
Palladium-catalyzed cyanation of aryl (pseudo)halides using a redox-active N–CN reagent†
Murugan
Dhanalakshmi
and
Pazhamalai
Anbarasan
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai – 600036, India. E-mail: anbarasansp@iitm.ac.in; Web: https://home.iitm.ac.in/anbarasansp/
First published on 6th June 2025
Abstract
An efficient palladium-catalyzed cyanation of aryl bromides has been demonstrated using a readily accessible and redox-active electrophilic cyanating reagent. The reaction was successfully extended to the cyanation of aryl iodides, triflates, and diazonium salts. Various functionalized aryl nitriles and drug intermediates were achieved in good to excellent yields. Important features of this reaction include wide functional group tolerance, a broad substrate scope, and a reductant-free approach. The preliminary mechanistic investigation and identification of oxidized imine supported the proposed plausible reaction mechanism.
Cyanation is an essential reaction in organic synthesis that involves incorporating a cyanide group into a molecule, resulting in the formation of nitrile derivatives.1 Particularly, aryl nitriles are highly valuable in pharmaceuticals, natural products, and industrial applications due to their wide utility.2 Representative aryl nitrile containing bioactive molecules are shown in Scheme 1. They also serve as versatile intermediates in organic synthesis, as they can be readily transformed into various functional groups, including carboxylic acids, amides, aldehydes, amines, and heterocycles. Due to the importance of aryl nitriles, numerous methods have been established for incorporating cyano groups into aromatic rings; among them, the prominent approach is the transition-metal-catalyzed cyanation of aryl (pseudo)halides.3
 | ||
| Scheme 1 Transition metal-catalyzed cyanation of aryl (pseudo)halides. | ||
Transition metal-catalyzed cyanation of aryl (pseudo)halides has been achieved by employing two types of cyanating reagents, which are nucleophilic and electrophilic CN sources (Scheme 1). Nucleophilic cyanating reagents have been explored well for the cyanation of aryl halides.4 However, these cyanating reagents are intrinsically toxic, produce large super-stoichiometric amounts of metal waste, and require careful regulation of cyanide concentration to ensure efficiency and reduce the catalyst loading. These challenges in conventional cyanating reagents drive the interest in developing safer and less toxic alternatives. One of the best alternatives is electrophilic cyanating reagents,5 which offer enhanced reactivity and selectivity, allowing cyanation to proceed under mild conditions. Although these cyanating reagents are well suited for the cyanation of aryl nucleophiles,6 their application in the cyanation of aryl (pseudo)halides is rather limited and requires additional reductants to promote the cyanation.
Cyanation with an electrophilic cyanating reagent, including a N–CN reagent,5a involves the generation of intermediate I, through the addition of aryl–M species to the cyano group of the reagent, followed by the elimination of aryl nitrile, which affords leaving-group bound metal II (Scheme 1). Species II generally undergo protodemetallation to afford a high-valent metal, which requires a reductant/additive to regenerate the active catalyst.7 This highlights the need for further developments in reagent design and process optimization. To achieve the regeneration of active catalyst from intermediate II, in the absence of an external reductant, and to continue our interest in the cyanation,8 we hypothesized to introduce the methylene group on the nitrogen, which can potentially undergo oxidation via β–H elimination to give M–H species. Next, the base-promoted reduction of M–H can regenerate the active catalyst (Scheme 1).9 The successful development of a redox-active electrophilic cyanating reagent would offer a new way to achieve aryl nitriles from aryl (pseudo)halides under environmentally benign conditions. Thus, we herein disclose the general and efficient palladium-catalyzed cyanation of aryl (pseudo)halides using a readily accessible redox-active N–CN reagent (Scheme 1).
To test our hypothesis, we synthesized a series of cyanating reagents 2a–2f containing methylene/methine groups and studied the palladium-catalyzed cyanation of p-bromoanisole 1a. After performing a series of studies, the optimized conditions were observed with an 83% yield of 3a when 1 equiv. of 1a was treated with 2 equiv. of 2a in the presence of 5 mol% of [Pd(cinnamyl)Cl]2, 30 mol% of nBu3P·HBF4 and 2 equiv. of KOMe at 140 °C (Table 1). The use of other N–CN reagents (2b–2f) in place of 2a gave inferior results. Among them, 2b, 2e, and 2f gave moderate yields.
