
Hypoelectronic titanaboranes: icosahedral and tetracapped tetrahedral clusters comprising bridging hydrides†
Subhash
Bairagi
 ,
Debipada
Chatterjee‡
,
Soumen
Giri‡
and
Sundargopal
Ghosh
*
,
Debipada
Chatterjee‡
,
Soumen
Giri‡
and
Sundargopal
Ghosh
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: sghosh@iitm.ac.in
First published on 3rd February 2025
Abstract
Unusual hypoelectronic closo-clusters, such as icosahedral [(Cp*Ti)3(B9H7)(μ3-H)(μ3-Cl)2] (1) and tetracapped tetrahedral [{(Cp*Ti)3B}(μ3-BH3)2(μ3-Te)2] (2), have been synthesized and structurally characterized. Clusters 1 and 2 represent the first examples of closo-titanaboranes having bridging hydrides, where the Ti⋯Ti interactions play a pivotal role in defining their shapes and electron counts. Furthermore, two face-fused clusters, [(Cp*Ti)2(μ-Se)(μ-SePh)(μ-η3:η3-B5H9R)] (3a: R = H; 3b: R = Ph), have been isolated that deviate from Mingos fusion formalism.
The electron-counting rules initially proposed by Wade and Williams1,2 for single-cage boranes, and later extended by Mingos and Jemmis3,4 for the single and fused polyhedral cages and their metal or main group derivatives, have been useful in understanding and classifying the structural and electronic properties of these compounds.5–9 Based on geometries and skeletal electron pairs (SEPs), Wade and Williams classified the boron clusters as closo [(n + 1) SEP], nido [(n + 2) SEP], arachno [(n + 3) SEP], and hypho [(n + 4) SEP]. The relationship among these cluster geometries was further illustrated by a Rudolph diagram.10 The successive removal of a vertex from a closo geometry, followed by the addition of bridging hydrogens (or 2e) to satisfy the electron count, generates nido, arachno, and hypho-geometries, respectively. Thus, closo-clusters, characterized by their high-symmetry spherical geometries, are inherently stable and typically do not require bridging hydrogens (or ligands) for stabilization.
Although closo-boron clusters are typically electronically saturated and obey the electron counting rules, a few hypoelectronic metallaboranes with such geometries have been reported.5,9a,11–13 For instance, Kennedy and co-workers observed metallaboranes with closo-geometries that deviate from spherical deltahedra, possessing n SEPs instead of the expected (n + 1) SEPs.5b,11 These ‘disobedient’ metallaborane frameworks are often classified as isocloso or hypercloso-deltahedra, which can be derived from the corresponding closo-deltahedra by diamond-square-diamond (DSD) rearrangements. Fehlner and co-workers reported a series of hypoelectronic closo-rhenaboranes [(Cp*Re)2BnHn] (n = 7–10, I, Chart 1), with (n − 2) SEP that can be generated from canonical [BnHn]2− (n = 9–12) shapes by carrying out two or more DSD rearrangements.12 Recently, we have isolated a series of hypoelectronic closo-osmaborane clusters [(Cp*Os)2BnHn] (n = 6–10, II) with (n − 1) SEP that show unusual less spherical deltahedral shapes.13 These findings illustrate that the formal SEP can vary widely from the canonical number, and as it decreases, the cluster shape deviates from that of a more spherical deltahedron. On the other hand, hypoelectronic metallaboranes may also result when they comprise early transition metal fragments. These clusters are structurally diverse and exhibit high metal coordination numbers and cross-cluster bonding.5b,9a,12
 | ||
| Chart 1 Examples of hypoelectronic closo (I, II) and hypho (III) metallaborane clusters. | ||
Although various metallaboranes have been synthesized over the past six decades, relatively few examples of those containing early transition metals are known.5,9,11–14 In this regard, group 4 metallaboranes are particularly scarce.15–18 Barton and co-workers obtained a few group 4 metallaboranes by reacting anionic boranes with metal halides.15 Using the same metal precursor, our group recently isolated zirconium and hafnium-guarded heptaborane and octaborane analogues.16 Very recently, we synthesized and structurally characterized a supraicosahedral 16-vertex hypho-titanaborane (III) using monoborane pyrolysis methodology.18 Encouraged by this achievement, we resumed our search for a closo-metallaborane cluster of titanium by varying the reaction conditions, which led to the isolation of the first two closo-titanaboranes with bridging hydrides.
The low-temperature reaction of [Cp*TiCl3] with [LiBH4] in toluene for 1 h, followed by thermolysis at 90 °C for 48 h in the presence of excess [BH3·THF], afforded 12-vertex titanaborane cluster [(Cp*Ti)3(B9H7)(μ3-H)(μ3-Cl)2] (1) along with cluster III (Scheme 1 and Fig. S1, ESI†).18 Compound 1 was isolated as a green solid in 12% yield. The 1H NMR spectrum of 1 exhibited two distinct Cp* proton signals at δ = 2.03 and 2.05 ppm in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, along with broad signals corresponding to terminal B–H protons in the downfield region and a broad resonance at δ = −5.67 ppm, attributed to μ3-H bridging hydride. The 11B{1H} NMR spectrum displayed five broad resonances at δ = 31.6, 40.2, 45.0, 52.3, and 66.4 ppm in 4:2:1:1:1 ratio. The IR spectrum featured a band at 2544 cm−1, characteristic of terminal B–H stretching vibrations. The mass spectrum displayed an isotopic distribution pattern at m/z 726.3257, corresponding to [M + H]+. Furthermore, solid-state X-ray structure analysis was performed on a suitable crystal grown at 5 °C from a CH2Cl2–hexane solution.
:1 ratio, along with broad signals corresponding to terminal B–H protons in the downfield region and a broad resonance at δ = −5.67 ppm, attributed to μ3-H bridging hydride. The 11B{1H} NMR spectrum displayed five broad resonances at δ = 31.6, 40.2, 45.0, 52.3, and 66.4 ppm in 4:2:1:1:1 ratio. The IR spectrum featured a band at 2544 cm−1, characteristic of terminal B–H stretching vibrations. The mass spectrum displayed an isotopic distribution pattern at m/z 726.3257, corresponding to [M + H]+. Furthermore, solid-state X-ray structure analysis was performed on a suitable crystal grown at 5 °C from a CH2Cl2–hexane solution.
 | ||
| Scheme 1 Synthesis of icosahedral cluster 1. | ||
The solid-state X-ray structure of 1 revealed a 12-vertex icosahedral geometry with one DSD rearrangement (Fig. 1). Interestingly, the presence of three Cp*Ti fragments in the icosahedral core significantly altered its geometry and SEP count. Although the connectivity pattern of 1 aligns with the hypercloso geometry, it possesses (n − 3) SEP and does not obey the Wade-Williams relationship.1,2 Jemmis and co-workers have postulated that transition metals preferentially occupy the highest-degree vertices due to their more diffuse d-orbitals, which is consistent with the structural features of 1.19 In particular, two Ti atoms occupy two vertices of degree six and form a cross-cluster Ti–Ti bond, while the third Ti atom is occupied at a degree-four vertex. Most boron atoms occupy degree-five vertices, except for B9, which is at a degree-four vertex due to the DSD rearrangement. The B9–B2 (1.61(2) Å) and B9–B3 (1.61(2) Å) bond distances are shorter than other B–B bonds, likely reflecting the reduced connectivity of the B9 vertex. All boron atoms in 1 feature terminal hydrogens except for B7 and B8, which are bonded to bridging Cl and H ligands, respectively. These bridging ligands further interact with Ti1–Ti2 and Ti2–Ti3 centers in μ3-fashion. The bulkier μ3-Cl ligand around the Ti1–Ti2 region resulted in a longer Ti1–Ti2 bond length (3.292 Å) as compared to the other Ti–Ti bonds (3.026(3) Å and 3.073(3) Å). Additionally, a μ3-Cl ligand bridges the Ti3 face. Intriguingly, an intramolecular Cp*–H⋯Cl interaction is observed, with an H⋯Cl distance of 2.399 Å, shorter than the typical van der Waals separation. This suggests the possibility of weak hydrogen bonding between a Cp*–H and the Cl1 atom (Fig. S1, ESI†). The Ti–B bond lengths in 1 (av. 2.369 Å) are comparable to those observed in related titanaboranes such as III18 (av. 2.409 Å), while the Ti–Ti bond distances (av. 3.130 Å) are significantly longer than those in [(Cp*Ti)2(μ-η6:η6-B6H6)(μ-H)6] (2.9307(12) Å).17 The B–B bond lengths (av. 1.79 Å) are within the typical range for titanaborane clusters;17,18 however, the anomalous B–B and Ti–B distances may be attributed to the presence of bridging ligands and one DSD rearrangement. Cluster 1 is the first example of a closo-icosahedron with bridging ligands.
 | ||
| Fig. 1 Molecular structure and atom labelling diagram of 1. Note that Cp* ligands attached to Ti atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) in 1: Ti–Ti3 3.073(3), Ti1–B1 2.513(16), Ti1–B9 2.174(15), B2–B9 1.61(2), B7–Cl1 1.854(19), Ti1–Cl1 2.543(5), B8–H1 1.19(3), Ti2–H1 1.95(13), Ti1–Ti2–Ti3 58.01(7), Ti1–B9–Ti3 91.3(6). | ||
The molecular structure of 1 also shows that the [Ti3B9] core is symmetrical with a symmetry plane containing Ti2, B5, B6, and B9 atoms. Its core geometry [Ti3B9] is comparable to that of recently reported metallaboranes [(Cp*M)2B10H10] (M = RhH or IrH with n SEP; M = Os with (n − 1) SEP).13 Similar to these clusters, the face in which the DSD rearrangement occurred is flattened in 1 (dihedral angle along B–Os–Os–B in II is 176.39°, along B–Ti–Ti–Ti in 1 is 173.30°). This flattening brings the Ti atoms closer, facilitating Ti–Ti bonding and forming an oblato-shape cluster. Among the closo 12-vertex clusters, 1 exhibits the lowest SEP count (n − 3), rendering it hypoelectronic and enabling the inclusion of bridging hydride and chloride ligands. While DSD-rearranged closo-metallaboranes such as M2B10 are known, 1 represents the first example of an M3B9 cluster with icosahedron geometry.
In metallaborane cluster chemistry, the choice of boron reagent plays a crucial role in isolating various metallaboranes with unique structural features. To explore this further, we modified the synthetic strategy by using [LiBH3TePh] as the monoborane reagent instead of [LiBH4]. As a result, the reaction of [Cp*TiCl3] with three equivalents of [LiBH3TePh] at −78 °C, followed by thermolysis at 80 °C for 18 h, yielded an 8-vertex closo-tetracapped tetrahedral cluster [{(Cp*Ti)3B}(μ3-BH3)2(μ3-Te)2] (2) as a yellow solid in 25% yield (Scheme 2). The 1H NMR spectrum of 2 displayed two Cp* resonances at δ = 2.04 and 2.09 ppm in 1:2 ratio, along with signals corresponding to the terminal BH proton (δ = 2.36 ppm) and bridging Ti–H–B proton (δ = −2.79 ppm). The 11B{1H} NMR spectrum exhibited two resonances: a peak in the upfield region at δ = −7.7 ppm, assigned to the two-BH3 boron, and a downfield resonance at δ = 54.0 ppm, attributed to the boride boron, indicating a stronger boron–metal interaction. The molecular ion peak at m/z 845.1155 in ESI-MS was in good agreement with the molecular formula [C30H51B3Te2Ti3].
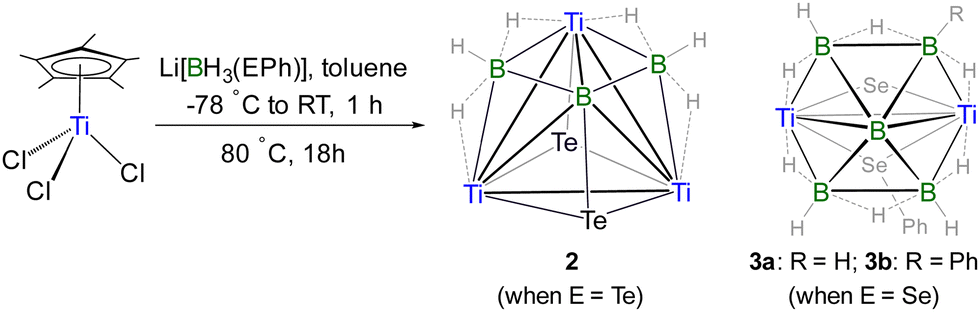 | ||
| Scheme 2 Synthesis of tetracapped tetrahedral cluster 2 and face-fused clusters 3a–b (Cp* ligands attached to Ti atoms in 2 and 3a–b are omitted for clarity; Ti = Cp*Ti). | ||
The solid-state X-ray structure of 2 revealed a [Ti3B] tetrahedral core with one boron and three titanium atoms occupying the corners of the tetrahedron, while two {BH3} fragments and two Te atoms capped its four faces (Fig. 2). For a tetrahedral [M3B] cluster, the SEP count is typically 6; however, 2 contains only 4 SEP.1,2 The [Ti3B] core geometry of 2 is comparable to that of other triply bridging borylene complexes, such as [(Cp*Ru)3(μ-H)3(μ3-BX)] (X = H, CN, OMe, OEt) with 4 SEP and [{(η5-C5H4Me)Mn}(CO)2{Pd(PCy3)}2(μ3-BX)] (X = Cl, tBu) with 3 SEP, where the borylene boron bears various substituents.20 In 2, three Ti atoms and one 'naked' B atom of the [Ti3B] tetrahedral core occupy the degree six vertices, while two B atoms and two Te atoms occupy the degree three vertices. The Ti–Ti (av. 3.077 (Å)) and Ti–B (av. 2.435 (Å)) bond lengths in 2 are consistent with single bond orders and are comparable to those observed in 1 and III.18 All the internal angles in 2 deviated from a regular tetrahedron due to the shorter Ti–B bonds compared to Ti–Ti bonds along with the capping on each face by boron and heteroatoms.
 | ||
| Fig. 2 Molecular structure and atom labelling diagram of 2 (left) and 3b (right). Note that Cp* ligands attached to Ti atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) in 2: Ti1–Ti2 2.94683(13), Ti2–B2 2.279(9), Ti2–B3 2.547(15), Ti1–Ti2–Ti3 63.787, Ti1–B2–Ti2 80.358; 3b: Ti1–Ti2 3.0281(7), Ti1–B3 2.271(4), Ti1–B3–Ti2 84.03(12). | ||
Alternatively, the core geometry [Ti3B3Te2] of 2 can be described as a cubane consisting of three Ti, three B, and two Te atoms. While cubane-type sulfido clusters have been extensively studied, their selenido and tellurido counterparts have received relatively less attention, and cluster 2 represents a rare example of a cubane geometry incorporating tellurium atoms.21 Although the qualitative shape of cubane clusters remains consistent, changes in M–M bond distances often reflect variations in the electronic structure due to the addition or removal of electrons.5b Earlier, Kennedy classified [(CpNi)4B4H4] and [(CpCo)4B4H4] as 68 and 64 cluster valence electrons (CVEs), respectively, featuring two and four metal–metal bonds (Chart 2).5a,22 He also predicted that a hypothetical 60-electron [(CpFe)4B4H4] cluster would adopt a cubane structure with six Fe–Fe bonds, forming a fully bonded metal tetrahedron. The 60 CVE metallaborane [(Cp*Mo)2(μ3-Se)2B2H(μ-H){Fe(CO)2}2Fe(CO)3], which features a cubane geometry with six M–M bonds, serves as a bridge among these clusters.23 In comparison, cluster 2 can be viewed as [M3E5] (E = main group fragment), derived from [M4E4] by replacing one of the metals with a boron atom. Therefore, cluster 2 should possess 50 CVEs with three M–M and three M–B bonds; however, the total number of CVEs available in 2 is 42. Unlike previously reported late transition metal cubane clusters,21–23 this electron deficiency arises from the use of early transition metal titanium and the replacement of one metal vertex with a main group element, boron. The deficiency is partially compensated by the presence of four Ti–H–B bridging hydrogens. However, the inclusion of boron in the [M3B] tetrahedral core significantly distorts the cubane geometry, leading to a structure better described as a tetracapped tetrahedron rather than a true cubane.
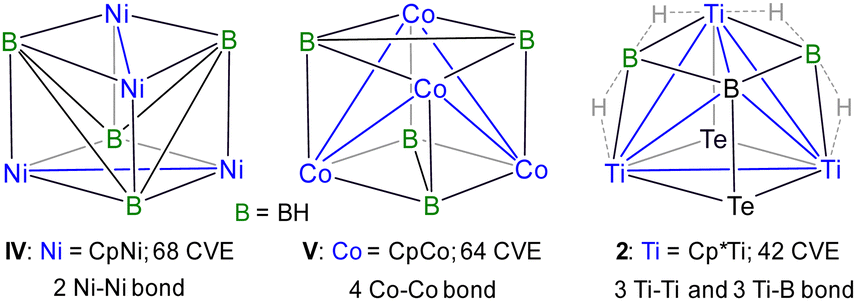 | ||
| Chart 2 Experimentally known cubane-type geometry, along with its CVE count and the number of bonds present in the central tetrahedral unit. | ||
After the effective isolation of the tetracapped tetrahedral cluster 2, we have tried to isolate its selenium analogue by replacing [LiBH3TePh] with [LiBH3SePh] under similar reaction conditions. Although the targeted selenium analogue of 2 was not obtained, the reaction yielded two face-fused clusters, [(Cp*Ti)2(μ-Se)(μ-SePh)(μ-η3:η3-B5H9R)] (3a: R = H, 22%; 3b: R = Ph, 17%, Scheme 2 and Scheme S3, ESI†). In 3a–b, two nido-square pyramidal [Ti2B3] geometries are fused through a common [Ti2B] triangle. Considering the Mingos fusion formalism, clusters 3a–b require 62 electrons but possess 60 CVEs. Therefore, clusters 3a–b can be classified as hypoelectronic clusters, which do not follow Mingos fusion formalism. Notably, similar to cluster 2, these clusters feature a central “naked” boron atom, attributed to its high connectivity of degree six. Alternatively, 3a–b can be described as a hypho-pentaborane cluster, where [B5H9R]3− is coordinated in a trihapto mode to each titanium center of the bimetallic template [(Cp*Ti)2(μ-SePh)(μ-Se)]3+. Although 3a–b resemble [(Cp2Zr)2B5H8]+,15 the presence of two bridging chalcogen ligands, which are intact with the titanium centers, allows the pentaborane ring to coordinate symmetrically to both metal centers (Fig. 2 and Fig. S5, S6, ESI†).
Density functional theory (DFT) studies show lower HOMO–LUMO gaps of 1.63 eV and 1.70 eV for 1 and 2, respectively, that indirectly suggest their reduced kinetic and thermodynamic stability as compared to III (3.45 eV).18 This reduced stability can be attributed to the change in the number of metal fragments, resulting in decreased SEP count relative to the parent clusters. Molecular orbital (MO) analysis of clusters 1 and 2 highlights strong Ti–Ti bonding interactions. For instance, the HOMO of 1 reveals significant bonding interactions between the d-orbitals of two Ti-atoms, indicating a Ti–Ti bond (Fig. 3a). In contrast, unlike cubane-type M4E4 geometries, the HOMO−1 of cluster 2 demonstrates triangular Ti3 bonding interactions facilitated by the overlap of d-orbitals of the Ti-centers (Fig. 3d). Natural bond orbital (NBO) analysis further supports the Ti–Ti bonding in clusters 1 and 2, with significant Wiberg bond indices (0.286, 0.421, and 0.535 for 1; 0.382, 0.438 and 0.630 for 2; Fig. 3b). Additionally, MO and NBO analyses of 1 and 2 reveal significant interactions between the Ti-centers and bridging ligands, which stabilize the clusters and influence their structural rearrangement, geometry, bonding orbitals, and electron count (Fig. 3c,e and f).
 | ||
| Fig. 3 Selected molecular orbitals (a), (d) and (e) and natural bond orbitals (b), (c) and (f) of clusters 1 (top) and 2 (bottom). Isocontour values: ±0.045 [e Bohr−3]1/2. | ||
In summary, this work describes the isolation of closo-titanaboranes 1 and 2 featuring bridging hydrides. Theoretical studies and electron counting formalisms demonstrated that the presence of Ti–Ti bonds and bridging ligands play a crucial role in the stabilization of these hypoelectronic clusters. These findings emphasize the importance of early transition metal fragments in facilitating higher connectivity for the synthesis of novel non-Wadean structures in boron cages.
We gratefully acknowledge the generous support of SERB-DST (Scheme No. CRG/2023/000189), India, and the CoE on Molecular Materials and Functions under the IoE scheme, IIT Madras. S. B. and D. C. thank IIT Madras, and S. G. thanks UGC for fellowships. We also thank Dr B. Varghese, Dr P. K. S. Antharjanam, and Dr E. J. Packium for X-ray structure analysis.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the CCDC under 2353840 (1), 2321603 (2), and 2385700 (3b).†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Wade, J. Chem. Soc. D., Chem. Commun., 1971, 792 RSC; (b) K. Wade, Inorg. Nucl. Chem. Lett., 1972, 8, 559 CrossRef CAS; (c) K. Wade, Adv. Inorg. Chem. Radiochem., 1976, 18, 1 Search PubMed.
- R. E. Williams, Inorg. Chem., 1971, 10, 210 CrossRef CAS.
- D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311 CrossRef CAS.
- E. D. Jemmis, M. M. Balakrishnarajan and P. D. Pancharatna, J. Am. Chem. Soc., 2001, 123, 4313 CrossRef CAS PubMed.
- (a) J. D. Kennedy, Prog. Inorg. Chem., 1986, 34, 211 CrossRef CAS; (b) T. P. Fehlner, J.-F. Halet and J.-Y. Saillard, Molecular Clusters A Bridge to Solid State Chemistry, Cambridge University Press, Cambridge, United Kingdom, 2007 CrossRef.
- (a) J. Zhang and Z. Xie, Chem. – Asian J., 2010, 5, 1742 CrossRef CAS PubMed; (b) R. N. Grimes, Carboranes, Elsevier, Oxford, 2016 Search PubMed.
- (a) G.-X. Jin, Coord. Chem. Rev., 2004, 248, 587 CrossRef CAS; (b) N. S. Hosmane, Boron Science: New Technologies and Applications, CRC, Boca Raton, FL, 2011 Search PubMed.
- (a) K. O. Kirlikovali, J. A. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132 RSC; (b) J. Bould and J. D. Kennedy, Chem. Commun., 2008, 2447 RSC.
- (a) S. Kar, A. N. Pradhan and S. Ghosh, Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O’hare, Elsevier, Amsterdam, 2022, 9, pp. 263 Search PubMed; (b) R. Borthakur, K. Saha, S. Kar and S. Ghosh, Coord. Chem. Rev., 2019, 399, 213021 CrossRef CAS; (c) K. Saha, D. K. Roy, R. D. Dewhurst, S. Ghosh and H. Braunschweig, Acc. Chem. Res., 2021, 54, 1260 CrossRef CAS PubMed.
- (a) R. W. Rudolph, Acc. Chem. Res., 1976, 9, 446 CrossRef CAS; (b) S. Kar, S. Bairagi, G. Joshi, E. D. Jemmis, H. Himmel and S. Ghosh, Acc. Chem. Res., 2024, 57, 2901 CrossRef CAS PubMed.
- (a) J. D. Kennedy, Disobedient Skeletons, in The Borane, Carborane, Carbocation Continuum, ed. J. Casanova, Wiley, New York, 1998, p. 85 Search PubMed; (b) J. D. Kennedy, Prog. Inorg. Chem., 1984, 32, 519 Search PubMed; (c) R. L. Johnston, D. M. P. Mingos and P. Sherwood, New J. Chem., 1991, 15, 831 CAS.
- (a) B. L. Guennic, H. Jiao, S. Kahlal, J.-Y. Saillard, J.-F. Halet, S. Ghosh, M. Shang, A. M. Beatty, A. L. Rheingold and T. P. Fehlner, J. Am. Chem. Soc., 2004, 126, 3203 CrossRef PubMed; (b) S. Ghosh, B. C. Noll and T. P. Fehlner, Angew. Chem., int. Ed., 2005, 44, 2916 CrossRef CAS PubMed.
- (a) K. Kar, S. Kar and S. Ghosh, Chem. Sci., 2024, 15, 4179 Search PubMed; (b) D. K. Roy, B. Mondal, P. Shankhari, R. S. Anju, K. Geetharani, M. Mobin and S. Ghosh, Inorg. Chem., 2013, 52, 6705 Search PubMed.
- (a) S. Aldridge, M. Shang and T. P. Fehlner, J. Am. Chem. Soc., 1998, 120, 2586 CrossRef CAS; (b) A. S. Weller and T. P. Fehlner, Organometallics, 1999, 18, 447 Search PubMed; (c) R. S. Anju, D. K. Roy, K. Geetharani, B. Mondal, B. Varghese and S. Ghosh, Dalton Trans., 2013, 42, 12828 Search PubMed.
- R. L. Thomas, N. P. Rath and L. Barton, J. Am. Chem. Soc., 1997, 119, 12358 Search PubMed.
- (a) A. De, Q. Zhang, B. Mondal, L. F. Cheung, S. Kar, K. Saha, L. Wang and S. Ghosh, Chem. Sci., 2018, 9, 1976 RSC; (b) S. Kar, S. Bairagi, G. Joshi, E. D. Jemmis and S. Ghosh, Chem. – Eur. J., 2021, 27, 15634 Search PubMed.
- S. Kar, S. Bairagi, A. Haridas, G. Joshi, E. D. Jemmis and S. Ghosh, Angew. Chem., Int. Ed., 2022, 61, e202208293 Search PubMed.
- S. Kar, S. Bairagi, J.-F. Halet and S. Ghosh, Chem. Commun., 2023, 59, 11676 Search PubMed.
- E. D. Jemmis, J. Am. Chem. Soc., 1982, 104, 7017 CrossRef CAS.
- (a) R. Okamura, K. Tada, K. Matsubara, M. Oshima and H. Suzuki, Organometallics, 2011, 20, 4772 CrossRef; (b) H. Braunschweig, C. Burschka, M. Burzler, S. Metz and K. Radacki, Angew. Chem., Int. Ed., 2006, 45, 4352 CrossRef CAS PubMed; (c) K. Yuvaraj, D. K. Roy, K. Geetharani, B. Mondal, V. P. Anju, P. Shankhari, V. Ramkumar and S. Ghosh, Organometallics, 2013, 32, 2705 Search PubMed.
- (a) E. I. Stiefel and K. Matsumoto, Transition Metal Sulfur Chemistry, Honolulu, Hawaii, 1995 Search PubMed; (b) R. Llusar, S. Uriel and C. Vicent, J. Chem. Soc., Dalton Trans., 2001, 2813 Search PubMed; (c) A. Thakur, S. Sao, V. Ramkumar and S. Ghosh, Inorg. Chem., 2012, 51, 8322 Search PubMed.
- (a) J. R. Bowser, A. Bonny, J. R. Pipal and R. N. Grimes, J. Am. Chem. Soc., 1979, 101, 6229 CrossRef CAS; (b) J. R. Pipal and R. N. Grimes, Inorg. Chem., 1979, 18, 257 CrossRef CAS.
- K. Geetharani, S. K. Bose, S. Sahoo and S. Ghosh, Angew. Chem., Int. Ed., 2011, 50, 3908 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2353840, 2321603, and 2385700. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06535b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |