
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-metallic nanoparticles: synthesis and their catalytic applications
Yuliang Chen,
Ahsan Zohaib,
Haobo Sun and
Shouheng Sun *
*
Department of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: ssun@brown.edu
First published on 21st July 2025
Abstract
Multi-metallic nanoparticles (MMNPs) have recently garnered significant interest due to their inclusion of different metal atoms within a single nanostructure. The interactions among these metal atoms induce novel properties in MMNPs, making them an ideal platform for exploring the complex interplay between structure and properties, particularly in terms of catalytic properties. This review summarizes recent advancements in the synthesis and catalytic studies of MMNPs. It begins by outlining the synthesis of MMNPs with well-defined structures, including solid solutions, intermetallics, composite core/shell structures, heterodimers, and high-entropy alloys. These MMNPs exhibit unique electronic and surface properties that are crucial for enhancing catalysis. Using representative examples, the review further highlights the promising applications of MMNPs in catalyzing important chemical reactions related to energy conversion and green chemistry, achieving high reaction efficiencies. Finally, the review discusses strategies for attaining atomic precision in the synthesis of MMNPs and optimizing their catalytic performance for a broader range of chemical reactions.
1. Introduction
Multi-metallic nanoparticles (MMNPs) discussed in this review are a class of nanomaterials composed of two or more distinct metal elements, forming either well-mixed alloy structures or phase-separated composites. The alloy structures can be further categorized into solid solutions, where different metal atoms randomly occupy positions within the crystal lattice, and intermetallic compounds, where metal atoms are arranged in a chemically ordered manner. In contrast, composite structures include core/shell and heterostructures, in which the constituent metals crystallize independently into distinct structural domains. MMNPs have garnered significant attention in recent years due to their unique structure-induced physical and chemical properties, particularly in catalysis.1,2 By incorporating multiple elements into a single NP, intermetallic interactions generate synergistic surface environments that facilitate multi-step chemical reactions with enhanced efficiency in both kinetics and energy usage. Moreover, the high degree of structural tunability makes MMNPs a promising platform for investigating structure–catalysis relationships, thereby enabling the rational design of NP parameters to optimize reaction performance.In this review, we summarize recent advances in the synthesis and catalytic applications of MMNPs. We begin with an overview of synthetic strategies for fabricating MMNPs, including conventional alloys, high-entropy alloys (HEAs), core/shell structures, and heterostructured configurations. Next, we highlight the catalytic roles of these MMNPs in representative reactions, including electrochemical reduction, electrochemical oxidation, and thermochemical processes. Finally, we discuss potential directions aimed at achieving improved control over the synthesis and properties of MMNPs to expand their applicability in catalysis.
MMNPs have recently become a prominent area of study and have been actively reviewed.2–5 However, these reviews often lack comprehensive coverage of MMNPs and provide limited discussion on NP-composition-dependent catalysis. This current review summarizes a broad range of MMNPs, particularly those studied in the past five years. Specifically, using representative examples, it covers recent advancements in MMNP synthesis and catalytic applications, including both electrocatalysis and thermocatalysis, which are closely related to highly efficient energy conversion and green chemistry.
2. Synthesis of MMNPs
MMNPs can be synthesized using various methods, including solution-phase reactions, laser ablation, and solvothermal techniques. Each method offers distinct advantages and limitations in controlling the NP composition, morphology, and crystal structure.2 Our focus here is on summarizing solution-phase synthesis. Based on their structural nature, MMNPs can have either a solid solution or an intermetallic structure. In the solid solution structure, constituent elements are randomly distributed throughout the crystal network, while in the intermetallic structure, atoms are orderly arranged within the crystal network. Both structural types are considered homogeneous. MMNPs can also form heterogeneous structures, in which atoms are organized in different crystal domains, as shown in core/shell and heterodimer structures. In this section, we comment on some common methods that have been explored to synthesize MMNPs, as summarized in Table 1.| MMNPs | Structure type | Synthetic approach | Synthetic conditions | Applications | Ref. |
|---|---|---|---|---|---|
| OAm: oleylamine; OAc: oleic acid; PVP: polyvinylpyrrolidone; ORR: oxygen reduction reaction; FAO: formic acid oxidation; MOR: methanol oxidation reaction. | |||||
| PtM (M = Co, Fe, Ni) | Solid solution | Co-reduction | Solution phase; reducing agents: superhydride/polyalcohol/borane; surfactants: trioctylphosphine, OAm, OAc | ORR (in 0.1 M HClO4) | 7, 18 and 19 |
| PdM (M = Co, Cu) | Solid solution | Co-reduction | Solution phase; reducing agent/surfactant: OAm | FAO (in 0.1 M HClO4 and 2 M HCOOH) | 9 |
| RuM (M = Pd, Ni, Cu) | Solid solution | Co-reduction | Solution phase; reducing agents: polyalcohol/superhydride; surfactants: PVP, OAm, OAc | NO/CO oxidation | 14–16 |
| PtCo–M (M = Mn, Fe, Ni, Cu, Zn) | Solid solution/intermetallic | Co-reduction/annealing | Solution phase; reducing agent: borane tert-butylamine. annealing at 650 °C under 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) vol% H2/Ar vol% H2/Ar |
ORR (in H2–O2 fuel cells) | 13 |
| PtCo–M (M = Ga, Pb, Sb, Cu) | Solid solution/intermetallic | Wet impregnation, annealing | Solid phase annealing at 1000 °C under 5vol% H2/Ar |
ORR (in 0.1 M HClO4) | 20 |
| B2-CuPd | Intermetallic | Halide-mediated co-reduction | Solution phase; reducing agent and surfactant: OAm | ORR (in 0.1 M HClO4) | 40 |
| L10-PtM/Pt (M = Co, Ni, Fe) | Core/shell | Dealloying | Surface etching in 0.1 M HClO4 and annealing at 400 °C under 5vol% H2/Ar |
ORR (in 0.1 M HClO4) | 19, 49 and 56 |
| Au/MPt (M = Fe, Co, Cu) | Core/shell | Seed-mediated growth | Solution phase; co-reduction of metal precursors and deposition of MPt over seeding Au NPs | MOR (in 0.1 M HClO4 + 0.1 M methanol) | 48 |
| Au/Pd NPs dimer | Dimer (Janus) | Polymer-mediated growth | Solution phase; growth of Pd onto Au NPs in amphiphilic block copolymers | — | 67 |
| FeCoNiPtRu | Solid solution | Co-reduction | Solution phase; reducing agent: polyalcohol; surfactant: OAm, OAc | — | 77 |
| PdCuPtNiCo | Solid solution | Core/shell diffusion | Annealing core/shell CuPd/PtNiCo at 600 °C | ORR (in 0.1 M KOH) | 76 |
| PtPdAuFeCoNiCuSn | Intermetallic | Annealing | Annealing HEA NPs at 1000 °C | — | 81 |
2.1. Synthesis of bi-/tri-metallic solid solution MMNPs
In conventional solution-phase synthesis, the formation of MMNPs alloys generally proceeds through sequential nucleation steps, which arise from the varying reduction potentials and/or decomposition chemistry of metal precursors. Solution-phase reduction reactions are frequently employed to synthesize bimetallic or trimetallic solid-solution NPs because they allow for precise control over NP growth conditions, including precursors, reducing agents, ligands, solvents, and reaction temperatures.6 Under common solution-phase synthetic conditions, solid-solution structured MMNPs are often formed. One widely used method for synthesizing solid-solution alloy NPs is the co-reduction of multiple metal precursors in the presence of a suitable reducing agent, as summarized in Table 1. An early example of this approach is the synthesis of monodisperse FePt NPs via the co-reduction of FeCl2 and Pt(acac)2 (acac = acetylacetonate), using lithium triethylborohydride (LiBEt3H, a superhydride) as the reducing agent, and oleic acid and oleylamine as stabilizing ligands.7 A high-boiling-point dioctyl ether solvent was used to dissolve the precursors, with the assistance of trioctylphosphine, oleic acid, and oleylamine, which formed complexes with the metal ions to facilitate their dissolution. LiBEt3H solution was then added to reduce both Fe(II) and Pt(II) ions, initiating nucleation and further growth of FePt NPs at the boiling point of the solvent (∼292 °C). NP stabilization was achieved through the coordination of oleate (RCOO−) and oleylamine (RNH2) ligands to the metal surface. This co-reduction method enabled excellent control over particle size, producing NPs as small as 4 nm, and allowed tuning of the Fe/Pt ratio by adjusting the molar ratios of FeCl2 and Pt(acac)2. However, the high reactivity of superhydride can pose challenges for handling and scale-up synthesis. Alternatively, polyalcohols like ethylene glycol were tested as milder reducing agents to initiate reduction reactions. However, their weak reducing power often led to the preferential reduction of noble metal precursors, resulting in noble-metal-rich NPs.8 To address this issue and enable easier control over NP composition, organic borohydrides such as borane morpholine and borane tert-butylamine have been employed as more effective reducing agents in the solution-phase synthesis of MMNPs.9Reductive thermal decomposition is another solution-phase reaction approach to MMNPs. Unlike co-reduction, which relies on metal salts as precursors, this approach involves the reduction and thermal decomposition of organometallic precursors. A well-known example is the synthesis of monodisperse FePt NPs through the combined decomposition of Fe(CO)5 and reduction of Pt(acac)2.10 This method utilizes thermally unstable, low-valent metal complexes that decompose swiftly upon heating, releasing metal atoms. This rapid decomposition shortens the nucleation time window, facilitating instantaneous nucleation and promoting the growth of uniform NPs, as demonstrated in the synthesis of Co NPs11 and Fe NPs12 via the decomposition of Co2(CO)8 and Fe(CO)5, respectively. Although this method is advantageous for producing monodisperse monometallic and bimetallic NPs, it is quite challenging to achieve the simultaneous decomposition and reduction of different precursors necessary for synthesizing MMNPs.
Solution-phase synthesis requires surfactants to stabilize NPs. However, these surfactants can be challenging to remove, which in turn hinders surface-sensitive catalytic applications. Recently, the solid-phase co-reduction method has emerged as a valuable alternative for synthesizing surfactant-free MMNPs.13 This method involves wet impregnation of a mixture of metal precursors onto solid supports, typically carbon, followed by high-temperature reaction in a reducing atmosphere (e.g., H2). In the process, the initial reduction of one metal forms nuclei that act as templates for the subsequent reduction and deposition of other metals. The elevated temperature also enhances atomic diffusion, promoting the formation of solid-solution alloy structures. This solid-phase co-reduction facilitates the large-scale synthesis of supported MMNPs, providing practical advantages over traditional solution-phase methods, especially regarding scalability and surfactant removal. This method has successfully been used to synthesize a wide range of MMNPs with diverse elemental compositions, including RuNi,14 PdRu,15 RuCu,16 RhCu,17 Pt–M (M = Fe, Co),18,19 and Pt–Co–M′ NPs (M′ = Fe, Ni, Zn, Cu, Ga, Pb, Sb).13,20
In summary, solution-phase co-reduction allows for precise control over NP sizes and compositions, particularly when using strong reducing agents. However, it often leads to compositional inhomogeneity due to varying metal reduction potentials and is prone to surfactant contamination. Reductive thermal decomposition produces highly monodisperse NPs, but its application to MMNPs is constrained by the difficulty of synchronizing precursor decomposition, which can result in phase segregation. The newly demonstrated direct solid-state reaction approach provides a surfactant-free, scalable method for producing supported MMNPs with improved alloying through high-temperature diffusion. The method may evolve as a practical approach to MMNPs if the issue of precise control over NP morphology can be resolved.
2.2. Synthesis of bi- and tri-metallic intermetallic MMNPs
An intermetallic structure is characterized by the ordered arrangement of metal atoms within the crystal lattice. This specific ordering aligns atoms along distinct crystallographic directions, making the structure ideal for examining d–d orbital interactions within each MMNP. These interactions provide enhanced chemical stability of metal components against oxidation and acid etching, compared to a disordered solid solution structure,21,22 making the structure promising for catalysis under high-temperature and corrosive conditions. However, conventional synthesis methods typically yield solid-solution NPs.23 These NPs must be annealed at a controlled high temperature to achieve the structural transition from solid-solution to intermetallic. This structural transition is generally governed by the Gibbs free energy change ΔG = ΔH − TΔS, where ΔH is enthalpy, ΔS is entropy, and T is temperature. The process leading to the formation of the ordered structure often has a negative ΔH due to stronger metal–metal bonding interactions associated with structure stabilization, which is also dependent on alloy compositions, and a negative ΔS related to the increase in atom ordering within the structure. This makes T and alloy compositions crucial parameters for promoting atom diffusion within the NP structure and achieving the desired degree of atom ordering.A common approach to synthesize intermetallic MMNPs is to anneal solid solution MMNPs with the appropriate alloy composition. Fig. 1(a) illustrates the general concept of this annealing-induced ordering process. For instance, intermetallic L10-PtM NPs (M = Fe,24 Co,19,25 Ni,26 Cu,27 Zn,28 Ga,29 Sn,30 etc.) are typically synthesized by annealing their solid-solution counterparts at 500–900 °C under a H2/Ar atmosphere.23 This method has also been extended to the synthesis of trimetallic L10-PtCoM NPs (M = Fe, Mn, Ni, Cu).20 A major challenge of this high-temperature annealing is the degradation of NP uniformity due to the surface energy-driven NP mobility, aggregation, and sintering.22 To mitigate aggregation, the MMNPs are often anchored on stable substrates or coated with stable protective shells (e.g., SiO2, MgO, and KCl),28,31–33 as illustrated in Fig. 1(a)-(i). For example, L10-FePt NPs have been prepared by annealing the MgO-coated FePt NPs. The structural transition occurs within the MgO shell, which is later removed by acid washing, yielding surfactant-free L10-FePt NPs.24 Similarly, a stable graphitic carbon shell can be formed by the decomposition of polydopamine around MMNPs to protect their structural transition.34 The sulfur (S)-doped carbon layer is also found to be effective in suppressing NP sintering.35
 | ||
| Fig. 1 Summary of (a) annealing-induced disorder-to-order transformation and (b) direct solution phase synthesis of intermetallic MMNPs. (c) The statistic of the number of free electrons, density of states (DOS) at the Fermi level and the relative metal bond strength (RMBS) of different Pt–M–M′ (100) surface. (d) The correlation between Ea (diffusion barrier), Evac (vacancy formation energy), and Ehop (hopping barrier energy) and RMBS. (e) Schematic illustration of the promoted ordering process in PtNiSn alloy. Reproduced from ref. 38 with permission from Springer Nature, copyright 2024. (f) The HAADF-STEM images of intermetallic FePt prepared from Cl− mediated solution synthesis. (g) The X-ray diffraction pattern of FePt under different Pt2+:Cl− molar ratio. Reproduced from ref. 39 with permission from American Chemical Society, copyright 2018. | ||
An alternative approach to stabilize NPs against uncontrolled aggregation during their high-temperature structure transition is to improve atom mobility within NPs. This allows the structural transition to occur at a lower temperature, minimizing the risk of NP aggregation. A common idea is to create defects within the NP structure to facilitate atom diffusion. This is achieved by alloying an atom that cannot integrate with the main structure during the high-temperature annealing process and can subsequently diffuse out of the internal structure, thereby creating defects that promote atomic diffusion, as illustrated in Fig. 1(a)-(ii). This has been applied to prepare various L10-MMNPs. For example, doping A1–FePt with Au, which has low surface energy and poor miscibility with Fe and Pt, facilitates Fe/Pt ordering at lower temperatures.24,36 Upon annealing, Au segregates to the NP surface, creating vacancies within the internal structure, lowering the FePt structure transition temperature. Similarly, L10-FeAgPt NPs are synthesized.37 Recently, this is further extended to alloying PtM (M = Ni, Co, Fe, Cu, Zn) with a low-melting-point metal M′ (∼15%), such as Sn, Ga, or In, to lower the ordering temperature to below 450 °C, enabling the formation of highly ordered L10-PtMM′ NPs without obvious NP aggregation.38 Structure studies indicate that M′ weakly binds to Pt or M within the structure (Fig. 1(c)) and can diffuse out easily when the structure is heated, leaving vacancies behind (Fig. 1(d)) and lowering the atom diffusion barrier (Fig. 1(e)). This leads to the formation of intermetallic structure below 450 °C.
Direct solution-phase synthesis has been investigated to produce dispersible intermetallic MMNPs while maintaining their NP shapes. This concept is summarized in Fig. 1(b). For example, intermetallic Pt3Sn nanocubes were synthesized by co-reducing PtCl2 and SnCl2·2H2O in 1-octadecene and dodecylamine at 240 °C.30 However, it is still challenging to extend this method to obtain anisotropic L10-ordered MMNPs. A more promising approach is to control NP growth kinetics in the presence of a halide ion to form the intermetallic structure, as outlined in Fig. 1(b)-(i). The halide ion (e.g., Br− or Cl−) can modulate metal deposition rates by forming strong metal–halide complexes, thereby favoring the formation of stoichiometric intermetallic phases.39,40 This is nicely demonstrated in the synthesis of L10-FePt NPs by co-reducing Fe(acac)2 and Pt(acac)2 in the presence of NH4Cl (Cl−/Pt2+ molar ratio = 3/1).39 The ordered structure is characterized by both TEM (Fig. 1(f)) and XRD (Fig. 1(g)). Intermetallic B2-CuPd NPs were also prepared by this halide-assisted method by co-reduction of Cu(acac)2 and PdBr2.40 The ordering can further be achieved directly from the solution phase synthesis condition by incorporating a 3rd metal element in the alloy structure, which lowers the ordering temperature and allows NPs to grow into an intermetallic structure (Fig. 1(b)-(ii)).41
2.3. Synthesis of MMNPs with heterostructures
Heterostructured MMNPs consist of distinct crystal domains of metals with well-defined interfaces. Depending on the domain interconnection, they can be classified as core/shell or heterodimer (heterotrimer, etc.) NPs. The coexisting components within these NPs frequently interact synergistically, resulting in novel or enhanced properties. For example, linking Au and Cu in the Au–Cu dimer structure greatly enhances their catalytic selectivity for electrochemical CO2 reduction to multi-carbon products. This improvement is due to synergistic domain interactions between Au and Cu, where CO is produced on Au and then spills over to Cu, where it is further reduced to multi-carbon products.42 It is now widely accepted that interfaces promote electronic and geometric effects, alter surface adsorption/desorption characteristics,43 and facilitate charge transfer44,45 – all crucial for catalytic enhancement. Therefore, precise structural control of heterostructures is essential for optimizing catalytic performance.Various methods have been developed for preparing core/shell and heterodimer MMNPs.43,46 The general synthetic ideas are summarized in Fig. 2(a) and (b). Core/shell NPs are commonly synthesized via one-pot reaction with controlled NP nucleation and growth kinetics, seed-mediated growth, and dealloying. In one-pot syntheses, metal salts with different reduction potentials are co-reduced, in which the easily reduced metal salt first reacts to form seeding NPs on which the second metal is deposited to form a shell, as demonstrated in the synthesis of core/shell Pd/Pt NPs from the co-reduction of Na2PdCl4 and Pt(acac)2.47 However, in this co-reduction process, it is difficult to prevent concurrent reduction and metal interdiffusion, which often result in NPs with indistinct core/shell boundaries. To prepare a core/shell NP structure with distinct core/shell boundaries, seed-mediated growth is preferred. This method begins with core NPs serving as seeds, around which the shell is grown without separate nucleation and growth processes. By precisely controlling the conditions for shell growth, core–shell interdiffusion can be minimized, allowing for better control over shell thickness. For example, Au/MPt (M = Fe, Co, Cu) core/shell NPs were synthesized by depositing MPt onto preformed Au NP seeds under conditions optimized exclusively for the formation of the MPt shell.48 The lattice mismatch between the core and shell structures can induce lattice strain around the shell,49 enabling the synthesis of uncommon crystal phases such as the 2H hexagonal phase (AB stacking) as seen in Rh/Ru,50 Pd/Rh,51 Au/Cu,52 and Au/Pd53 core/shell structures. When lattice mismatch exceeds ∼5%, epitaxial growth becomes challenging.54 Nonetheless, core/shell structures with significant mismatch can still be synthesized by reducing core size and controlling deposition kinetics, as demonstrated in the synthesis of Rh/Au NPs (7.2% mismatch) (Fig. 2(c)–(e)), where 4 nm Rh nanocubes serve as seeds and Au shell is deposited around these seeds by a slow ascorbic acid-induced reduction of HAuCl4. NaOH and KBr may be further added to the reaction solution to control the Au growth kinetics (Fig. 2(f)).55
 | ||
| Fig. 2 Summary of the synthetic approaches for (a) core/shell and (b) heterodimer (also called Janus) structures. The HAADF-STEM image of (c) Rh/Au structure with larger lattice distortion, (d) The Rh/Au core/shell NP with the inset showing a model in the same orientation, and (e) the corresponding EDX elemental mapping. (f) The effect of KBr, NaOH concentration on the formation of Rh/Au core/shell structure. Reproduced from ref. 55, licensed under CC BY 4.0.(g) Schematic illustration of the synthesis of dimer structures from amphiphilic-block-copolymers capped Au NPs. (h) The TEM image of a typical Au–Pd dimer NPs. Reproduced from ref. 67 with permission from Wiley, copyright 2017. | ||
Core/shell NPs can also be synthesized by selectively etching reactive metals from the surface of alloy NPs. For instance, acid treatment of L10-FePt removes surface Fe, producing a thin (<1 nm) Pt shell.32 This method has been applied to prepare CoPt/Pt,19 NiPt/Pt,56 and CuPt/Pt49 NPs. As the etching creates surface defects around the shell structure, the core/shell NPs are often annealed at a proper temperature to repair the surface defects and to smooth the shell structure for core stabilization and catalysis studies. The annealing must be carefully controlled to avoid interdiffusion between the core and shell structures.57
Unlike core/shell structured MMNPs, heterodimer MMNPs consist of individual NP domains that are attached side-by-side. In solution-phase syntheses, forming heterodimers is challenging when the involved metals have similar atomic sizes, valences, and lattice constants, which favor alloy formation, or when they have highly dissimilar properties, which favor independent nucleation and growth.58 As a result, the seed-mediated growth method is often employed for the pre-designed growth of new NPs on existing NPs. For example, in the synthesis of Au–Fe3O4 and FePt–Fe3O4 NPs, Fe is controlled to nucleate asymmetrically on Au or FePt seeds before being oxidized by air to form iron oxide.59,60 This method has been applied to prepare many other heterostructured NPs.61,62 Alternatively, heterodimer NPs can be prepared by controlling the deposition kinetics of the second NPs. By slowing the deposition rate, selective growth can be directed toward specific sites on the surface of the core NPs. This approach is demonstrated in the synthesis of Pd–Ag and Pd–Au heterodimers, where slow injection of the Ag (or Au) precursor leads to nucleation on a single facet of Pd nanocubes, forming a dimer structure. Conversely, a faster injection results in uniform deposition over all Pd facets, yielding a core/shell structure.63,64
Surfactants anchored on the surface of seeding NPs can significantly influence the deposition of second NPs. This method utilizes the binding affinity of surfactants on different facets of the seeding NPs, permitting new growth only on the facet that is loosely occupied by surfactants, thereby promoting directional growth. In the synthesis of Au–Ag dimers, bifunctional ligands containing both –SH and –COOH groups guided Ag deposition, resulting in an Au sphere–Ag wire dimer structure.65 Similarly, a polymer mask can be applied to selectively coat the surface of seeding NPs, enabling preferential growth of a new nanostructure, as demonstrated in the synthesis of Au–Pd (Fig. 2(g) and (h)),66,67 and Au–Ag heterodimers.68 Annealing-induced phase separation offers another method for forming heterodimers. This approach was used in the synthesis of Pt–Pb dimer NPs by annealing core/shell PtPb/Pt NPs.69
2.4. Synthesis of MMNPs of high-entropy alloys
MMNPs containing more than 3 metal components can be viewed as an extension of the alloy structures discussed in previous sections. However, due to the challenges of incorporating multiple metal elements into a single NP structure and the inherent complexity of these multicomponent systems, these NPs require special attention. In this review, we follow the literature tradition and refer to the MMNPs comprising five or more elements as high-entropy alloy (HEA) NPs.70 Recently, significant efforts have been focused on developing reliable synthesis strategies for producing HEA NPs.71,72High-temperature pyrolysis is a popular method for producing HEA NPs. This technique involves the rapid pyrolysis of mixed metal precursors using laser, microwave, or electrical heating, followed by quenching to produce NPs with the desired elemental compositions.71 Plasmonic ablation is employed to fragment bulk HEA materials into isolated NPs.73,74 Conventional material fabrication techniques like mechanical alloying and electrodeposition are tested, but fail to achieve the desired NP morphology and composition controls. Solution-phase methods developed for bi- and tri-metallic NP syntheses have also been adapted for the synthesis of HEA NPs.75–77 Co-reduction of metal precursors in solution from a non-equilibrium state at elevated temperatures can lead to the formation of 4.7 nm FeCoNiPtRu HEA NPs.77 However, as the number of constituent elements increases, the complexity of synthesis grows substantially. Variations in reduction potentials, reaction kinetics, and elemental miscibility can cause sequential rather than simultaneous reduction, leading to phase-separated structures.78 To address this issue, solution-phase synthesis is often combined with solid-state annealing. In this approach, MMNPs are first prepared from solution phase synthesis, and then annealed at high temperatures to promote atomic diffusion into HEA, as demonstrated in the synthesis PdCuPtNiCo HEA NPs from annealing core/shell CuPd/PtNiCo NPs at 600 °C (Fig. 3(a) and (b)).76 This core/shell design allows sequential incorporation of multiple elements, thus bypassing the need for simultaneous reduction of five elements. This approach has been extended to synthesize other HEA NPs incorporating up to 14 elements.79 Nevertheless, overcoming the ‘mixing’ barrier during annealing to form the HEA structure remains a significant challenge. A promising solution is to incorporate a low-melting-point element, such as Ga, which has favorable mixing enthalpies with other metals and provides a strong thermodynamic driving force for homogenization. This has enabled the synthesis of HEA NPs that contain elements of a broad range of atomic radii (1.24–1.97 Å) and melting points (303–3683 K).80
 | ||
| Fig. 3 (a) Scheme of the synthetic process of PdCuPtNiCo HEA NPs. (b) STEM-EDS elemental mapping images of the HEA NP in (a). Reproduced from ref. 76 with permission from Royal Society of Chemistry, copyright 2019. (c) Schematic of the disorder-to-order phase transition from a multi-elemental disordered solid-solution NP to an ordered intermetallic NP. (d) The long-range order (LRO) within the NP as a function of NP size. The insets schematically show the microstructural evolution as a function of the NP size. Reproduced from ref. 81, licensed under CC BY-NC 4.0. | ||
Controlled annealing of HEA NPs can induce disorder-to-order transitions, leading to the formation of high-entropy intermetallic (HEI) NPs (Fig. 3(c)).81 This transformation is highly size-dependent: NPs smaller than 5 nm tend to achieve full ordering, while larger NPs exhibit only partial ordering or form core/shell-like heterostructures (Fig. 3(d)). This highlights the close relationship between structural ordering and high surface energy of NPs. This strategy has been successfully applied to synthesize HEI NPs of PtIrFeCoCu82 and PtRhFeNiCu.83
3. Catalytic properties of MMNPs
The integration of multiple metallic components in MMNPs, particularly on their surfaces, leads to unique physical and chemical properties that differ from those of their single-component counterparts.2,84 This section highlights their catalytic properties.Catalytic activity is dependent on a catalyst's ability to adsorb and activate reactants, and to desorb products, following the Sabatier principle.85 MMNPs provide an optimal platform for tuning the electronic structure of active sites to match the activation orbitals of adsorbates, and for designing atomic arrangements that encourage desired binding modes. In MMNPs with fewer than three elements, the electronic structure is mainly influenced by ligand and strain effects, while the ensemble effect dictates the binding configurations of intermediates.86 To correlate electronic structure with adsorption energy, the d-band center is commonly used as a descriptor: a higher d-band center relative to the Fermi level strengthens adsorption, while a lower center weakens it (Fig. 4(a)).87 The ligand effect arises from electron transfer between surface and subsurface atoms, modifying the d-band center. Lattice mismatch introduces strain: tensile strain narrows the d-band and shifts it upward, enhancing adsorption, while compressive strain has the opposite effect. The ensemble effect, arising from variations in local atomic arrangements, also influences adsorption strength and configuration, thereby affecting catalytic kinetics. HEA NPs, which incorporate a larger number of elements, possess several distinctive features that set them apart from traditional bi- or trimetallic NPs: the high-entropy effect, sluggish diffusion, lattice distortion, and the cocktail effect (Fig. 4(b)).88 The high-entropy effect, stemming from the mixing entropy of five or more elements, stabilizes the structure, especially under extreme conditions.89 Sluggish diffusion refers to slower atomic mobility due to varying diffusion barriers and potential energy landscapes, which further enhance phase stability.90 Lattice distortion, resulting from atomic radius mismatches, introduces tunable strain that can alter catalytic properties.91,92 The cocktail effect captures the synergistic electronic and structural interactions among the constituent elements, further enhancing performance.93 With a wide range of surface compositions, HEA NPs offer a spectrum of active sites, each uniquely contributing to overall activity.94 Collectively, these effects underpin the exceptional catalytic performance and durability of HEA NPs.
 | ||
| Fig. 4 (a) The local density of state projected onto an adsorbate state interacting with the d bands. Reproduced from ref. 21 with permission from American Chemical Society, copyright 2019. (b) Schematic illustration of the distinct properties of HEA NPs. | ||
4. MMNPs for electrochemical reduction reactions
4.1. Electrochemical CO2 reduction
The electrochemical CO2 reduction reaction (CO2RR) is gaining significant attention as a sustainable route for producing value-added chemicals and renewable energy carriers. NP catalysts are promising for CO2RR due to their high surface area, abundant active sites, and tunable electronic structure. Depending on catalyst composition and applied reduction potential, CO2RR can yield single-carbon (C1) products, such as formate (HCOO−) and carbon monoxide (CO), or multi-carbon (C2+) products like ethylene and ethanol. However, the sluggish initial reduction of CO2 to HCOO*/COOH*—the rate-determining step—limits the subsequent formation of CO* or CO.95 Moreover, in aqueous electrolytes, the competing hydrogen evolution reaction (HER) often compromises selectivity. Alloying NPs offers a strategy to improve CO2 adsorption and activation while suppressing HER.Noble metal NPs such as Au,96 Ag,97 Pd98 NPs are effective for CO2RR to CO, which proceeds via COOH* stabilization and CO* desorption. However, pure metals exhibit a scaling relationship where stronger COOH* binding can hinder CO desorption or promote HER, thereby reducing overall efficiency.98 Alloying can break this trade-off by balancing COOH* stabilization and CO* desorption while suppressing HER. For instance, Pd strongly binds CO, impeding its desorption.98 Alloying Pd with Au downshifts the d-band center, weakening the Pd–CO bond and facilitating CO release at lower overpotentials.99 Similar improvements are reported for PdAg and PdCu alloys.100,101 Ternary alloys offer further tunability. PdCuZn nanosheets, for example, exhibit a faradaic efficiency for CO (FECO) of 96% at −0.35 V (vs. RHE), outperforming PdZn (65%) and PdCu (90%) due to a weakened Pd–CO bond that favors linear *CO adsorption over the strongly bound triple-bonded *CO on pure Pd.102 In Au-based alloys, selectivity gains are less pronounced, but Au3Cu1 NPs show a 30% increase in mass activity compared to pure Au.103 Crystal structure also impacts catalytic performance. Ordered intermetallic AuCu NPs (Fig. 5(a)) enhance CO production while suppressing HER.104 DFT studies reveal this ordering lowers the activation barrier for COOH* formation and destabilizes H* adsorption, leading to >80% FECO (Fig. 5(b)). These intermetallics also exhibit enhanced stability under reaction conditions.105
 | ||
| Fig. 5 (a) TEM images showing atomic ordering transformation of AuCu NPs. Scale bar: 20 nm. (b) CO2RR performance of AuCu NPs with different degrees of ordering (−0.77 V vs. RHE, 0.1 M KHCO3 solution) (d: disordered, o: ordered, i1 and i2 are intermediate states). Reproduced from ref. 104 with permission from American Chemical Society, copyright 2017. (c) Cu nanorods (NRs) with surface alloyed with Au. Scale bar: 50 nm (d) The Au-atomic-percentage dependent product distribution in CO2RR. Reproduced from ref. 109 with permission from American Chemical Society, copyright 2024. (e) Co-axial heterostructured fcc-2H-fcc Au/Cu. (f) Heterodimer fcc-2H-fcc Au–Cu structure. (g) Core/shell fcc-2H-fcc Au/Cu. (h) CO2RR performance of different heterostructures. JNSs: Janus (heterodimer) structures, CAH: co-axial heterostructures, CSNs: core/shell nanostructures. Reproduced from ref. 52 with permission from Wiley, copyright 2024. (i) Crystal structure and morphology dependent CO2RR performance of CuPd NPs. Reproduced from ref. 112 with permission from American Chemical Society, copyright 2017. | ||
C2+ product formation depends on efficient CO coupling, which requires moderate CO binding to ensure adequate surface coverage without inhibiting CO–CO interactions. Cu-based NPs are widely used for this purpose due to optimal CO* binding strength. Alloying Cu with CO-producing metals like Au,103 Ag,106 and Ga107 tunes CO* adsorption and promotes *CO spillover, boosting C2+ yields, as demonstrated by CuAu NPs with a low Au content (∼3%).108 Interestingly, Cu nanorods stained with 2% Au exhibit enhanced selectivity for n-propanol (18.2% FE). This improvement is likely due to Au staining on Cu (100) surfaces, which facilitates further CO coupling to the existing C–C coupling intermediates (Fig. 5(c) and (d)).109
Core/shell structures or heterodimers are even better catalyst candidates for elective CO2RR to C2+ products. Cu catalysis of the core/shell AgAu/Cu is tuned from a dominant CO product to 77% (FE) C2+ products, which is attributed to enhanced CO* adsorption on the Cu shell due to the core-induced shell strain and electronic effects.110 Crystal structure control in the heterodimers is another important parameter to tune CO2RR towards C–C coupling products, as demonstrated in 4H- or 2H-structure control of the catalyst in core/shell type Au–Cu heterodimer structures with the C2+ products FE reaching 84.3% (Fig. 5(e)–(h)).52,111 Similarly, heterodimer Pd–Cu or Ag–Cu favors C2+ formation.112–114 As a comparison, the intermetallic PdCu structure is more selective towards C1 products (Fig. 5(i)). Given the multi-step nature of CO2RR, catalysts with diverse active sites can accelerate different steps of the reaction. HEA NPs, with their compositional diversity, offer this versatility.115 Recently, machine learning was applied to model reaction pathways and identify ideal compositions,116,117 and PdCuAuAgBiIn HEA NPs were found to be active for CO2RR to formate (98.1% FE).118
Stabilizing catalysts for CO2R remains challenging. Under reaction conditions, NP catalysts tend to undergo structural reconstruction, complicating the correlation between structure and activity. For instance, an initial AgCu alloy NP can disintegrate into smaller Cu- and Ag-rich domains during the reduction process.119 Numerous designs have been proposed to stabilize Cu-based catalysts, though with limited success. One approach involves alloying Cu with Ga, a more oxophilic metal, to strengthen the Cu bond under reduction conditions and protect it from rapid oxidation.120
4.2. Oxygen reduction reaction
The electrocatalytic oxygen reduction reaction (ORR) is a critical process in energy conversion applications such as membrane fuel cells and metal–air batteries. However, its sluggish kinetics hinder practical implementation, necessitating the development of efficient catalysts. Pt-based catalysts remain state-of-the-art, with extensive research dedicated to improving their activity and overcoming kinetic limitations. Theoretical studies and in situ experiments suggest that both associative and dissociative mechanisms occur on Pt surfaces, involving multiple oxygenated intermediates (*O, *OH, *OOH) during O2 reduction.121,122 Among the Pt catalysts studied, monometallic Pt suffers from strong oxygen adsorption, which limits electron transfer and reduces catalytic activity.123 Alloying Pt with first-row transition metals such as Co, Fe, and Ni has proven effective in modifying the surface electronic structure and weakening oxygen adsorption, thereby enhancing ORR activity. MMNPs based on Pt alloys, such as PtFe,18 PtCo,19 and PtCoNi,124 have demonstrated impressive improvements in ORR activity compared to commercial Pt/C catalysts. These improvements arise from strain and ligand effects introduced by additional elements: the compressive strain from smaller metal atoms and their electronic interactions with Pt downshift the Pt d-band center, weakening Pt–OH adsorption and accelerating ORR kinetics.125 This alloying strategy has also been extended to rare-earth metals such as La, Tb, and Tm, which induce pronounced lattice strain and further optimize the adsorption of reaction intermediates, achieving even higher intrinsic activity than alloys with first-row transition metals.126Despite their high activity, Pt-based MMNPs face stability challenges due to oxidation and leaching of non-noble metals under corrosive ORR conditions, particularly in solid-solution structures. Surface reconstruction and changes in NP size, composition, and morphology can compromise long-term performance.21 To mitigate these issues, intermetallic Pt-based MMNPs with core/shell architectures, such as CoPt/Pt,19 PtZn/Pt,127 and PtFe/Pt,32 have been developed, exhibiting both superior stability and activity. Strong d–d coupling between Pt and 3d transition metals stabilizes the non-noble components, while the strained Pt shell offers corrosion resistance and boosts catalytic efficiency. For example, L10-FePt NPs with a Pt shell, synthesized via MgO coating, annealing, and acid etching, exhibited exceptional ORR activity and robust stability in acidic media.32 Similarly, L10-CoPt NPs with a thin Pt shell (Fig. 6(a) and (b)) retained 95% of their Co content after 24 hours of ORR operation (Fig. 6(c)), whereas their A1-phase (solid-solution) counterparts lost 34% within just 7 hours.19 L10-CoPt NPs outperform L10-FePt/Pt ones for ORR in 0.1 M HClO4 (Fig. 6(d)), likely due to the Co-induced lattice reduction and strong ligand effects. Beyond traditional Pt-transition metal alloys, Pt-dichalcogenide alloys are also promising ORR catalysts. For instance, defective PtSe2 alloys undergo structural reconstruction during long-term ORR operation to form a PtSe2/Pt core/shell structure. This transformation significantly enhances its catalytic performance, achieving a mass activity 4.53 times greater than the commercial Pt catalyst even after 126000 accelerated durability cycles. The PtSe2 core provides excellent structural stability, while the reconstructed Pt surface maintains high activity.128
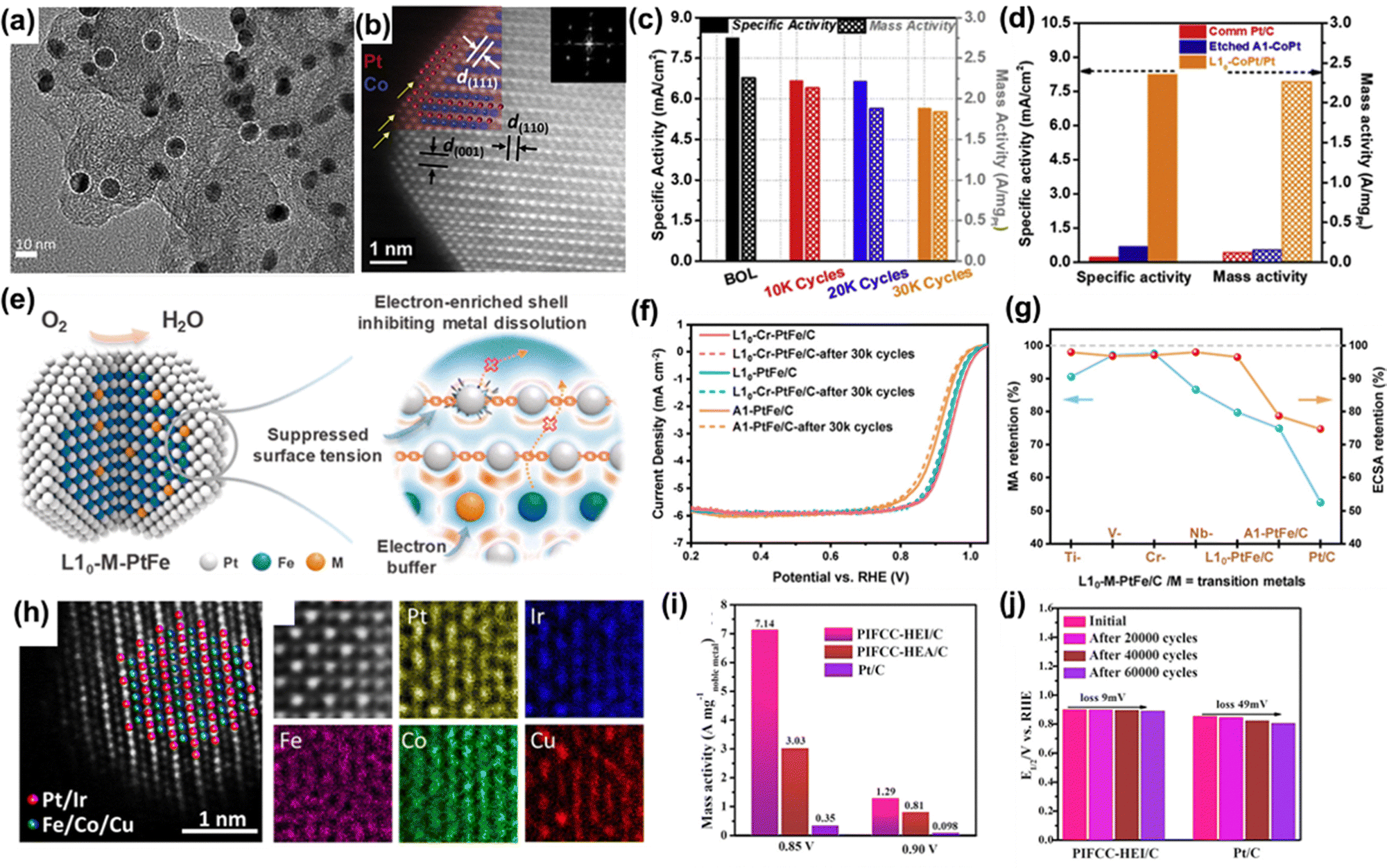 | ||
| Fig. 6 (a) TEM image of L10-CoPt/Pt NPs. (b) HAADF-STEM image of L10-CoPt with a thin Pt shell. (c) The retention of mass & specific activity of L10-CoPt/Pt in the ADT test. (d) The activity comparison of commercial Pt, etched A1-CoPt, and L10-CoPt/Pt. Reproduced from ref. 19 with permission from Elsevier Inc, copyright 2018. (e) Schematic illustration of Cr doping as an electron buffer to stabilize L10-PtFe NPs. (f) ORR LSV polarization curve of the PtFe and PtFeCr catalysts. (g) Retention in mass activity and electrochemical active surface area (ECSA) after ADT test with L10-M-PtFe. Reproduced from ref. 130 with permission from American Chemical Society, copyright 2024. (h) HAADF-STEM of PtIrFeCoCu (PIFCC) HEI NPs and the corresponding EDX mapping. (i) ORR performance comparison of the HEA NP and commercial Pt catalysts. (j) Shift in half-wave potential (E1/2) after ADT test with PIFCC HEI NPs. Reproduced from ref. 82 with permission from American Chemical Society, copyright 2023. | ||
Incorporating additional elements into MMNPs offers greater flexibility to tune properties and improve ORR stability. As an extension of L10-CoPt NPs, ternary L10-CoMPt NPs (M = Mn, Fe, Ni, Cu, Zn) have been investigated both theoretically and experimentally. Intermediates’ adsorption was evaluated based on the compatibility of anisotropic strain induced by the Pt surface and the adsorbates. Among the variants, L10-CoNiPt exhibited the highest ORR activity.20 Incorporating Rh in the PtRhCu/Pt core/shell structure effectively suppresses Pt dissolution in the ORR process, maintaining excellent stability after 30000 potential cycles.129 Further alloying Ti, V, Cr, or Nb with L10-PtFe NPs can provide electron buffers, suppressing surface polarization of Pt shells under harsh electrochemical conditions (Fig. 6(e)). The L10-PtFeCr NPs exhibited only a 2.5% drop in mass activity and a 2.9% loss in electrochemically active surface area after 30000 accelerated durability tests (ADT), compared favorably to L10-PtFe (3.5% loss in mass activity and 21.2% losses in surface area) (Fig. 6(f)). Alloying with Ti, V, or Nb also resulted in higher mass activity retention after ADT (Fig. 6(g)).130
HEI NPs are being explored as next-generation ORR catalysts due to their tunable electronic structures and excellent robustness. For example, HEI PtFeCoNiCu NPs demonstrated a mass activity 15.8 times that of commercial Pt.131 HEI PtIrFeCoCu NPs (Fig. 6(h)) achieved an even higher mass activity of 7.14 A mg−1 noble metal at −0.85 V, compared to 3.63 A mg−1 noble metal for the corresponding HEA counterpart (Fig. 6(i)). Importantly, these HEI NPs retained 99% of their activity after 80 hours of continuous operation, with only a 9 mV shift in half-wave potential (Fig. 6(j)). Their outstanding performance is attributed to a unique (001) surface that enhances Pt-heteroatom interactions and significantly lowers the d-band center, thereby optimizing oxygen adsorption.82 Recent theoretical studies suggest that non-Pt elements in HEI NPs may also participate in the four-electron ORR pathway by facilitating O–O bond cleavage in *OOH intermediates.94 However, this mechanism remains speculative, given that non-noble metals are unstable under acidic ORR conditions. Machine learning has increasingly been employed to design HEI catalysts by optimizing surface strain and formation energy. For instance, computational predictions identified Pt(FeCoNiCu)3 as an optimal candidate, which was experimentally validated to exhibit superior ORR performance (4.09 A mg−1 Pt) compared to PtCu3 (0.96 A mg−1 Pt), Pt(CoCu)3 (1.77 A mg−1 Pt), and Pt(FeCoCu)3 (3.98 A mg−1 Pt).132 The enhanced specific activity of Pt(FeCoNiCu)3 correlates strongly with optimized surface strain, while its excellent stability stems from sluggish diffusion of non-Pt atoms, which increases the alloy's formation energy. These advancements underscore the growing potential of MMNPs for practical ORR applications. Continued efforts in multi-metallic catalyst design, aided by computational modeling and machine learning, are expected to further improve catalytic activity and durability, bringing ORR catalysts closer to commercial viability.
5. MMNPs for electrochemical oxidation reaction
5.1. Alcohol oxidation reaction
The alcohol oxidation reaction (AOR) is widely applied in organic synthesis, energy conversion and storage, and fuel cells. Among these, direct alcohol fuel cells are particularly attractive due to alcohols’ high energy density and ease of storage and transport. Common alcohols such as methanol, ethanol, and ethylene glycol undergo direct oxidation in fuel cells to generate electricity. This energy conversion efficiency is critically dependent on Pt- or Pd-based catalysts.133During the AOR, for example, methanol oxidation reaction (MOR), CO-based intermediates frequently form and can poison the catalyst due to the strong binding between Pt (Pd) and CO.134 Alloying Pt or Pd with oxophilic elements such as Fe, Co, Ni, Cu, Ru, or Sn helps remove oxygenated intermediates from active sites, thereby mitigating the detrimental CO effects.135–137 Nevertheless, the catalysis remains sluggish due to the additional dehydrogenation steps required to break the C–H bonds.138 Recent studies have shown that MMNPs can overcome these issues and accelerate MOR kinetics. For instance, Pt18Ni26Fe15Co14Cu27 HEA NPs (Fig. 7(a)) show exceptional mass activity (15.04 A mg−1 Pt) in catalyzing MOR (Fig. 7(b)).139
 | ||
| Fig. 7 (a) TEM image and the corresponding EDX mapping of Pt18Ni26Fe15Co14Cu27 NPs (scale bar, 5nm). (b) MOR performance of the HEA NP and commercial Pt catalysts. (c) The PDOSs of the Pt18Ni26Fe15Co14Cu27 HEA NPs. Reproduced from ref. 139 with permission from Springer Nature, copyright 2020. (d) TEM image of PdAu NPs. (e) Composition-dependent mass activity of PdAu NPs in EOR. (f) Composition-dependent products selectivity of PdAu NPs. Reproduced from ref. 143 with permission from American Chemical Society, copyright 2019. (g) TEM image of Au/Pt nanowires. (h) High resolution TEM of an Au/Pt nanowire, showing the stepped surface. Scale bar: 2 nm. (i) Schematic illustration of promoted C–C cleavage in EOR on Au/Pt step sites. (j) DFT calculations on activation energy of C–C bond cleavage on different Pt, Au and AuPt facets. Reproduced from ref. 147 with permission from American Chemical Society, copyright 2023. | ||
Density functional theory (DFT) calculations reveal that Co and Ni 3d orbitals contribute to electron depletion, while Cu, Fe, and Co 3d orbitals enhance electron transfer (Fig. 7(c)), thereby facilitating oxidation steps. This synergy underscores the critical role of multi-metallic composition in MOR catalysis.
Ethanol, with its lower toxicity and higher energy density compared to methanol, is another promising fuel. However, its oxidation involves more complex C–C bond cleavage and multi-step dehydrogenation, making complete conversion to CO2 challenging. It is now generally believed that the ethanol oxidation reaction (EOR) proceeds via three main pathways: (i) the direct C1 (12e−) pathway, where ethanol undergoes full oxidation to CO2 via C–C bond cleavage without forming CO; (ii) the indirect C1 (12e−) pathway, in which ethanol partially dehydrogenates, forming CO intermediates before they are oxidized to CO2; and (iii) the C2 (4e−) pathway, where ethanol is oxidized to acetate without C–C cleavage. Although Pt and Pd are widely used for EOR, they suffer from poor selectivity and catalyst poisoning due to sluggish C–C cleavage.133 The removal of poisonous species requires *OH involvement, which is difficult to achieve on Pd or Pt surfaces.140 Alloying with oxophilic metals improves *OH adsorption and CO tolerance, but often favors the C2 pathway.141,142 To enhance C–C cleavage, alloying Pt or Pd with Au,143 Ag,144 Rh,145 and Ir146 has been extensively studied. While Au itself is inactive for EOR, Au-rich PdAu NPs (Au:Pd = 66:34) (Fig. 7(d)) can show high mass activity of 19.5 A mg−1 Pd in 1 M KOH + 1 M EtOH (Fig. 7(e)), favoring the C1 pathway (Fig. 7(f)). This enhancement is attributed to the optimized Pd–Pd bond length and electron transfer from Pd to Au, which weakens CO adsorption.143 The modulation of Pd sites is further demonstrated in core/shell fcc-2H/fcc Au/Pd nanorods, which exhibit enhanced EOR catalysis with superior mass activity (6.82 A mg−1 Pd) and specific activity (13.77 mA cm−2Pd).53 The epitaxial growth of the Pd shell on Au induces Pd lattice expansion, which promotes *OH adsorption and facilitates complete ethanol oxidation. Furthermore, undercoordinated Pt sites along ultrathin (2 nm) Au nanowires (NWs) (Fig. 7(g) nad (h)) are found to be highly active for C–C bond cleavage via the direct C1 pathway (Fig. 7(i)), demonstrating an unprecedented mass activity of 196.9 A mg−1 Pt (Fig. 7(j)).147 The structure is generally active for catalyzing the primary alcohol, including methanol, ethanol, and n-propanol. In another example, core/shell structured Au/PtIr NPs with a stretched PtIr shell exhibit improved ethanol dissociation and C1 intermediate dehydrogenation. Without the Au core, the PtIr alloy favors the indirect pathway, leading to CO formation and catalyst deactivation.148
Stability is a major concern for alloy catalysts under oxidative conditions, particularly in acidic media. To address this issue, intermetallic L10-PtCoAu NPs were introduced as a stable catalyst for AOR in 0.1 M HClO4. The catalyst has a mass activity of 1.55 A mg−1 Pt (vs. 0.45 A mg−1 Pt for Pt/C) and retains its high performance with only a 1.6% loss after 10000 potential cycles, demonstrating much improved stability than the commercial PtRu NP catalyst (52.4% drop after the same potential cycling).149 The enhanced performance is attributed to the stable L10-PtCo core and the CO-resistant AuPt alloy shell.
HEA MMNPs composed of Ir, Os, Pd, Pt, Rh, and Ru were also found to favor the C1 pathway and deliver record-high EOR activity.150 Incorporating oxophilic elements such as Fe, Ni, and Cu into the HEA NPs further promotes *OH adsorption and C–C cleavage, as demonstrated by PtRhFeNiCu NPs with excellent CO tolerance.83 Similar improvements in EOR catalysis have been observed in 15-element HEA NPs.151 However, it is extremely difficult to unravel the specific roles these metals play in the catalysis. Given that intermetallic structures are significantly more robust than solid solution structures, HEI NPs are expected to be more promising catalysts for EOR. Indeed, a recent study shows that HEI PtPdAuFeCoNiCuSn NPs outperform their HEA counterparts in both activity and stability.152
5.2. Oxygen evolution reaction
The oxygen evolution reaction (OER), a critical half-reaction in water splitting and metal–air batteries, involves the oxidation of water molecules to generate oxygen gas.116 Like the ORR, OER also suffers from sluggish kinetics due to its complex four-electron oxidation process. Noble metals such as Ir and Ru,153 along with first-row transition metals like Co, Ni, and Fe,154,155 and their corresponding oxides and hydroxides156 have been studied as effective OER catalysts. OER typically follows two mechanistic pathways: the adsorbate evolution mechanism (AEM) and the lattice oxygen-mediated mechanism (LOM). In the AEM, the reaction advances through sequential oxidation steps involving surface-bound intermediates such as *OH, *O, and *OOH. The catalytic activity is determined by the adsorption energies of these intermediates, which must be precisely balanced—excessively strong or weak binding can impede the reaction rate. Various MMNPs have been studied to optimize these adsorption energies in line with the d-band theory. For example, Ru1−xMx (M = Co, Fe, Ni) NPs demonstrate improved OER performance compared to pure Ru, due to the incorporation of electron-rich 3d metals that modulate the electronic structure. Among these NPS, Ru0.7Co0.3 exhibits the highest activity.157 However, a major limitation in AEM-based catalysis is the linear scaling relationship between the adsorption energies of OER intermediates, which restricts the simultaneous optimization of all reaction steps.158 To overcome this challenge, HEA NPs have been studied as a promising class of OER catalysts.159 Their diverse atomic environments allow different reaction intermediates to adopt distinct adsorption configurations, effectively breaking the scaling relationship. For instance, HEA CoFeNiGaZn NPs exhibit superior OER activity compared to bimetallic and ternary analogues. The combined ligand and strain effects shift the d-band center, while the adsorption free energy difference (ΔGO – ΔGOH) at the Ga sites closely aligns with the apex of the OER activity volcano plot, indicating optimized energetics.160Despite their high activity, MMNP-based OER catalysts encounter ongoing durability issues due to metal dissolution in severe oxidative environments. This instability is often linked to the LOM pathway, where lattice oxygen is involved in the reaction. Although LOM can boost activity, it may also lead to catalyst degradation due to lattice oxygen loss. Reducing lattice oxygen participation by weakening the binding of oxygen species can slow oxygen diffusion and improve catalyst stability.161 A recent study demonstrates that Ru-diluted PtCu alloys achieve both low overpotential (220 mV at 10 mA cm−2) and remarkable stability. In this system, weaker oxygen binding on the PtCu surface prevents overoxidation of isolated Ru atoms, thereby reducing dissolution and promoting the AEM pathway.162
Overall, MMNPs provide significant benefits for OER catalysis, such as adjustable electronic structures, high intrinsic conductivity, and multifunctional active sites. However, their surfaces frequently experience oxidation or hydroxylation under OER conditions, resulting in oxide/hydroxide layers that may act as the true catalytic species.163,164 Therefore, the future design of robust OER catalysts should integrate these structural transformations into both experimental evaluations and theoretical models to more accurately represent the dynamic nature of electrocatalyst surfaces during operation.
6. MMNPs for thermal catalysis under green chemistry conditions
Green chemistry focuses on creating sustainable solutions to environmental issues by designing chemical products and processes that reduce the use and generation of hazardous substances. Catalysis plays a crucial role in this process. It involves reducing energy consumption, minimizing chemical waste, utilizing renewable resources, and developing safer, more efficient reaction pathways.165 MMNPs are promising catalyst candidates for green chemistry applications due to their high and adjustable activity, solid-state nature, and ease of separation from reaction media. Their multi-component composition enables precise tuning of catalytic efficiency and facilitates tandem reactions with high selectivity.The dehydrogenation of hydrogen-rich small molecules has been extensively studied for the release of H2 under green chemistry conditions. Two notable molecules in these studies are formic acid (HCOOH, FA)166 and ammonia borane (NH3BH3, AB).167 FA has a hydrogen content of 4.38 wt% and can be easily obtained from biomass-derived syngas synthesis168 or directly from CO2 reduction reactions.169 Au, Pd, and Pt NPs show catalytic activity in FA dehydrogenation to produce H2 and CO2. However, their efficiency is often limited by low hydrogen production rates and poor selectivity due to the competing dehydration pathway (HCOOH → CO + H2O). The key steps in FA dehydrogenation involve C–H bond cleavage and hydrogen (H) atom desorption for H2 recombination. Although Pd exhibits high hydrogen dissociation activity, its strong hydrogen adsorption and susceptibility to CO poisoning hinder the reaction rate. Alloying Pd with a weaker hydrogen-binding metal, such as Ag or Au, in bimetallic NPs effectively enhances FA dehydrogenation while mitigating CO poisoning due to the weak CO-binding affinity of Ag or Au.170,171 For example, Ag42Pd58 NPs showed significantly higher catalytic activity (TOF = 382 h−1) for aqueous FA dehydrogenation at 323 K compared to Pd NPs (TOF = 8.3 h−1).172 Further engineering the NPs into a core/shell AgPd/Pd structure substantially improved its activity (TOF = 21500 h−1) under similar conditions,173 suggesting that beyond electronic effects,174,175 strain in the thin Pd shell also contributes significantly to the catalysis enhancement. However, the promotional effects of metal on the alloy catalysis are not that simple: Ag may function not only as an electronic modifier but also as an active site in AgPd NPs.176 Further atomic-level mechanistic studies are needed to elucidate FA dehydrogenation pathways on MMNP surfaces and, more importantly, to develop an economically viable catalyst and process for dehydrogenation. Currently, catalyst deactivation, CO contamination from competing dehydration pathways, and the need for efficient gas–liquid separation systems complicate the scale-up process. While FA itself is relatively affordable ($0.40 to $0.70 per kg), the overall viability of the process depends on developing cost-effective, durable catalysts and integrating FA production from renewable CO2 sources to create a sustainable and economically competitive hydrogen supply chain.
Similar alloying and structural modification strategies have also been applied to study NP catalysis for AB dehydrogenation via hydrolysis (NH3BH3 + 2H2O → NH4+ + BO2− + 3H2) or methanolysis (NH3BH3 + 4CH3OH → NH4+ + B(OCH3)4− + 3H2). AB is another attractive hydrogen storage material with high hydrogen content (∼19 wt%), low toxicity, and air stability.177 Pd-based NPs are commonly studied as active catalysts for AB methanolysis, and alloying Pd with Cu, Ni, or Fe has been shown to accelerate hydrogen production rates.178,179 Alloying likely modifies BH3NH3 adsorption properties, facilitating B–N bond cleavage, a critical step in AB dehydrogenation.180 Recently, non-noble alloy NP catalysis for AB dehydrogenation has gained much attention.181 Recent focus has been on Ni-based alloy NPs, such as CuNi NPs.182,183 Despite this progress, this process is hardly commercially viable, primarily due to the high cost of AB ($50 to $300 per kg). Consequently, this reaction is more suitable for niche applications requiring high gravimetric energy density and compact hydrogen release systems, especially for direct hydrogenation reactions.
Hydrogen generated in situ via dehydrogenation can be directly used in tandem hydrogenation reactions catalyzed by MMNPs. This approach is both safe and environmentally friendly as it avoids the need for hydrogen separation and high-pressure conditions. Tandem hydrogenation is more generally applied in biomass conversion, pollutant remediation, and the synthesis of value-added chemicals.184–186 For example, CuNi NPs can catalyze not only AB methanolysis but also hydrogenation of nitro and nitrile compounds (R-NO2/R-CN) into primary amines (R-NH2/RCH2NH2) with nearly quantitative yield.182 CoNi NPs are also active for catalyzing this tandem reaction.187 However, using AB for hydrogenation reactions does have its limitations – the other reaction products NH3 and borates may interfere with subsequent hydrogenation steps if not removed. On the other hand, the presence of NH3 can be further utilized as a N-source for C–N bond formation, as demonstrated in AgPd NP catalyzed AB hydrogenation of o-nitroacetophenone, which not only hydrogenates the nitro group, but also promotes nitrogenation of the acetyl group, forming quinazoline.188 MMNPs have also been found to be active for catalyzing the tandem semihydrogenation of phenylacetylene to styrene189 and the hydrodehalogenation of halogenated aromatics.186
Compared to AB, FA is a better agent for the hydrogenation reaction, as its dehydrogenation yields only gaseous products (CO2 and H2) without any liquid/solid residue accumulation in the reaction mixture. AgPd NPs can efficiently catalyze FA dehydrogenation and subsequent hydrogenation of nitroarenes with near-quantitative yields under mild conditions (1 atm, 60–80 °C).186,190 These NPs further promote condensation of hydrogenated products with aldehydes, forming heterocycles such as benzoxazoles and quinazolines.
Tandem green chemistry strategies have also been extended to polymer synthesis. A notable example is the synthesis of poly(benzoxazole) (PBO) via sequential hydrogenation and condensation using AuPd NPs as a catalyst (Fig. 8(a) and (b)).191,192 PBO is a rigid and thermally stable polymer, and has found potential applications in ballistic fibers and thermomechanical components.193 Conventional PBO synthesis relies on polyphosphoric acid, a highly corrosive medium, and the main source of polymer contamination that is detrimental to PBO's hydrolytic stability.194 This limitation can be addressed by using AuPd alloy NPs to catalyze the FA dehydrogenation, selective hydrogenation of 1,5-diisopropoxy-2,4-dinitrobenzene, and Schiff-base condensation with terephthalaldehyde (Fig. 8(c)), yielding PBO precursor that can be further converted to PBO via annealing (Fig. 8(d)). The resulting PBO shows much enhanced hydrolytic and thermal stability compared to commercial PBO (Zylon).192 Core/shell B2-CuPd/Pd NPs (Fig. 8(e) and (f)) are also found to be efficient for catalyzing the tandem reactions. More importantly, they show the desired selective hydrogenation of –NO2 over –CHO (Fig. 8(g)), making it possible to synthesize PBO in a one-pot reaction.195
 | ||
| Fig. 8 (a) TEM image of AuPd NPs and (b) AuPd/C. Scale bar: 2 nm, inset 1 nm (c) One-pot synthesis of PBO precursor using diisopropoxy-2,4-dinitrobenzene and terephthalaldehyde. HCOOH is used as a hydrogen source. (d) Transformation of PBO precursor to PBO upon annealing. Reproduced from ref. 192 with permission from American Chemical Society, copyright 2021. (e) TEM images of B2-CuPd NPs with a thin Pd shell. (f) HAADF-STEM image of B2-CuPd. (g) Summary of activity of B2-CuPd with thin Pd shell in catalyzing hydrogenation of nitrobenzene, benzaldehyde, and the Schiff base condensation process. Reproduced from ref. 195 with permission from American Chemical Society, copyright 2024. | ||
The examples above demonstrate that the modular catalytic properties of MMNPs enable complex chemical reactions to occur in a single pot, highlighting their transformative potential in green chemistry.
7. Conclusions
This review summarizes the advances in MMNPs research, including NP synthesis and applications in representative electrocatalysis and thermocatalysis. Binary and tertiary MMNPs are used as examples to illustrate synthetic and catalytic concepts before extending to multicomponent high-entropy alloys. The MMNPs discussed are categorized into three primary structural types: solid solutions, intermetallics, and heterostructures. Solid solution alloys have different metal atoms randomly distributed in the crystal structure. They provide broad compositional tunability and flexible electronic structures, allowing for property tailoring to specific catalytic reactions. Intermetallic structure features metal atoms arranged in a chemically ordered fashion with well-defined stoichiometry, which contributes to their excellent structural robustness and catalytic site uniformity. These characteristics make them highly effective in specific catalytic applications where uniformity and stability are crucial. Heterostructured MMNPs capitalize on synergistic effects at different NP interfaces, facilitating unique interactions between different materials, leading to improved performance that surpasses the capabilities of individual components.Three structural features of MMNPs provide clear guidance for future catalyst design. For systems requiring fine-tuned activity under mild reaction conditions, solid solution NPs should be selected as catalysts. Their broad compositional tunability and flexible electronic structures allow for precise optimization of catalytic activity and selectivity. However, their chemical instability must be managed to ensure long-term durability. For applications demanding structural robustness, intermetallic NPs are the preferred choice. Their chemically ordered atomic arrangements and well-defined stoichiometry contribute to excellent structural stability and uniform catalytic sites. This makes them highly effective in environments where durability and consistency are critical, despite their limited compositional flexibility and more demanding synthesis requirements. For processes requiring multifunctional or interfacial catalysis, heterostructured NPs are the best candidates. These NPs leverage synergistic effects at interfaces, combining high activity with enhanced durability, leading to superior catalytic performance, although precise interface engineering and a deeper mechanistic understanding are necessary to fully exploit their potential. These structure–property correlations offer a comprehensive framework for rational MMNPs design tailored to specific catalytic reactions. By understanding and leveraging the distinct advantages and limitations of each structural type, one can develop more effective and specialized catalysts for a wide range of applications.
Despite the promise of MMNPs, the inherent complexity of multi-component systems makes precise design and mechanistic understanding challenging. The role of individual elements in promoting activity or stability is not yet fully understood, especially in high-entropy systems. Synthetic control over phase purity, elemental distribution, and surface termination also remains a significant hurdle. Machine/deep learning and first-principles modeling should be more broadly applied to accelerate theoretical screening of complex MMNPs and predict optimal compositions and structures. It is also important to develop high-throughput and automated synthesis/testing platforms to experimentally validate theoretical predictions and construct comprehensive structure–activity databases of MMNPs. Advanced in situ/operando characterization techniques should be widely adopted to track structural and electronic evolution during reactions to establish direct links between structure and function. By integrating computational design, advanced characterization, and synthetic innovation, the field is well-positioned to unlock next-generation materials for sustainable electrochemical and chemical technologies.
Beyond the conventional MMNPs summarized in this review, new concepts such as single-atom alloys,196 intermetallic single-atom alloys,197 and HEI structures82,83 have recently been pursued. These architectures retain the benefits of traditional MMNPs while introducing high densities of isolated active sites or enhanced entropy-driven stability. For example, intermetallic single-atom alloys integrate isolated active atoms within robust intermetallic matrices, achieving high performance and loading, which is a challenge in traditional single-atom systems.198 However, these novel materials require clearer structural definitions and controlled synthesis strategies to fully unlock their catalytic potential.
In all, MMNPs have emerged as a versatile and powerful platform for catalysis, offering unparalleled compositional flexibility and adjustable surface properties. This flexibility allows for the precise tuning of catalytic activity and stability, making MMNPs highly adaptable to various catalytic processes. By manipulating the electronic structure, atomic arrangement, and surface coordination environments, researchers can achieve specific catalytic behaviors tailored to desired reactions. This precise control presents a unique opportunity to delve into and exploit nanoscale structure–property relationships, providing deeper insights into how these properties influence catalytic performance. Understanding these relationships can lead to the development of more efficient and effective catalysts, paving the way for advancements in various industrial applications. However, the complexity of these manipulations requires sophisticated techniques and a thorough understanding of the underlying principles, highlighting the need for continued research and innovation in the field.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
This work was supported in part by the NSF under grants of CHE-2102290, CBET-2324345, and by Toyota.Notes and references
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- S. Akbayrak, S. Özkar, H. Kim, T. Y. Yoo, M. S. Bootharaju, J. H. Kim, D. Y. Chung and T. Hyeon, Adv. Sci., 2022, 9, 2104054 CrossRef PubMed.
- B. A. Yusuf, W. Yaseen, J. Xie, A. A. Babangida, A. I. Muhammad, M. Xie and Y. Xu, Nano Energy, 2022, 104, 107959 CrossRef CAS.
- Z. Cui, W. Jiao, Z. Huang, G. Chen, B. Zhang, Y. Han and W. Huang, Small, 2023, 19, 2301465 CrossRef CAS PubMed.
- D. J. Loevlie, B. Ferreira and G. Mpourmpakis, Acc. Chem. Res., 2023, 56, 248–257 CrossRef CAS PubMed.
- H. Duan, D. Wang and Y. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- S. Sun, S. Anders, T. Thomson, J. E. E. Baglin, M. F. Toney, H. F. Hamann, C. B. Murray and B. D. Terris, J. Phys. Chem. B, 2003, 11, 5419–5425 CrossRef.
- M. Nakaya, Y. Tsuchiya, K. Ito, Y. Oumi, T. Sano and T. Teranishi, Chem. Lett., 2004, 33, 130–131 CrossRef CAS.
- V. Mazumder, M. Chi, M. N. Mankin, Y. Liu, Ö. Metin, D. Sun, K. L. More and S. Sun, Nano Lett., 2012, 12, 1102–1106 CrossRef CAS PubMed.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed.
- V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115–2117 CrossRef CAS PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- R.-Y. Shao, X.-C. Xu, Z.-H. Zhou, W.-J. Zeng, T.-W. Song, P. Yin, A. Li, C.-S. Ma, L. Tong, Y. Kong and H.-W. Liang, Nat. Commun., 2023, 14, 5896 CrossRef CAS PubMed.
- G. Chen, S. Desinan, R. Rosei, F. Rosei and D. Ma, Chem. – Eur. J., 2012, 18, 7925–7930 CrossRef CAS PubMed.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 18, 1864–1871 CrossRef PubMed.
- B. Huang, H. Kobayashi, T. Yamamoto, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2017, 139, 4643–4646 CrossRef CAS PubMed.
- T. Komatsu, H. Kobayashi, K. Kusada, Y. Kubota, M. Takata, T. Yamamoto, S. Matsumura, K. Sato, K. Nagaoka and H. Kitagawa, Chem. – Eur. J., 2017, 23, 57–60 CrossRef CAS PubMed.
- S. Guo and S. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Joule, 2019, 3, 124–135 CrossRef CAS.
- J. Li, S. Sharma, K. Wei, Z. Chen, D. Morris, H. Lin, C. Zeng, M. Chi, Z. Yin, M. Muzzio, M. Shen, P. Zhang, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2020, 142, 19209–19216 CrossRef CAS PubMed.
- J. Li and S. Sun, Acc. Chem. Res., 2019, 52, 2015–2025 CrossRef CAS PubMed.
- J. Guan, D. Dong, N. A. Khan and Y. Zheng, Chem. Commun., 2024, 60, 1811–1825 RSC.
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed.
- J. Kim, Y. Lee and S. Sun, J. Am. Chem. Soc., 2010, 132, 4996–4997 CrossRef CAS PubMed.
- D. Zhang, B. Gökce and S. Barcikowski, Adv. Funct. Mater., 2023, 13, 10988–11000 Search PubMed.
- B. M. Leonard, Q. Zhou, D. Wu and F. J. DiSalvo, Chem. Mater., 2011, 23, 1136–1146 CrossRef CAS.
- A. C. Foucher, S. Yang, D. J. Rosen, R. Huang, J. B. Pyo, O. Kwon, C. J. Owen, D. F. Sanchez, I. I. Sadykov, D. Grolimund, B. Kozinsky, A. I. Frenkel, R. J. Gorte, C. B. Murray and E. A. Stach, J. Am. Chem. Soc., 2023, 145, 5410–5421 CrossRef CAS PubMed.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, J. Am. Chem. Soc., 2021, 4, 1179–1201 Search PubMed.
- H. Lv, Y. Zheng, Y. Wang, J. Wang, B. Liu and Z.-A. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202304420 CrossRef CAS PubMed.
- H.-S. Chen, T. M. Benedetti, V. R. Gonçales, N. M. Bedford, R. W. J. Scott, R. F. Webster, S. Cheong, J. J. Gooding and R. D. Tilley, J. Am. Chem. Soc., 2020, 142, 3231–3239 CrossRef CAS PubMed.
- X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 CrossRef CAS PubMed.
- J. Li, Z. Xi, Y.-T. Pan, J. S. Spendelow, P. N. Duchesne, D. Su, Q. Li, C. Yu, Z. Yin, B. Shen, Y. S. Kim, P. Zhang and S. Sun, J. Am. Chem. Soc., 2018, 140, 2926–2932 CrossRef CAS PubMed.
- H. Chen, D. Wang, Y. Yu, K. A. Newton, D. A. Muller, H. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2012, 134, 18453–18459 CrossRef CAS PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y.-H. Chung, H. Kim, B. S. Mun, K.-S. Lee, N.-S. Lee, S. J. Yoo, D.-H. Lim, K. Kang, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS PubMed.
- T.-W. Song, C. Xu, Z.-T. Sheng, H.-K. Yan, L. Tong, J. Liu, W.-J. Zeng, L.-J. Zuo, P. Yin, M. Zuo, S.-Q. Chu, P. Chen and H.-W. Liang, Nat. Commun., 2022, 13, 6521 CrossRef CAS PubMed.
- S. Zhang, S. Guo, H. Zhu, D. Su and S. Sun, J. Am. Chem. Soc., 2012, 134, 5060–5063 CrossRef CAS PubMed.
- H. Wang, P. Shang, J. Zhang, M. Guo, Y. Mu, Q. Li and H. Wang, Chem. Mater., 2013, 25, 2450–2454 CrossRef CAS.
- J. Liang, Y. Wan, H. Lv, X. Liu, F. Lv, S. Li, J. Xu, Z. Deng, J. Liu, S. Zhang, Y. Sun, M. Luo, G. Lu, J. Han, G. Wang, Y. Huang, S. Guo and Q. Li, Nat. Mater., 2024, 23, 1259–1267 CrossRef CAS PubMed.
- W. Lei, J. Xu, Y. Yu, W. Yang, Y. Hou and D. Chen, Nano Lett., 2018, 18, 7839–7844 CrossRef CAS PubMed.
- K. Jiang, P. Wang, S. Guo, X. Zhang, X. Shen, G. Lu, D. Su, X. Huang, C. Wang, D. P. Chen, X. Sang, R. R. Unocic and S. E. Skrabalak, ACS Nano, 2016, 10, 6345–6353 CrossRef PubMed.
- J. K. Mathiesen, E. D. Bøjesen, J. K. Pedersen, E. T. S. Kjær, M. Juelsholt, S. Cooper, J. Quinson, A. S. Anker, G. Cutts, D. S. Keeble, M. S. Thomsen, J. Rossmeisl and K. M. Ø. Jensen, Small Methods, 2022, 6, 2200420 CrossRef CAS PubMed.
- H. Li, J. Lai, Z. Li and L. Wang, ACS Catal., 2019, 2, 1370–1380 Search PubMed.
- J. Qiu, Q. N. Nguyen, Z. Lyu, Q. Wang and Y. Xia, Adv. Mater., 2021, 34, 2102591 CrossRef PubMed.
- Y. Song, K. Liu and S. Chen, Langmuir, 2012, 28, 17143–17152 CrossRef CAS PubMed.
- B. Ni, P. He, W. Liao, S. Chen, L. Gu, Y. Gong, K. Wang, J. Zhuang, L. Song, G. Zhou and X. Wang, Small, 2018, 14, 1703749 CrossRef PubMed.
- C. Wang, Y. Shi, D. Qin, Y. Xia, Y. Song, K. Liu and S. Chen, Nanoscale Horiz., 2023, 8, 1194–1204 RSC.
- M. Zhou, H. Wang, A. O. Elnabawy, Z. D. Hood, M. Chi, P. Xiao, Y. Zhang, M. Mavrikakis and Y. Xia, Chem. Mater., 2019, 31, 1370–1380 CrossRef CAS.
- X. Sun, D. Li, S. Guo, W. Zhu and S. Sun, Nanoscale, 2016, 8, 2626–2631 RSC.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- M. Zhao, Z. Chen, Z. Lyu, Z. D. Hood, M. Xie, M. Vara, M. Chi and Y. Xia, J. Am. Chem. Soc., 2019, 141, 7028–7036 CrossRef CAS PubMed.
- S.-I. Choi, A. Young, S. R. Lee, C. Ma, M. Luo, M. Chi, C.-K. Tsung and Y. Xia, Nanoscale Horiz., 2019, 4, 1232–1238 RSC.
- Y. Ma, M. Sun, H. Xu, Q. Zhang, J. Lv, W. Guo, F. Hao, W. Cui, Y. Wang, J. Yin, H. Wen, P. Lu, G. Wang, J. Zhou, J. Yu, C. Ye, L. Gan, D. Zhang, S. Chu, L. Gu, M. Shao, B. Huang and Z. Fan, Adv. Mater., 2024, 36, 2402979 CrossRef CAS PubMed.
- X. Zhou, Y. Ma, Y. Ge, S. Zhu, Y. Cui, B. Chen, L. Liao, Q. Yun, Z. He, H. Long, L. Li, B. Huang, Q. Luo, L. Zhai, X. Wang, L. Bai, G. Wang, Z. Guan, Y. Chen, C.-S. Lee, J. Wang, C. Ling, M. Shao, Z. Fan and H. Zhang, J. Am. Chem. Soc., 2021, 144, 547–555 CrossRef PubMed.
- F.-R. Fan, D.-Y. Liu, Y.-F. Wu, S. Duan, Z.-X. Xie, Z.-Y. Jiang and Z.-Q. Tian, J. Am. Chem. Soc., 2008, 8, 6949–6951 CrossRef PubMed.
- V. D. Pawlik, A. Janssen, Y. Ding and Y. Xia, J. Phys. Chem. C, 2024, 128, 1377–1385 CrossRef CAS PubMed.
- X. Lyu, Y. Jia, X. Mao, D. Li, G. Li, L. Zhuang, X. Wang, D. Yang, Q. Wang, A. Du and X. Yao, Adv. Mater., 2020, 32, 2003493 CrossRef CAS PubMed.
- M. Vara, L. T. Roling, X. Wang, A. O. Elnabawy, Z. D. Hood, M. Chi, M. Mavrikakis and Y. Xia, ACS Nano, 2017, 11, 4571–4581 CrossRef CAS PubMed.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- S. Liu, S. Guo, S. Sun and X.-Z. You, Nanoscale, 2015, 7, 4890–4893 RSC.
- W. Yang, W. Lei, Y. Yu, W. Zhu, T. A. George, X.-Z. Li, D. J. Sellmyer and S. Sun, J. Mater. Chem. C, 2015, 3, 7075–7080 RSC.
- M. N. Tahir, F. Natalio, M. A. Cambaz, M. Panthöfer, R. Branscheid, U. Kolb and W. Tremel, Nanoscale, 2013, 5, 9944 RSC.
- A. Mendoza-Garcia and S. Sun, Adv. Funct. Mater., 2016, 26, 3809–3817 CrossRef CAS.
- J. Zeng, C. Zhu, J. Tao, M. Jin, H. Zhang, Z.-Y. Li, Y. Zhu and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 2354–2358 CrossRef CAS PubMed.
- X. Qiu, V. Pawlik, S. Zhou, J. Tao and Y. Xia, J. Am. Chem. Soc., 2023, 145, 13400–13410 CrossRef CAS PubMed.
- Y. Feng, Y. Wang, J. He, X. Song, Y. Y. Tay, H. H. Hng, X. Y. Ling and H. Chen, J. Am. Chem. Soc., 2015, 137, 7624–7627 CrossRef CAS PubMed.
- J. Feng, F. Yang, X. Wang, F. Lyu, Z. Li and Y. Yin, Adv. Mater., 2019, 31, 1900789 CrossRef PubMed.
- B. Liu, S. Thanneeru, A. Lopes, L. Jin, M. McCabe and J. He, Small, 2017, 13, 1700091 CrossRef PubMed.
- H. Duan, H. Tan and J. He, Nano Res., 2025, 18, 94907147 CrossRef.
- Q. Wang, Z. L. Zhao and M. Gu, Small, 2019, 15, 1903122 CrossRef CAS PubMed.
- Y. Y. Chen, T. Duval, U. D. Hung, J. W. Yeh, H. C. Shih, J. Li, S. Sharma, K. Wei, Z. Chen, D. Morris, H. Lin, C. Zeng, M. Chi, Z. Yin, M. Muzzio, M. Shen, P. Zhang, A. A. Peterson, S. Sun, T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2005, 47, 2257–2279 CAS.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu and M. Rehwoldt, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed.
- W. Ji, W. Wang, H. Wang, J. Zhang, Y. Wang, F. Zhang and Z. Fu, Intermetallics, 2015, 56, 24–27 CrossRef CAS.
- H. Lin, M. Muzzio, K. Wei, P. Zhang, J. Li, N. Li, Z. Yin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2019, 9, 18547–18558 Search PubMed.
- J. Johny, Y. Li, M. Kamp, O. Prymak, S.-X. Liang, T. Krekeler, M. Ritter, L. Kienle, C. Rehbock, S. Barcikowski and S. Reichenberger, Nano Res., 2022, 15, 4807–4819 CrossRef CAS.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2020, 11, 12731–12736 RSC.
- Y. Chen, X. Zhan, S. L. A. Bueno, I. H. Shafei, H. M. Ashberry, K. Chatterjee, L. Xu, Y. Tang and S. E. Skrabalak, Nanoscale Horiz., 2021, 6, 231–237 RSC.
- C. Moreira Da Silva, H. Amara, F. Fossard, A. Girard, A. Loiseau and V. Huc, Nanoscale, 2022, 14, 9832–9841 RSC.
- P.-C. Chen, X. Liu, J. L. Hedrick, Z. Xie, S. Wang, Q.-Y. Lin, M. C. Hersam, V. P. Dravid and C. A. Mirkin, Science, 2016, 352, 1565–1569 CrossRef CAS PubMed.
- S. L. A. Bueno, A. Leonardi, N. Kar, K. Chatterjee, X. Zhan, C. Chen, Z. Wang, M. Engel, V. Fung and S. E. Skrabalak, ACS Nano, 2022, 16, 18873–18885 CrossRef CAS PubMed.
- G. Cao, J. Liang, Z. Guo, K. Yang, G. Wang, H. Wang, X. Wan, Z. Li, Y. Bai, Y. Zhang, J. Liu, Y. Feng, Z. Zheng, C. Lu, G. He, Z. Xiong, Z. Liu, S. Chen, Y. Guo, M. Zeng, J. Lin and L. Fu, Nature, 2023, 619, 73–77 CrossRef CAS PubMed.
- M. Cui, C. Yang, S. Hwang, M. Yang, S. Overa, Q. Dong, Y. Yao, A. H. Brozena, D. A. Cullen, M. Chi, T. F. Blum, D. Morris, Z. Finfrock, X. Wang, P. Zhang, V. G. Goncharov, X. Guo, J. Luo, Y. Mo, F. Jiao and L. Hu, Sci. Adv., 2022, 8, eabm4322 CrossRef CAS PubMed.
- G. Feng, F. Ning, Y. Pan, T. Chen, J. Song, Y. Wang, R. Zou, D. Su and D. Xia, J. Am. Chem. Soc., 2023, 145, 11140–11150 CrossRef CAS PubMed.
- D. Wang, Z. Chen, Y. Wu, Y. Huang, L. Tao, J. Chen, C. Dong, C. V. Singh and S. Wang, Sci. China Mater., 2021, 64, 2454–2466 CrossRef CAS.
- Z. Ma, J. Mohapatra, K. Wei, J. P. Liu and S. Sun, Chem. Rev., 2023, 123, 3904–3943 CrossRef CAS PubMed.
- M. Che, Catal. Today, 2013, 218–219, 162–171 CrossRef CAS.
- H. Lin, Y. Liu, J. Deng, L. Jing, Z. Wang, L. Wei, Z. Wei, Z. Hou, J. Tao and H. Dai, Environ. Sci. Adv., 2025, 4, 33–56 CAS.
- S. Bhattacharjee, U. V. Waghmare and S.-C. Lee, Sci. Rep., 2016, 6, 35916 CrossRef CAS PubMed.
- L. Sun, K. Wen, G. Li, X. Zhang, X. Zeng, B. Johannessen and S. Zhang, ACS Mater. Au, 2024, 4, 547–556 CrossRef CAS PubMed.
- N. Hashimoto, K. Mori, H. Yoshida, N. Kamiuchi, R. Kitaura, R. Hirasawa and H. Yamashita, Nano Lett., 2024, 24, 7063–7068 CrossRef CAS PubMed.
- A. Mehta and Y. Sohn, Mater. Res. Lett., 2021, 9, 239–246 CrossRef CAS.
- E. Lotfi-Khojasteh, H. Elmkhah, M. Nouri and P. H. Mayrhofer, Adv. Eng. Mater., 2024, 26, 2301934 CrossRef CAS.
- H. Kuang, Z. Xu, X. Tan, K. Yu and C. Chen, Small, 2024, 20, 2308421 CrossRef CAS PubMed.
- J. Hao, F. Ma, Y. Chen, S. Lu, F. Duan, M. Du, C. Wang, W. Zhang and H. Zhu, New J. Chem., 2024, 48, 511–514 RSC.
- T. Chen, C. Qiu, X. Zhang, H. Wang, J. Song, K. Zhang, T. Yang, Y. Zuo, Y. Yang, C. Gao, W. Xiao, Z. Jiang, Y. Wang, Y. Xiang and D. Xia, J. Am. Chem. Soc., 2023, 146, 1174–1184 CrossRef PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, ACS Catal., 2018, 8, 1510–1519 CrossRef CAS.
- X. Yuan, L. Zhang, L. Li, H. Dong, S. Chen, W. Zhu, C. Hu, W. Deng, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2019, 141, 4791–4794 CrossRef CAS PubMed.
- R. Lin, X. Ma, W.-C. Cheong, C. Zhang, W. Zhu, J. Pei, K. Zhang, B. Wang, S. Liang, Y. Liu, Z. Zhuang, R. Yu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nano Res., 2019, 12, 2866–2871 CrossRef CAS.
- Y. Mun, S. Lee, A. Cho, S. Kim, J. W. Han and J. Lee, Appl. Catal., B, 2019, 246, 82–88 CrossRef CAS.
- S. Wei, Y. Xu, T. Song, H. Dai, F. Li, X. Gao, Y. Zhai, S. Gong, R. Li, X. Zhang and K. Chan, J. Am. Chem. Soc., 2025, 147, 4219–4229 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- S. Kuang, M. Li, X. Chen, H. Chi, J. Lin, Z. Hu, S. Hu, S. Zhang and X. Ma, Chin. Chem. Lett., 2023, 34, 108013 CrossRef CAS.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu, J.-L. Luo, A. N. Kuhn, H. Zhao, U. O. Nwabara, X. Lu, M. Liu, Y. Pan, W. Zhu, P. J. A. Kenis and H. Yang, Adv. Funct. Mater., 2021, 31, 2101668 CrossRef.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, Adv. Funct. Mater., 2021, 31, 2101668 CrossRef.
- Y. Zhao, Y. Wang, Z. Yu, C. Song, J. Wang, H. Huang, L. Meng, M. Liu and L. Liu, ACS Nano, 2025, 19, 4505–4514 CrossRef CAS PubMed.
- S. Jeong, C. Huang, Z. Levell, R. X. Skalla, W. Hong, N. J. Escorcia, Y. Losovyj, B. Zhu, A. N. Butrum-Griffith, Y. Liu, C. W. Li, D. Reifsnyder Hickey, Y. Liu and X. Ye, J. Am. Chem. Soc., 2024, 146, 4508–4520 CrossRef CAS PubMed.
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rümmerli, L. Xu, Y. Peng, S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi, P. J. A. Kenis, V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi, R. Buonsanti, A. N. Kuhn, H. Zhao, U. O. Nwabara, X. Lu, M. Liu, Y. Pan, W. Zhu, P. J. A. Kenis and H. Yang, Adv. Funct. Mater., 2021, 60, 2508–2518 CAS.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS.
- S. Zhang, B. Zhang, S. Yang, T. Shao, X. Li, R. Cao and M. Cao, ACS Appl. Nano Mater., 2025, 8, 1893–1902 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 109, 2106715 CrossRef.
- H. Li, X. Li, P. Wang, Z. Zhang, K. Davey, J. Q. Shi and S.-Z. Qiao, J. Am. Chem. Soc., 2024, 146, 22850–22858 CrossRef CAS PubMed.
- Z. W. Chen, Z. Gariepy, L. Chen, X. Yao, A. Anand, S.-J. Liu, C. G. Tetsassi Feugmo, I. Tamblyn and C. V. Singh, ACS Catal., 2022, 12, 14864–14871 CrossRef CAS.
- H. Li, H. Huang, Y. Chen, F. Lai, H. Fu, L. Zhang, N. Zhang, S. Bai and T. Liu, Adv. Mater., 2023, 35, 2209242 CrossRef CAS.
- P.-C. Chen, C. Chen, Y. Yang, A. L. Maulana, J. Jin, J. Feijoo and P. Yang, J. Am. Chem. Soc., 2023, 145, 10116–10125 CrossRef CAS PubMed.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- V. Tripković, E. Skúlason, S. Siahrostami, J. K. Nørskov and J. Rossmeisl, Electrochim. Acta, 2010, 55, 7975–7981 CrossRef.
- J. Zhao, J. Lian, Z. Zhao, X. Wang and J. Zhang, Nano-Micro Lett., 2023, 15, 19 CrossRef CAS.
- Z. Zhao, C. Chen, Z. Liu, J. Huang, M. Wu, H. Liu, Y. Li and Y. Huang, Adv. Mater., 2019, 31, 1808115 CrossRef PubMed.
- T. Wang, J. Liang, Z. Zhao, S. Li, G. Lu, Z. Xia, C. Wang, J. Luo, J. Han, C. Ma, Y. Huang and Q. Li, Adv. Energy Mater., 2019, 9, 1803771 CrossRef.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velazquez-Palenzuela, V. Tripkovic, J. Schiotz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- J. Liang, Z. Zhao, N. Li, X. Wang, S. Li, X. Liu, T. Wang, G. Lu, D. Wang and B.-J. Hwang, Adv. Energy Mater., 2020, 10, 2000179 CrossRef CAS.
- W. Niu, S. Pakhira, G. Cheng, F. Zhao, N. Yao, J. L. Mendoza-Cortes and B. E. Koel, Nat. Mater., 2024, 23, 1704–1711 CrossRef CAS PubMed.
- Z. An, H. Li, X. Zhang, Z. Xia, H. Zhang, W. Chu, S. Yu, S. Wang and G. Sun, ACS Catal., 2024, 14, 2572–2581 CrossRef CAS.
- X. Liu, Y. Wang, J. Liang, S. Li, S. Zhang, D. Su, Z. Cai, Y. Huang, L. Elbaz, Q. Li, W.-W. Zhan, Q.-L. Zhu and Q. Xu, J. Am. Chem. Soc., 2024, 146, 2033–2042 CrossRef CAS PubMed.