|
|
||
|---|---|---|
| Entry | Deviation from optimized conditions | Yieldb (%) |
| a Reaction conditions: 1a (50 mg, 0.26 mmol, 1 equiv.), 2a (104 mg, 0.53 mmol, 2 equiv.), [Pd(cinnamyl)Cl]2 (6.9 mg, 5 mol%), nBu3P·HBF4 (23 mg, 30 mol%), KOMe (55 mg, 2 equiv.), diglyme (2 mL for 0.26 mmol), 140 °C, 16 h. b All are isolated yields. | ||
| 1 | None | 83 |
| 2 | Pd(OAc)2 instead of [Pd(cinnamyl)Cl]2 | 23 |
| 3 | [Pd(allyl)Cl]2 instead of [Pd(cinnamyl)Cl]2 | 24 |
| 4 | Pd(CH3CN)4(BF4)2 instead of [Pd(cinnamyl)Cl]2 | 36 |
| 5 | Cy3P·HBF4 instead of nBu3P·HBF4 | 48 |
| 6 | t Bu3P·HBF4 instead of nBu3P·HBF4 | — |
| 7 | (o-Tolyl)3P instead of nBu3P·HBF4 | 17 |
| 8 | X-phos instead of nBu3P·HBF4 | 30 |
| 9 | NaOMe instead of KOMe | 25 |
| 10 | K2SO4 instead of KOMe | 52 |
| 11 | K2CO3 instead of KOMe | 46 |
| 12 | DMSO instead of diglyme | 20 |
| 13 | CH3CN instead of diglyme | 73 |

Palladium catalysts like Pd(OAc)2, [Pd(allyl)Cl]2, and Pd(CH3CN)4(BF4)2 were also examined, but all of them gave low yields compared to [Pd(cinnamyl)Cl]2 (Table 1, entries 2–4). When electron-rich and bulky phosphines, such as Cy3P·HBF4, tBu3P·HBF4, (o-tolyl)3P, and X-phos were employed, the formation of 3a was observed in most of the cases, except with bulky tBu3P·HBF4 (Table 1, entries 5–7). Replacement of KOMe with NaOMe led to a significant decrease in the yield of 3a, which suggests that the potassium ion is crucial for this reaction. This observation was further supported by the reaction with K2SO4 and K2CO3 (Table 1, entries 10 and 11). Employing other solvents like DMSO gave only 20% yield, and acetonitrile afforded a comparable yield (Table 1, entries 12 and 13).
Upon successfully optimizing the reaction conditions, we focused on studying the scope of (hetero)aryl bromides (Scheme 2). Initially, para-, meta-, and ortho-bromotoluenes were subjected to the optimized conditions, affording 3b, 3c, and 3d in good yields, respectively. This observation reveals that the present conditions tolerate substitution on different positions of the aryl group. tert-Butyl and phenyl substituted aryl bromides gave products 3e and 3f in ∼70% yield. A 1 mmol scale reaction of bromobiphenyl also gave 3f in a comparable yield.
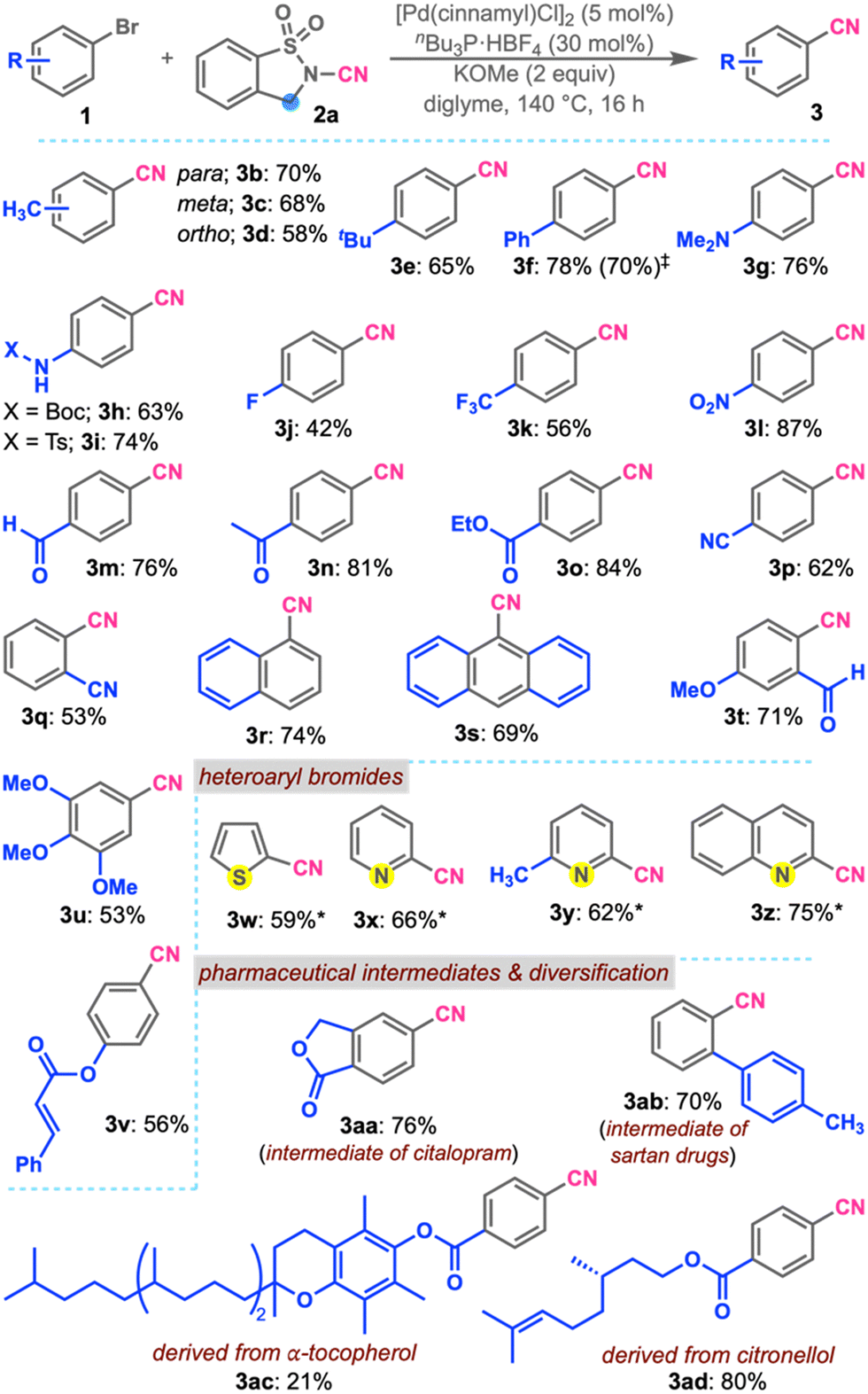 | ||
Scheme 2 Substrates scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reactions were performed in acetonitrile. b 1 mmol scale. Reactions were performed in acetonitrile. b 1 mmol scale. | ||
Aryl bromides substituted with strongly electron-donating (Me2N) and moderately electron-donating (BocNH and TsNH) groups, which are relatively poor substrates for oxidative addition, underwent smooth reactions, affording 3g, 3h, and 3i in ∼70% yield. Besides, cyanobenzenes bearing moderately and strongly electron-withdrawing substituents like fluoro-, trifluoromethyl- and nitro-groups (3j, 3k, and 3l) were obtained in good yields. Oxidatively unstable formyl and base-sensitive acetyl and ethoxycarbonyl groups were well tolerated, affording 3m, 3n, and 3o in 76, 81, and 84% yields, respectively. Interestingly, the formation of dicyanobenzenes 3p and 3q was observed in good yields. Sterically demanding substrates, due to the presence of peri-proton(s), 1-bromonaphthalene, and 9-bromoanthracene, were successfully converted to 3r and 3s in 74 and 69% yields, respectively. Formation of 3t and 3u having multiple substituents was also achieved in good yields. The reaction was compatible with conjugated ester and resulted in the formation of 3v in 56% yield. Since heteroaromatic systems are well-known ligands and can coordinate with metals to quench catalytic activity, heterocyclic bromides such as 2-bromothiophene were subjected to the reaction in acetonitrile, affording 3w in 59% yield. Similarly, strongly basic pyridine derivatives were successfully transformed into products 3x and 3y in good yields. Synthesis of 2-cyanoquinoline 3z was also accomplished in 75% yield. These studies reveal that the present conditions show high compatibility with various functional groups and heterocycles. Subsequently, the synthesis of pharmaceutical intermediates and late-stage functionalization were examined. The synthesis of cyanoarene 3aa, the key intermediate for citalopram,10 was achieved in 76% through the palladium-catalyzed cyanation of the corresponding bromide with 2a. Similarly, cyanation of o-tolylbromobenzene furnished the cyano compound 3ab, the common intermediate for many sartan drugs.11 Considering late-stage functionalization, aryl bromides derived from α-tocopherol and citronellol were successfully converted to products 3ac and 3ad in good yields.
Following the successful demonstration of the cyanation of aryl bromides, we next aimed to study the cyanation of aryl iodides and pseudohalides. Interestingly, cyanation of p-iodoanisole under the optimized conditions at reduced temperature (120 °C) in acetonitrile furnished 3a in 92% yield (Scheme 3). Similarly to the earlier observations, tert-butyl-, phenyl-, trifluoromethyl-, and nitro-substituted cyanoarenes 3e, 3f, 3k, and 3l were synthesized in excellent yields. Base-sensitive esters and Lewis acid-sensitive acetals were well tolerated under the cyanation conditions to afford 3o, 3ae, and 3af. Sterically challenging substrates were also successfully transformed into the cyanated products 3ag and 3ah. Dicyanation of 4,4′-diiodobiphenyl was also achieved in good yield. Following aryl iodides, aryl triflates were subjected to the reaction conditions. To our delight, all the aryl triflates furnished the cyano compounds 3a, 3e, 3k, and 3l in slightly lower yields than aryl halides. Interestingly, the use of aryldiazonium salts led to a marginal improvement in the cyanation and afforded the cyanoarenes 3a, 3l, and 3p in good yields.
 | ||
| Scheme 3 Scope of aryl iodides and pseudohalides. | ||
Having successfully demonstrated the generality of the present cyanation, we next focused our attention on the control experiments. The reaction of 1a and 2a under the optimized conditions in the absence of a palladium catalyst did not furnish 3a, which suggests that the present reaction is catalyzed by palladium (Scheme 4).
 | ||
| Scheme 4 Preliminary mechanistic investigation. | ||
In the absence of nBu3P, only a small amount of 3a was observed, which reveals the vital role of nBu3P in stabilizing the Pd catalyst. Similar results were observed when the reaction was performed in the absence of KOMe. These studies establish the key role of [Pd(cinnamyl)Cl]2, nBu3P, and KOMe in the present reaction.
Next, the standard reaction was performed with TEMPO to investigate the possible involvement of radicals, which afforded 3a in 41% yield. Besides, 2a alone was treated with TEMPO, which did not give any product, and 2a was fully recovered. These observations provide evidence for the non-radical mechanism. Finally, to understand the fate of 2a after the reaction, the standard reaction was performed with K2SO4, which gave the imine 4 in 45% yield along with 3a. The formation of 4 supports our hypothesis of the redox active cyanating reagent.
Based on the preliminary mechanistic investigation and literature precedent,7a–c the plausible reaction mechanism for the present reaction has been proposed. Catalytically active catalyst A could be generated from [Pd(cinnamyl)Cl]2 and nBu3P, which upon reaction with 1 would give intermediate Bvia oxidative addition. Coordination of 2a to B, followed by 1,2-migratory insertion, would give intermediate D. Elimination of aryl nitrile 3 from D would generate palladium species E. β-Hydride elimination in E would form imine 4 and Pd–H species F, which upon base-mediated reduction would regenerate the active catalyst A (Scheme 5).
 | ||
| Scheme 5 Plausible mechanism. | ||
In conclusion, a general and efficient palladium-catalyzed cyanation of aryl bromides has been achieved by employing a new electrophilic cyanating reagent, which is readily accessible and redox-active. The optimized conditions demonstrated excellent compatibility with diverse sterically and electronically different functional groups. It was successfully extended to the cyanation of aryl iodides, triflates, and diazonium salts, which allows access to various aryl nitriles in good to excellent yields. Notably, the present reaction could be used for the synthesis of drug intermediates and their diversification, and it avoids the requirement of reductants and toxic cyanating reagents, and the generation of metal waste. The preliminary mechanistic investigation was performed, including the isolation of the imine intermediate, and also the plausible reaction mechanism was proposed.
We thank the DST-SERB, New Delhi, India (Project No. SB/SJF/2020-21/15) for financial support through the SwarnaJayanti Fellowship. We also thank the DST-FIST for HRMS support and IITM for access to other instrumentation facilities.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (b) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed; (c) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC.
- (a) J. I. Raats, G. Falkson and H. C. Falkson, J. Clin. Oncol., 1992, 10, 111 CrossRef CAS PubMed; (b) Citalopram and escitalopram in Meyler's Side Effects of Drugs, ed. J. K. Aronson, Elsevier, Oxford, 16th edn, 2016, p. 383 Search PubMed; (c) C. Davis, Pericyazine in xPharm: The Comprehensive Pharmacology Reference, ed. S. J. Enna and D. B. Bylund, Elsevier, New York, 2007, p. 1 Search PubMed.
- (a) M. Dhanalakshmi and P. Anbarasan, Transition-Metal-Catalyzed C–CN Cross-Coupling, The Chemical Transformations of C1 Compounds, 2022, pp. 1337 Search PubMed; (b) U. S. Kanchana, T. V. Mathew and G. Anilkumar, J. Organomet. Chem., 2020, 920, 121337 CrossRef CAS; (c) T. Najam, S. S. A. Shah, K. Mehmood, A. U. Din, S. Rizwan, M. Ashfaq, S. Shaheen and A. Waseem, Inorg. Chim. Acta, 2018, 469, 408 CrossRef CAS.
- (a) C. Yang and J. M. Williams, Org. Lett., 2004, 6, 2837 CrossRef CAS PubMed; (b) D. M. Tschaen, R. Desmond, A. O. King, M. C. Fortin, B. Pipik, S. King and T. R. Verhoeven, Synth. Commun., 1994, 24, 887 CrossRef CAS; (c) P. E. Maligres, M. S. Waters, F. Fleitz and D. Askin, Tetrahedron Lett., 1999, 40, 8193 CrossRef CAS; (d) B. Mariampillai, D. Alberico, V. Bidau and M. Lautens, J. Am. Chem. