
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuneable stimuli-responsive behaviour, spectroscopic signatures and redox properties of indolo[3,2-b]carbazole-based diradicals†
Irene
Badía-Domínguez
a,
Deliang
Wang
b,
Rosie
Nash
 c,
Víctor Hernández
Jolín
a,
David
Collison
d,
Muralidharan
Shanmugam
d,
Hongxiang
Li
*be,
František
Hartl
*c and
M. Carmen
Ruiz Delgado
*a
c,
Víctor Hernández
Jolín
a,
David
Collison
d,
Muralidharan
Shanmugam
d,
Hongxiang
Li
*be,
František
Hartl
*c and
M. Carmen
Ruiz Delgado
*a
aDepartment of Physical Chemistry, University of Málaga, Campus de Teatinos s/n, Málaga, 229071, Spain. E-mail: carmenrd@uma.es
bKey Laboratory of Synthetic and Self-assembly Chemistry for Organic Functional Materials, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
cDepartment of Chemistry, University of Reading, Whiteknights, Reading RG6 6DX, UK. E-mail: f.hartl@reading.ac.uk
dPhoton Science Institute and Department of Chemistry, The University of Manchester, Manchester M13 9PL, UK
eKey Laboratory for Advanced Materials, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: lihongxiang@ecust.edu.cn
First published on 9th September 2024
Abstract
During the last decade, there has been an increasing interest in the rationalisation of how structural changes stabilise (or destabilise) diradical systems. Demonstrated herein is that indolocarbazole (ICz) diradicals, substituted with dicyanomethylene (DCM) groups, are useful motifs for dynamic covalent chemistry by self-assembling from isolated monomers to cyclophane structures. The comparison of ICz-based systems substituted with DCM groups in para- or meta-positions (p-ICz-CN and m-ICz-CN) and their short-chain carbazole analogues (p-Cz-CN and m-Cz-CN) may identify new potential design strategies for stimuli-responsive materials. The principal objectives of this investigation are the elucidation of (i) the connection between diradical character and the cyclophane stability, (ii) the spatial disposition of the cyclophane structures, (iii) the monomer/cyclophane interconversion both in solution and solid state in response to external stimuli and (iv) the impact that the different π-conjugation and electronic communication between the DCM terminals exerts on the electronic adsorption of the diradicals and their redox behavior. The spontaneous nature of the cyclophane structure is supported by the negative relative Gibbs free energies calculated at 298 K and experimentally by UV-Vis and Raman spectroscopy of the initial yellow solid powder. The conversion to monomeric species having diradical character was demonstrated by variable-temperature (VT) EPR, UV-Vis, Raman and IR measurements, resulting in appreciable chromic changes. In addition, electrochemical oxidation and reduction convert the cyclophane dimer (m-ICz-CN)2 to the monomer monocations and dianions, respectively. This research demonstrates how the chemical reactivity and physical properties of π-conjugated diradicals can be effectively tuned by subtle changes in their chemical structures.
1. Introduction
Open-shell organic conjugated materials quickly attracted the attention of scientists because of their new chemical and physical properties with great potential in flexible organic electronics,1–4 spintronics,5,6 stimuli-responsive materials,7–17 spin-crossover materials18–21 or molecular self-assembly.22,23 Open-shell organic materials, known as radicals (classified as monoradicals, diradicals and other high-spin radicals), are small organic molecules with unpaired electrons. These molecules can build cyclophane structures by forming C–C σ-bonds between radical centres longer than typical C(sp3)–C(sp3) bonds.24–26 Therefore, these systems can show reversible association–dissociation behaviour by switching between closed-shell molecules and unpaired electron systems. These electronic spin-state changes would result in (thermo-, mechano-, photo-)chromic behaviour caused by an external stimulus, which makes it a very attractive tool for the design of stimuli-responsive materials. Thanks to this property, organic mono- and diradicals have emerged as essential building blocks in dynamic covalent chemistry (DCC).27–31Some of us have recently demonstrated how external stimuli impact the molecular structure and supramolecular organization, and thus upon the resulting optical and electronic properties of a family of carbazole (Cz) and indolocarbazole-based (ICz) diradicals with dicyanomethylene (DCM) groups.7,10,14,15,32 The beginning of the interest in this research was based on a Cz-based system substituted with dicyanomethylene (DCM) groups, para-connected (p-Cz-CN).7,10 This molecule formed long σ-bonds between the radical centres of the four different monomer units, resulting in a tetrameric cyclophane. The formation/dissociation of these long C–C bonds implies an equilibrium between the tetrameric structure and the isolated monomers in response to external conditions, giving rise to strong chromic changes (i.e., from the white colour corresponding to the cyclophane to purple ascribed to the diradical monomer). The next research line was based on a similar Cz-based compound with DCM groups meta-substituted (m-Cz-CN) which created cyclophanes structures (i.e., a trimer and a tetramer).7,10 The comparison of these two Cz-based systems (p-Cz-CN and m-Cz-CN) represented a magnificent test case to study the impact of the substitution pattern on the formation of stimuli-responsive cyclophanes.14 Later on, the influence of the elongation of the conjugated backbone was investigated through the characterization of an indolo[3-2,b]carbazole (ICz) compound substituted with DCM groups para-linked (p-ICz-CN).15 This molecule showed a dimeric cyclophane structure with two co-facial ICz units. Comparing the p-ICz-CN compound with its short-chain analogue p-Cz-CN, the extension of the conjugated chain increased the diradical character leading to stronger cyclophane structure with shorter C–C bonds and attractive π–π interactions.
Herein, a new ICz compound containing the terminal DCM groups in the meta-positions (m-ICz-CN) was synthesized and characterized to demonstrate how its chemical reactivity and physical properties are modulated by DCM substitution when compared to its para-substituted homologue (p-ICz-CN) (Fig. 1). This work aims to experimentally reveal the differences previously identified at the theoretical level between the m-ICz-CN and p-ICz-CN systems.32 Interestingly, the meta substitution pattern implies an unusual closed-shell (CS) molecule together with the presence of a zwitterionic resonance structure which will be determinant for the molecular and optoelectronic properties.33–36 The main objectives of this study are the investigation of (i) the connection between diradical character and the cyclophane stability, (ii) the spatial disposition of the cyclophane structures, (iii) the monomer/cyclophane interconversion both in solution and solid state in response to external stimuli and (iv) how the difference in π-conjugation and electronic communication between the DCM termini affects the electronic absorption and the redox behavior. To this end, a combined experimental and theoretical approach was developed to reach the proposed goals by linking electronic, vibrational (IR and Raman) and EPR spectroscopic and spectro-electrochemical techniques with density functional theory (DFT) calculations.
 | ||
| Fig. 1 Structures of (a) m-ICz-CN and (b) p-ICz-CN open-shell (OS) diradicals and their closed-shell (CS) quinoid/zwitterion resonance structures. The cyclophane dimeric arrangements of (a) m-ICz-CN and (b) p-ICz-CN are also shown. | ||
2. Results and discussion
2.1 Synthesis and experimental characterization
The detailed synthesis of (m-ICz-CN)2 is shown in Scheme S1 (ESI†). Based on the presence of the zwitterionic resonance structure of the m-ICz-CN monomer, a higher reactivity compound was expected which could promote a more feasible formation of a dimeric cyclophane structure. HPLC (high performance liquid chromatography) and GPC (gel permeation chromatography) results have shown that (m-ICz-CN)2 has a similar retention time as (p-ICz-CN)2,15 indicating that they exhibit similar aggregated structures (Fig. S1a and b, ESI†). In addition, the calculated BET surface area (Fig. S1c, ESI†) through the nitrogen adsorption–desorption isotherm has a value of 4.7 m2 g−1. This is in line with the creation of a reduced cavity structure in the aggregate, pointing out the formation of a cyclophane structure where two ICz units are co-facially coupled. Variable-temperature 1H NMR spectra showed that (m-ICz-CN)2 displayed clear hyperfine peak splitting at ambient temperature; as the temperature increased, the hyperfine splitting of the signals disappeared (Fig. S2, ESI†). The original 1H NMR spectrum was recovered when the solution was cooled back to ambient temperature. These results prove the formation of cyclophane dimer (m-ICz-CN)2 at ambient conditions (Fig. S3, ESI†), which tends to dissociate into diradical monomers at high temperatures (Fig. S2, ESI†). Mass spectrometry is an useful analytical tool for measuring the mass-to-charge ratio (m/z) of one or more molecules present in a sample which allows the assignment of possible structures to the detected fragmentation patterns.37 The mass spectrum of m-ICz-CN displays two main peaks (Fig. S4, ESI†); one peak is associated with the monomer structure (m/z 830) and the other one corresponds to double the molecular weight of the monomer (m/z 1660), in consonance with the co-facial coupling between the two diradical monomers giving rise to the dimeric cyclophane, (m-ICz-CN)2.2.2 DFT-calculations
 | ||
Fig. 2 (a) Potential energy curve (PES) scan of (p-ICz-CN)2 and (m-ICz-CN)2 in gas phase computed at the (U)M06-2X/6-31G** level. Here RCC is the length of the C–C σ-bonds formed between two ICz units. (b) Lateral and top view of the DFT-calculated global minimum structure for (m-ICz-CN)2 (left) and (p-ICz-CN)2 (right) displaying the distances of the closest C–C σ-bonds and π–π interactions between the co-facially oriented monomers. The N-substituted alkyl groups have been omitted for clarity. Free energies of formation  of the (m-ICz-CN)2 and (p-ICz-CN)2 are also shown. of the (m-ICz-CN)2 and (p-ICz-CN)2 are also shown. | ||
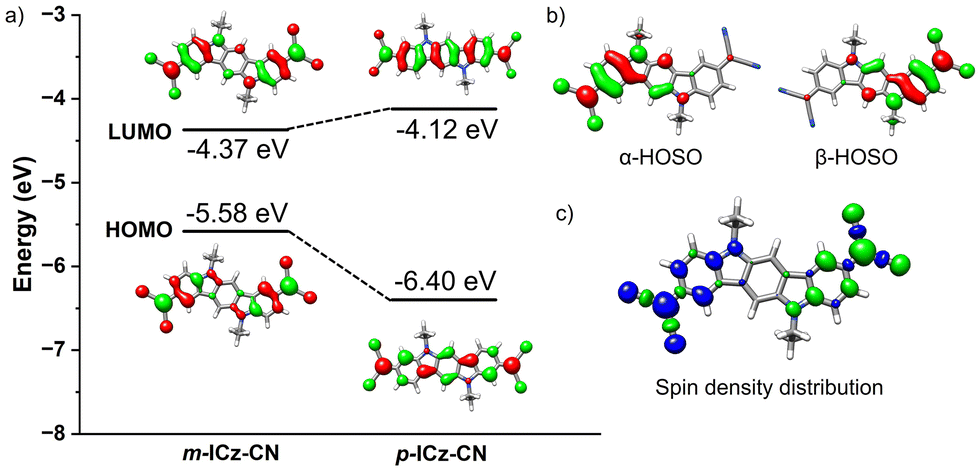 | ||
| Fig. 3 (a) DFT-calculated (M06-2X/6-31G**) molecular orbital diagram of m-ICz-CN and p-ICz-CN. (b) Calculated highest-occupied single orbitals (HOSO) of α and β electrons for m-ICz-CN. (c) Spin density distribution in the diradical singlet ground state of m-ICz-CN at the (U)M06-2X/6-31G** level of theory with an isosurface value of 0.03 a.u. The blue and green surfaces represent α and β spin densities, respectively. | ||
The investigation of the molecular orbitals of the open-shell (OS) singlet state is useful to get additional insight into the nature of the diradicals.38,39 As shown in Fig. 3b, the highest-occupied single orbital (HOSO) of α and β electrons in the m-ICz-CN system can be classified as singlet disjoint diradicals where the two unpaired electrons reside on different parts of the molecule with almost no overlap between them (see Fig. S7, ESI,† for p-ICz-CN). Interestingly, the resulting spin density distribution in the m-ICz-CN system is located exclusively on the external parts of the conjugated backbone. Consequently, the two unpaired electrons in m-ICz-CN are less electronically coupled because of the disruption of the effective π-conjugation.32,36 This coincides with the trend of the calculated diradical character shown below.
As seen in Fig. 4a the diradical character has been calculated using two methods (diradical character, y0, and fractional orbital density, NFOD). In all cases, the FOD plots show that the spatial distribution of the unpaired electrons is highly delocalized over the whole molecule with a strong contribution from the central carbon atoms of the DCM groups. However, the NFOD values can be categorized as (i) mild for p-Cz-CN (NFOD ∼ 1.3), (ii) moderate for m-Cz-CN (NFOD ∼ 1.6) and (iii) pronounced for p-ICz-CN and m-ICz-CN with the longest conjugated cores (NFOD ∼ 1.9 and 2, respectively). Thus, the diradical character can remarkably be tuned upon increasing the chain length (i.e., y0 = 0.59 in m-Cz-CNvs. y0 = 0.83 in m-ICz-CN) and when going from the para- to meta-substitution (i.e., y0 = 0.61 in p-ICz-CNvs. y0 = 0.83 in m-ICz-CN).
 | ||
| Fig. 4 (a) Isocontour plot of the FOD density (σ = 0.005 e bohr−3) and predicted NFOD values together with the diradical character y0 values of four DCM-substituted diradicals (p-Cz-CN,10,14m-Cz-CN,7,14p-ICz-CN15 and m-ICz-CN). (b) Comparison of the singlet–triplet gap (ΔES–T), (c) the HL gap and (d) the energy difference between OS and CS states (ΔEOS–CS) of p-Cz-CN,10,14m-Cz-CN,7,14p-ICz-CN15 and m-ICz-CN compounds at (U)M06-2X/6-31G** level of theory. | ||
As seen in Fig. 4b–d, DFT-calculated diradical character values are in consonance with the trend predicted for the physical parameters within the series (i.e., the larger diradical character, the larger ΔEOS–CS, the smaller ΔES–T and smaller HL gap values). Overall, these data reveal that the extension of the molecular size and the meta-substitution promote the diradical state. Thus, the calculations reveal a stronger diradical character of m-ICz-CN and more reactive radical sites compared to p-ICz-CN. Note that the OS singlet ground state of ICz-based systems is supported by negative Jab values (see Table S1, ESI†) revealing a lower interaction between the unpaired electrons in line with the large diradical character.
2.3 Cyclophane/monomer interconversion in solution
 | ||
| Fig. 5 (a) UV-Vis-NIR absorption of (m-ICz-CN)2 in o-DCB as a function of temperature. (b) UV-Vis-NIR spectral changes accompanying conversion of dimeric cyclophane (p-ICz-CN)2 to p-ICz-CN upon heating in o-DCB from 300 to 410 K. | ||
TD-DFT calculations support (i) the experimental UV-Vis-NIR spectral evolution when going from the dimeric cyclophanes to the isolated monomers (Fig. S9, ESI†), and (ii) the red-shifting of the maximum absorption band on going from the para- to the meta-substituted compound. These results are in good agreement with the HOMO–LUMO gap decreasing on going from (m-ICz-CN)2 to m-ICz-CN (Fig. S12, ESI†). Interestingly, the UV-Vis absorption spectrum of the brominated precursor m-ICz-Br resembles that of the aggregate (m-ICz-CN)2 (Fig. S9, ESI†). This is in line with the similar energy levels and topologies of the frontier molecular orbitals of m-ICz-Br and (m-ICz-CN)2 (Fig. S10–S12, ESI†), which in both cases exhibit an aromatic ICz backbone.
The magnetic properties of the dissolved monomers were investigated in o-dichlorobenzene (Fig. 6) and toluene (Fig. S16, ESI†) by variable-temperature (VT) EPR spectroscopy in the temperature range of 300–410 K and 300–380 K, respectively. The diradical resonance forms of both monomers exhibit in o-dichlorobenzene an unresolved singlet signal at g = 2.0028. The signals are very weak at ambient temperature, in line with the dominant diamagnetic dimeric cyclophane structure in the solution revealed by VT NMR and UV-Vis-NIR spectroscopies. The intensity of the EPR signal of p-ICz-CN grows much faster with increasing temperature compared to m-ICz-CN until 390 K (Fig. 6). This difference reflects the stronger C–C σ-bonds in (m-ICz-CN)2 (Fig. 2). Above 390 K the EPR signal of m-ICz-CN starts to grow abruptly faster. In less polar toluene, the broad EPR signal of p-ICz-CN shows a partly resolved hyperfine structure. This signal was growing in intensity with temperature less steeply compared to when heating in o-dichlorobenzene. For m-ICz-CN the difference became even larger and its EPR signal remained very weak up to 380 K (Fig. 6).
 | ||
| Fig. 6 (Left) Variable-temperature, cw-EPR spectra of diradical (a) m-ICz-CN and (c) p-ICz-CN generated from dimer cyclophane (m-ICz-CN)2 and (p-ICz-CN)2, respectively, in o-dichlorobenzene upon gradual temperature elevation from 300 K to 410 K. Experimental conditions: microwave power 23 dB (1 mW), modulation amplitude 1 G, time constant 82 ms, conversion time ∼12 ms, sweep time 30 s, receiver gain 30 dB and an average microwave frequency of 9.441 GHz. (Right) T-Dependent relative intensities of EPR signals of diradicals (b) m-ICz-CN and (d) p-ICz-CN measured as fluid solution in o-dichlorobenzene. | ||
These observations comply qualitatively well with the outcomes of the corresponding VT UV-Vis-NIR absorption measurements. The dissolved cyclophane-type dimers (p-ICz-CN)2 and (m-ICz-CN)2 remain stable at ambient temperature. Stronger thermal activation is required for (m-ICz-CN)2 to break the C–C σ-bonds and the π–π interaction between the monomer chains. The fluid solution spectra of p-ICz-CN and m-ICz-CN in o-dichlorobenzene and in toluene were both simulated with the same isotropic g = 2.0028 within experimental error and are shown in Fig. S16 and 17 in ESI.† The spectrum of p-ICz-CN in toluene shows poorly resolved hyperfine couplings due to the unknown number of 14N and 1H nuclei, which are not included in the simulation to avoid over-parameterisation.
 | ||
| Fig. 7 Cyclic voltammogram of (a) 0.2 mM m-ICz-Br and (b) 0.2 mM (m-ICz-CN)2 in CH2Cl2/Bu4NPF6 at 298 K; v = 100 mV s−1, Pt disk microelectrode. | ||
| Compound | E(ox.)/V | E(red.)/V | Ref. |
|---|---|---|---|
| a In dichloromethane/Bu4NPF6 at T = 298 K, Ep values vs. E1/2(Fc+/Fc), unless stated otherwise. b E 1/2 value, reversible redox reaction; ΔEp = 80–100 mV. c Beyond the electrode potential limit of the electrolyte. d Poorly resolved broad waves. e In o-dichlorobenzene/Bu4NPF6 at T = 393 K. f Unresolved 2e− step. g Closely spaced 1e− steps. | |||
| m-ICz-Br | 0.38ab | —c | This work |
| p-ICz-Br | 0.45ab | —c | 15 |
| (m-ICz-CN)2 | 0.84ae | This work | |
| (p-ICz-CN)2 | 0.63ab | 15 | |
| m-ICz-CN | 0.63e | −0.75ef | This work |
| p-ICz-CN | 0.70e | −0.46/−0.67beg | 15 |
| [m-ICz-CN]+ | —c | 0.57a | This work |
| [m-ICz-CN]2− | −0.45af | —c | This work |
| −0.61ef | —c | This work | |
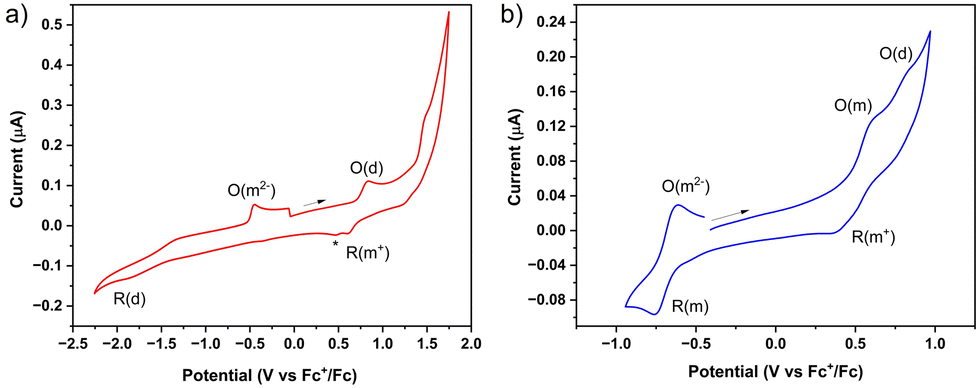 | ||
| Fig. 8 Cyclic voltammograms of (a) 0.2 mM (m-ICz-CN)2 in CH2Cl2/Bu4NPF6 at 293 K; v = 100 mV s−1, Pt disk microelectrode, and (b) ca. 0.2 mM m-ICz-CN in o-dichlorobenzene/Bu4NPF6 at 393 K; v = 100 mV s−1, Pt disk microelectrode. Abbreviations: d = dimer (m-ICz-CN)2, m = monomer m-ICz-CN, O = oxidation, R = reduction. | ||
The electrochemical reduction of (m-ICz-CN)2 at ambient temperature in the negative potential region is poorly defined, similar to the behaviour reported15 for (p-ICz-CN)2. The reason behind this observation is the thermal equilibrium between (m-ICz-CN)2 and m-ICz-CN that still exists in dichloromethane and in o-dichlorobenzene at ambient temperature despite being strongly shifted towards the dimer. The cyclic voltammogram of (m-ICz-CN)2 (Fig. 8a) shows a broad reduction wave (R(d)) around −1.9 V. Passing this wave and scanning back to the starting point leads to the appearance of a new oxidation wave at −0.45 V that is ascribed to the oxidation of the fully reduced monomer [m-ICz-CN]2−. This assignment, which complies with the reduction behaviour of (p-ICz-CN)2,15 receives support hereinafter from UV-Vis spectroelectrochemistry.
Cyclic voltammetry in o-dichlorobenzene at T = 393 K provides an important insight into the redox behaviour of monomer m-ICz-CN and supports the assignments made above for the cyclovoltammetric responses at ambient temperature where both the oxidation and reduction of (m-ICz-CN)2 triggers dissociation of the cyclophane C–C σ-bonds. Based on the EPR study under these conditions, the major component present in the electrolyte is monomer m-ICz-CN. This is also clearly reflected in the cyclic voltammogram in Fig. 8b (recorded in with a CV cell thermostatted at 393 K), which shows (m-ICz-CN)2 only as a minor component (the anodic wave O(d) at 0.84 V). The dominant oxidation wave O(m) at 0.63 V belongs to m-ICz-CN, being less positively shifted compared to that of p-ICz-CN under the same conditions (Table 1) due to the higher-lying HOMO of m-ICz-CN (−5.58 eV vs. −6.40 V calculated for p-ICz-CN). A very important new feature in the high-temperature cyclic voltammogram of dominant m-ICz-CN is its reduction wave R(m) at −0.75 V, which is negatively shifted in comparison to the reduction of p-ICz-CN (Table 1) in line with the higher-lying LUMO (−4.37 eV (meta) vs. −4.12 eV (para)). The scan-reversal oxidation wave O(m2−) at −0.61 V belongs to [m-ICz-CN]2− converting directly back to neutral m-ICz-CN, as confirmed by UV-Vis spectroelectrochemistry in the following section. The striking difference between the unresolved two-electron reduction of the radical centres in m-ICz-CN to [m-ICz-CN]2− and the stepwise one-electron reduction of p-ICz-CN to [p-ICz-CN]− and [p-ICz-CN]2− (Table 1) can be explained by considering the very limited electronic communication between the radical halves of m-ICz-CN through the central benzene ring, visualized by the LUMO characters in Fig. 3.
 | ||
| Fig. 9 UV-Vis absorption spectral changes resulting from oxidation of m-ICz-Br (blue) to [m-ICz-Br]+ (red.). Conditions: in CH2Cl2/Bu4NPF6 at T = 298 K; Pt minigrid in an OTTLE cell. | ||
Changes in the UV-Vis absorption spectra accompanying the oxidation and reduction of (m-ICz-CN)2 were recorded in parallel with the thin-layer cyclic voltammogram at a Pt mesh (Fig. S13, ESI†). The latter reproduced all key features in the conventional cyclic voltammogram of the dimer cyclophane at a Pt microelectrode (Fig. 10). The oxidation of (m-ICz-CN)2 may result either in stable [(m-ICz-CN)2]+ (as observed15 for (p-ICz-CN)2), or monomer radical cation [m-ICz-CN]+. The experimental spectra recorded at the oxidation wave O(d) are shown in Fig. 10a, revealing smooth conversion to a single product. TDDFT calculations (Fig. S14 (top) and Table S3, ESI†) allow its assignment to stable [m-ICz-CN]+, in particular by the presence of medium-intensity absorption between 400–500 nm, seen in the experimental spectrum as shoulders at the intense structured band at 380 nm. Only very weak absorption in this region is predicted for [(m-ICz-CN)2]+ (Fig. S13, ESI†). similar to that reported15 for stable [(p-ICz-CN)2]+.
 | ||
| Fig. 10 UV-Vis absorption spectral changes resulting from (a) oxidation of (m-ICz-CN)2 (blue) to [m-ICz-CN]+ (purple), and (b) single-step reduction of (m-ICz-CN)2 (blue) to [m-ICz-CN]2− (green). Conditions: in CH2Cl2/Bu4NPF6 at T = 298 K; Pt minigrid in an OTTLE cell. | ||
The electrochemical reduction of (m-ICz-CN)2 within the OTTLE cell at the wave R(d) is shifted to a less negative electrode potential compared to the conventional CV response (Fig. 10a). This has been ascribed15 to a dynamic behaviour of the dimer cyclophane that co-exists with the corresponding diradical monomer, m-ICz-CN. No observable response of m-ICz-CN in the conventional CV at a less negative reduction potential (much below −1 V) complies with the solution VT EPR data and stronger C–C σ bonds in (m-ICz-CN)2. UV-Vis spectral changes accompanying the single-step reduction of (m-ICz-CN)2 at R(d) are presented in Fig. 10b. The single product exhibits characteristic medium-intensity absorption in the visible region between 400–550 nm and two strong structured bands in the UV region. This pattern complies with TDDFT data calculated for the doubly reduced monomer [m-ICz-CN]2− (Table S3, middle, ESI†). The UV-Vis absorption spectrum of [m-ICz-CN]2− also closely resembles that of [p-ICz-CN]2− published elsewhere.15 Reverse oxidation of [m-ICz-CN]2− at O(m2−) (Fig. S13, ESI†) fully restored the electronic absorption of parent (m-ICz-CN)2 in a single step, alike the stepwise reoxidation of [p-ICz-CN]2−. As discussed above, the stronger diradical nature of m-ICz-CN not only results in the higher stability of (m-ICz-CN)2 (alike the other redox states of the cyclophane dimer) but also imposes the instability of the singly reduced (‘mixed-valence’) radical anions [m-ICz-CN]− that rapidly disproportionate to [m-ICz-CN]2− and reducible parent m-ICz-CN. The likely zwitterionic resonance structure of m-ICz-CN, in contrast to the more π-conjugated quinoidal resonance form of p-ICz-CN,15 prevents sufficient electronic communication between the radical termini in m-ICz-CN and separation of the two one-electron reduction steps, as observed15 for the series [m-ICz-CN]n− (n = 0–2).
2.4 Cyclophane/monomer interconversion in solid-state
 | ||
| Fig. 11 IR spectrum as a yellow KBr pellet at RT (top), after heating (middle) and back to RT (down) for (a) (p-ICz-CN)2 and (b) (m-ICz-CN)2 compounds. | ||
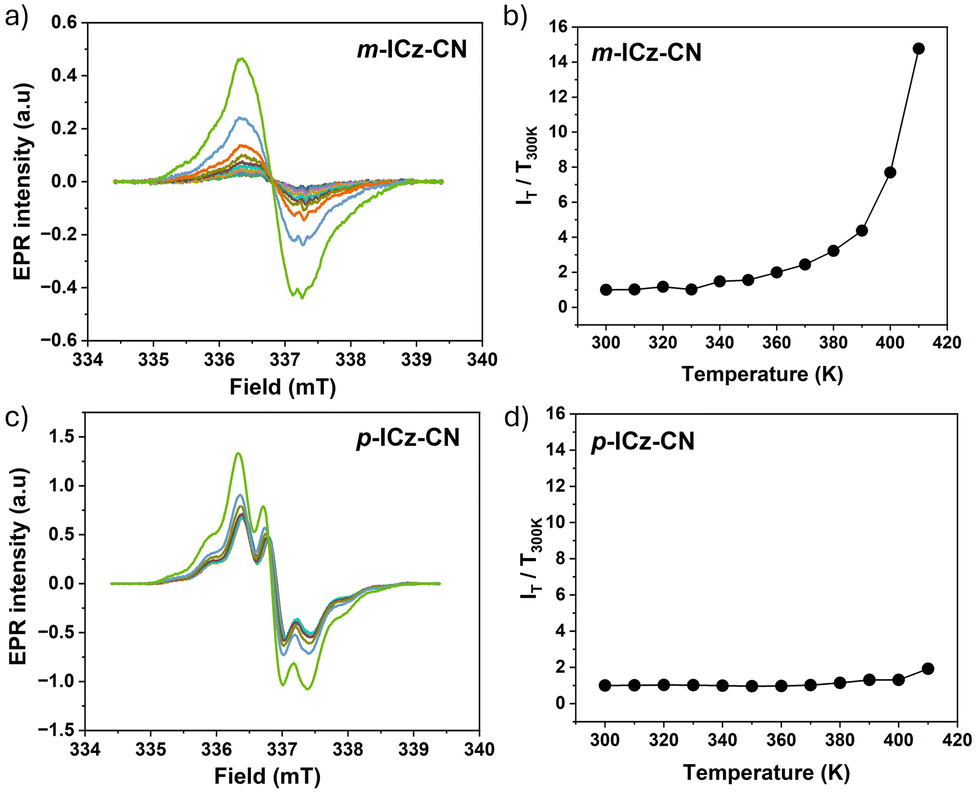 | ||
| Fig. 12 Variable temperature, cw-EPR spectra of diradical (a) m-ICz-CN and (c) p-ICz-CN generated from dimer cyclophane (m-ICz-CN)2 and (p-ICz-CN)2, respectively, in solid state upon gradual temperature elevation from 300 K to 410 K. Experimental conditions: see Fig. 6. T-Dependent relative intensities of EPR signals of diradicals (b) m-ICz-CN and (d) p-ICz-CN measured as polycrystalline powder. | ||
 | ||
| Fig. 13 (a) Top: FT-Raman spectra at λexc = 1064 nm of the yellow powder of (m-ICz-CN)2 (black line) and its brominated precursor m-ICz-Br (black dots). Bottom: Raman spectrum at λexc = 785 nm of isolated diradical m-ICz-CN in the solid state at 410 K. (b) Theoretical Raman spectra of (m-ICz-CN)2 (top) and m-ICz-CN (down) at the M06-2X/6-31G** level. | ||
Interestingly, in the range between 2300 cm−1 and 2180 cm−1 (related to the CN stretching modes), the band at 2250 cm−1 undergoes a downshift to 2205 cm−1 upon heating, corresponding to the formation of the isolated diradical m-ICz-CN (Fig. S15, ESI†); this is in line with the IR spectroscopic data. Overall, the Raman spectral evolution reveals that the cyclophane/monomer transformation involves a loss of aromaticity of the ICz units and the appearance of a more conjugated scenario of the CN groups.
3. Conclusions
In this work, the different structural and electronic parameters that control the cyclophane formation from ICz-based diradical systems are investigated in depth. Subtle modifications of the chemical structure (i.e., DCM substitution position, core elongation) are found to significantly affect the dynamic covalent chemistry behaviour. Theoretical data reveal that: (i) the elongation of the conjugated core and the meta-substitution provoke less interaction between the unpaired electrons resulting in a large diradical character, and (ii) meta-substituted ICz-based systems can form more stable cyclophanes when compared to their para-substituted analogues. All of these factors are shown to influence the cyclophane/monomer transformation. Interestingly, the dynamic ICz-based cyclophane/monomer transformation is accompanied by a colour change from light yellow to deep green, although a less reversible dissociation is found in the ICz-based diradicals when compared to the shorter Cz-based analogues. On the other hand, the effect of the linkage position is key to understand how the external stimuli affect the C–C bond dissociation. The meta substitution pattern favours the presence of a zwitterionic resonance structure in m-ICz-CN, resulting in a less favoured dissociation towards the monomer in solution compared to the para-substituted system. However, a more feasible C–C bond dissociation is found in the solid state when comparing meta vs. para, since a more restricted conformational space is created in solid state and intermolecular interactions might play a significant role. This is in line with a recent theoretical study that reveals that resonance structure and the diradical character can be modulated even in regions dominated by the dispersion forces.48 This cyclophane/monomer conversion has successfully been followed by NMR, vibrational spectroscopy and EPR techniques. In addition, the redox properties of the isomeric cyclophane dimers (at ambient temperature) and diradical monomers (at elevated temperature) confirm the differences in electronic structures and frontier orbital energies. The dissociation of the C–C σ-bonds in (m-ICz-CN)2 upon both oxidation and reduction was also revealed by UV-Vis spectro-electrochemistry. To summarize, this comprehensive study helps to evaluate the stimuli-responsive properties of diradical-based systems, providing new strategies for the design of novel dynamic covalent chemistry materials.4. Methodology
4.1 Experimental details
Commercial reagents were used without further purification and anhydrous solvents were purified by standard distillation methods prior to use. NMR spectra were recorded on an Agilent 400 spectrometer. Mass spectra were recorded on a Orbitrap Q-Exactive high-resolution mass spectrometer and atmospheric-pressure ionization system using heated electrospray H-ESI (Thermo Scientific). Elemental analyses were performed on an Elementar Vario EL III elemental analyzer. Gel permeation chromatography results were recorded on a PolymerLaboratories GPC 220 instrument, using standard polystyrene (PS) as the reference. High-performance liquid chromatography measurements were carried out on an Agilent ZORBAX Eclipse XDB-C18 apparatus. Nitrogen adsorption–desorption isotherm was measured at 77.3 K using a micromeritics ASAP2020 HD88 surface area and porosity analyzer. The samples were degassed at 393 K for 12 h under vacuum (10−5 bar) before analysis. The surface areas were calculated in the relative pressure (P/P0) range from 0.05 to 0.30.The characteristics of the analyses performed by high-performance liquid chromatography (HPLC) are as follows. Chromatographic column: cortex C18, 2.7 μm, 4.6 × 100 mm mobile phase: water (A), acetonitrile (B) gradient elution: 0 min 50% B, 10 min 100% B, 15 min 100% B. Flow rate: 0.6 mL min−1 temperature: ambient temperature. Sample preparation: we take an appropriate amount of the sample, dissolve it in 200 μL of chloroform, and add 800 μL of acetonitrile. We filter the organic phase through a membrane filter and proceed to HPLC analysis.
UV-Vis-NIR spectra were measured on an Agilent 8453 diode-array spectrophotometer between 190–1100 nm, with a resolution of 1 nm. A Varian Cary 5000 spectrophotometer was used in the UV-Vis-NIR range (170–3300 nm), with a PbSmart detector and 0.4 nm resolution. Variable-temperature UV-Vis-NIR absorption spectra were recorded using an optical cryostat Optistat DN (Oxford Instruments). IR spectra were carried out in the spectral range 4000–400 cm−1 using a Bruker Vertex 70 FT-IR spectrometer, which consists of a KBr beam splitter and a DLaGTS detector. A spectral resolution of 4 cm−1 and 64 scans were used to record the spectra. The compound was studied dispersed in KBr pellets (transmission). For variable-temperature FT-IR measurements, the Specac P/N 2100 accessory was used, which allows the recording of both solid and liquid samples. It has a Graseby Specac automatic temperature control system and can work in the range of 103 to 523 K.
Raman measurements were performed in an FT-Raman Bruker spectrometer which makes use of a continuous-wave Nd-YAG laser working at 1064 nm for excitation and a germanium detector operating at liquid nitrogen temperature. Raman radiation performs the detection in a back-scattering geometry, forming an angle of 180° between the incident radiation and the scattered radiation with a standard spectral resolution of 4 cm−1. Variable-temperature Raman spectra were registered by using a Bruker Senterra dispersive Raman microscope which makes use of two integrated lasers, with a wavelength of 785 nm. The detection system is a charge couple device (CCD) that is thermoelectrically cooled to 208 K. It also consists of a Neon lamp, used for laser calibration. For thermo-spectroscopic measurements, the Linkam FTIR600 accessory was used, which allows to access a thermal range between 93 and 873 K. Cyclic voltammetry (CV) was performed under an inert argon atmosphere with a heart-shaped single-compartment glass cell containing a coiled Ag wire pseudo-reference electrode, a 0.4-mm diameter Pt microdisc working electrode, and a coiled Pt wire counter electrode. The cell was held inside an earthed Faraday cage and connected to a Metrohm-Autolab PGSTAT302N potentiostat. The internal standard ferrocenium/ferrocene (Fc+/Fc) was added prior to the final scans. The sample solutions were 0.2 mM in analyte dissolved in dichloromethane containing 10−1 M tetrabutylammonium hexafluorophosphate (TBAH). UV-Vis spectroelectrochemistry was performed using an optically transparent thin-layer electrochemical (OTTLE) cell49 equipped with a Pt minigrid working electrode. The sample solution contained 2 × 10−1 M TBAH as the supporting electrolyte and 1 mM redox active compounds in dichloromethane. The course of the spectroelectrochemical experiment was monitored by thin-layer cyclic voltammetry (v = 2 mV s−1), with a potential control by a PalmSens EmStat3 potentiostat operated with the PSTrace5 software. UV-Vis spectra were recorded with a Scinco S-3100 diode-array spectrophotometer covering 200–1100 nm.
All EPR samples were prepared either as a polycrystalline powder or as fluid solutions in o-dichlorobenzene/toluene solvents. Microwave power saturation experiments were performed with reference p-ICz-CN (a solid-state sample) and TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) in toluene (data not shown). From the analysis, non-saturating experimental conditions were chosen for all EPR experiments, which are: microwave power of 23 dB (1.0 mW) and modulation amplitude (MA) of 1 G for both fluid solutions and polycrystalline powders at room/high temperature. For quantitative/qualitative analysis of the polycrystalline powders, both p-ICz-CN (ca. 0.34 mg) and m-ICz-CN (ca. 0.4 mg) were loaded into a 1.3-mm (OD)/1-mm (ID) glass-capillary tube at ca. 21-mm height. This glass capillary tube was inserted into a 4.0-mm (OD)/3-mm (ID) quartz EPR tube (Wilmad LabGlass) for subsequent measurements. The fluid solutions in o-dichlorobenzene contained ca. 0.4 mg of p-ICz-CN in 20 μL and ca. 1.1 mg of m-ICz-CN in 50 μL. The fluid solutions in toluene contained an undetermined amount of p-ICz-CN soluble in 70 μL and ca. 0.82 mg of m-ICz-CN in 40 μL. Also, the solutions were loaded into a 1.3-mm (OD)/1-mm (ID) glass-capillary tube at ca. 21-mm height. The optimum position of the samples (both powders and solutions) inside the sample cavity (resonator) and the temperature at the sample were calibrated before the actual EPR measurements. Note that in addition to the diradical resonance forms of both monomers which exhibit in o-dichlorobenzene an unresolved singlet signal at g = 2.0028, an extremely weak, sharp radical signal is also observed at 3371 G with g = 2.0005 and its contribution to the overall spectrum is estimated to be less than one percent (see Fig. S17, ESI†). This weak sharp EPR signal is likely to belong to a trace paramagnetic impurity in the quartz tube(s) used for the measurements.
The RT and elevated temperatures X-band measurements were achieved on an EMXmicro EPR spectrometer using the Bruker ER414VT variable temperature accessory, connected to an ER 4112SHQ X-band resonator equipped with cooling side plates (ER4112SHQE-1004). The optimum conditions used for recording the spectra are: microwave power 23 dB (1 mW), modulation amplitude 1 G, time constant 82 ms, conversion time ∼12 ms, sweep time 30 s, receiver gain 30 dB and an average microwave frequency of 9.441 GHz. All experimental parameters were kept constant, except sample concentration and/or weight. All spectra were multi-scanned and averaged (ca. 30 scans) to obtain good S/N ratio. The analysis of the continuous wave EPR spectra and simulations were performed using EasySpin toolbox (5.2.36) for the Matlab program package.50 The observed polycrystalline powder and fluid solutions spectra looks similar to that of the previously reported open-shell monomer, p-ICz-CN of EPR-silent cyclophane dimer, (p-ICz-CN)2.15
4.2 Theoretical details
All geometry optimizations were carried out in the gas-phase, using the Gaussian 16 package51 based on the density functional theory (DFT). In all calculations, the long alkyl N-substituents were replaced by ethyl groups. Herein, we have made use of the exchange–correlation functional M06-2X52 and the 6-31G (d,p)53,54 basis set. This functional has been parameterized so that the electronic correlation for medium-range distances (5 Å or less) is enough to describe the non-covalent interactions within many complexes.55 This fact allows that the functional M06-2X has widely been used to solve problems of non-covalent interactions, and so far, it has been recognized as one of the best in the description of the interactions of π–π stacking and the study of molecular aggregates (dimeric species).56 The broken-symmetry solution was simulated for OS singlet species and triplet state form using the keyword guess = mix and unrestricted wavefunction. To evaluate the importance of the solvent on the pancake dimer structural properties, the geometries optimized were obtained in toluene, chloroform and o-DCB by using the PCM method57 (see Table S2, ESI†). The effective electron exchange interaction (Jab) which reflects the overlap integral between the two nearly energetically degenerate molecular orbitals was calculated by using eqn (1): | (1) |
The spin correction for ΔES–T was computed by eqn (2).
 | (2) |
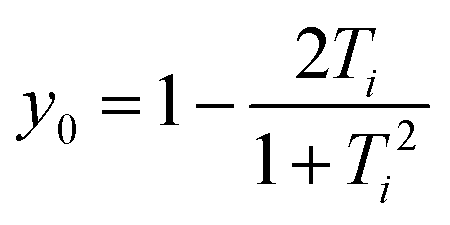 | (3) |
 | (4) |
Second, the fractional orbital density (FOD) method incorporating a strong correlation effect has also been used to calculate the NFOD values by the ORCA 4.0 program.71 An analysis based on FOD was performed, whose main characteristics is to provide the localization of “hot” electrons (strongly correlated and chemically active) in a molecule.72,73 This method gives a quantitative description of the OS diradical form in the ground state. We used the FT-TPSS74/def2-TZVP75 level at a default electronic temperature (Tel) of 5000 K in order to calculate the number of “hot” electrons, NFOD. The isocontour value of the ρFOD plots was fixed to 0.005 e bohr−3.
Author contributions
V. H. J., I. B. D. and M. C. R. D: DFT-calculations together with UV-Vis absorption and Raman/IR spectra at RT and T-dependent. R. N. and F. H.: redox properties and spectro-electrochemistry experiments. D. C. and M. S.: T-dependent EPR experiments. H. L. and D. W.: synthesis and characterization of the sample. All authors have read and agreed to the published version of the manuscript.Data availability
The data that support the findings of this study are available within the article and the ESI.† Details about methodology, experimental procedures, characterization data, theoretical calculations, EPR studies and NMR spectra are included.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work at the University of Málaga was funded by MICINN/AEI/10.13039/501100011033 (project PID2022-139548NB-I00) and by Junta de Andalucía (FQM-159). The authors would like to thank the computer resources, technical expertise and assistance provided by the SCBI (Supercomputing and Bioinformatics) center and the vibrational spectroscopy (EVI) lab of the Research Central Services (SCAI) of the University of Málaga. We thank EPSRC (EP/W014521/1, EP/X034623/1, EP/V035231/1, NS/A000055/1) and the University of Manchester for access to the EPR Spectroscopy National Research Facility. The work at SIOC was supported by National Natural Science Foundation of China (grants no. 21875279, 22075080 and 21790362). The spectro-electrochemical studies were fully funded by Spectroelectrochemistry Reading (a spinout company of the University of Reading led by F. H.).References
- X. Hu, W. Wang, D. Wang and Y. Zheng, The electronic applications of stable diradicaloids: present and future, J. Mater. Chem. C, 2018, 6, 11232 RSC.
- Z. Cui, A. Abdurahman, X. Ai and F. Li, Stable luminescent radicals and radical-based LEDs with doublet emission, CCS Chem., 2020, 2, 1129–1145 CrossRef CAS.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, 6,12-Diarylindeno[1,2-b]fluorenes: Syntheses, Photophysics, and Ambipolar OFETs, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- A. M. Zeidell, L. Jennings, C. K. Frederickson, Q. Ai, J. J. Dressler, L. N. Zakharov, C. Risko, M. M. Haley and O. D. Jurchescu, Organic Semiconductors Derived from Dinaphtho-Fused s-Indacenes: How Molecular Structure and Film Morphology Influence Thin-Film Transistor Performance, Chem. Mater., 2019, 31, 6962–6970 CrossRef CAS.
- A. Sánchez-Grande, J. I. Urgel, A. Cahlík, J. Santos, S. Edalatmanesh, E. Rodríguez-Sánchez, K. Lauwaet, P. Mutombo, D. Nachtigallová, R. Nieman, H. Lischka, B. de la Torre, R. Miranda, O. Gröning, N. Martín, P. Jelínek and D. Écija, Diradical organic one-dimensional polymers synthesized on a metallic surface, Angew. Chem., Int. Ed., 2020, 59, 17594–17599 CrossRef PubMed.
- W. Zeng, H. Phan, H. Yamada, J. Ding, J. Wu, T. S. Herng, T. Y. Gopalakrishna, N. Aratani and Z. Zeng, Rylene ribbons with unusual diradical character, Chem, 2017, 2, 81–92 CAS.
- T. Kobashi, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2016, 128, 8776–8780 CrossRef.
- J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS.
- K. Okino, S. H. Hira, Y. Inoue, D. Sakamaki and S. Seki, N-Substituted Dicyanomethylphenyl Radicals: Dynamic Covalent Properties and Formation of Stimuli-Responsive Cyclophanes by Self-Assembly, Angew. Chem., Int. Ed., 2017, 56, 16597–16601 CrossRef CAS PubMed.
- D. Wang, C. Capel Ferrón, J. Li, S. Gámez-Valenzuela, R. Ponce Ortiz, J. T. López Navarrete, V. Hernández Jolín, X. Yang, M. Peña Álvarez, V. García Baonza, F. Hartl, M. C. Ruiz Delgado and H. Li, New Multiresponsive Chromic Soft Materials: Dynamic Interconversion of Short 2,7-Dicyanomethylenecarbazole-Based Biradicaloid and the Corresponding Cyclophane Tetramer, Chem. – Eur. J., 2017, 23, 13776–13783 CrossRef CAS PubMed.
- R. Zhang, J. P. Peterson, L. J. Fischer, A. Ellern and A. H. Winter, Effect of Structure on the Spin–Spin Interactions of Tethered Dicyanomethyl Diradicals, J. Am. Chem. Soc., 2018, 140, 14308–14313 CrossRef CAS PubMed.
- H. Yokoi, S. Hiroto and H. Shinokubo, Reversible σ-bond formation in bowl-shaped π-radical cations: the effects of curved and planar structures, J. Am. Chem. Soc., 2018, 140, 4649–4655 CrossRef CAS.
- B. Adinarayana, D. Shimizu, K. Furukawa and A. Osuka, Stable radical versus reversible σ-bond formation of (porphyrinyl) dicyanomethyl radicals, Chem. Sci., 2019, 10, 6007–6012 RSC.
- I. Badía-Domínguez, J. Carlos Sancho-García, J. T. López Navarrete, V. Hernández Jolín, H. Li, D. Sakamaki, S. Seki and M. C. Ruiz Delgado, Formation of Cyclophane Macrocycles in Carbazole-Based Biradicaloids: Impact of the Dicyanomethylene Substitution Position, ACS Omega, 2019, 4, 4761–4769 CrossRef.
- I. Badía-Domínguez, M. Peña Álvarez, D. Wang, A. Perez Guardiola, Y. Vida, S. Rodríguez González, J. T. López Navarrete, V. Hernández Jolín, J. C. Sancho García, V. García Baonza, R. Nash, F. Hartl, H. Li and M. C. Ruiz Delgado, Dynamic Covalent Properties of a Novel Indolo[3,2-b]carbazole Diradical, Chem. – Eur. J., 2021, 27, 5509–5520 CrossRef PubMed.
- L. Moshniaha, M. Żyła-Karwowska, P. J. Chmielewski, T. Lis, J. Cybińska, E. Gońka, J. Oschwald, T. Drewello, S. M. Rivero, J. Casado and M. Stepień, Aromatic nanosandwich obtained by σ-dimerization of a nanographenoid π-radical, J. Am. Chem. Soc., 2020, 142, 3626–3635 CrossRef CAS.
- T. Xu, J. Zhu, Y. Han and C. Chi, Carbon-centered radical based dynamic covalent chemistry for stimuli-responsive chromic materials, J. Mater. Chem. C, 2023, 11, 7957 RSC.
- M. R. Geraskina, A. T. Buck and A. H. Winter, An organic spin crossover material in water from a covalently linked radical dyad, J. Org. Chem., 2014, 79, 7723–7727 CrossRef CAS.
- M. Juetten, A. Buck and A. Winter, A radical spin on viologen polymers: organic spin crossover materials in water, Chem. Commun., 2015, 51, 25 RSC.
- R. Zhang, A. Ellern and A. Winter, Steric Hindrance Favors σ Dimerization over π Dimerization for Julolidine Dicyanomethyl Radicals, J. Org. Chem., 2022, 87, 1507–1511 CrossRef CAS PubMed.
- S. Mishra, M. Vilas-Varela, L. A. Lieske, R. Ortiz, S. Fatayer, I. Rončević, F. Albrecht, T. Frederiksen, D. Peña and L. Gross, Bistability between π-diradical open-shell and closed-shell states in indeno[1,2-a]fluorene, Nat. Chem., 2024, 16, 755–761 CrossRef CAS.
- Y. Jin, C. Yu, R. Denman and W. Zang, Recent advances in dynamic covalent chemistry, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- S. Dong and Z. Li, Recent progress in open-shell organic conjugated materials and their aggregated states, J. Mater. Chem. C, 2022, 10, 2431–2449 RSC.
- D. Beaudoin, O. Levasseur-Grenon, T. Maris and J. D. Wuest, Building Giant Carbocycles by Reversible C–C Bond Formation, Angew. Chem., Int. Ed., 2016, 55, 894–898 CrossRef CAS PubMed.
- L. Yuan, Y. Han, T. Tao, H. Phan and C. Chi, Formation of a Macrocycles-in-a-Macrocycle Superstructure with All-gauche Conformation by Reversible Radical Association, Angew. Chem., Int. Ed., 2018, 130, 9161–9165 CrossRef.
- M. Liu, X. Yang, Q. Sun, T. Wang, R. Pei, X. Yang, Y. Zhao, L. Zhao, G. Frenking and X. Wang, Lewis Acid-Mediated Radical Stabilization and Dynamic Covalent Bonding: Tunable, Reversible and Photocontrollable, Angew. Chem., Int. Ed., 2023, 62, e202300068 CrossRef CAS PubMed.
- S. Rowan, S. Cantrill, G. Cousins, J. K. Sanders and J. F. Stoddart, Dynamic covalent chemistry, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- Y. Jin, C. Yu, R. Denman and Z. Wang, Recent advances in dynamic covalent chemistry, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- J. Winne, L. Leibler and F. E. Du Prez, Dynamic covalent chemistry in polymer networks: a mechanistic perspective, Polym. Chem., 2019, 10, 6091–6108 RSC.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Dynamic covalent chemistry approaches toward macrocycles, molecular cages, and polymers, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS.
- L. You, Dual reactivity based dynamic covalent chemistry: mechanisms and applications, Chem. Commun., 2023, 59, 12943–12958 RSC.
- I. Badía-Domínguez, S. Canola, V. Hernández Jolín, J. T. López Navarrete, J. C. Sancho-García, F. Negri and M. C. Ruiz Delgado, Tuning the diradical character of indolocarbazoles: impact of structural isomerism and substitution position, J. Phys. Chem. Lett., 2022, 13, 6003–6010 CrossRef.
- D. Sakamaki, S. Yano, T. Kobashi, S. Seki, T. Kurahashi, S. Matsubara, A. Ito and K. Tanaka, A Triphenylamine with Two Phenoxy Radicals Having Unusual Bonding Patterns and a Closed-Shell Electronic State, Angew. Chem., Int. Ed., 2015, 127, 8385–8388 CrossRef.
- X. Hu, H. Chen, L. Zhao, M. Miao, X. Zeng and Y. Zeng, Nitrogen-coupled blatter diradicals: the fused versus unfused bridges, J. Mater. Chem. C, 2019, 7, 10460–10464 RSC.
- F. Miao, Y. Ji, B. Han, S. Quintero, H. Chen, G. Xue, L. Cai, J. Casado and Y. Zheng, Asymmetric and zwitterionic Blatter diradicals, Chem. Sci., 2023, 14, 2698–2705 RSC.
- G. Xue, X. Hu, H. Chen, L. Ge, W. Wang, J. Xiong, F. Miao and Y. Zheng, Understanding the nature of quinoidal and zwitterionic states in carbazole-based diradicals, Chem. Commun., 2020, 56, 5143–5146 RSC.
- J. Mehara and J. Roithová, Identifying reactive intermediates by mass spectrometry, Chem. Sci., 2020, 11, 11960–11972 RSC.
- Z. D. Pozun, X. Su and K. D. Jordan, Establishing the ground state of the disjoint diradical tetramethyleneethane with quantum Monte Carlo, J. Am. Chem. Soc., 2013, 135, 13862–13869 CrossRef CAS PubMed.
- P. Ravat and M. Baumgarten, “Tschitschibabin type biradicals”: benzenoid or quinoid?, Phys. Chem. Chem. Phys., 2015, 17, 983–991 RSC.
- P. Rapta, L. Kress, P. Hapiot and L. Dunsch, Dimerization of thianthrene radical cations as studied by in situ ESR and UV-Vis-NIR voltammetry at different temperatures, Phys. Chem. Chem. Phys., 2002, 4, 4181–4185 RSC.
- J. Casado, S. Patchkovskii, M. Z. Zgierski, L. Hermosilla, C. Sieiro, M. M. Oliva and J. T. L. Navarrete, Raman Detection of “Ambiguous” Conjugated Biradicals: Rapid Thermal Singlet-to-Triplet Intersystem Crossing in an Extended Viologen, Angew. Chem., Int. Ed., 2008, 120, 1465–1468 CrossRef.
- E. V. Canesi, D. Fazzi, L. Colella, C. Bertarelli and C. Castiglioni, Tuning the quinoid versus biradicaloid character of thiophene-based heteroquaterphenoquinones by means of functional groups, J. Am. Chem. Soc., 2012, 134, 19070–19083 CrossRef CAS.
- J. Romanova, V. Liégeois and B. Champagne, Resonant Raman spectra of molecules with diradical character: multiconfigurational wavefunction investigation of neutral viologens, Phys. Chem. Chem. Phys., 2014, 16, 21721–21731 RSC.
- H. Hayashi, J. E. Barker, A. Cárdenas Valdivia, R. Kishi, S. N. Macmillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S. I. Kato, M. M. Haley and J. Casado, Monoradicals and Diradicals of Dibenzofluoreno[3,2-b]fluorene Isomers: Mechanisms of Electronic Delocalization, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS.
- J. Lee, H. Hwang, S. Chae and J. M. Kim, A reversibly mechanochromic conjugated polymer, Chem. Commun., 2019, 55, 9395–9398 RSC.
- R. P. Ortiz, J. Casado, S. R. González, V. Hernández, J. T. López Navarrete, P. M. Viruela, E. Ortí, K. Takimiya and T. Otsubo, Quinoidal Oligothiophenes: Towards Biradical Ground-State Species, Chem. – Eur. J., 2010, 16, 470–484 CrossRef PubMed.
- K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. Ruiz Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, Stable organic diradicals based on fused quinoidal oligothiophene imides with high electrical conductivity, J. Am. Chem. Soc., 2020, 142, 4329–4340 CrossRef CAS.
- K. Tada, T. Kawakami and Y. Hinuma, Model calculations for the prediction of the diradical character of physisorbed molecules: p-benzyne/MgO and p-benzyne/SrO, Phys. Chem. Chem. Phys., 2023, 25, 29424 RSC.
- M. Krejčík, M. Daněk and F. Hartl, Simple construction of an infrared optically transparent thin-layer electrochemical cell: applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholate), J. Electroanal. Chem. Interfacial Electrochem., 1991, 317, 179–187 CrossRef.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Tomasi and M. Persico, Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- K. Yamaguchi, Y. Takahara, T. Fueno and K. Nasu, Ab initio MO calculations of effective exchange integrals between transition-metal ions via oxygen dianions: nature of the copper-oxygen bonds and superconductivity, Jpn. J. Appl. Phys., 1986, 26, L1362 CrossRef.
- T. Saito, S. Nishihara, S. Yamanaka, Y. Kitagawa, T. Kawakami, S. Yamada, H. Isobe, M. Okumura and K. Yamaguchi, Symmetry and broken symmetry in molecular orbital description of unstable molecules IV: comparison between single- and multi-reference computational results for antiaromtic molecules, Theor. Chem. Acc., 2011, 130, 749 Search PubMed.
- K. Tada, H. Koga, M. Okumura and S. Tanaka, Estimation of spin contamination error in dissociative adsorption of Au2 onto MgO(001) surface: first application of approximate spin projection (AP) method to plane wave basis, Chem. Phys. Lett., 2018, 701, 103 CrossRef CAS.
- K. Tada, Y. Kitagawa, T. Kawakami, M. Okumura and S. Tanaka, Electron density-based estimation of diradical character: an easy scheme for DFT/plane-wave calculations, Chem. Lett., 2021, 50, 392 CrossRef CAS.
- K. Yamaguchi, in Self-Consistent Field: Theory and Applications, ed. R. Carbo and M. Klobukowski, Elsevier, Amsterdam, The Netherands, 1990, p. 727 Search PubMed.
- T. D. Crawford, E. Kraka, J. F. Stanton and D. Cremer, Problematic p-benzyne: orbital instabilities, biradical character, and broken symmetry, J. Chem. Phys., 2001, 114, 10638–10650 CrossRef CAS.
- V. Barone, I. Cacelli, A. Ferretti, S. Montic and G. Prampolini, Singlet–triplet energy gap of a diarylnitroxide diradical by an accurate many-body perturbative approach, Phys. Chem. Chem. Phys., 2011, 13, 4709–4714 RSC.
- A. Das, T. Müller, F. Plasser and H. Lischka, Polyradical Character of Triangular Non-Kekulé Structures, Zethrenes, p-Quinodimethane-Linked Bisphenalenyl, and the Clar Goblet in Comparison: An Extended Multireference Study, J. Chem. Phys., 2016, 120, 1625–1636 CrossRef CAS.
- A. J. da Silva Filho, L. da Cruz Dantas and O. L. de Santana, Diradicalar Character and Ring Stability of Mesoionic Heterocyclic Oxazoles and Thiazoles by Ab Initio Mono and Multi-Reference Methods, Molecules, 2020, 25, 4524 CrossRef CAS.
- S. Markovič, J. Durdević, S. Jeremić and I. Gutman, Diradical character of some fluoranthenes, J. Serb. Chem. Soc., 2010, 75, 1241–1249 CrossRef.
- S. Canola, J. Casado and F. Negri, The double exciton state of conjugated chromophores with strong diradical character: insights from TDDFT calculations, Phys. Chem. Chem. Phys., 2018, 20, 24227–24238 RSC.
- M. Nakano, R. Kishi, T. Nitta, T. Kubo, K. Nakasuji, K. Kamada, K. Ohta, B. Champagne, E. Botek and K. Yamaguchi, Second hyperpolarizability (γ) of singlet diradical system: dependence of γ on the diradical character, J. Phys. Chem. A, 2005, 109, 885–891 CrossRef CAS PubMed.
- K. Yamaguchi, The electronic structures of biradicals in the unrestricted Hartree–Fock approximation, Chem. Phys. Lett., 1975, 33, 330–335 CrossRef CAS.
- F. Neese, The ORCA program system, Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. Pérez-Guardiola, M. E. Sandoval-Salinas, D. Casanova, E. San-Fabián, A. J. Pérez-Jiménez and J. C. Sancho-Garcia, The role of topology in organic molecules: origin and comparison of the radical character in linear and cyclic oligoacenes and related oligomers, Phys. Chem. Chem. Phys., 2018, 20, 7112–7124 RSC.
- S. Grimme and A. Hansen, A practicable real-space measure and visualization of static electron-correlation effects, Angew. Chem., Int. Ed., 2015, 54, 12308–12313 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02729a |
| This journal is © the Owner Societies 2024 |