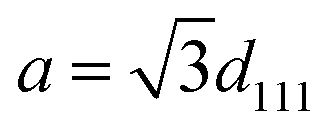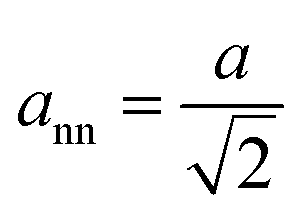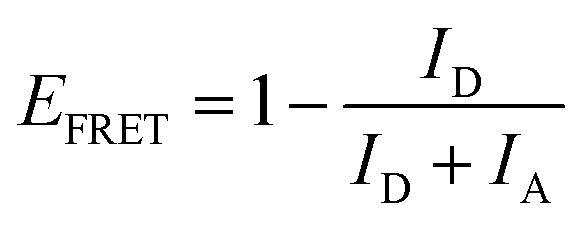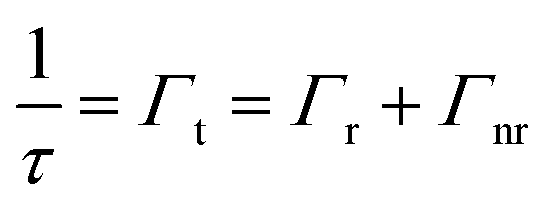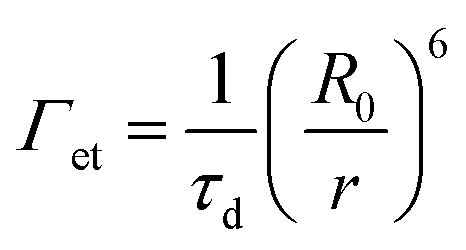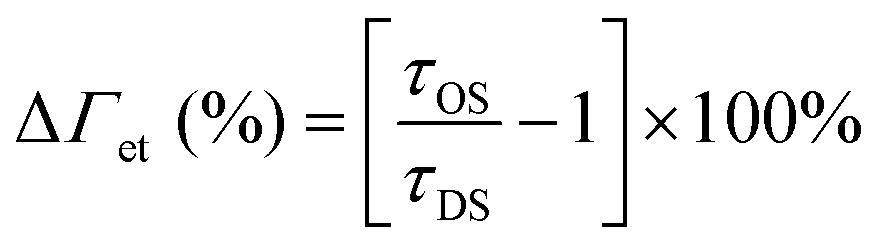DOI:
10.1039/D4TC03973D
(Paper)
J. Mater. Chem. C, 2025,
13, 1694-1703
The intersection of field-limited density of states and matter: nanophotonic control of fluorescence energy transfer†
Received
18th September 2024
, Accepted 20th November 2024
First published on 25th November 2024
Abstract
Understanding the unresolved connection between a structured environment and Förster resonance energy transfer (FRET) is critical in the realm of quantum light–matter interactions, especially for quantum technology applications. This crucial topic was explored by copolymerizing three emitters capable of energy transfer within two nanoparticle series (n1 and n2) that self-assembled into a crystalline colloidal array. Upon excitation, sequential energy transfer between the copolymerized derivatives of anthracene, naphthalimide, and rhodamine B within n1 and n2 resulted in emission spanning the visible spectrum. Nanophotonic control over the photoluminescence of n1 and n2 assembled in an ordered structure was demonstrated by red-shifting the partial photonic bandgap of the ordered structure through the emission spectra of the copolymerized emitters, which was achieved by dilution with deionized water. Nanophotonic manipulation of the energy transfer between the two FRET pairs copolymerized within n1 and n2 was observed, revealing insights in the context of light–matter interactions. Specifically, nanophotonic control over photoluminescence, energy transfer efficiency, and decay kinetics was demonstrated by strategic placement of the partial photonic bandgap.
1 Introduction
Förster resonance energy transfer (FRET) is a well-known nonradiative energy transfer process that occurs between two quantum emitters, an excited-state donor and a ground-state acceptor.1 FRET is a resonant dipole–dipole interaction and is, therefore, the dominant energy transfer mechanism when emitters are within nanometer proximity to each other. FRET is a vital mechanism in photosynthesis2 and is the basis of some photovoltaic,3,4 lighting,5 and biomedical sensing6 applications. Systems that exploit FRET can be controlled by engineering the spectral characteristics of the emitters or the relative orientations of their dipole moments;1,7 however, FRET control by a structured environment remains a highly debated and relevant topic8–20 with the potential to revolutionize high-priority technologies, such as quantum computing and communications.21
Nanophotonic control over quantum states of matter represents a pivotal area of research within the ongoing second quantum revolution, offering unprecedented capabilities in manipulating quantum phenomena at the nanoscale.21 While it is well established that a structured environment, such as a photonic crystal, can be utilized to control the spontaneous emission of light (i.e., radiative decay) of an emitter,22,23 the ability to control and manipulate the interactions between multiple emitters, such as the nonradiative transfer of energy between a donor/acceptor FRET pair, by these means remains unresolved. Andrew and Barnes' pioneering work on this subject suggested a linear relationship between the local density of optical states (LDOS) and the energy transfer rate between a donor/acceptor pair of emitters,8 which was later theoretically supported.9 In the past decade, additional support for the linear dependence of FRET on the LDOS has been reported in the literature.10–12 However, some reports provide evidence of contradicting relationships between FRET and the LDOS, including a quadratic dependence on the LDOS13 or complete independence.16,18–20
Photonic crystals describe a class of periodic dielectric materials that exhibit a photonic bandgap corresponding to wavelengths where propagation through the crystal is forbidden.24–26 Photonic crystals are commonly embedded with emitters and exploited for their control over the LDOS of the emitter. In the case of a photonic crystal exhibiting a complete photonic bandgap, the LDOS at the bandgap frequency range is significantly diminished in all directions. However, in the case of a photonic crystal exhibiting an incomplete, or partial, photonic bandgap, the LDOS at the bandgap frequency range is inhibited only in certain directions. This work focuses on photonic crystals composed of highly-ordered colloidal nanoparticles known as crystalline colloidal arrays (CCAs), which rely on the spontaneous self-assembly of electrically charged nanoparticles into three-dimensional periodic arrays driven by repulsive Coulombic interactions.27,28 The minimum energy configuration of the nanoparticles is a face-centered cubic (FCC) structure at the high particle concentrations employed in this work.29 While a CCA does not exhibit a complete photonic bandgap due to the low refractive index contrast between the nanoparticles and surrounding medium, a CCA possesses a partial photonic bandgap (i.e., stop-band) in the visible regime, which can be approximated by Bragg's Law.30 It is well-known that the stop-band of the CCA, which corresponds to the observed reflectance of the liquid system and is denoted as the rejection wavelength (λrw), can be dynamically shifted across the full visible spectrum by a change in interparticle spacing or refractive index.31 CCAs have been employed as precursors for various photoluminescent and optoelectronic materials32,33 and have been utilized for investigations of quantum light–matter interactions in nanostructured environments.34–39 Notably, control over nonradiative decay mechanisms, such as intersystem crossing, has been demonstrated in a sterically-packed colloidal crystal exhibiting a partial photonic bandgap.39 Specifically, the suppressed emission intensity of an embedded emitter by the photonic bandgap effect has been suggested to increase the likelihood of nonradiative decay, thereby facilitating direct manipulation of luminescence.39
Motivated by the unresolved dependence, if any, of FRET on the photonic bandgap effect, two polystyrene-based nanoparticle series (n1 and n2) copolymerized with three FRET-pairing emitters were synthesized. Notably, n2 was synthesized using 100× less emitter content than n1 such that the decay kinetics of each emitter could be investigated using the n2 nanoparticles. Both n1 and n2 spontaneously self-assembled into CCAs, allowing for precise control over the partial photonic bandgap frequency range of the liquid structures without chemically or geometrically altering the copolymerized emitters. Specifically, the partial photonic bandgap of the liquid structures could be red-shifted across the full visible spectrum by dilution with deionized water, which increased the interparticle spacing. In both nanoparticle series, an anthracene derivative (AMMA) was copolymerized to play the role of the initial donor in the FRET system. In addition to AMMA, naphthalimide and rhodamine B derivatives (NMMA and RMMA, respectively) were copolymerized within the nanoparticles, extending the emission to the red region of the visible spectrum through two sequential transfers of energy. Anthracene, naphthalimide, and rhodamine B derivatives were chosen due to their known FRET-pairing with each other.40 Upon nanoparticle excitation with ultraviolet (UV) light, the AMMA donor transfers energy to NMMA, which acts as an acceptor in the AMMA/NMMA FRET pair. Subsequently, NMMA acts as a donor and transfers energy to RMMA, which acts as an acceptor in the NMMA/RMMA FRET pair. Steady state photoluminescence (PL) measurements of n1 and n2 assembled into an ordered structure (OS) were collected as the λrw of each structure was shifted across the emission spectrum. Additionally, the decay kinetics of n2 were investigated as the λrw was shifted to overlap the emission attributed to each emitter copolymerized within the nanoparticles. Specifically, as the λrw coincided with the emission attributed to each emitter, the lifetime was monitored at the λrw as well as at the peaks attributed to the corresponding donor and/or acceptor emitters. In this way, the photonic bandgap effect on the decay kinetics of the donor and acceptor in each FRET pair was observed. Notably, the reference systems utilized to quantify the photonic bandgap effect on the steady state and time-resolved spectra of the liquid structures with various λrw positions were fabricated such that nanoparticle density and emitter content were constant for a precise assessment of only photonic effects.
2 Experimental
2.1 Reagents and solvents
Inhibitors were removed from styrene (99% Alfa Aesar), propargyl acrylate (96% Alfa Aesar), and divinylbenzene (80% Aldrich) by passing over alumina basic prior to use. All other commercial reagents were used without further purification. A Nanopure System supplied deionized (DI) water at a resistivity of ca. 18.2 MΩ cm. Anthracen-9-ylmethyl methacrylate (AMMA), 2-(1,3-dioxo-6-(piperidin-1-yl)-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl methacrylate (NMMA), and N-(6-(diethylamino)-9-(2-((2-(methacryloyloxy)ethoxy)carbonyl)phenyl)-3H-xanthen-3-ylidene)-N-ethylethanaminium chloride (RMMA) were synthesized according to previously described methods.41 Additionally, N-(9-(2-((2-(2-(2-azidoethoxy)ethoxy)ethoxy)carbonyl)phenyl)-6-(diethylamino)9,9a-dihydro-3H-xanthen-3-ylidene-N-ethylethanaminium) (azRhod) was synthesized according to previously described methods.42
2.2 Synthesis
2.2.1 Nanoparticle series n1.
Monodisperse n1 nanoparticles with three copolymerized emitters were synthesized using a general emulsion polymerization procedure43,44 previously described.41 Briefly, 25% (3.68 mL) of a 0.3% (w/w) solution of each emitter (AMMA, NMMA, and RMMA) in styrene (32.1 mmol styrene, 36.2 μmol AMMA, 25.5 μmol NMMA, 18.0 μmol RMMA) and propargyl acrylate (0.32 mL, 2.9 mmol) were added to 50 mL of DI water in a 250 mL four-neck round bottom flask equipped with a dropping funnel, condenser, stir bar, nitrogen inlet/outlet, and thermocouple. The remaining 75% (11.04 mL) of the solution of AMMA, NMMA, and RMMA in styrene (96.4 mmol styrene, 108.6 μmol AMMA, 76.4 μmol NMMA, 54.0 μmol RMMA) and propargyl acrylate (0.96 mL, 8.7 mmol) were added to the dropping funnel and the apparatus was purged with nitrogen and stirred for 40 minutes at room temperature. After purging, an aqueous 29% (w/w) sodium dodecyl sulfate (SDS, 99% Aldrich) solution (0.18 mL, 0.62 mmol) and divinylbenzene (0.10 mL, 0.7 mmol) were injected into the round bottom flask followed by an increase in temperature to 50 °C. Once stabilized at 50 °C, a 0.8 mL aqueous solution of 1.24 M disodium phosphate (ACS Grade Acros Organics) and a 1 mL aqueous solution of 0.82 M potassium persulfate (ACS Grade Fisher Scientific) were injected into the round bottom flask followed by a temperature increased to 70 °C. Once the temperature stabilized, the dropping funnel contents were added dropwise to round bottom flask at a rate of ca. 10 drops in 20 seconds. After dropping approximately 50% of the contents of the dropping funnel (6 mL) into the round bottom flask, divinylbenzene (0.24 mL, 1.7 mmol) was injected dropwise to the round bottom flask. After the remaining 50% of the dropping funnel contents were added to the round bottom flask, the reaction proceeded for 2.5 hours. After 2.5 hours, the contents of the round bottom flask were gravity filtered through filter paper (P5 Fisherbrand) and placed in dialysis bags (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MWCO). The product was dialyzed against DI water for 1–2 weeks, with water changed once a day. The product was then stored in a Nalgene container with an excess of mixed bed ion-exchange resin (Bio-Rad AB-501-X8(D)). The hydrodynamic particle size and zeta potential of the n1 nanoparticles after dialysis was 125 ± 8 nm and −47.6 ± 1.8 mV, respectively.
000 MWCO). The product was dialyzed against DI water for 1–2 weeks, with water changed once a day. The product was then stored in a Nalgene container with an excess of mixed bed ion-exchange resin (Bio-Rad AB-501-X8(D)). The hydrodynamic particle size and zeta potential of the n1 nanoparticles after dialysis was 125 ± 8 nm and −47.6 ± 1.8 mV, respectively.
2.2.2 Nanoparticle series n2.
Monodisperse n2 nanoparticles with 100× less emitter content than n1 were synthesized using a modified version of the emulsion polymerization procedure used to synthesize the n1 nanoparticles. Modifications were made to the solution of emitters in styrene, where a 0.003% (w/w) solution of each emitter (AMMA, NMMA, and RMMA) in styrene (128.5 mmol styrene, 1.45 μmol AMMA, 1.02 μmol NMMA, 0.72 μmol RMMA total) was utilized. After dialysis the product was then stored in a Nalgene container with an excess of mixed bed ion-exchange resin. The hydrodynamic particle size and zeta potential of the n2 nanoparticles after dialysis was 133 ± 11 nm and −49.4 ± 1.6 mV, respectively.
2.2.3 Blank polystyrene-based nanoparticles.
Monodisperse “blank” polystyrene-based nanoparticles were synthesized using a modified version of the emulsion polymerization procedure used to synthesize the n1 and n2 nanoparticles. Modifications were made to the styrene monomer, where no emitters were added to styrene (128.5 mmol) prior to polymerization such that no emitters were copolymerized within the nanoparticles. After dialysis, the product was then stored in a Nalgene container with an excess of mixed bed ion-exchange resin. The hydrodynamic particle size and zeta potential of the “blank” polystyrene-based nanoparticles after dialysis was 132 ± 10 nm and −52.7 ± 1.5 mV, respectively.
2.2.4 Rhodamine B-clicked polystyrene-based nanoparticles.
A copper(I)-catalyzed azide/alkyne cycloaddition (CuAAC) click reaction was utilized to attach azide-modified rhodamine B (azRhod) to the surface of the “blank” polystyrene-based nanoparticles described above. Briefly, “blank” polystyrene-based nanoparticles (195 mg) in deionized water (1 mL) were added to a J-KEM mini-reactor tube equipped with a stir bar followed by aqueous solutions of copper sulfate pentahydrate (6.8 mM, 0.5 mL) and sodium ascorbate (17.0 mM, 0.5 mL). Subsequently, a solution of azRhod in tetrahydrofuran (1.7 mM, 1 mL) was added to the J-KEM tube and the reaction was allowed to run for 24 hours in the dark under nitrogen at 28 °C with stirring. After 24 hours, resulting nanoparticles were cleaning via centrifugation at 4500 rpm for 10 minutes multiple times (ca. 10) with deionized water and tetrahydrofuran to remove the catalyst and any unattached dye.
2.3 Nanoparticle characterization
A Coulter N4Plus Dynamic Light Scatter (DLS) system was used to measure the hydrodynamic diameter of the nanoparticles. A Brookhaven Instruments Corp. ZetaPlus Zeta Potential Analyzer was used to measured the zeta potential of the nanoparticles.
2.4 Optical characterization
Absorbance spectra was collected using a PerkinElmer Lambda 900 UV-vis/NIR spectrophotometer (cf. Fig. S1 and S6, ESI†). Photoluminescence excitation (PLE) spectra, photoluminescence (PL) spectra, and fluorescence decay profiles via a time-correlated single photon counting (TCSPC) technique were collected using a Horiba Jobin-Yvon Fluorolog 3-222 Tau/TCSPC spectrofluorometer. PL spectra presented in Fig. 1 and 3 were collected using a 370 nm excitation, 2 nm entrance/exit slit width, and 0.5 second integration time. PL spectra integrated in Fig. 2 were collected using a 370 nm excitation, 2 nm entrance/exit slit width, and 0.2 second integration time. PL spectra presented in Fig. 4A–D were collected using a pulsed 370 nm NanoLED source with a short pass filter (Horiba UG1 Short Pass Filter), 2 nm exit slit width, and 0.5 second integration time. PLE/PL spectra presented in Fig. S2 (ESI†) were collected using emission/excitation wavelengths indicated, 2 nm entrance/exit slit width, and a 0.5 second integration time. PL spectra integrated in Fig. S3c (ESI†) were collected using a 366 nm excitation, 2 nm entrance/exit slit width, and a 0.2 second integration time. PL spectra integrated in Fig. S3f (ESI†) were collected using a 403 nm excitation, 2 nm entrance/exit slit width, and a 0.2 second integration time. PL spectra presented in Fig. S5 (ESI†) were collected using a 370 nm or 400 nm excitation, 2 nm entrance/exit slit width, and a 0.5 second integration time. PL spectra presented in Fig. S7 (ESI†) were collected using a 530 nm excitation, 2 nm entrance/exit slit width, and a 0.5 second integration time. PL spectra presented in Fig. S8 (ESI†) were collected using a 370 nm excitation, 2 nm entrance/exit slit width, and a 0.5 second integration time. A Horiba Jobin-Yvon NanoLED 370 nm pulsed light source with a short pass filter (Horiba UG1 Short Pass Filter) operating at a frequency of 1 MHz was utilized as the excitation source for all TCSPC measurements (cf.Fig. 4, Fig. S4, S9–S13 and Tables S1–S6, ESI†), which was run in reverse mode and using a TAC range of 100 ns, coaxial delay of 50 ns, and sync delay of 116 ns. A peak present of 10000 counts (P10k) and a bandpass width of 2 nm at the monitored wavelength was employed for all TCSPC measurements. Additionally, it was ensured that α < 2% for all samples. A Horiba Jobin-Yvon DAS6 fluorescence decay analysis software was utilized to fit fluorescence decay profiles and best fits were determined by the lowest χ2 value. An Ocean Optics bifurcated fiber optic bundle attached to an Ocean Optics USB2000 fiber coupled spectrometer, where the input arm of the fiber bundle was attached to a white light source (Ocean Optics LS-1-CAL) and the output arm was attached to the spectrometer, was utilized to collect reflectance spectra of the nanoparticles assembled in an ordered structure (OS) (cf.Fig. 1E–G and 3E–G). It should be noted that significant scattering of reflectance spectra in the blue region of the visible spectrum occurs (cf.Fig. 1E and 3E). The incident light for all reflectance spectra was normal to the sample surface at the [111] plane of the OS. A Canon Rebel Ti1 was utilized to collect photographs of the OS (cf.Fig. 2D and E), where ultraviolet (UV) excitation was supplied by a Spectroline B-100 black light lamp (long wave ultraviolet, 365 nm).
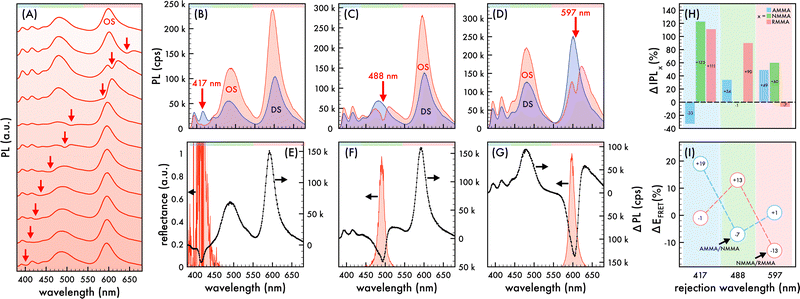 |
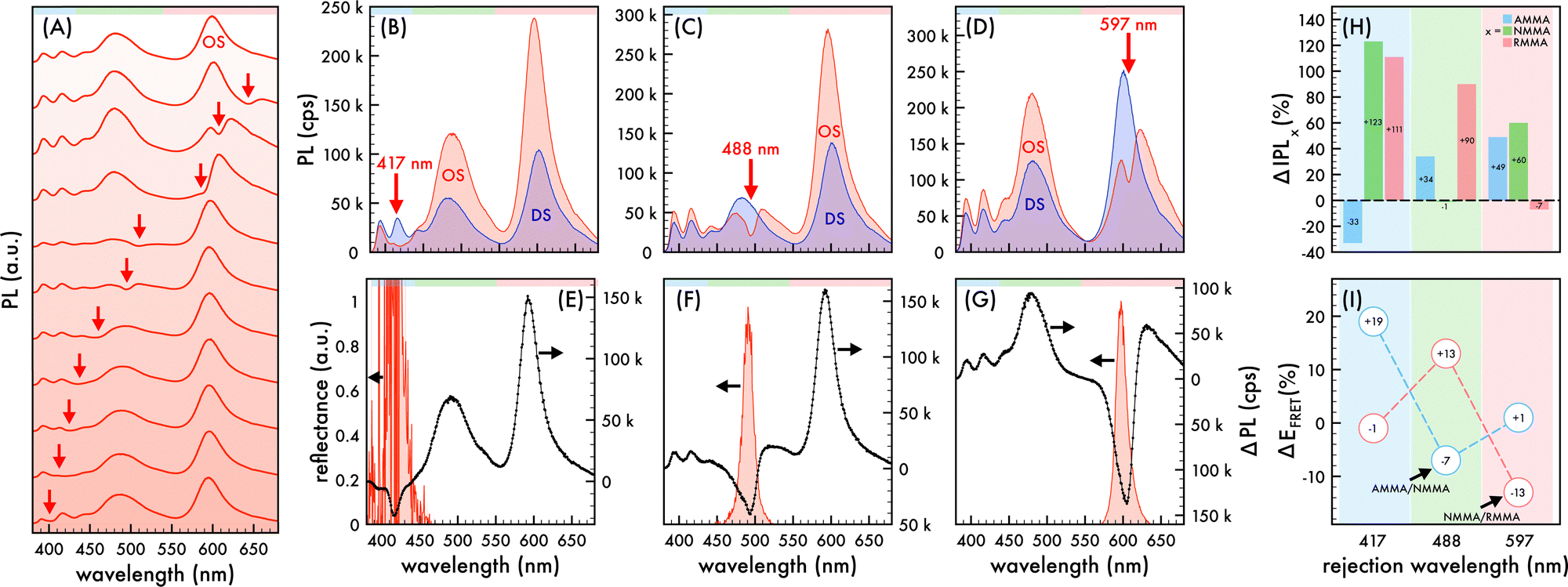
| | Fig. 1 Nanophotonic manipulation of n1 photoluminescence and FRET efficiency. (A) Photoluminescence (PL) spectra of n1 nanoparticles assembled in an ordered structure (OS) as the rejection wavelength (λrw) was shifted through the visible spectrum. PL spectra of n1 assembled in an OS (red) compared to that of corresponding disordered structures (DS, blue) as the λrw (denoted by a red arrow) was shifted to overlap AMMA, NMMA, and RMMA emission at (B) 417 nm, (C) 488 nm, and (D) 597 nm, respectively. A 370 nm excitation wavelength was utilized to obtain PL spectra presented in (A)–(D). Difference in PL spectra (ΔPL, black dotted line) of n1 in an OS compared to that of the corresponding DS as the λrw was shifted to (E) 417 nm, (F) 488 nm, and (G) 597 nm. The observed reflectance peak of each OS is presented in (E)–(G). The spectral regions attributed to AMMA (blue), NMMA (green), and RMMA (red) emission are lightly shaded at the top of (A)–(G) as a visual aid. (H) Change (%) in integrated PL (ΔIPL) attributed to AMMA (blue), NMMA (green), and RMMA (red) emission of n1 assembled in an OS compared to that of the corresponding DS as the λrw was shifted to 417 nm, 488 nm, and 597 nm. (I) Change (%) in observed Förster resonance energy transfer (FRET) efficiency (ΔEFRET) of the AMMA/NMMA (blue circles) and NMMA/RMMA (red circles) FRET pairs in n1 assembled in an OS compared to that of the corresponding DS as the λrw was shifted to 417 nm, 488 nm, and 597 nm. As a visual aid, the emitter overlapped at each λrw position is lightly shaded in (H) and (I). | |
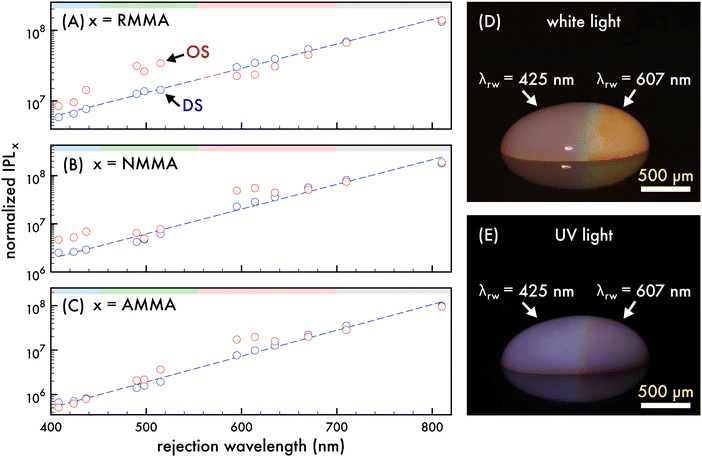 |
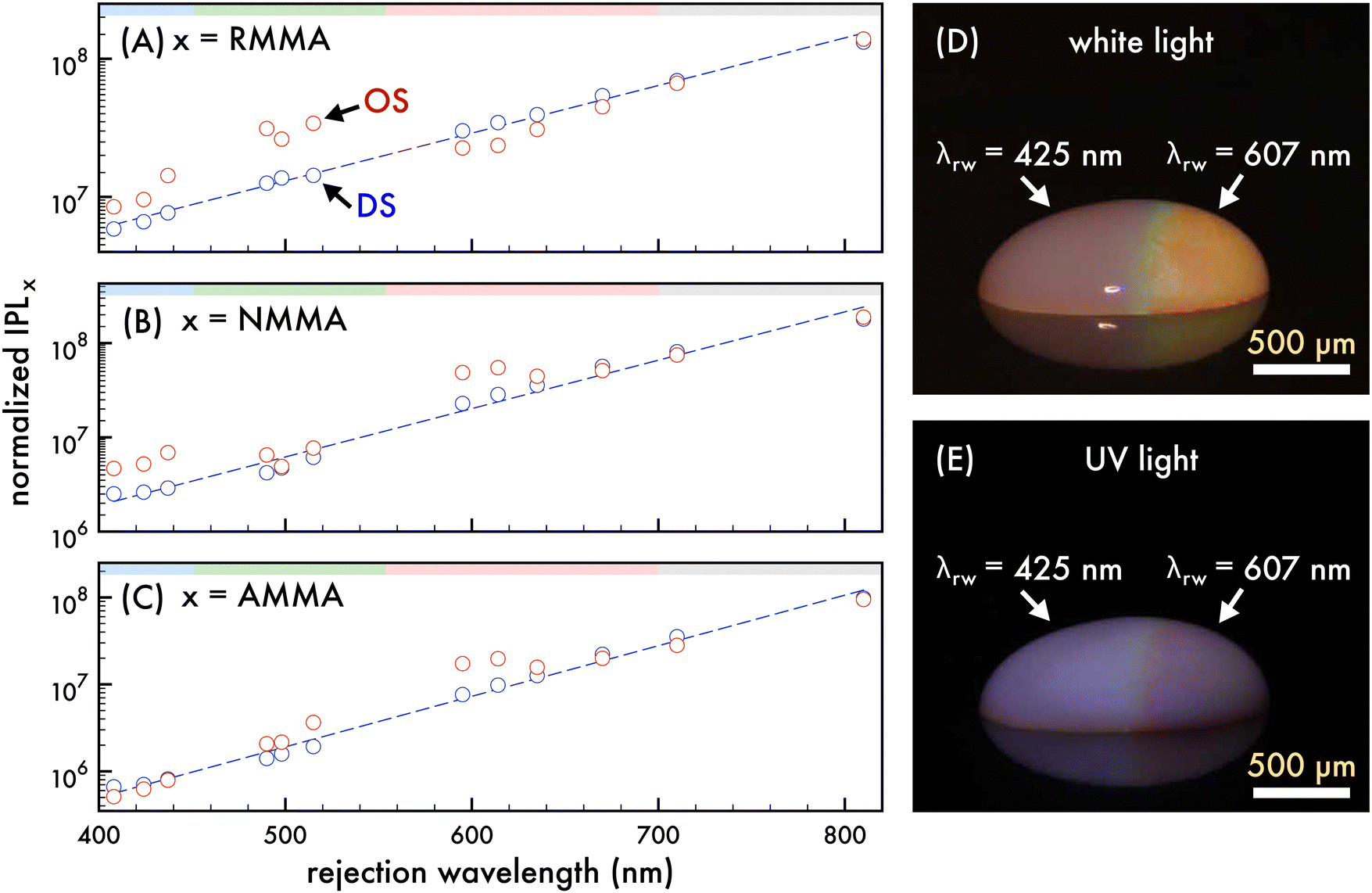
| | Fig. 2 Comparison of the integrated photoluminescence of n1 assembled in an ordered structure and corresponding disordered structure with dilution. Normalized integrated photoluminescence (IPL) of the n1 ordered structure (OS, red circles) and disordered structure (DS, blue circles) emission attributed to (A) RMMA, (B) NMMA, and (C) AMMA as the rejection wavelength (λrw) of the OS was shifted through the visible spectrum by dilution with deionized water. A 370 nm excitation wavelength was utilized to obtain PL spectra presented in (A)–(C) and the IPL was normalized by the concentration of nanoparticles in each dilution. The spectral regions attributed to AMMA (blue), NMMA (green), RMMA (red) emission are lightly shaded at the top of (A)–(C) as a visual aid. A trendline for the DS IPL (blue dashed line) is included in (A)–(C) to aid the eye. Photographs of a ca. 500 μL droplet the n1 OS under (D) white light and (E) UV light exposure, where the λrw of the left and right half of the OS droplet was at 425 nm and 607 nm, respectively. Scale bars are approximate. | |
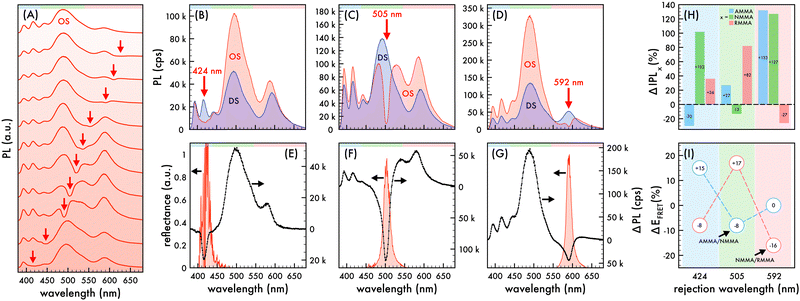 |
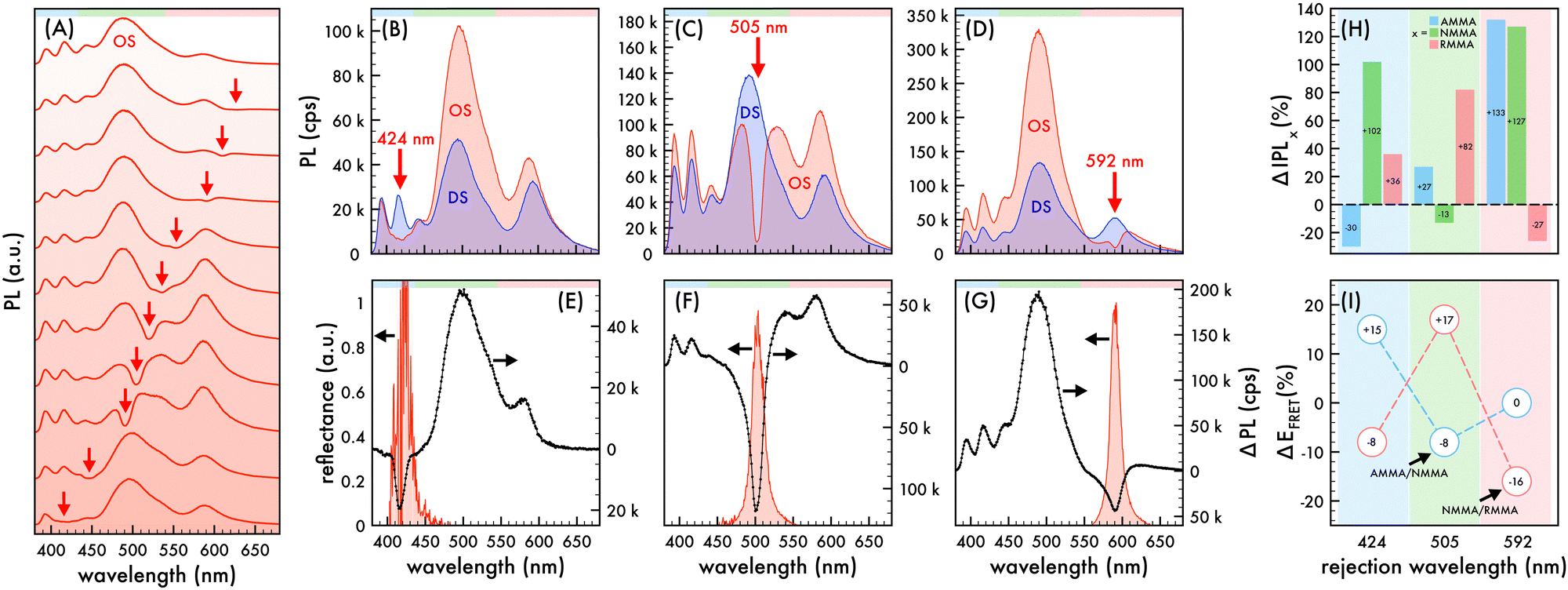
| | Fig. 3 Nanophotonic manipulation of n2 photoluminescence and FRET efficiency. (A) Photoluminescence (PL) spectra of n2 nanoparticles assembled in an ordered structure (OS) as the rejection wavelength (λrw) was shifted through the visible spectrum. PL spectra of n2 assembled in an OS (red) compared to that of corresponding disordered structures (DS, blue) as the λrw (denoted by a red arrow) was shifted to overlap AMMA, NMMA, and RMMA emission at (B) 424 nm, (C) 505 nm, and (D) 592 nm, respectively. A 370 nm excitation wavelength was utilized to obtain PL spectra presented in (A)–(D). Difference in PL spectra (ΔPL, black dotted line) of n2 in an OS compared to that of the corresponding DS as the λrw was shifted to (E) 424 nm, (F) 505 nm, and (G) 592 nm. The observed reflectance peak of each OS is presented in (E)–(G). The spectral regions attributed to AMMA (blue), NMMA (green), and RMMA (red) emission are lightly shaded at the top of (A)–(G) as a visual aid. (H) Change (%) in integrated PL (ΔIPL) attributed to AMMA (blue), NMMA (green), and RMMA (red) emission of n2 assembled in an OS compared to that of the corresponding DS as the λrw was shifted to 424 nm, 505 nm, and 592 nm. (I) Change (%) in observed Förster resonance energy transfer (FRET) efficiency (ΔEFRET) of the AMMA/NMMA (blue circles) and NMMA/RMMA (red circles) FRET pairs in n2 assembled in an OS compared to that of the corresponding DS as the λrw was shifted to 424 nm, 505 nm, and 592 nm. As a visual aid, the emitter overlapped at each λrw position is lightly shaded in (H) and (I). | |
3 Results and discussion
Two series of polystyrene-based nanoparticles containing three copolymerized emitters, AMMA, NMMA, and RMMA, were synthesized using a general emulsion polymerization procedure (cf. Experimental). The photophysical properties of the individual emitters are presented in the ESI† (cf. Fig. S1–S6 and Table S1). Briefly, the AMMA emitter exhibits blue emission upon UV excitation. The absorption spectrum of the NMMA emitter has adequate overlap with the emission spectrum of AMMA such that, when in close proximity, energy transfer occurs between the donor AMMA and the acceptor NMMA. Subsequently, the absorption spectrum of the RMMA emitter has adequate overlap with the emission spectrum of NMMA such that, when in close proximity, energy transfer occurs between the donor NMMA and the acceptor RMMA.
Series n1 was synthesized using 0.3% (w/w) of each emitter to styrene while a more emitter dilute series, n2, was synthesized using 100× less emitter content (0.003% (w/w) to styrene). Both n1 and n2 spontaneously self-assembled into a CCA due to their monodispersity and excellent colloidal stability (cf. ESI†). The incorporation and arrangement of the emitters in n1 was confirmed and assessed in the ESI† (cf. Fig. S6 and S7, ESI†). Due to the very small emitter content of n2, the absorbance spectrum of n2 in aniline did not reveal any discernible peaks such that the number of emitter molecules per particle could be estimated. Nonetheless, the incorporation of all three emitters is evident in the PL spectra of n2 discussed later (cf.Fig. 3 and Fig. S8, ESI†).
3.1 Nanophotonic manipulation of spectral properties and FRET efficiency
Nanophotonic manipulation of the PL spectra of n1 and n2 was explored by shifting the observed λrw of each nanoparticle series assembled into an ordered structure (OS) through their respective emission. It is important to note that due to the large quantity of emitters copolymerized in n1, the n1 OSs utilized for all PL measurements were composed of 1.36% (v/v) of n1 in “blank” polystyrene-based nanoparticles that contained no copolymerized emitters (cf. ESI†). Due to the size and stability similarities of n1 and the “blank” nanoparticles, it was assumed that there was no preferential ordering of the particles during mixing. Therefore, the probability of n1 nanoparticle proximity to other n1 nanoparticles within the OS lattice was determined by its volume fraction. At this n1 dilution, the probability that two n1 nanoparticles were located next to each other was 0.000185, or 1 in 5405. PL spectra of n1 and n2 assembled in an OS were obtained at the (111) face of the FCC crystal, which aligns parallel to the wall of a cuvette when the liquid arrays are placed within a cuvette.45,46 Thus, the observed λrw of the OS can be ascribed to the {111} stop-band. The interplanar spacing (d111) of each nanoparticle series assembled into an OS could be estimated using Bragg's law and the refractive index of the system.47| | 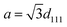 | (1) |
Using the estimated d111-spacing, the unit cell parameter (a) could be determined by eqn (1), which could subsequently be utilized to approximate the nearest neighbor distance between nanoparticles in the OS (ann) (cf.eqn (2)).| | 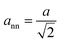 | (2) |
It should be noted that the ann of electrostatically-stabilized CCAs can be significantly larger than the diameter of the assembled nanoparticles. As the λrw of the electrostatically-stabilized n1 nanoparticles in the OS shifted from 417 nm to 800 nm, the d111-spacing increased from 152 nm to 292 nm and the surface-to-surface distance of the nearest neighbor particles ranged from 55 nm to 226 nm. As the λrw of the electrostatically-stabilized n2 nanoparticles in the OS shifted from 424 nm to 800 nm, the d111-spacing increased from 155 nm to 300 nm and the surface-to-surface distance of the nearest neighbor particles ranged from 57 nm to 234 nm. Upon UV excitation, n1 and n2 exhibited detectable emission attributed to AMMA, NMMA, and RMMA (cf.Fig. 1 and 3). In both n1 and n2, three small finger-like peaks attributed to AMMA were observed at 394 nm, 416 nm, and 444 nm. The maximum emission of n1 was at 602 nm and attributed to RMMA while a smaller emission peak attributed to NMMA was detected at 482 nm. The maximum emission of n2 was at 492 nm and attributed to NMMA while a smaller emission peak attributed to RMMA was detected at 592 nm. As shown in Fig. 1A and 3A, the PL spectral characteristics of n1 and n2 assembled in an OS were heavily influenced by the stop-band position, where the λrw (denoted by a red arrow) could be shifted through the entire PL spectrum of each nanoparticle series and a suppressed emission at the λrw coincidence was observed. The notable emission suppression at the λrw coincidence is indicative of the modified LDOS and resulting inhibited photon emission at the stop-band frequency. When the probability of radiative processes at the {111} stop-band is diminished by the photonic bandgap effect, it is hypothesized that the probability for nonradiative processes, such as FRET, is increased.
To explore the hypothesized phenomenon, the partial photonic bandgap effect on the PL characteristics of n1 and n2 and the energy transfer efficiency (EFRET) between the two FRET pairs copolymerized within n1 and n2 was investigated. To that end, the PL spectra of n1 and n2 assembled in an OS are presented in Fig. 1B–G and 3B–G, respectively, as the λrw of each liquid structure was shifted through the spectral regimes attributed to each copolymerized emitter. At each stop-band frequency, the PL spectra of the OS was compared to that of a disordered reference structure, referred to as the DS. Precisely comparable DSs were fabricated for each stop-band frequency of n1 and n2 assembled in an OS by adding a small amount of an ionic impurity (NaCl) to the OS.45 In this way, the long-range order of the nanoparticles in the OS was disrupted while maintaining particle density and emitter content for a controlled investigation of purely photonic effects.
3.1.1 Nanoparticle series n1.
When the λrw of the n1 OS was shifted to overlap the emission of the AMMA donor at 417 nm (cf.Fig. 1B and E), the integrated emission over the spectral range corresponding to the full width at half maximum (FWHM) of the reflectance peak exhibited a decrease of −74% compared to that of the DS. However, the integrated emission over the entire PL spectrum of the n1 OS exhibited a significant enhancement of +94%. The change in integrated PL intensity (ΔIPL) attributed to each emitter in the OS is presented in Fig. 1H, where an overall −33% decrease in emission attributed to AMMA was detected when the λrw was at 417 nm. Concurrently, a +123% increase in emission attributed to the NMMA acceptor/donor was observed due to the increase in energy transfer from the AMMA donor to the intermediate NMMA acceptor/donor.| | 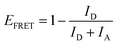 | (3) |
Additionally, a +111% increase in emission attributed to the RMMA acceptor was observed, attributed to a cascaded energy transfer to RMMA through the NMMA intermediate. The EFRET of each donor/acceptor pair was estimated by eqn (3), where ID and IA denote the integrated intensity of the donor and acceptor, respectively. The change in EFRET (ΔEFRET) of the OS in reference to the corresponding DS at each λrw condition is presented in Fig. 1I. The EFRET of the AMMA/NMMA pair exhibited the largest increase of +19% owing to an increased amount of energy transferred from AMMA to NMMA when AMMA photon emission at the stop-band frequency was inhibited. While the intensity of RMMA emission was enhanced due to the cascaded energy transfer through the intermediate NMMA emitter, the EFRET of the NMMA/RMMA pair exhibited a slight decrease of −1% due to the greater enhancement of n1 emission attributed to NMMA compared to that of RMMA. Next, when the λrw of the n1 OS was shifted to overlap the spectral regime attributed to the NMMA acceptor/donor emission at 488 nm (cf.Fig. 1C and F), a −53% decrease in PL intensity at the λrw and a +54% increase in total PL intensity was observed. The stop-band overlapping NMMA emission resulted in a −1% decrease in overall NMMA emission while the emission attributed to the AMMA donor and RMMA acceptor exhibited a +34% and +90% increase, respectively. The EFRET of the AMMA/NMMA pair decreased by −7%, owing to the inhibited acceptor emission and decreased amount of energy able to be transferred from AMMA to NMMA. However, the EFRET of the NMMA/RMMA pair increased by +13%, owing to the inhibited donor emission and enhanced amount of energy transferred from NMMA to RMMA. When the λrw of the n1 OS was shifted to overlap the spectral regime attributed to the RMMA acceptor emission at 597 nm (cf.Fig. 1D and G), a −52% decrease in PL intensity at the λrw and a +21% increase in total PL intensity was observed. The stop-band overlapping RMMA emission resulted in a −7% decrease in overall RMMA emission accompanied by a +49% and +60% increase in the AMMA donor and the NMMA acceptor/donor emission, respectively, attributed to the suppressed energy transfer from the initial donor AMMA and intermediate acceptor/donor NMMA to the final acceptor RMMA. The EFRET of the AMMA/NMMA pair increased slightly by +1% due to the greater enhancement of NMMA emission compared to that of RMMA. The EFRET of the NMMA/RMMA pair exhibited the largest decrease of −13% due to the inhibited final acceptor emission and decreased amount of energy transferred to RMMA. Overall, the observed changes in spectral properties of the n1 OS and EFRET between the emitters copolymerized within the n1 nanoparticles support the previously hypothesized photonic bandgap effect. Coupling the partial photonic bandgap of the n1 OS to its emission resulted in a modified LDOS and inhibited photon emission at the λrw frequency. When the radiative decay of the excited-state donors (AMMA and NMMA) was suppressed, the efficiency of energy transfer to the respective acceptors (NMMA and RMMA) was enhanced, attributed to the increased probability of the donor to decay via nonradiative processes, such as FRET. On the other hand, when the radiative decay of the excited state acceptors (NMMA and RMMA) was suppressed, the efficiency of energy transfer from the respective donors was inhibited, attributed to the suppressed photon emission of the acceptor at the stop-band frequency.
To ascertain that the spectral trends observed were a result of the photonic bandgap effect, the integrated PL spectra attributed to each copolymerized emitter in the n1 OS was compared to that of a corresponding DS as the λrw of the OS was shifted from ca. 400 nm to 800 nm (cf.Fig. 2A–C). In this case, a n1 DS was fabricated at the same initial particle density of the n1 OS and was diluted by deionized water similar to the OS such that the particle densities of the OS and DS were closely matched. The sample volume and optically sampled area of the OS and DS remained invariant through the dilution, resulting in a decreasing number of nanoparticles optically sampled with each dilution. Thus, the integrated PL was normalized by the number of nanoparticles in the optically sampled area at each dilution. The decreased nanoparticle density associated with each subsequent dilution resulted in decreased scattering from the colloid. Due to this, the integrated PL attributed to each copolymerized emitter in the DS exhibited an positive increment with decreasing particle density. Conversely, the integrated PL attributed to each copolymerized emitter in the OS did not exhibit this same trend and instead exhibited fluctuations of increased or decreased integrated PL intensity compared to that of the DS, depending on the stop-band frequency. Dips below the trendlines fit to the integrated PL of the emitters in the DS were observed in the integrated PL of the emitters in the OS as the λrw was shifted through the emission spectrum. When the λrw overlapped the emission of the donor (AMMA or NMMA), the emission of the respective acceptor (NMMA or RMMA) was enhanced. Conversely, when the λrw overlapped the emission of the acceptor (NMMA or RMMA), the emission of the respective donor (AMMA or NMMA) was enhanced. Additionally, when the λrw of the n1 OS was red-shifted past the emission range of the nanoparticles, the integrated PL attributed to each emitter in the OS and DS was closely matched, indicating no photonic bandgap effect on OS emission when the λrw was not coupled to the emission. The nanophotonic amplification and suppression of FRET between the copolymerized emitters in the n1 OS and resulting enhanced or inhibited PL emission are visually exemplified in Fig. 2D and E, where photographs of a droplet of the n1 OS under white light and UV excitation are presented. The λrw of the left half of the droplet is at 425 nm, exhibiting an enhanced total emission and a visually brighter optical output when excited with a UV source. The λrw of the right half of the droplet is at ca. 607 nm, resulting in a suppressed total emission and a visually dimmer optical output when excited with a UV source.
3.1.2 Nanoparticle series n2.
In order to confirm that the trends observed in the n1 OS were maintained in a system with less emitter content, a similar investigation utilizing the n2 nanoparticles was performed. To that end, the λrw of n2 assembled in an OS was shifted to overlap the spectral regimes attributed to AMMA, NMMA, and RMMA emission as shown in Fig. 3B–G. The resultant changes in integrated PL intensity attributed to each emitter and EFRET between each FRET pair in the OS compared to that of a corresponding DS are presented in Fig. 3H and I, respectively. When the λrw of the n2 OS was shifted to overlap AMMA emission at 424 nm (cf.Fig. 3B and E), the integrated PL of the OS exhibited a decrease of −63% at the λrw and an increase of +59% over the entire emission spectrum. The emission attributed to AMMA decreased by −30% while that of NMMA and RMMA increased by +102% and +36%, respectively. The EFRET of the AMMA/NMMA pair exhibited a +15% enhancement, owing to the enhanced energy transfer from AMMA to NMMA. However, the EFRET of the NMMA/RMMA pair exhibited a −8% decrease due to the ca. 3× larger enhancement of NMMA emission compared to that of RMMA. When the λrw of the n2 OS was shifted to overlap NMMA emission at 505 nm (cf.Fig. 3C and F), the integrated PL of the OS exhibited a decrease of −78% at the λrw and an increase of +16% over the entire emission spectrum. The emission attributed to NMMA decreased by −13% while that of AMMA and RMMA increased by +27% and +82%, respectively. The EFRET of the AMMA/NMMA pair exhibited a −8% decrease, owing to the suppressed energy transfer from AMMA to NMMA, while that of the NMMA/RMMA pair exhibited the largest increase of +17%, owing to the enhanced energy transfer from NMMA to RMMA. Lastly, when the λrw of the n2 OS was shifted to overlap RMMA emission at 592 nm (cf.Fig. 3D and G), the integrated PL of the OS exhibited a decrease of −74% at the λrw and an increase of +94% over the entire emission spectrum. The emission attributed to RMMA decreased by −27% while that of AMMA and NMMA increased by +133% and +127%, respectively. The EFRET of the AMMA/NMMA pair exhibited no change due to an approximately proportional increase in AMMA and NMMA emission. However, the EFRET of the NMMA/RMMA pair exhibited the largest decrease of −16%, owing to the suppressed energy transfer to RMMA. Overall, trends observed in the n1 OS were maintained in the n2 OS, despite a substantial reduction of dye content in n2. These results provide strong support for the significant impact of the partial photonic bandgap on the interactions between multiple emitters.
3.2 Nanophotonic manipulation of FRET in time-resolved fluorescence measurements
To ascertain that the trends observed in the steady-state measurements were indicative of decay process modulations, a time-correlated single photon counting (TCSPC) technique was utilized to evaluate the photonic bandgap effect on the fluorescence lifetime of the three emitters copolymerized within the nanoparticle building blocks of the n2 OS. It is important to note that fluorescence lifetime measurements were performed using an undiluted array of colloidal nanoparticles. Thus, the n2 nanoparticles with 100× less dye content than the n1 nanoparticles were utilized such that changes in decay kinetics of each emitter could be discerned. The measured fluorescence lifetime (τ), or total decay rate (Γt), is comprised of both radiative (Γr) and nonradiative (Γnr) decay components (cf.eqn (4)). It is well known through Fermi's golden rule that the rate of radiative decay is proportional to the LDOS at the emission frequency and, thus, it can be predicted that the radiative decay rate is decreased at the stop-band frequency.| | 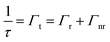 | (4) |
A decreased radiative decay rate at the stop-band frequency should result in an increased fluorescence lifetime if the nonradiative decay rate is unaltered by the photonic bandgap effect. However, if the photonic bandgap enhances the probability of nonradiative decay (e.g., FRET processes), the measured lifetime at the photonic bandgap may exhibit a decrease ascribed to the much higher rate of nonradiative decay processes compared to that of radiative decay processes.
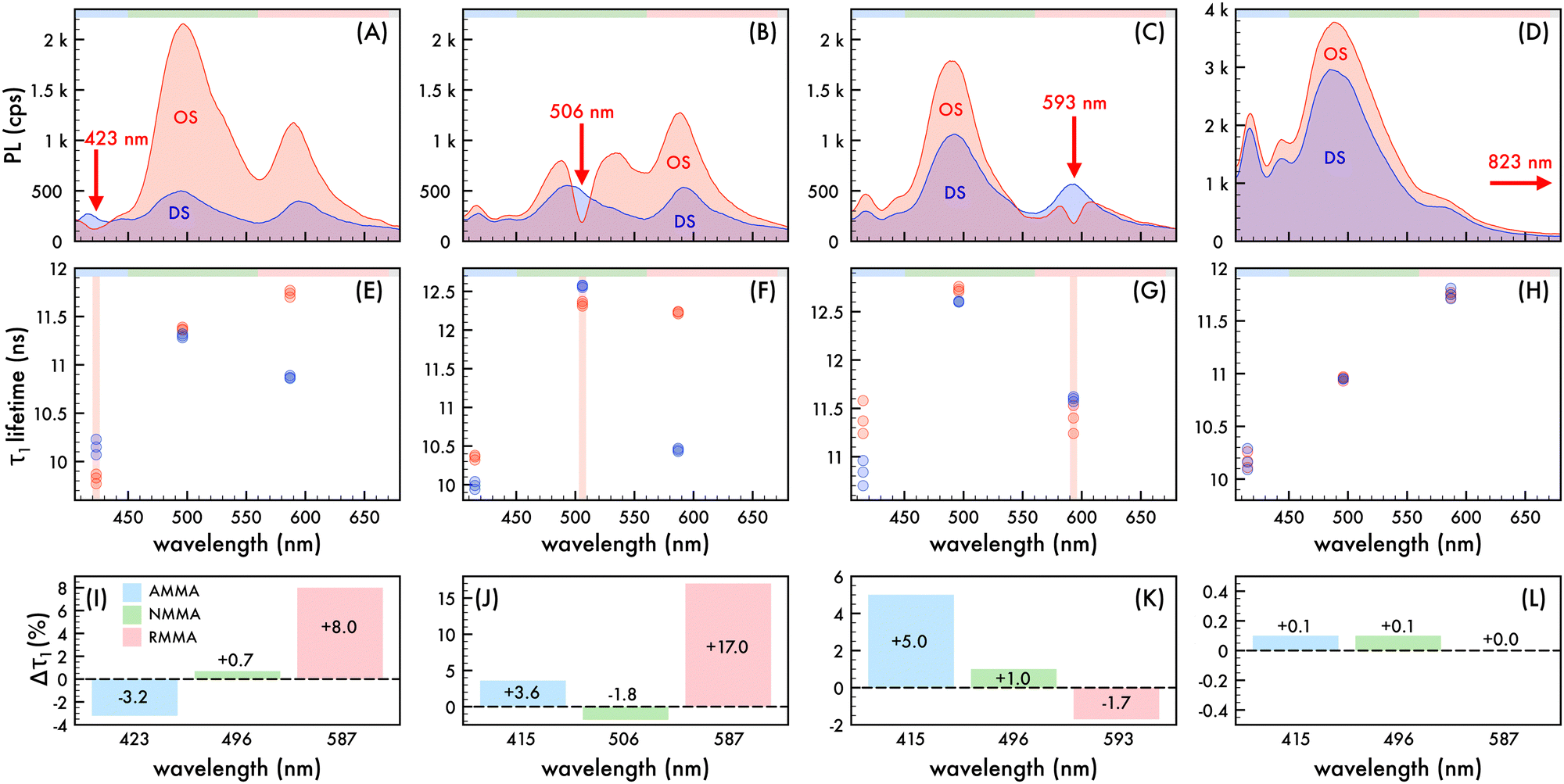
To explore the photonic bandgap modulation of decay kinetics, the lifetime of the n2 OS was monitored at wavelengths corresponding to AMMA, NMMA, and RMMA emission as the λrw was red-shifted across the entire emission spectrum (cf.Fig. 4). Precisely comparable reference DSs were fabricated for each λrw condition of the OS as previously described such that the nanoparticle density and emitter content were invariant for a controlled assessment of only photonic effects. All collected decay profiles of n2 in an OS and DS exhibited nonexponential behavior and were adequately modeled by assuming two or three decay components (τ1–3) (cf. Fig. S9–S12 and Tables S2–S5, ESI†), identified using a multi-exponential least-squares fitting procedure. The multi-exponential characteristics of the decay of each emitter are ascribed to different sub-ensembles of emitter molecules, each subject to varying nonradiative decay rates, in addition to contributions from the polystyrene-based host polymer. It was assumed that the greatest lifetimes with the largest contribution to the decay profile (τ1), which range from approximately 9.8 ns to 12.8 ns, were due to the copolymerized emitters. The components with shorter lifetimes (cf. Fig. S13, ESI†), which are less than ca. 8 ns, were ascribed to the host material (cf. Table S6, ESI†) and excluded from further analysis. The spectral properties of the n2 OS and corresponding DS as the λrw was shifted through the emission of the nanoparticles are presented in Fig. 4A–D. The average measured fluorescence lifetime attributed to each emitter at each λrw position is presented in Fig. 4E–H, where the average value was obtained from three lifetime acquisitions at different locations in the structure. Additionally, the highest and lowest observed lifetime at each wavelength is presented. When the λrw overlapped the n2 OS emission attributed to the AMMA donor at 423 nm (cf.Fig. 4I), the lifetime attributed to AMMA at 423 nm exhibited a −3.2% decrease, owing to the decreased probability of photon emission at the bandgap frequency. The lifetime attributed to the acceptor/donor NMMA and acceptor RMMA exhibited a +0.7% and +8.0% increase, respectively, owing to the enhanced probability of energy transfer between the AMMA/NMMA and NMMA/RMMA pairs through the cascaded energy transfer and, thus, enhanced probability of photon emission by NMMA and RMMA. Shifting the λrw of the n2 OS to overlap the emission attributed to NMMA at 506 nm (cf.Fig. 4J) resulted in a −1.8% decrease in lifetime attributed to the NMMA acceptor/donor. The lifetime attributed to the AMMA donor increased by +3.6%, owing to the increased probability of radiative emission from AMMA when energy transfer from AMMA to NMMA was suppressed. Additionally, the largest increase in RMMA lifetime was observed (+17.0%), attributed to the greater probability of NMMA to decay nonradiatively and transfer energy to RMMA, resulting in a greater probability of RMMA to emit a photon. Shifting the λrw of the n2 OS to overlap the emission attributed to RMMA at 593 nm (cf.Fig. 4K), the RMMA acceptor lifetime decreased by −1.7%. The lifetime of the AMMA donor and NMMA acceptor/donor exhibited an increase of +1.0% and +5.0%, respectively, owing to the suppressed energy transfer from AMMA and NMMA, resulting in a greater probability of AMMA and NMMA to emit a photon. Finally, by shifting the λrw of the n2 OS outside of the emission spectrum of the nanoparticles at 823 nm (cf.Fig. 4L), the lifetime of each emitter showed no significant change compared to that of the DS, indicating that the lifetime modulations were a result of the photonic bandgap effect.
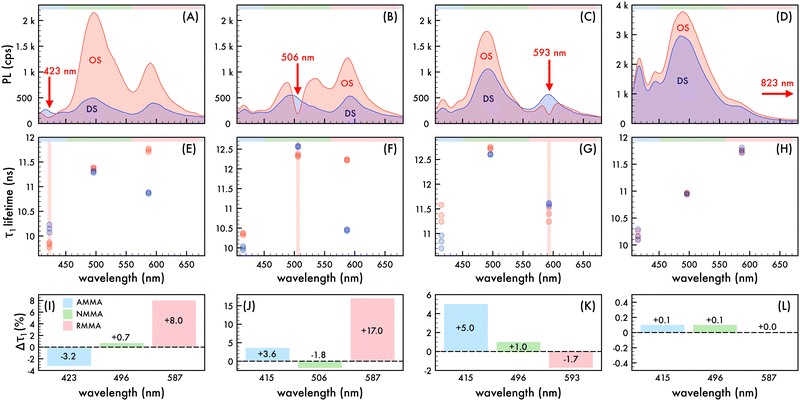 |
| | Fig. 4 Nanophotonic manipulation of n2 decay kinetics. Photoluminescence (PL) of n2 assembled in an ordered structure (OS, red) as the rejection wavelength (λrw) was shifted to (A) 423 nm, (B) 506 nm, (C) 593 nm, and (D) 823 nm compared to the corresponding disordered structure (DS, blue). A 370 nm excitation wavelength was utilized to obtain PL spectra presented in (A)–(D). Average (n = 3) measured lifetime (τ1) of spectral regions attributed to each emitter in the OS (red circles) as the λrw was shifted to (E) 423 nm, (F) 506 nm, (G) 593 nm, and (H) 823 nm compared to the corresponding DS (blue circles). The spectral regions attributed to AMMA (blue), NMMA (green), RMMA (red) emission are lightly shaded at the top of (A)–(H) as a visual aid. Change (%) in lifetime (Δτ1) attributed to AMMA (blue), NMMA (green), and RMMA (red) as the λrw was shifted to (I) 423 nm, (J) 506 nm, (K) 593 nm, and (L) 823 nm. | |
Förster's rate equation (cf.eqn (5)) can be utilized to estimate the energy transfer rate (Γet) between a donor/acceptor pair, where r is the distance between the donor and acceptor, R0 is the Förster distance, and τd is the lifetime of the donor without the acceptor.
| | 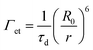 | (5) |
In this case, the influence of the photonic bandgap on the
Γet between the two donor/acceptor pairs in the n
2 nanoparticles can be estimated by replacing
τd with the observed lifetime of each donor (AMMA and NMMA) with (
τOS) and without (
τDS) stop-band coupling. In this way, changes in
Γet (Δ
Γet) can be predicted by
eqn (6), where the
R0 and
r are assumed to remain unchanged between measurements of the OS and DS.
| |  | (6) |
Through this relation, when the
λrw was within the emission of AMMA, the
Γet of the AMMA/NMMA pair increased by +3.3% and that of NMMA/RMMA decreased by −0.7%. When the
λrw was within the emission of NMMA, the
Γet of the AMMA/NMMA pair decreased by −3.5% and that of NMMA/RMMA increased by +1.9%. When the
λrw was within the emission of RMMA, the
Γet of the AMMA/NMMA pair decreased by −4.7% and that of NMMA/RMMA decreased by −1.0%. Finally, when the
λrw was out of the emission of the n
2 nanoparticles, the AMMA/NMMA pair and NMMA/RMMA pair exhibited no significant change in
Γet (−0.1%).
4 Conclusions
These results present significant evidence that the nonradiative energy transfer between multiple interacting emitters can be manipulated and controlled by the photonic bandgap effect. Specifically, the direction and efficiency of FRET was modulated in two FRET pairs, exhibiting a cascaded energy transfer, that were copolymerized within two series of nanoparticles assembled into CCAs. As evidenced by steady-state PL and time-correlated fluorescence measurements, the probability and efficiency of FRET between a donor/acceptor pair was enhanced by coinciding the partial photonic bandgap with the donor emission. Conversely, the probability and efficiency of FRET between a donor/acceptor pair was suppressed by coinciding the partial photonic bandgap with the acceptor emission. Both cases exhibited significant modifications in the spectral properties of the nanoparticles, energy transfer efficiency of each FRET pair, and decay kinetics of the interacting emitters, emanating from the optical confinement of the emitters within a nanostructured environment and ability to manipulate their likelihood of decay by radiative and nonradiative pathways using the partial photonic bandgap of the CCA. These results support that the modified LDOS at the partial bandgap frequency enhances nonradiative processes, such as FRET, and a nanostructured environment, such as a CCA, can be exploited for strategic control over the quantum interactions between multiple emitters. These findings have profound implications for both advancing the understanding of basic quantum light–matter interactions and serving as a versatile tool across various quantum technologies, including computing, communication, and sensing applications.
Author contributions
H. J. contributed to the conceptualization, methodology, investigation, data curation, visualization, writing – original draft, and writing – review & editing. Y. B. contributed to the resources and methodology. S. F. contributed to the conceptualization, methodology, data curation, visualization, writing – original draft, and writing – review & editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the Gregg-Graniteville Foundation and the National Science Foundation (OIA-1632881) for financial support.
References
- T. Forster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- R. van Grondelle, J. P. Dekker, T. Gillbro and V. Sundstrom, Biochim. Biophys. Acta, 1994, 1187, 1–65 CrossRef.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 Search PubMed.
- S. Buhbut, S. Itzhakov, E. Tauber, M. Shalom, I. Hod, T. Geiger, Y. Garini, D. Oron and A. Zaban, ACS Nano, 2010, 4, 1293–1298 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 Search PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- D. L. Andrews and J. Rodriguez, J. Chem. Phys., 2007, 127, 084509 Search PubMed.
- P. Andrew and W. L. Barnes, Science, 2000, 290, 785–788 Search PubMed.
- H. T. Dung, L. Knoll and D. G. Welsch, Phys. Rev. A: At., Mol., Opt. Phys., 2002, 65, 043813 Search PubMed.
- P. Ghenuche, J. de Torres, S. B. Moparthi, V. Grigoriev and J. Wenger, Nano Lett., 2014, 14, 4707–4714 CrossRef CAS PubMed.
- D. Weeraddana, M. Premaratne, S. D. Gunapala and D. L. Andrews, J. Chem. Phys., 2017, 147, 074117 Search PubMed.
- S. Patra, J.-B. Claude and J. Wenger, ACS Photonics, 2022, 9, 2109–2118 CrossRef CAS.
- T. Nakamura, M. Fujii, S. Miura, M. Inui and S. Hayashi, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045302 CrossRef.
- B. Kolaric, K. Baert, M. Van der Auweraer, R. A. L. Vallee and K. Clays, Chem. Mater., 2007, 19, 5547–5552 CrossRef CAS.
- L. Gonzalez-Urbina, K. Baert, B. Kolaric, J. Perez-Moreno and K. Clays, Chem. Rev., 2012, 112, 2268–2285 CrossRef CAS PubMed.
- M. J. A. de Dood, J. Knoester, A. Tip and A. Polman, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115102 CrossRef.
- F. T. Rabouw, S. A. den Hartog, T. Senden and A. Meijerink, Nat. Commun., 2014, 5, 3610 CrossRef.
- C. Blum, N. Zijlstra, A. Lagendijk, M. Wubs, A. P. Mosk, V. Subramaniam and W. L. Vos, Phys. Rev. Lett., 2012, 109, 203601 Search PubMed.
- M. Wubs and W. L. Vos, New J. Phys., 2016, 18, 053037 CrossRef.
- G. Rosolen, B. Maes, P. Y. Chen and Y. Sivan, Phys. Rev. B, 2020, 101, 155401 CrossRef CAS.
- B. Kolaric, B. Maes, K. Clays, T. Durt and Y. Caudano, Adv. Quantum Technol., 2018, 1, 1800001 CrossRef.
- K. H. Drexhage, J. Lumin., 1970, 1(2), 693–701 Search PubMed.
- W. L. Barnes, J. Mod. Opt., 1998, 45, 661–669 CrossRef CAS.
- T. Okubo, Prog. Polym. Sci., 1993, 18, 481–517 CrossRef CAS.
- E. Yablonovitch, Phys. Rev. Lett., 1987, 58, 2059–2062 CrossRef CAS PubMed.
- S. John, Phys. Rev. Lett., 1987, 58, 2486–2489 CrossRef CAS PubMed.
- P. A. Hiltner, Y. S. Papir and I. M. Krieger, J. Phys. Chem., 1971, 75, 1881–1886 CrossRef CAS.
- P. A. Hiltner and I. M. Krieger, J. Phys. Chem., 1969, 73, 2386 CrossRef CAS.
- N. Clark, A. Hurd and B. Ackerson, Nature, 1979, 281, 57–60 CrossRef CAS.
- P. Rundquist, P. Photinos, S. Jagannathan and S. Asher, J. Chem. Phys., 1989, 91, 4932–4941 CrossRef CAS.
- C. López, Adv. Mater., 2003, 15, 1679–1704 CrossRef.
- J. R. Lawrence, Y. Ying, P. Jiang and S. H. Foulger, Adv. Mater., 2006, 18, 300–303 CrossRef CAS.
- J. R. Lawrence, G.-H. Shim, P. Jiang, M. Han, Y. Ying and S. H. Foulger, Adv. Mater., 2005, 17, 2344–2349 CrossRef CAS.
- J. Martorell and N. M. Lawandy, Phys. Rev. Lett., 1990, 65, 1877–1880 CrossRef CAS PubMed.
- B. Tong, P. John, Y. Zhu, Y. Liu, S. Wong and W. Ware, J. Opt. Soc. Am. B, 1993, 10, 356–359 CrossRef CAS.
- M. Megens, J. E. G. J. Wijnhoven, A. Lagendijk and W. L. Vos, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 4727–4731 CrossRef CAS.
- K. Shibata, H. Kimura, A. Tsuchida and T. Okubo, Colloid Polym. Sci., 2006, 285, 127–133 CrossRef CAS.
- M. Khokhar, Priya and R. V. Nair, Phys. Rev. A, 2020, 102, 013502 CrossRef CAS.
- L. González-Urbina, J. Perez-Moreno, K. Clays and B. Kolaric, Mol. Phys., 2016, 114, 2248–2252 CrossRef.
- M. K. Burdette, Y. P. Bandera, G. M. Gray and S. H. Foulger, Adv. Opt. Mater., 2019, 7, 1801142 CrossRef.
- S. Mell, H. W. Jones, Y. P. Bandera and S. H. Foulger, Langmuir, 2022, 38, 10089–10097 CrossRef CAS PubMed.
- M. K. Burdette, H. W. Jones, Y. Bandera and S. H. Foulger, Opt. Mater. Express, 2019, 9, 1416–1429 CrossRef CAS.
- M. E. Woods, J. S. Dodge, I. M. Krieger and P. E. Pierce, J. Paint Technol., 1968, 40, 541 CAS.
- Y. S. Papir, M. E. Woods and I. M. Krieger, J. Paint Technol., 1970, 42, 571–578 CAS.
- Y. Monovoukas and A. P. Gast, J. Colloid Interface Sci., 1989, 128, 533–548 CrossRef CAS.
- Y. Monovoukas and A. P. Gast, Phase Transform., 1990, 21, 183 CrossRef CAS.
- S. H. Foulger, P. Jiang, A. Lattam, D. Smith and J. Ballato, Langmuir, 2001, 17, 6023–6026 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Yuriy
Bandera
ab and
Stephen H.
Foulger
ab,
Yuriy
Bandera
ab and
Stephen H.
Foulger