The scale bar is 30 μm. See also Fig. S21 and S22 (ESI†). | ||
Numerous mitochondria-targeting probes, including tetramethylrhodamine ethyl ester (TMRE) or MitoTracker™ probes, are cationic to take advantage of mitochondria's negative membrane potential (150 to 180 mV, with the matrix being negative relative to the intermembrane space). To validate the role of the mitochondrial membrane potential in the accumulation of this peptide, we employed the mitochondrial uncoupler and protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP).62 HeLa cells were treated with CCCP for 5 min before staining with either the peptide, TMRE or MitoTracker™ Red CMXRos. As shown in Fig. 2(B), there was a strong reduction in mitochondrial staining for the three compounds upon incubation with the uncoupler. The observed behaviour of the control probes aligns with previously reported literature,63–65 suggesting that the peptide is electrostatically driven to mitochondria in a membrane potential-dependent manner. Studies in the presence of organelle markers confirmed that, in the absence of mitochondrial potential, the peptide is distributed throughout the cytosol and the nucleus, with a reduced accumulation in the mitochondria and an increased accumulation in the nucleus. The concentration of the signal in the nucleolus was also observed, similar to previous observations for other guanidinium-rich peptides66 (Fig. 2(B) and Fig. S21, S22, ESI†).
Peptide stability
To evaluate the resistance of the peptide to enzymatic degradation, we examined its structural integrity after incubation in the presence of a peptidase, trypsin (Fig. 3(A) and Fig. S23, ESI†). A non-enzymatically degradable internal standard (acetamidobenzoic acid, ABA) was added to the samples as an internal control. Two conventional peptides containing cationic residues, the cell-penetrating peptide R8 and the cytotoxic peptide KLAK,67 were used as controls for trypsin activity. Control peptides exhibited immediate degradation upon enzyme addition, evidenced by the complete disappearance of their peaks and the emergence of low retention time peaks corresponding to peptide fragments after 5 min of incubation. In contrast, γ-CC maintained its original peak even after 24 h of incubation with the enzyme. These results confirm the peptide resistance against enzymatic degradation. Encouraged by these findings, we next investigated whether the peptide maintained its bioavailability and structural integrity in the presence of fetal bovine serum (FBS). Serum contains several peptidases and proteases that can degrade peptides, as well as proteins that may hinder peptide–cell interaction.68,69 We investigated the effect of 10% FBS during the incubation of cells with γ-SCC. Confocal microscopy images revealed efficient mitochondrial accumulation (Fig. 3(B) and Fig. S24A, ESI†), similar to the one observed under no serum conditions (Fig. 1(B) and Fig. S13, ESI†), providing robust evidence of the peptide's activity in the presence of serum. Subsequently, the γ-SCC peptide was dissolved in complete DMEM (10% FBS) and subjected to a 7-day incubation at 37 °C, after which we analysed its targeting performance (Fig. 3(B) and Fig. S24B, ESI†). Remarkably, even after one week of incubation, the peptide retained its activity and accumulated in the mitochondria. This stability is likely attributed to the presence of non-natural amino acids. Unlike conventional peptides, which often have short half-lives in serum due to the presence of multiple proteases, ranging from minutes to hours,13,14γ-SCC displayed prolonged resistance against enzymatic hydrolysis, presenting opportunities for applications that require sustained integrity in biological environments.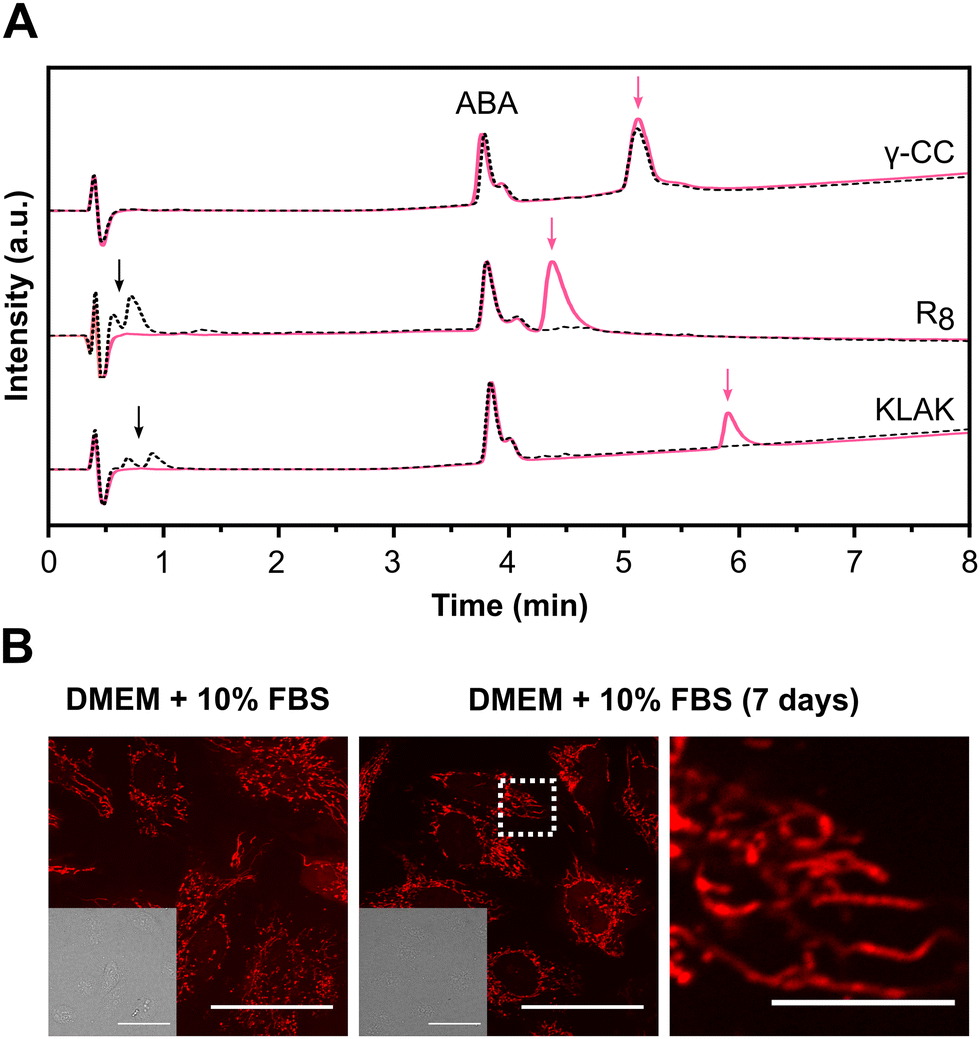 | ||
| Fig. 3 Evaluation of peptide stability against enzymatic degradation. (A) γ-CC and control peptides, R8 and KLAK, normalised chromatograms before (pink lines) and after (black dotted lines) incubation with trypsin (24 h incubation for γ-CC, 5 min for control peptides). Pink arrows indicate intact peptide peaks; black arrows indicate fragmented peptides resulting from enzymatic activity. Internal standard ABA is present in all chromatograms and was used as a reference for normalisation. See also Fig. S23 (ESI†). (B) Peptide intracellular distribution after incubation with cells in the presence of 10% FBS or after preincubation of the peptide in complete DMEM (10% FBS) for 7 days before addition to the cells (γ-SCC concentration: 25 μM). A representative cellular region is magnified (right). Differential interference contrast (DIC) images are provided in insets. The scale bar is 50 μm (10 μm for the magnified image). See also Fig. S24 (ESI†). | ||
Tracking mitochondria dynamics and pH
The simple labelling protocol and the robust staining of the developed peptidomimetic open avenues for multiple applications in mitochondrial research. Two distinct applications have been explored, highlighting the versatility and utility of this novel mitochondria-targeting probe.Mitochondria exhibit a remarkably dynamic nature, continuously altering their shape and size through intricate fusion and fission events, while being actively transported to specific subcellular locations.70 These dynamic processes play crucial roles in mammalian development, apoptosis, and various diseases, underscoring the importance of real-time tracking methodologies.70 In this context, our labelled peptide enabled real-time tracking of mitochondrial dynamics within living cells. HeLa cells were incubated with the peptide and visualised over time using confocal microscopy. Mitochondrial images were acquired at 5-second intervals, forming videos that captured dynamic episodes, including fission events (Fig. 4(A) and Video S1, ESI†), as well as mitochondrial movement throughout the cytoplasm (Fig. 4(B) and Video S2, ESI†). Mitochondrial dynamics studies often require frequent image acquisition over extended time intervals, and γ-SCC exhibited excellent mitochondrial residence and resistance to photobleaching, maintaining sustained fluorescence under demanding imaging conditions, with performance comparable to that of the most commonly used mitochondria-targeting fluorophores (Fig. S25 and Videos S3–S5, ESI†).
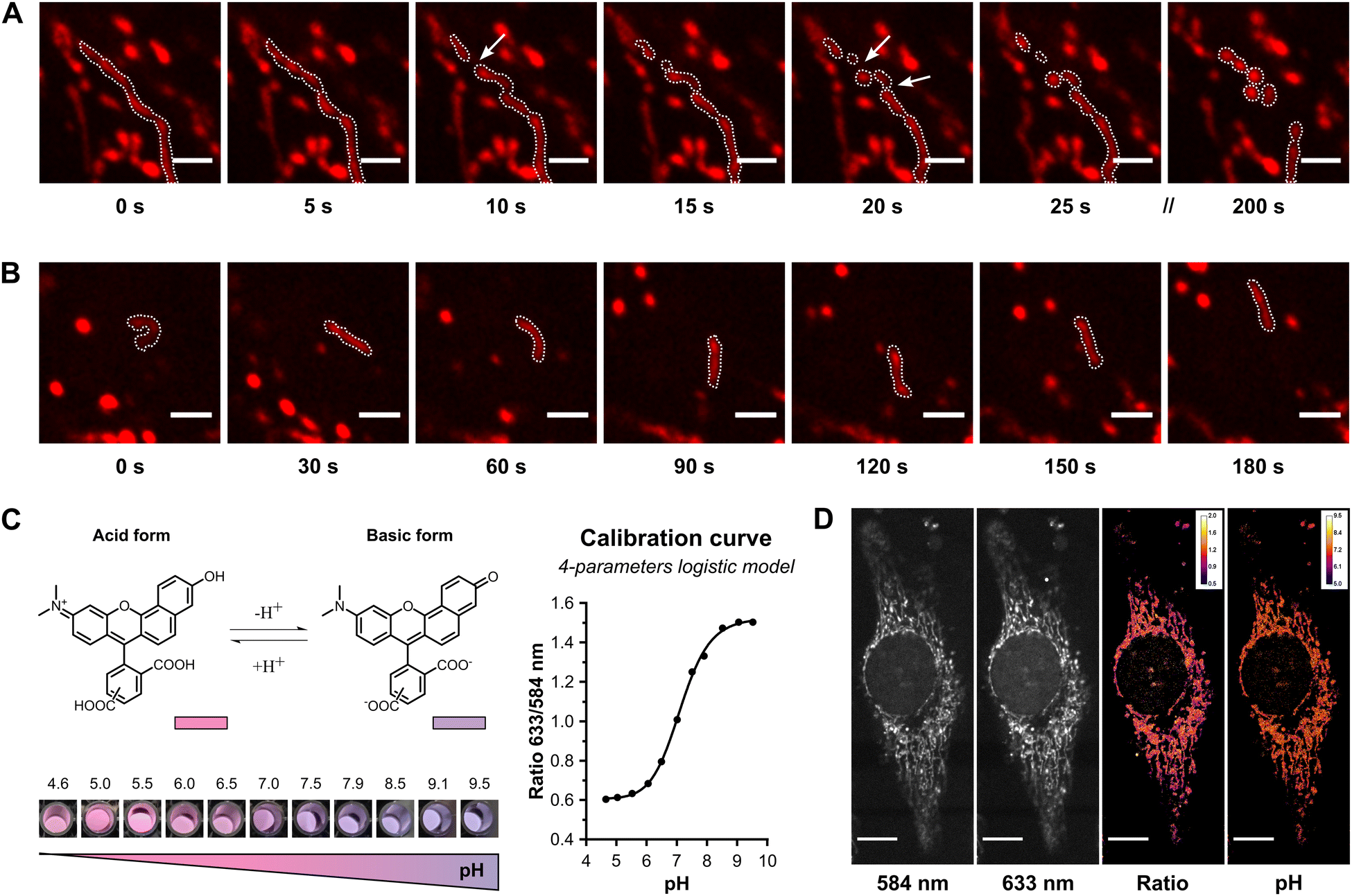 | ||
| Fig. 4 Real-time tracking of mitochondria dynamics and pH monitoring. (A) Confocal microscopy images of a mitochondria fission event over a period of 200 s, with images acquired at 5-second intervals. Arrows indicate several fission points in the original elongated structure. (B) Observation of the mitochondria movement over a period of 180 s. For A and B, the scale bars are 2 μm. Cells were labelled with 25 μM γ-SCC. (C) The ratiometric SNARF probe exhibits a large emission spectral shift in response to pH variations, with a maximum emission peak at 584 nm for the acidic form (pink colour) and at 633 nm for the basic form (purple colour). To correlate the emission properties with pH, a calibration curve was generated by plotting the emission ratio at wavelengths of 633 and 584 nm against pH, fitting the data to a 4-parameter logistic model. (D) Image analysis for measuring mitochondrial pH. The images of a HeLa cell incubated with 25 μM γ-SCC at the two maximum emissions, the ratio between them, and the calculated pH image applying the calibration curve are shown. The scale bar is 10 μm. See also Fig. S27 (ESI†). | ||
Mitochondrial pH is a vital parameter determining the rate of essential cellular functions, such as metabolism, membrane potential, apoptosis, and ROS production.71,72 The mitochondrial matrix presents a slightly basic pH, typically between 7.5 and 8.2, due to the pumping of protons to the intermembrane space.73 This value can be altered using metabolic stressors and other stimuli, decreasing in the presence of mitochondrial uncouplers,74,75 proton pump inhibitors,74 redox species75,76 or starvation,74,77 and increasing in response to ATP synthase inhibitors such as oligomycin.74,78 Temperature can also impact mitochondrial pH, with a pH close to 8 at 40 °C that decreases to about 7.3 when the temperature changes.79 Furthermore, mitochondrial pH can also vary among different regions from the same cell,73,80 with some authors reporting the coexistence of regions with pH values of 8 with values as low as 4.63.77 Thus, analysing mitochondrial pH serves as a proxy for both mitochondrial and cellular functions. The selected SNARF probe, acting as a dual emission system, exhibits a large emission spectral shift in response to pH variations, making it suitable for monitoring pH. Previous attempts to measure mitochondrial pH using SNARF required coincubation with mitochondria-specific dyes, such as DiOC6 or MitoTracker™ Green, to compensate for the lack of inherent mitochondrial targeting, or a careful optimisation of the selective loading based on the efflux of the cytosolic dye by anion transporters.81–83 In contrast, our system specifically targets this ratiometric probe to mitochondria, presenting a streamlined and efficient approach for consistently measuring mitochondrial pH. To test this, HeLa cells incubated with the labelled peptide were imaged at the emission maxima of the acidic phenol form (584 nm) and the basic phenolate form (633 nm). To determine pH values at each image point, a calibration curve was generated with γ-SCC diluted in buffer and acquired under the same conditions (Fig. 4(C) and Fig. S26, ESI†). Following this calibration, the ratio at each pixel of the cell micrographs was calculated, translating it into a pH value by solving the obtained 4-parameter logistic model (Fig. 4(D) and Fig. S27, ESI†). Our system determined a median mitochondrial pH value of approximately 7.2, which is in range with mitochondrial pH values.73,84–86
Experimental
Synthesis and characterisation
Non-natural amino acids, Boc-protected guanidinylated Fmoc-protected cis-γ-amino-L-proline amino acid VI and Fmoc-protected (1S,3R)-γ-CBAA were synthesised as detailed in the ESI.† These amino acids were characterised by polarimetry, melting point determination, IR spectroscopy, 1H-NMR and 13C-NMR spectroscopy, and HRMS. All peptides were synthesised using manual Fmoc solid phase peptide synthesis using a Rink Amide resin. After synthesis, the peptides were purified by RP-HPLC and characterised by analytical HPLC, as well as MS. Detailed synthesis protocols and characterisation results are provided in the ESI.†Cell culture
A549, CHO, HeLa, HepG2, and Vero cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; 4500 mg l−1 glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate) supplemented with 10% FBS and 1% penicillin–streptomycin–glutamine mix (Fisher) (complete DMEM). They were maintained at 37 °C, 5% CO2, and 95% humidity in an INCO 108 incubator (Memmert). The day before an experiment, cells were seeded on the corresponding 96-well plate at a density of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per ml (100 μl per well).
000 cells per ml (100 μl per well).
Intracellular distribution and colocalization
To observe the intracellular distribution of the peptide in different cell lines, cells were incubated with γ-SCC at final concentrations of 25, 50, and 75 μM in DMEM (85 μl per well) for 1 h. Afterwards, the peptide was removed and cells were washed prior to incubation in complete DMEM for 6 h. Alternatively, mitochondria were stained with 50 nM MitoTracker™ Red CMXRos for 25 min. Nuclei were stained with 1 μM Hoechst 33342 for 20 min at the end of the incubation.For colocalization experiments, HeLa cells were treated with 25 μM γ-SCC in DMEM (85 μl per well) for 1 h. Afterwards, cells were washed and incubated in complete DMEM for 6 h and, subsequently, mitochondria were stained with 100 nM BioTracker 488 Green Mitochondria Dye, the ER with 2 μM ER-Tracker Blue-White DPX, lysosomes with 75 nM LysoTracker Green DND-26 or nuclei with 1 μM Hoechst for 20 min.
In all cases, imaging was performed using Fusion software (Andor) with a Dragonfly spinning disk confocal microscope mounted on a Nikon Eclipse Ti-E, equipped with an Andor Sona sCMOS digital camera. Hoechst was excited with the 405 nm laser and the emission was recorded at 450/50 nm; for γ-SCC and MitoTracker™ Red CMXRos, an excitation of 561 nm and an emission of 620/60 nm were used; BioTracker 488 Green and LysoTracker Green DND-26 were excited at 488 nm and the emission was collected at 525/50 nm; Hoechst was excited at 405 nm and the emission was collected at 450/50 nm; the ER-tracker was excited at 405 nm and the emission was collected at 525/50 nm. Images were processed with ImageJ v1.52b.87 Colocalization coefficients were calculated to assess the degree of colocalization between γ-SCC and the organelles. These colocalization parameters were calculated from several representative images (Fig. S14 and S15, ESI†) with ImageJ using the plugin BIOP JACoP.87–89
Internalisation and targeting mechanisms
To investigate the internalisation mechanism of the peptide, HeLa cells were pre-treated with K+PBS (140 mM KCl, 30 mM Na2HPO4, 1.76 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgCl2, pH 7.4) or, alternatively, with endocytic inhibitors, including a mixture of sodium azide and 2-deoxy-D-glucose (10 mM each, in HKR), dynasore (80 μM), EIPA (50 μM), and chlorpromazine (30 μM), for 30 min in DMEM unless otherwise specified. Then, γ-SCC (50 μM) was added in the presence of the same inhibitors for 1 h. Cells were subsequently washed with PBS and trypsinised, and trypsin was neutralised with PBS containing 2% FBS and 5 mM EDTA. SNARF fluorescence was measured using a Guava easyCyte BG HT flow cytometer, exciting with a green laser (532 nm) and collecting the emission at 620/52 nm (Orange-G channel). Data were analysed using InCyte v. 3.2 (GuavaSoft, Millipore). Cells with typical FSC/SSC values were selected and the median fluorescence intensity was extracted for each sample. Values were normalised to the uptake of the γ-SCC control (100%) after blank subtraction (untreated cells).To study the mitochondria-targeting mechanism, HeLa cells were pre-treated with 50 μM CCCP for 5 min to ensure mitochondrial membrane potential depolarisation. Afterwards, this solution was replaced by γ-SCC (50 μM), TMRE (200 nM) or MitoTracker™ Red CMXRos (50 nM) diluted in DMEM containing 50 μM CCCP (50 μl per well) and incubated for 1 h in the case of the peptide and 25 min in the case of the control probes. Cells were immediately imaged using confocal microscopy.
Peptide stability
The resistance of γ-CC to enzymatic hydrolysis was evaluated by incubating the peptide in the presence of the protease trypsin. As controls, two cationic peptides, R8 and KLAK, were tested parallelly. The peptides γ-CC (50 μM), R8 (500 μM), and KLAK (100 μM) were dissolved in PBS containing 100 μM 4-acetamidobenzoic acid (ABA), an internal standard not degradable by trypsin and, before adding the protease, peptides were analysed by RP-HPLC. Trypsin was then added to the peptide solutions at a concentration ratio of 50:1 (peptide:enzyme). Following 5 min of incubation, the control peptides R8 and KLAK were again analysed by RP-HPLC, while γ-CC was analysed after 24 h of incubation. Absorbance data at 220 nm were normalised to the maximum intensity value of the ABA peak for plotting.
Additionally, to assess the activity and stability of γ-SCC in the presence of fetal bovine serum (FBS), γ-SCC was diluted (25 μM) in complete DMEM containing 10% FBS. Then, HeLa cells were incubated with the peptide solution for 1 h. In parallel, an aliquot of γ-SCC was stored in complete DMEM for 7 days at 37 °C before being added to the cells under the same conditions. After incubation, cells were washed and incubated in complete DMEM for an additional 6 h before imaging by confocal microscopy.
Measuring mitochondrial pH
For the calibration curve, γ-SCC was dissolved at 50 μM in a buffer (10 mM MES, 10 mM HEPES, 20 mM glucose, 1 mM CaCl2, 1 mM MgCl2, 135 mM KCl, and 20 mM NaCl) adjusted to pH values from 4.6 to 9.5. Samples were imaged using a Leica Stellaris 8 FALCON confocal microscope (λex = 561 nm; λem = 584/30 nm and 633/30 nm). Six z-plane images were acquired per pH and the pixel intensity ratio of 633 nm to 584 nm was calculated for each plane, averaged per pH, and plotted using GraphPad Prism 6 to create a calibration curve. Then, HeLa cells were incubated with 25 μM γ-SCC in DMEM (85 μl per well) for 1 h. After washing and 6 h of incubation in complete DMEM, cells were imaged using a Leica Stellaris 8 FALCON confocal microscope. Images were processed with ImageJ v1.52b,87 dividing the 633 nm by 584 nm images to create ratio images, which were converted to pH values using the calibration curve. A mask created with the default threshold was applied to the resulting pH images to exclude the values in the background regions, and the median mitochondrial pH was calculated from the image intensities.Conclusions
Mitochondria-targeting carriers typically contain sequences with both cationic (e.g., arginine and lysine) and hydrophobic (e.g., phenylalanine and cyclohexylalanine) residues.40 The cationic residues aid to direct the peptide to the negatively charged mitochondria, while the hydrophobic residues facilitate its transport across mitochondrial hydrophobic membranes.41,90 The amphiphilic peptidomimetic γ-SCC, characterised by a non-canonical hybrid peptide backbone, has proven to be a promising and biocompatible candidate for targeted mitochondrial applications. The amphiphilic design, which incorporates alternating hydrophobic and cationic domains, has demonstrated a robust synergy between its structure and functionality, with a behaviour similar to other mitochondrial targeted peptides,40 despite the differences in the peptide backbone structure. Notably, the integration of non-natural amino acids has provided the peptide with remarkable stability. This is evidenced by its resistance to enzymatic degradation and bioavailability in the presence of trypsin or serum, showcasing its potential for extended applications. The mechanism of internalisation involves an energy-dependent endocytic pathway and direct translocation, eventually accumulating within mitochondria due to their characteristic membrane potential. Exploring the versatility of our system, preliminary applications for real-time tracking of mitochondrial dynamics and pH monitoring have shown promising results. These applications highlight the potential of our system in advancing mitochondrial research methodologies. In the broader context, this study highlights the biocompatibility and stability of γ,γ-peptidomimetics, and their potential to overcome the inherent limitations associated with conventional peptides. By combining the principles of biomimicry and rational design, γ,γ-peptidomimetics offer a powerful approach for developing innovative therapeutics and molecular tools capable of addressing challenging biological targets and diseases.Data availability
Data supporting this article have been included as part of the Supplementary information. Raw data of the main figures are available at Zenodo at https://doi.org/10.5281/zenodo.14617753.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M. thanks the Spanish AEI (PCI2019-103400, PID2020-117143RB-I00, and PID2023-152181OB-I00), the Xunta de Galicia (Centro singular de investigación de Galicia accreditation 2023–2027, ED431G 2023/03, and ED431F 2023/12, the Oportunius Program (GAIN)), and the European Regional Development Fund (ERDF). Y. F.-C. and P. F.-G. thank AEI (FPU21/04747; PRE2021-096972) and M. P. and I. M.-M. thank Xunta de Galicia (ED481A-2017/142 and ED481A-2024-049) for their predoctoral fellowships. D. T.-M. thanks the Generalitat de Catalunya for his predoctoral FI-SDUR fellowship (2020 FISDUR 00055). O. I. and D. T.-M. are grateful for the financial support of the Spanish AEI (PID2022-139826OB-I00) and the Generalitat de Catalunya (2020 FI-SDUR 00055). M. R. acknowledges support from CIBER BBN(CB06/01/0074) and the Generalitat de Catalunya (2021SGR00230). All authors thank Dr Irene Lostalé-Seijo for her assistance and comments. The authors would like to thank Rebeca Menaya-Vargas for her technical assistance in the maintenance of cell lines. The table of contents and Fig. 1(A) contain elements created with https://BioRender.com.Notes and references
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- N. Svensen, J. G. A. Walton and M. Bradley, Trends Pharmacol. Sci., 2012, 33, 186–192 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- B. M. Cooper, J. Iegre, D. H. O’Donovan, M. Ölwegård Halvarsson and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1480–1494 RSC.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Adv. Drug Delivery Rev., 2017, 110–111, 112–126 CrossRef CAS PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed.
- Cell penetrating peptides: methods and protocols, ed. Ü. Langel, Humana Press, New York, NY, 3rd edn, 2022 Search PubMed.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- M. Lei, A. Jayaraman, J. A. Van Deventer and K. Lee, Annu. Rev. Biomed. Eng., 2021, 23, 339–357 CrossRef CAS PubMed.
- P. Lingasamy and T. Teesalu, in Bio-Nanomedicine for Cancer Therapy, ed. F. Fontana and H. A. Santos, Springer International Publishing, Cham, 2021, vol. 1295, pp. 29–48 Search PubMed.
- C. P. Cerrato and Ü. Langel, Expert Opin. Drug Delivery, 2022, 19, 133–146 CrossRef CAS PubMed.
- S. Kim, H. Y. Nam, J. Lee and J. Seo, Biochemistry, 2020, 59, 270–284 CrossRef CAS PubMed.
- M. Pazo, G. Salluce, I. Lostalé-Seijo, M. Juanes, F. Gonzalez, R. Garcia-Fandiño and J. Montenegro, RSC Chem. Biol., 2021, 2, 503–512 RSC.
- M. Werle and A. Bernkop-Schnürch, Amino Acids, 2006, 30, 351–367 CrossRef CAS PubMed.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454–462 CrossRef CAS PubMed.
- Y.-D. Wu and S. Gellman, Acc. Chem. Res., 2008, 41, 1231–1232 CrossRef CAS PubMed.
- I. Avan, C. D. Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43, 3575 RSC.
- D. R. Goldsmith and C. M. Perry, Drugs, 2003, 63, 1679–1693 CrossRef CAS PubMed.
- D. Althuon, F. Rönicke, D. Fürniss, J. Quan, I. Wellhöfer, N. Jung, U. Schepers and S. Bräse, Org. Biomol. Chem., 2015, 13, 4226–4230 RSC.
- T. Zhao, X. Liu, S. Singh, X. Liu, Y. Zhang, J. Sawada, M. Komatsu and K. D. Belfield, Bioconjugate Chem., 2019, 30, 2312–2316 CrossRef CAS PubMed.
- R. F. Hirschmann, K. C. Nicolaou, A. R. Angeles, J. S. Chen and A. B. Smith, Acc. Chem. Res., 2009, 42, 1511–1520 CrossRef CAS PubMed.
- M. M. Gomari, S. Abkhiz, T. G. Pour, E. Lotfi, N. Rostami, F. N. Monfared, B. Ghobari, M. Mosavi, B. Alipour and N. V. Dokholyan, Mol. Med., 2022, 28, 146 CAS.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- N. Molchanova, P. R. Hansen and H. Franzyk, Molecules, 2017, 22, 1430 CrossRef PubMed.
- A. D. Cunningham, N. Qvit and D. Mochly-Rosen, Curr. Opin. Struct. Biol., 2017, 44, 59–66 CrossRef CAS PubMed.
- K. G. Sreejalekshmi and P. D. Nair, J. Biomed. Mater. Res., Part A, 2011, 96A, 477–491 CrossRef CAS PubMed.
- L. Oliver-Cervelló, H. Martin-Gómez, C. Gonzalez-Garcia, M. Salmeron-Sanchez, M.-P. Ginebra and C. Mas-Moruno, Front. Bioeng. Biotechnol., 2023, 11, 1192436 CrossRef PubMed.
- G. S. Gorman, P. F. Chinnery, S. DiMauro, M. Hirano, Y. Koga, R. McFarland, A. Suomalainen, D. R. Thorburn, M. Zeviani and D. M. Turnbull, Nat. Rev. Dis. Primer, 2016, 2, 16080 CrossRef PubMed.
- N. Sun, R. J. Youle and T. Finkel, Mol. Cell, 2016, 61, 654–666 CrossRef CAS PubMed.
- A. Dhir, S. Dhir, L. S. Borowski, L. Jimenez, M. Teitell, A. Rötig, Y. J. Crow, G. I. Rice, D. Duffy, C. Tamby, T. Nojima, A. Munnich, M. Schiff, C. R. de Almeida, J. Rehwinkel, A. Dziembowski, R. J. Szczesny and N. J. Proudfoot, Nature, 2018, 560, 238–242 CrossRef CAS PubMed.
- G. S. Ling, G. Crawford, N. Buang, I. Bartok, K. Tian, N. M. Thielens, I. Bally, J. A. Harker, P. G. Ashton-Rickardt, S. Rutschmann, J. Strid and M. Botto, Science, 2018, 360, 558–563 CrossRef CAS PubMed.
- D. C. Wallace, Nat. Rev. Cancer, 2012, 12, 685–698 CrossRef CAS PubMed.
- W. I. Sivitz and M. A. Yorek, Antioxid. Redox Signaling, 2010, 12, 537–577 CrossRef CAS PubMed.
- C. Lopez-Crisosto, C. Pennanen, C. Vasquez-Trincado, P. E. Morales, R. Bravo-Sagua, A. F. G. Quest, M. Chiong and S. Lavandero, Nat. Rev. Cardiol., 2017, 14, 342–360 CrossRef CAS PubMed.
- A. Federico, E. Cardaioli, P. Da Pozzo, P. Formichi, G. N. Gallus and E. Radi, J. Neurol. Sci., 2012, 322, 254–262 CrossRef CAS PubMed.
- P. A. Gammage, C. Viscomi, M.-L. Simard, A. S. H. Costa, E. Gaude, C. A. Powell, L. Van Haute, B. J. McCann, P. Rebelo-Guiomar, R. Cerutti, L. Zhang, E. J. Rebar, M. Zeviani, C. Frezza, J. B. Stewart and M. Minczuk, Nat. Med., 2018, 24, 1691–1695 CrossRef CAS PubMed.
- S. R. Bacman, S. L. Williams, M. Pinto, S. Peralta and C. T. Moraes, Nat. Med., 2013, 19, 1111–1113 CrossRef CAS PubMed.
- S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 CrossRef CAS PubMed.
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 CrossRef CAS PubMed.
- L. F. Yousif, K. M. Stewart and S. O. Kelley, ChemBioChem, 2009, 10, 1939–1950 CrossRef CAS PubMed.
- K. G. Asfaw, Q. Liu, J. Maisch, S. W. Münch, I. Wehl, S. Bräse, I. Bogeski, U. Schepers and P. Nick, Sci. Rep., 2019, 9, 9839 CrossRef CAS PubMed.
- H. Y. Nam, J.-A. Hong, J. Choi, S. Shin, S. K. Cho, J. Seo and J. Lee, Bioconjugate Chem., 2018, 29, 1669–1676 CrossRef CAS PubMed.
- O. Illa, J.-A. Olivares, N. Gaztelumendi, L. Martínez-Castro, J. Ospina, M.-Á. Abengozar, G. Sciortino, J.-D. Maréchal, C. Nogués, M. Royo, L. Rivas and R. M. Ortuño, Int. J. Mol. Sci., 2020, 21, 7502 CrossRef CAS PubMed.
- R. Gutiérrez-Abad, D. Carbajo, P. Nolis, C. Acosta-Silva, J. A. Cobos, O. Illa, M. Royo and R. M. Ortuño, Amino Acids, 2011, 41, 673–686 CrossRef PubMed.
- E. Gorrea, D. Carbajo, R. Gutiérrez-Abad, O. Illa, V. Branchadell, M. Royo and R. M. Ortuño, Org. Biomol. Chem., 2012, 10, 4050 RSC.
- J. Farrera-Sinfreu, L. Zaccaro, D. Vidal, X. Salvatella, E. Giralt, M. Pons, F. Albericio and M. Royo, J. Am. Chem. Soc., 2004, 126, 6048–6057 CrossRef CAS PubMed.
- O. Illa, J. Ospina, J.-E. Sánchez-Aparicio, X. Pulido, M. Á. Abengozar, N. Gaztelumendi, D. Carbajo, C. Nogués, L. Rivas, J.-D. Maréchal, M. Royo and R. M. Ortuño, Int. J. Mol. Sci., 2021, 22, 5092 CrossRef CAS PubMed.
- J. Farrera-Sinfreu, E. Giralt, S. Castel, F. Albericio and M. Royo, J. Am. Chem. Soc., 2005, 127, 9459–9468 CrossRef CAS PubMed.
- J. Fominaya, J. Bravo and A. Rebollo, Ther. Delivery, 2015, 6, 1171–1194 CrossRef CAS PubMed.
- S. Pujals and E. Giralt, Adv. Drug Delivery Rev., 2008, 60, 473–484 CrossRef CAS PubMed.
- G. Gasparini, E.-K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389–10402 RSC.
- M. Vazdar, J. Heyda, P. E. Mason, G. Tesei, C. Allolio, M. Lund and P. Jungwirth, Acc. Chem. Res., 2018, 51, 1455–1464 CrossRef CAS PubMed.
- Q. Xiao, X. Dong, F. Yang, S. Zhou, M. Xiang, L. Lou, S. Q. Yao and L. Gao, Chem. – Eur. J., 2021, 27, 14721–14729 CrossRef CAS PubMed.
- E. Bankell, X. Liu, M. Lundqvist, D. Svensson, K. Swärd, E. Sparr and B.-O. Nilsson, Biochem. Biophys. Rep., 2022, 29, 101192 CAS.
- B. Chen, J.-J. Hu, H. Ouyang, W. Zhang, J. Dai, L. Xu, F. Xia and X. Lou, Anal. Chem., 2023, 95, 12903–12912 CrossRef CAS PubMed.
- G. Appiah Kubi, Z. Qian, S. Amiar, A. Sahni, R. V. Stahelin and D. Pei, Angew. Chem., Int. Ed., 2018, 57, 17183–17188 CrossRef CAS PubMed.
- M. T. Jeena, L. Palanikumar, E. M. Go, I. Kim, M. G. Kang, S. Lee, S. Park, H. Choi, C. Kim, S.-M. Jin, S. C. Bae, H. W. Rhee, E. Lee, S. K. Kwak and J.-H. Ryu, Nat. Commun., 2017, 8, 26 CrossRef CAS PubMed.
- P. Chandra Saha, R. S. Das, T. Chatterjee, M. Bhattacharyya and S. Guha, Bioconjugate Chem., 2020, 31, 1301–1306 CrossRef CAS PubMed.
- E. D. Schwoebel, T. H. Ho and M. S. Moore, J. Cell Biol., 2002, 157, 963–974 CrossRef CAS PubMed.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS PubMed.
- P. G. Heytler and W. W. Prichard, Biochem. Biophys. Res. Commun., 1962, 7, 272–275 CrossRef CAS PubMed.
- D. L. Farkas, M. D. Wei, P. Febbroriello, J. H. Carson and L. M. Loew, Biophys. J., 1989, 56, 1053–1069 CrossRef CAS PubMed.
- M. Poot, Y. Z. Zhang, J. A. Krämer, K. S. Wells, L. J. Jones, D. K. Hanzel, A. G. Lugade, V. L. Singer and R. P. Haugland, J. Histochem. Cytochem., 1996, 44, 1363–1372 CrossRef CAS PubMed.
- M. I. Sánchez, Y. Vida, E. Pérez-Inestrosa, J. L. Mascareñas, M. E. Vázquez, A. Sugiura and J. Martínez-Costas, Sci. Rep., 2020, 10, 3528 CrossRef PubMed.
- R. M. Martin, G. Ter-Avetisyan, H. D. Herce, A. K. Ludwig, G. Lättig-Tünnemann and M. C. Cardoso, Nucleus, 2015, 6, 314–325 CrossRef CAS PubMed.
- M. M. Javadpour, M. M. Juban, W.-C. J. Lo, S. M. Bishop, J. B. Alberty, S. M. Cowell, C. L. Becker and M. L. McLaughlin, J. Med. Chem., 1996, 39, 3107–3113 CrossRef CAS PubMed.
- R. Böttger, R. Hoffmann and D. Knappe, PLoS One, 2017, 12, e0178943 CrossRef PubMed.
- X. Lai, J. Tang and M. E. H. ElSayed, Expert Opin. Drug Delivery, 2021, 16, 1467–1482 CrossRef CAS PubMed.
- S. A. Detmer and D. C. Chan, Nat. Rev. Mol. Cell Biol., 2007, 8, 870–879 CrossRef CAS PubMed.
- M. F. C. Abad, G. Di Benedetto, P. J. Magalhães, L. Filippin and T. Pozzan, J. Biol. Chem., 2004, 279, 11521–11529 CrossRef CAS PubMed.
- J. Aklima, T. Onojima, S. Kimura, K. Umiuchi, T. Shibata, Y. Kuraoka, Y. Oie, Y. Suganuma and Y. Ohta, Front. Cell Dev. Biol., 2021, 9, 692776 CrossRef PubMed.
- J. Santo-Domingo and N. Demaurex, J. Gen. Physiol., 2012, 139, 415–423 CrossRef CAS PubMed.
- L. T. Wilson, W. J. Tipping, C. Wetherill, Z. Henley, K. Faulds, D. Graham, S. P. Mackay and N. C. O. Tomkinson, Anal. Chem., 2021, 93, 12786–12792 CrossRef CAS PubMed.
- X. Liu, L. Wang, T. Bing, N. Zhang and D. Shangguan, ACS Appl. Bio Mater., 2019, 2, 1368–1375 CrossRef CAS PubMed.
- Y. Chen, C. Zhu, J. Cen, Y. Bai, W. He and Z. Guo, Chem. Sci., 2015, 6, 3187–3194 RSC.
- M. H. Lee, N. Park, C. Yi, J. H. Han, J. H. Hong, K. P. Kim, D. H. Kang, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 14136–14142 CrossRef CAS PubMed.
- S. Matsuyama, J. Llopis, Q. L. Deveraux, R. Y. Tsien and J. C. Reed, Nat. Cell Biol., 2000, 2, 318–325 CrossRef CAS PubMed.
- L. Cao, Z. Zhao, T. Zhang, X. Guo, S. Wang, S. Li, Y. Li and G. Yang, Chem. Commun., 2015, 51, 17324–17327 RSC.
- A. R. Sarkar, C. H. Heo, L. Xu, H. W. Lee, H. Y. Si, J. W. Byun and H. M. Kim, Chem. Sci., 2016, 7, 766–773 RSC.
- M. vandeVen, C. Balut, S. Baron, I. Smets, P. Steels and M. Ameloot, in Live Cell Imaging: Methods and Protocols, ed. D. B. Papkovsky, Humana Press, Totowa, NJ, 2010, pp. 275–309 Search PubMed.
- A. Takahashi, Y. Zhang, V. E. Centonze and B. Herman, Biotechniques, 2001, 30, 804–815 CrossRef CAS PubMed.
- K. Baysal, G. P. Brierley, S. Novgorodov and D. W. Jung, Arch. Biochem. Biophys., 1991, 291, 383–389 CrossRef CAS PubMed.
- D. Poburko, J. Santo-Domingo and N. Demaurex, J. Biol. Chem., 2011, 286, 11672–11684 CrossRef CAS PubMed.
- A. M. Porcelli, A. Ghelli, C. Zanna, P. Pinton, R. Rizzuto and M. Rugolo, Biochem. Biophys. Res. Commun., 2005, 326, 799–804 CrossRef CAS PubMed.
- M. Benčina, Sensors, 2013, 13, 16736–16758 CrossRef PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- S. Bolte and F. P. Cordelières, J. Microsc., 2006, 224, 213–232 CrossRef CAS PubMed.
- EPFL's BioImaging And Optics Platform (BIOP), BIOP JACoP https://github.com/BIOP/ijp-jacop-b.
- C. Ma, F. Xia and S. O. Kelley, Bioconjugate Chem., 2020, 31, 2650–2667 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01626b |
| This journal is © The Royal Society of Chemistry 2025 |