- T. Chen, F. Ning, J. Qi, G. Feng, Y. Wang, J. Song, T. Yang, X. Liu, L. Chen and D. Xia, iScience, 2023, 26, 105890 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, C. Chen, J. Zhang, W. Tan, Z. Xu, Z. Zhong, L. Du, H. Song, S. Liao, Y. Zhu, Z. Zhou and Z. Cui, Angew. Chem., Int. Ed., 2024, 136, e202411123 CrossRef.
- X. Fu, C. Wan, Y. Huang and X. Duan, Science, 2003, 125, 3680–3681 Search PubMed.
- S. M. M. Ehteshami and S. H. Chan, Electrochim. Acta, 2013, 93, 334–345 CrossRef CAS.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Mater. Chem. A, 2018, 6, 8531–8536 RSC.
- T. Frelink, W. Visscher and J. A. R. van Veen, Surf. Sci., 1995, 335, 353–360 CrossRef CAS.
- J. Creus, J. De Tovar, N. Romero, J. García-Antón, K. Philippot, R. Bofill, X. Sala, B. Jiang, Y. Guo, J. Kim, A. E. Whitten, K. Wood, K. Kani, A. E. Rowan, J. Henzie and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 2323–2330 RSC.
- T. Holm, S. Sunde, F. Seland, D. A. Harrington, J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu, S. Sun, L. Hui, Y. Xue, C. Xing, Y. Liu, Y. Du, Y. Fang, H. Yu, C. Zhang, F. He and Y. Li, Nano Energy, 2019, 323, 134764 CAS.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- R. Kavanagh, X.-M. Cao, W.-F. Lin, C. Hardacre and P. Hu, Angew. Chem., Int. Ed., 2012, 51, 1572–1575 CrossRef CAS PubMed.
- L. Wang, W. Gao, Z. Liu, Z. Zeng, Y. Liu, M. Giroux, M. Chi, G. Wang, J. Greeley, X. Pan and C. Wang, ACS Catal., 2018, 8, 35–42 CrossRef CAS.
- W. Du, K. E. Mackenzie, D. F. Milano, N. A. Deskins, D. Su and X. Teng, ACS Catal., 2012, 2, 287–297 CrossRef CAS.
- H. Lin, M. Muzzio, K. Wei, P. Zhang, J. Li, N. Li, Z. Yin, D. Su and S. Sun, ACS Appl. Energy Mater., 2019, 2, 8701–8706 CrossRef CAS.
- W. Huang, X. Kang, C. Xu, J. Zhou, J. Deng, Y. Li and S. Cheng, Adv. Mater., 2017, 29, 1703057 CrossRef PubMed.
- S. Luo, L. Zhang, Y. Liao, L. Li, Q. Yang, X. Wu, X. Wu, D. He, C. He and W. Chen, Adv. Mater., 2021, 33, 2008508 CrossRef CAS PubMed.
- W. Zhang, Y. Yang, B. Huang, F. Lv, K. Wang, N. Li, M. Luo, Y. Chao, Y. Li and Y. Sun, Adv. Mater., 2019, 31, 1805833 CrossRef PubMed.
- K. Wei, H. Lin, X. Zhao, Z. Zhao, N. Marinkovic, M. Morales, Z. Huang, L. Perlmutter, H. Guan, C. Harris, M. Chi, G. Lu, K. Sasaki and S. Sun, J. Am. Chem. Soc., 2023, 145, 19076–19085 CrossRef CAS PubMed.
- Z. Liang, L. Song, S. Deng, Y. Zhu, E. Stavitski, R. R. Adzic, J. Chen and J. X. Wang, J. Am. Chem. Soc., 2019, 141, 9629–9636 CrossRef CAS PubMed.
- J. Li, S. Z. Jilani, H. Lin, X. Liu, K. Wei, Y. Jia, P. Zhang, M. Chi, Y. J. Tong, Z. Xi and S. Sun, J. Am. Chem. Soc., 2019, 58, 11527–11533 CAS.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- H. Minamihara, K. Kusada, T. Yamamoto, T. Toriyama, Y. Murakami, S. Matsumura, L. S. R. Kumara, O. Sakata, S. Kawaguchi, Y. Kubota, O. Seo, S. Yasuno and H. Kitagawa, J. Am. Chem. Soc., 2023, 145, 17136–17142 CrossRef CAS PubMed.
- M. Cui, C. Yang, S. Hwang, M. Yang, S. Overa, Q. Dong, Y. Yao, A. H. Brozena, D. A. Cullen, M. Chi, T. F. Blum, D. Morris, Z. Finfrock, X. Wang, P. Zhang, V. G. Goncharov, X. Guo, J. Luo, Y. Mo, F. Jiao and L. Hu, Electrochim. Acta, 2022, 8, eabm4322 CAS.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 137, 7071–7074 CrossRef CAS PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed.
- H. Wang, Y. Yang, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 4608–4616 CrossRef CAS.
- M. J. Craig, G. Coulter, E. Dolan, J. Soriano-López, E. Mates-Torres, W. Schmitt, M. García-Melchor, Y. Da, R. Jiang, Z. Tian, X. Han, W. Chen and W. Hu, Nat. Commun., 2019, 10, 4993 CrossRef PubMed.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845 CrossRef CAS.
- L. Sharma, N. K. Katiyar, A. Parui, R. Das, R. Kumar, C. S. Tiwary, A. K. Singh, A. Halder and K. Biswas, Nano Res., 2022, 1–8 CAS.
- Y. Da, R. Jiang, Z. Tian, X. Han, W. Chen and W. Hu, SmartMat, 2023, 4, e1136 CrossRef CAS.
- S. Li, Y. Yang, S. Huang, H. Li, J. Lai, Z. Li, L. Wang, C. Bianchini and P. K. Shen, Science, 2021, 2, 304–313 Search PubMed.
- Y. Sun, X. Zhang, M. Luo, X. Chen, L. Wang, Y. Li, M. Li, Y. Qin, C. Li, N. Xu, G. Lu, P. Gao and S. Guo, Adv. Mater., 2018, 30, 1802136 CrossRef PubMed.
- J. Park, J. Kim, Y. Yang, D. Yoon, H. Baik, S. Haam, H. Yang and K. Lee, Adv. Sci., 2016, 3, 1500252 CrossRef PubMed.
- P. Anastas, N. Eghbali, V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- Z. Li, Q. Xu, C. Yu, X. Guo, Z. Yin, Z. Zhao, X. Li, J. Robinson, M. Muzzio, C. J. Castilho, M. Shen, Y. Yuan, J. Wang, J. Antolik, G. Lu, D. Su, O. Chen, P. Guduru, C. T. Seto and S. Sun, Matter, 2017, 50, 1449–1458 CAS.
- D. Sun, V. Mazumder, Ö. Metin and S. Sun, ACS Catal., 2016, 6, 6892–6905 CrossRef.
- S. Prodinger, M. F. K. Verstreken and R. F. Lobo, ACS Sustainable Chem. Eng., 2020, 8, 11930–11939 CrossRef CAS.
- Y. Song, K. Liu, S. Chen, L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Nat. Commun., 2012, 11, 3633 Search PubMed.
- Y. Yang, H. Xu, D. Cao, X. C. Zeng and D. Cheng, ACS Catal., 2018, 9, 781–790 CrossRef.
- E. Doustkhah, M. Hasani, Y. Ide and M. H. N. Assadi, ACS Appl. Nano Mater., 2020, 3, 22–43 CrossRef CAS.
- S. Zhang, Ö. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS PubMed.
- B.-S. Choi, J. Song, M. Song, B. S. Goo, Y. W. Lee, Y. Kim, H. Yang and S. W. Han, ACS Catal., 2019, 9, 819–826 CrossRef CAS.
- J. Cheng, X. Gu, P. Liu, H. Zhang, L. Ma and H. Su, Appl. Catal., B, 2017, 218, 460–469 CrossRef CAS.
- W.-Y. Yu, G. M. Mullen, D. W. Flaherty and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 11070–11078 CrossRef CAS PubMed.
- M. Karatok, H. T. Ngan, X. Jia, C. R. O’Connor, J. A. Boscoboinik, D. J. Stacchiola, P. Sautet and R. J. Madix, J. Am. Chem. Soc., 2023, 145, 5114–5124 CrossRef CAS PubMed.
- S. Akbayrak, S. Özkar, H. Kim, T. Y. Yoo, M. S. Bootharaju, J. H. Kim, D. Y. Chung and T. Hyeon, Adv. Sci., 2018, 43, 18592–18606 CAS.
- D. Sun, V. Mazumder, Ö. Metin and S. Sun, ACS Catal., 2012, 2, 1290–1295 CrossRef CAS.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen, J. Rossmeisl, Z. Zhao, C. Chen, Z. Liu, J. Huang, M. Wu, H. Liu, Y. Li and Y. Huang, Catal. Sci. Technol., 2016, 6, 6137–6143 RSC.
- J. Li, S. M. Kathmann, H.-S. Hu, G. K. Schenter, T. Autrey and M. Gutowski, Inorg. Chem., 2010, 49, 7710–7720 CrossRef CAS PubMed.
- S. Özkar, Chem. Mater., 2020, 45, 7881–7891 Search PubMed.
- C. Yu, J. Fu, M. Muzzio, T. Shen, D. Su, J. Zhu and S. Sun, Chem. Mater., 2017, 29, 1413–1418 CrossRef CAS.
- X. Su and S. Li, Int. J. Hydrogen Energy, 2021, 46, 14384–14394 CrossRef CAS.
- M. Muzzio, H. Lin, K. Wei, X. Guo, C. Yu, T. Yom, Z. Xi, Z. Yin and S. Sun, ACS Sustainable Chem. Eng., 2020, 8, 2814–2821 CrossRef CAS.
- X. Guo, C. Yu, Z. Yin, S. Sun and C. T. Seto, ChemSusChem, 2018, 11, 1617–1620 CrossRef CAS PubMed.
- C. Yu, X. Guo, Z. Xi, M. Muzzio, Z. Yin, B. Shen, J. Li, C. T. Seto and S. Sun, J. Am. Chem. Soc., 2017, 139, 5712–5715 CrossRef CAS PubMed.
- S. Cheng, Y. Liu, Y. Zhao, X. Zhao, Z. Lang, H. Tan, T. Qiu and Y. Wang, Dalton Trans., 2019, 48, 17499–17506 RSC.
- C. Yu, X. Guo, M. Shen, B. Shen, M. Muzzio, Z. Yin, Q. Li, Z. Xi, J. Li, C. T. Seto and S. Sun, Angew. Chem., Int. Ed., 2017, 57, 451–455 CrossRef PubMed.
- Y. Liu, Q. Wang, L. Wu, Y. Long, J. Li, S. Song and H. Zhang, Nanoscale, 2019, 11, 12932–12937 RSC.
- C. Yu, X. Guo, B. Shen, Z. Xi, Q. Li, Z. Yin, H. Liu, M. Muzzio, M. Shen, J. Li, C. T. Seto and S. Sun, J. Mater. Chem. A, 2018, 6, 23766–23772 RSC.
- Z. Li, Q. Xu, C. Yu, X. Guo, Z. Yin, Z. Zhao, X. Li, J. Robinson, M. Muzzio, C. J. Castilho, M. Shen, Y. Yuan, J. Wang, J. Antolik, G. Lu, D. Su, O. Chen, P. Guduru, C. T. Seto and S. Sun, Matter, 2019, 1, 1631–1643 CrossRef.
- M. Shen, C. Yu, H. Guan, X. Dong, C. Harris, Z. Xiao, Z. Yin, M. Muzzio, H. Lin, J. R. Robinson, V. L. Colvin, S. Sun, K. Sun, T. Cheng, L. Wu, Y. Hu, J. Zhou, A. Maclennan, Z. Jiang, Y. Gao, W. A. Goddard and Z. Wang, J. Am. Chem. Soc., 2021, 143, 2115–2122 CrossRef CAS PubMed.
- M. Afshari, D. J. Sikkema, K. Lee and M. Bogle, Polym. Rev., 2008, 48, 230–274 CrossRef CAS.
- E. S. Park, J. Sieber, C. Guttman, K. Rice, K. Flynn, S. Watson and G. Holmes, Anal. Chem., 2009, 81, 9607–9617 CrossRef CAS PubMed.
- M. Shen, A. Afshar, N. Sinai, H. Guan, C. Harris, B. Rubenstein and S. Sun, ACS Nano, 2023, 18, 178–185 CrossRef PubMed.
- X. Li, P. Shen, Y. Luo, Y. Li, Y. Guo, H. Zhang and K. Chu, Angew. Chem., Int. Ed., 2022, 134, e202205923 CrossRef.
- Q. Gao, B. Yao, H. S. Pillai, W. Zang, X. Han, Y. Liu, S.-W. Yu, Z. Yan, B. Min, S. Zhang, H. Zhou, L. Ma, H. Xin, Q. He and H. Zhu, Nat. Synth., 2023, 2, 624–634 CrossRef CAS.
- M. Xie, S. Tang, Z. Li, M. Wang, Z. Jin, P. Li, X. Zhan, H. Zhou and G. Yu, J. Am. Chem. Soc., 2023, 145, 13957–13967 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |