
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencesyn-1,2-Diaminobenzocyclobutenes from [2+2] cycloaddition of 2-imidazolones with arynes†
Haseeb
Ur Rehman Shah
,
Qi
Li
and
Christopher R.
Jones
 *
*
Department of Chemistry, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: c.jones@qmul.ac.uk
First published on 24th September 2024
Abstract
Formal [2+2] cycloaddition of arynes with 2-imidazolones affords syn-1,2-diaminobenzocyclobutenes. The transformation can also be conducted as a one-pot, three-stage process direct from simple propargyl amines and isocyanates to afford the new stereochemically defined benzocyclobutene frameworks.
The bicyclo[4.2.0]octa-1,3,5-triene structural motif (commonly known as benzocyclobutene, BCB) occurs in many bioactive natural products and pharmaceutically active compounds (Scheme 1a).1 BCBs also represent attractive building blocks for generating diverse carbo- and hetero-cyclic frameworks and in the preparation of polymeric materials. This is due to the thermally activated electrocyclic ring-opening of cyclobutene that reveals valuable o-quinonedimethide intermediates. These reactive species are trapped via intra- and inter-molecular Diels–Alder reactions in the synthesis of natural products and drug-like scaffolds,2 as well as undergoing a range of polymerization reactions to afford high-performance resins with broad applications in materials chemistry.3 BCBs have been prepared using many different methods, including 1,4-elimination from o-difunctionalised arenes,4a,b Parham cyclisation,4c [2+2+2] cyclotrimerisations,4d photochemical cycloadditions,4e extrusion reactions4f and cyclopropene ring expansion.4g
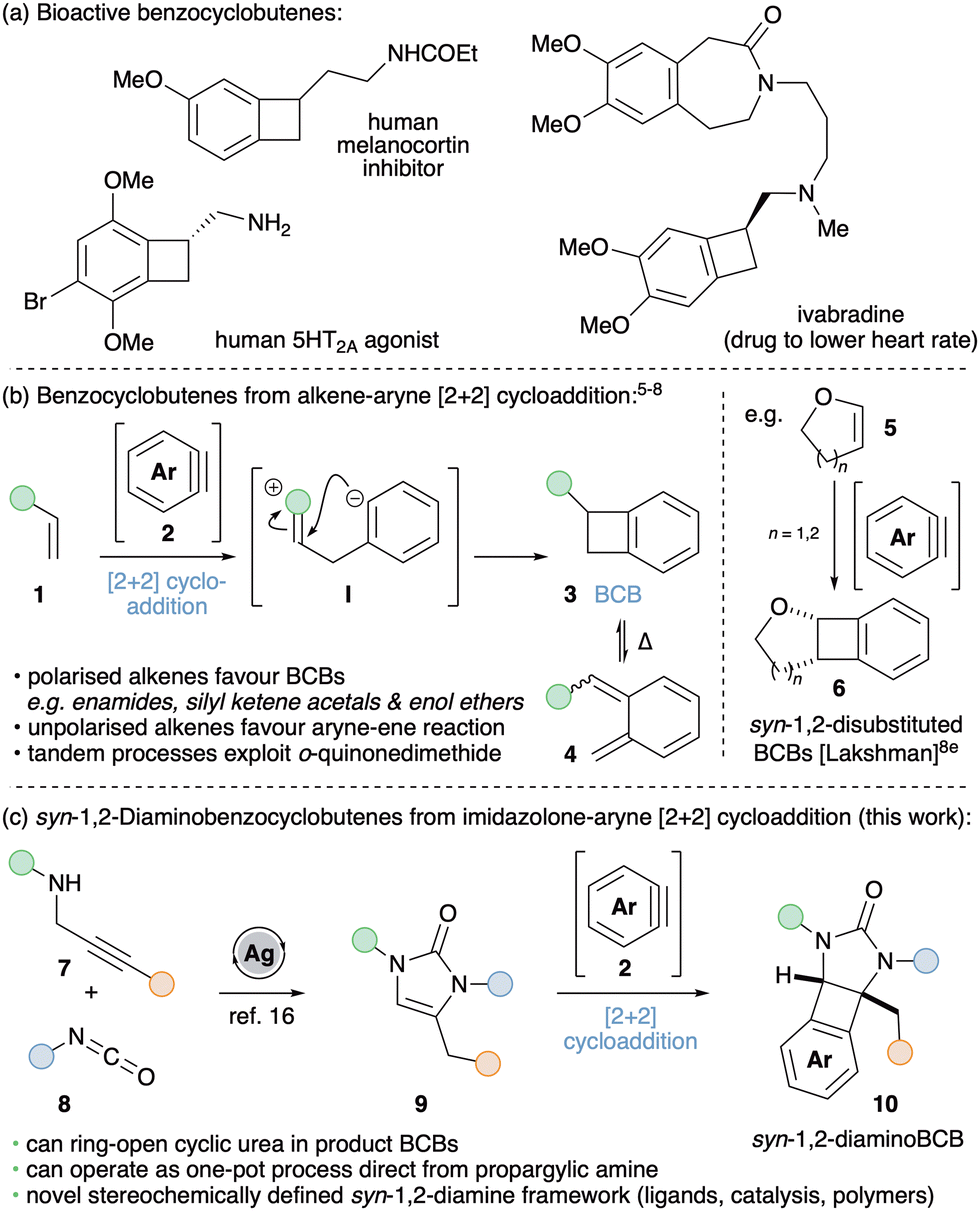 | ||
| Scheme 1 (a) Benzocyclobutenes in medicinal chemistry. (b) and (c) Synthetic approaches to BCBs via formal [2+2] cycloaddition between alkenes and arynes. | ||
Conceptually, the simplest approach to accessing BCBs 3 is through formal [2+2] cycloaddition between alkenes 1 and arynes 2 (Scheme 1b).5–8 Arynes are versatile reactive intermediates that rapidly afford valuable benzenoid and heterocyclic frameworks.9 They have experienced a recent resurgence in interest due to the advent of aryne precursors that act under mild conditions, such as the o-trimethylsilylaryl triflates (oSATs),10 hexadehydro-Diels–Alder reaction of polyalkynes11 and the development of arenes bearing onium ion leaving groups.12 With a few exceptions,5 the reaction between simple alkenes and arynes typically affords BCBs in low yields as a result of competing ene reactions.6 However, more polarised alkenes favour cycloaddition; the introduction of a single nitrogen atom proving successful with enamide and enamine-type coupling partners.7 For example, Hsung and co-workers reported an elegant ring-expansion methodology used for the total synthesis of chelidonine and norchelidonene that operates via a tandem aryne-enamide [2+2] cycloaddition – electrocyclic ring-opening – intramolecular Diels–Alder reaction.7b,c Silyl ketene acetals and enol ethers have also shown to be productive substrates for BCB formation; more specifically cis-enol ethers underwent preferential [2+2] cycloaddition whereas trans-enol ethers favoured ene reaction.8 Lakshman and co-workers exploited these observations by employing cyclic enol ethers 5 to furnish a range of stereochemically defined syn-1,2-disubstituted BCBs 6 in good yields with a range of arynes accessed via oSAT precursors.8e
Given our interest in the chemistry of arynes,13 we postulated that suitably protected cyclic 1,2-diaminosubstituted alkenes (9) would give rise to syn-1,2-diaminoBCBs (10) via formal [2+2] cycloaddition with arynes (Scheme 1c). Inspired by the previous work of Lakshman with cyclic enol ethers8e and reports using mono-substituted cyclic enamine equivalents,7e,f our strategy would afford novel stereochemically defined 1,2-diamino frameworks. Chiral 1,2-diamines are important structural motifs in bio- and pharmaceutically active compounds.14a,b They also act as privileged ligands in asymmetric transition metal catalysis and are key components of chiral organocatalysts.14c,d It follows that methods to access new 1,2-diamino motifs are of significant synthetic interest. Furthermore, the development of a differently functionalized BCB derivative should offer exciting opportunities in polymer research.
To test our hypothesis, 2-imidazolones 9 were selected as the cyclic 1,2-diaminosubstituted alkenes.15 These N-heterocycles are easily prepared from commercially available propargylic amines and isocyanates, as reported by Van der Eycken.16 In addition to the synthetic accessibility of these 1,2-diaminosubstituted alkenes, the urea moiety was chosen with a view to attenuating potential competing aryne reactivity at nitrogen. As such, N-phenyl-N-methyl-2-imidazolone 11a was exposed to 2-trimethylsilylphenyl triflate 12a under a variety of common aryne-forming conditions (see Table 1 for selected optimisation studies). Pleasingly, treatment with CsF in acetonitrile afforded the desired syn-1,2-diaminoBCB 13aa as the major product in 41% yield (entry 1). A small amount of starting material was recovered, along with a by-product identified as benzodiazepine 14aa.17 This is proposed to form via aryne insertion into either of the C–N sigma bonds of the urea; analogous ring-expansion having previously been described with the related saturated cyclic urea, N,N-dimethylimidazolidone.18 Increasing the reaction temperature, the molar ratio of CsF to oSAT 12a and the number of equivalents of 12a all resulted in reduced BCB formation and a more complex reaction mixture (entries 2–4). Employing different salts and/or additives to modify fluoride solubility did not improve BCB formation; instead, higher levels of benzodiazepine were observed (entries 5–10). With the rate of aryne formation seemingly playing a key role in determining product selectivity, slow addition of precursor 12a was performed. Whilst BCB 13aa remained the major product, the yield from dropwise addition was lowered compared to adding in one portion (entry 11 vs. entry 1). Finally, altering the reaction concentration did not have a significant effect on the amount of BCB produced, however the relative quantities of benzodiazepine increased (entries 12 and 13).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Fluoride source | Additive | Solvent | T (°C) | Yieldb (%) | ||
| 11a | 13aa | 14aa | |||||
| a Reaction conditions: oSAT 12a (1.0 equiv.), fluoride source (3.0 equiv.), additive (3.0 equiv.), solvent [0.15 M], 14 h, N2 atmosphere. b 1H NMR yield vs. CH2Br2 internal standard. c 5.0 equiv. of CsF. d 3.0 equiv. of oSAT 12a & 9.0 equiv. of CsF. e 1.0 equiv. of oSAT 12a added dropwise via syringe pump over 1 h. f 0.03 M in MeCN. g 0.3 M in MeCN. | |||||||
| 1 | CsF | — | MeCN | 50 | 12 | 41 | 7 |
| 2 | CsF | — | MeCN | 70 | 9 | 24 | 1 |
| 3c | CsF | — | MeCN | 50 | 23 | 32 | 14 |
| 4d | CsF | — | MeCN | 50 | 11 | 12 | 2 |
| 5 | CsF | 18-Crown-6 | MeCN | 50 | 12 | 12 | 35 |
| 6 | KF | 18-Crown-6 | THF | 50 | 4 | 14 | 14 |
| 7 | TBAF | — | THF | 50 | 20 | 12 | 22 |
| 8 | TBAT | — | THF | 50 | 37 | 20 | 46 |
| 9 | CsF | — | PhMe/MeCN (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
110 | 61 | 8 | 8 |
| 10 | CsF | — | PhMe/MeCN (3:1) |
50 | 8 | 21 | 19 |
| 11e | CsF | — | MeCN | 50 | 11 | 21 | 12 |
| 12f | CsF | — | MeCN | 50 | 14 | 43 | 29 |
| 13g | CsF | — | MeCN | 50 | 9 | 36 | 22 |

With optimised conditions in hand for the formation of BCBs and having established that there is a fine balance between competing reaction pathways, attention turned to investigating the influence of substitution around the 2-imidazolone framework (Scheme 2). Firstly, the effect of the N-substituent introduced by the isocyanate starting material was studied. Pleasingly, N-aryl moieties with electron-withdrawing groups (p-NO211b, p-F 11e) and electron-donating groups (p-OMe 11c, p-Me 11d, m-OMe 11f, o-OMe 11g) all afforded the corresponding BCBs in similar yields to the parent N-Ph derivative 13aa. Interestingly, the difference between m-OMe (13fa, 22%) and o-OMe (13ga, 57%) derivatives suggests that sterics at nitrogen do not play a key role in product selectivity. The transformation was also amenable to N-alkyl imidazolones, affording N-ethyl BCB 13ha and N-benzyl BCB 13ia in 27% and 39% yields respectively. Next, several modifications were made to the imidazolone scaffold via the propargylic amine component. Changing from an N-methyl to N-benzyl imidazolone (11j) had little effect on the yield, with N-benzyl BCB 13ja obtained in 39% yield. Likewise, replacing the exocyclic methyl group on the imidazolone alkene with a larger substituent did not significantly alter the yield of the [2+2] cycloaddition; the benzyl BCB 13ka isolated in 32% yield. Finally, the transformation was attempted with a fully substituted imidazolone 11l. Intriguingly this afforded no trace of the expected BCB 13la, instead the corresponding regioisomeric benzodiazepines were produced in a separable 1.2:1.0 ratio (combined 55% NMR yield, see ESI†). This observation offered an insight into the mechanism of the [2+2] cycloaddition, suggesting that initial attack of the aryne occurs at the unsubstituted end of the imidazolone alkene. When the aryne–alkene approach is too hindered, in the case of tetra-substituted alkene 11l, attack at nitrogen takes precedence and leads to formal aryne insertion into the N–C(O) sigma bonds.
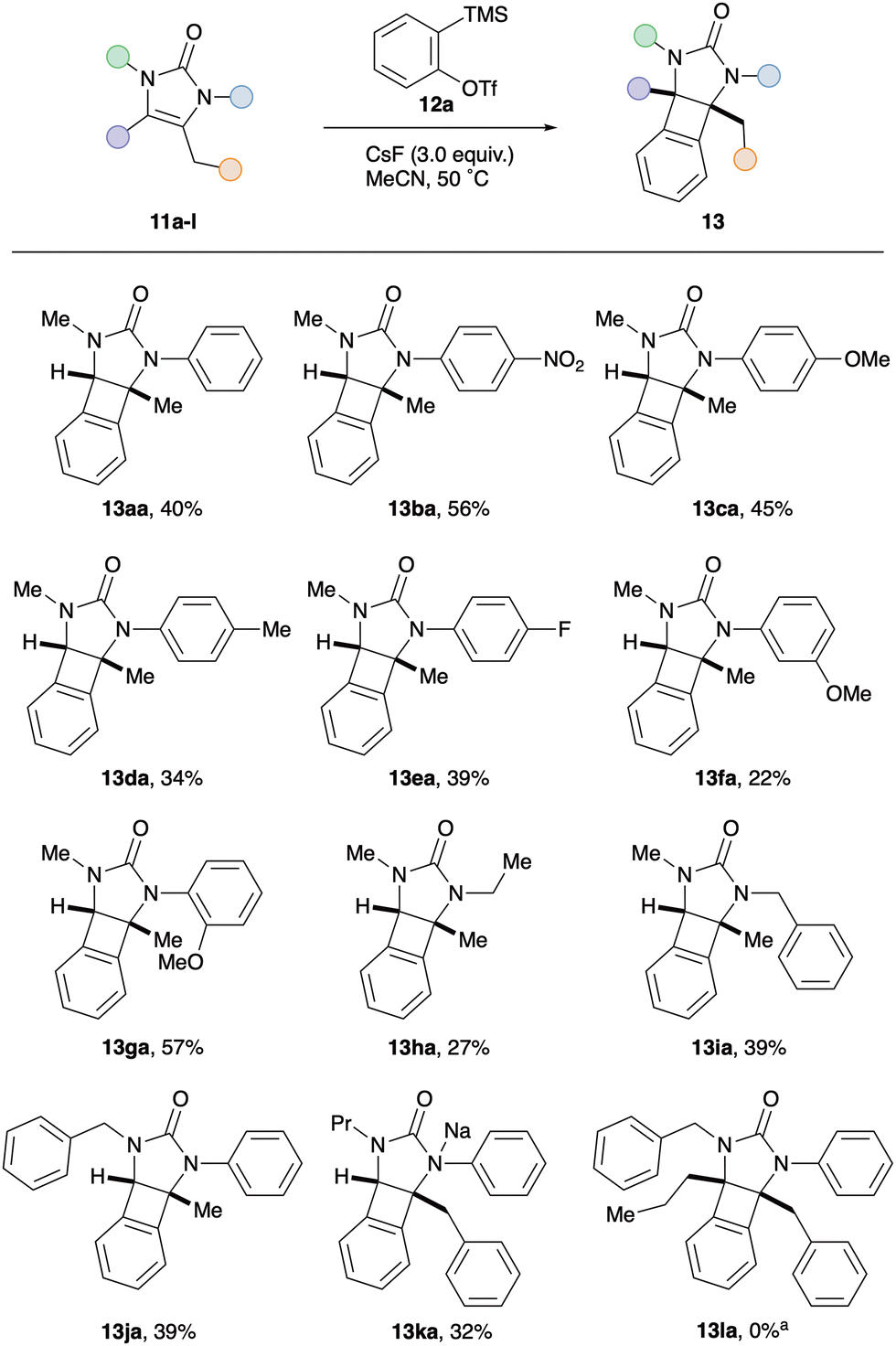 | ||
| Scheme 2 Synthesis of syn-1,2-diaminoBCBs 13 with different 2-imidazolone derivatives 11a–l. Reaction conditions: imidazolone 11 (1.0 equiv.), oSAT 12a (1.0 equiv.), CsF (3.0 equiv.), MeCN (0.15 M), 50 °C, 14 h, N2 atmosphere. Yields of isolated products throughout. aAfforded 1.2:1.0 ratio of corresponding regioisomeric benzodiazepines (55% combined NMR yield). | ||
We next investigated the effect of substitution on the aryne (Scheme 3). Electron-donating and withdrawing groups (12b–f) typically afforded the corresponding arene-functionalised BCBs 13 in comparable yields to benzyne precursor 12a. It is noteworthy that the most electron deficient aryne (3,4-difluoro, 12d) proved to be the most proficient coupling partner overall, furnishing BCB 13ad in excellent yield (83%). Finally, when imidazolone 11a was exposed to unsymmetrical o-methoxy aryne precursor 12f, o-Me BCB 13af was isolated as a single regioisomer in 38% yield. Given the established preference for nucleophilic attack at the distal position of o-methoxy aryne, the exclusive formation of BCB 13af suggests that the imidazolone reacts at the least substituted end of the alkene. This observation supports the previous mechanistic insight drawn from the tetrasubstituted alkene 11j (see Scheme 2).
 | ||
Scheme 3 Synthesis of syn-1,2-diaminoBCBs 13 with different aryne derivatives 12a–f. Reaction conditions: imidazolone 11 (1.0 equiv.), oSAT 12 (1.0 equiv.), CsF (3.0 equiv.), MeCN (0.15 M), 50 °C, 14 h, N2 atmosphere. Yields of isolated products throughout. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Single regioisomer observed. Single regioisomer observed. | ||
Having synthesised a range of syn-1,2-diaminoBCBs 13 from the corresponding 2-imidazolones 11, we postulated whether the stereochemically defined BCBs could be accessed directly from propargyl amines 7 and isocyanates 8. This would streamline the process by removing the isolation and purification of intermediate imidazolones. To this end, N-methyl propargyl amine 15 and phenyl isocyanate 16 were subjected to the urea-formation – cyclisation procedure described by Van der Eycken (Scheme 4a).16 Upon complete conversion to the imidazolone 11a, CsF and oSAT 12a were added and the reaction heated at 50 °C for 14 hours. Pleasingly, the desired BCB 13aa was isolated in 36% yield from propargyl amine 15via the one-pot, three-stage process. This compares favourably to performing two separate steps, first to isolate the imidazolone (94%) and then to generate the BCB (41%). Finally, the cyclic urea within the product BCBs could be ring-opened via a two-step reduction and acetylation procedure, to reveal the general syn-1,2-diaminoBCB framework 17 for future applications (Scheme 4b).
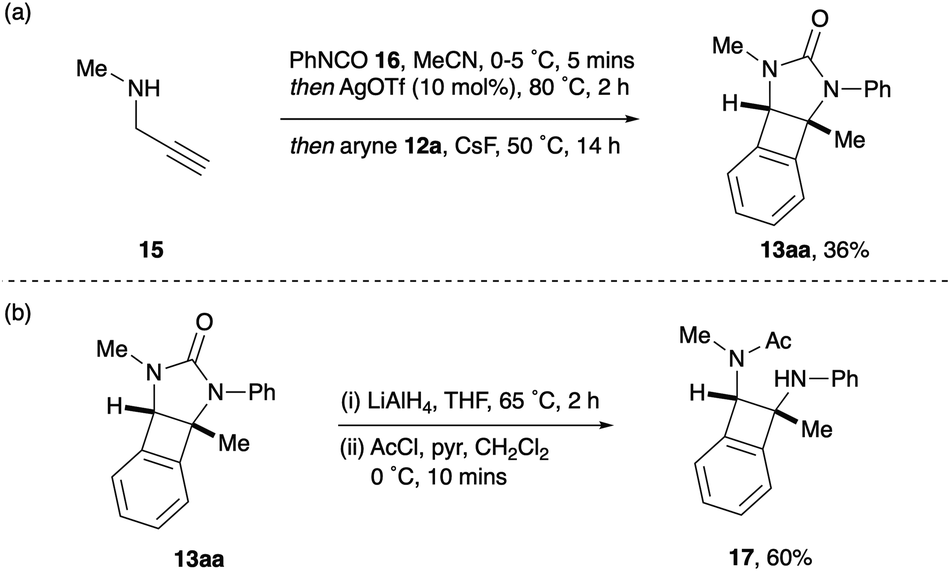 | ||
| Scheme 4 (a) One-pot formation of BCBs from propargyl amine. (b) Imidazolidone ring-opening. | ||
In conclusion, the formal [2+2] cycloaddition of 2-imidazolones and arynes has been achieved, delivering novel syn-1,2-diaminoBCBs. The transformation can also be conducted as a one-pot, three-stage process direct from commercially available propargylic amines and isocyanates, offering simple access to imidazolone substrate variation. Overcoming competing reactivity at nitrogen, in addition to other deleterious reaction pathways, proved a challenge for the methodology and accounts for the moderate yields typically observed. Nevertheless, these new stereochemically defined BCB frameworks offer attractive synthetic potential for use in asymmetric catalysis – as chiral ligands for transition metals or key components in organocatalysts – and as building blocks for both the synthesis of complex molecules and the preparation of polymeric materials.
We are grateful to the Higher Education Commission of Pakistan (studentship to HS) and China Scholarship Council (studentship to QL) for financial support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Selected reviews: (a) G. Mehta and S. Kotha, Tetrahedron, 2001, 57, 625 CrossRef CAS; (b) A. K. Sadana, R. K. Saini and W. E. Billups, Chem. Rev., 2003, 103, 1539 CrossRef CAS PubMed; (c) S. Kotha, K. Lahiri and Y. Tangella, Asian J. Org. Chem., 2021, 10, 3166 CrossRef CAS.
- Selected examples: (a) Y. Matsuya, S. Masuda, T. Itoh, T. Murai and H. Nemoto, J. Org. Chem., 2005, 70, 6898 CrossRef CAS PubMed; (b) Z. X. Ma, J. B. Feltenberger and R. P. Hsung, Org. Lett., 2012, 14, 2742 CrossRef CAS PubMed; (c) M. Mazurais, C. Lescot, P. Retailleau and P. Dauban, Eur. J. Org. Chem., 2014, 66 CrossRef CAS; (d) P. Maurina, D. Moraledab, H. Pellissier, R. Rodriguez and M. Santelli, Synlett, 2015, 725 Search PubMed.
- Selected examples: (a) M. F. Farona, Prog. Polym. Sci., 1996, 21, 505 CrossRef CAS; (b) J. Yang, Y. Huang and K. Cao, in Polymerisation, ed. A. De Silva Gomes, IntechOpen, 2012, vol. 9, p. 201 Search PubMed; (c) C. Pugh, J. S. Baker and W. K. Storms, Synlett, 2014, 148 CAS.
- Selected examples: (a) H. Finkelstein, Chem. Ber., 1959, 92, xxxvii CrossRef; (b) M. P. Cava and D. R. Napier, J. Am. Chem. Soc., 1956, 78, 500 CrossRef CAS; (c) M. S. Sardessai and H. N. Abramson, Org. Prep. Proced. Int., 1991, 23, 419 CrossRef CAS; (d) S. Kotha and P. Khedkar, J. Org. Chem., 2009, 74, 5667 CrossRef CAS PubMed; (e) Y. Wada, T. Tago, K. Sugata and J. Nishimura, J. Org. Chem., 1992, 57, 5955 CrossRef CAS; (f) H. Higuchi, T. Otsubo, F. Ogura, H. Yamaguchi, Y. Sakata and S. Misumi, Bull. Chem. Soc. Jpn., 1982, 55, 182 CrossRef CAS; (g) P. Muller, G. Bernardinelli, Y. Jacquier and A. Ricca, Helv. Chim. Acta, 1989, 72, 1618 CrossRef.
- (a) Z. Chen, X. Han, J. H. Liang, J. Yin, G. A. Yu and S. H. Liu, Chin. Chem. Lett., 2014, 25, 1535 CrossRef CAS; (b) X. Que, Y. Yan and Z. Qiu, Eur. Polym. J., 2018, 103, 410 CrossRef CAS.
- Y. Yang and C. R. Jones, Synthesis, 2022, 5042 CAS.
- (a) M. E. Kuehne, J. Am. Chem. Soc., 1962, 84, 837 CrossRef CAS; (b) J. B. Feltenberger, R. Hayashi, Y. Tang, E. S. C. Babiash and R. P. Hsung, Org. Lett., 2009, 11, 3666 CrossRef CAS PubMed; (c) Z.-X. Ma, J. B. Feltenberger and R. P. Hsung, Org. Lett., 2012, 14, 2742 CrossRef CAS PubMed; (d) A. Picard, S. Ciblat and Y. K. Ramtohul, Synlett, 2020, 507 CAS; (e) X. Cheng, N. Yang, W. Zeng, L. Wang, P. Li and H. Li, Chem. Commun., 2021, 57, 7047 RSC; (f) D. F. Dewez, C. Diacofotaki and G. Evano, Org. Chem. Front., 2021, 8, 6699 RSC.
- (a) R. V. Stevens and G. S. Bisacchi, J. Org. Chem., 1982, 47, 2393 CrossRef CAS; (b) P. Maurin, M. Ibrahim-Ouali and M. Santelli, Tetrahedron Lett., 2001, 42, 8147 CrossRef CAS; (c) T. Hamura, Y. Ibusuki, H. Uekusa, T. Matsumoto, J. S. Siegel, K. K. Baldridge and K. Suzuki, J. Am. Chem. Soc., 2006, 128, 10032 CrossRef CAS PubMed; (d) C. Pugh, J. S. Baker and W. K. Storms, Synlett, 2014, 148 CAS; (e) V. R. Yedulla, P. Pradhan, L. Yang and M. K. Lakshman, Eur. J. Org. Chem., 2015, 750 CrossRef CAS PubMed.
- Selected reviews: (a) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215 CrossRef CAS; (b) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (c) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191 RSC; (d) R. W. Hoffmann and K. Suzuki, Angew. Chem., Int. Ed., 2013, 52, 2655 CrossRef CAS PubMed; (e) D. Pérez, D. Peña and E. Guitían, Eur. J. Org. Chem., 2013, 5981 CrossRef; (f) J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (g) O. J. Diamond and T. J. Marder, Org. Chem. Front., 2017, 4, 891 RSC; (h) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044 RSC; (i) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892 CrossRef CAS PubMed.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211 CrossRef CAS.
- T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208 CrossRef CAS PubMed.
- Selected examples: (a) M. Lanzi, T. Rogge, T. S. Truong, K. N. Houk and J. Wencel-Delord, J. Am. Chem. Soc., 2023, 145, 345 CrossRef CAS PubMed; (b) R. A. Roberts, B. E. Metze, A. Nilova and D. R. Stuart, J. Am. Chem. Soc., 2023, 145, 3306 CrossRef CAS PubMed; (c) B. E. Metze, R. A. Roberts, A. Nilova and D. R. Stuart, Chem. Sci., 2023, 14, 13885 RSC; (d) O. Smith, M. J. Hindson, A. Sreenithya, V. Tataru, R. S. Paton, J. W. Burton and M. D. Smith, Nat. Synth., 2024, 3, 58 CrossRef.
- (a) P. Trinchera, W. Sun, J. E. Smith, D. Palomas, R. Crespo-Otero and C. R. Jones, Org. Lett., 2017, 19, 4644 CrossRef CAS PubMed; (b) F. I. M. Idiris, C. E. Majesté, G. B. Craven and C. R. Jones, Chem. Sci., 2018, 9, 2873 RSC; (c) W. Sun, P. Trinchera, N. Kurdi, D. Palomas, R. Crespo-Otero, S. Afshinjavid, F. Javid and C. R. Jones, Synthesis, 2018, 4591 CAS; (d) Y. Yang, Y. Xu and C. R. Jones, Eur. J. Org. Chem., 2019, 5196 CrossRef CAS; (e) E. A. Neal, A. Y. R. Werling and C. R. Jones, Chem. Commun., 2021, 57, 1663 RSC; (f) W. Sun, M. Uttendorfer, F. I. M. Idiris, A. W. Y. Werling, K. Siddiq and C. R. Jones, Chem. Commun., 2023, 59, 11823 RSC.
- Selected examples: (a) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580 CrossRef CAS; (b) S. R. Saibabu Kotti, C. Timmons and G. Li, Chem. Biol. Drug Des., 2006, 67, 101 CrossRef CAS PubMed; (c) J.-C. Kizirian, Chem. Rev., 2008, 108, 140 CrossRef CAS PubMed; (d) A. Mishra and S. Hanessian, Synthesis, 2024, 56, 2747 CrossRef CAS.
- Selected reviews: (a) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091 CrossRef CAS PubMed; (b) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56 CrossRef.
- V. A. Peshkov, O. P. Pereshivkov, S. Sharma, T. Meganathan, V. S. Parmar, D. S. Ermalot’ev and E. V. Van der Eycken, J. Org. Chem., 2011, 76, 2867 CrossRef PubMed.
- Unable to isolate a pure sample of 14aa for unequivocal characterisation, due to apparent instability of trisubstituted alkene in product. However, benzodiazepines 14la and 14la′ (containing a tetrasubstituted alkene and obtained from the reaction of imidazolone derivative 11l with aryne 2a) were isolable and fully characterised (see ESI† for details). Benzodiazepine 14aa was therefore proposed by analogy with the NMR spectroscopic data of 14la and 14la′ and supported by mass spectrometric data. Note: assignment of regioisomers was not possible for 14aa.
- (a) H. Yoshida, E. Shirakawa, Y. Honda and T. Hiyama, Angew. Chem., Int. Ed., 2002, 41, 3247 CrossRef CAS; (b) N. Saito, K. I. Nakamura, S. Shibano, S. Ide, M. Minami and Y. Sato, Org. Lett., 2013, 15, 386 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04023f |
| This journal is © The Royal Society of Chemistry 2024 |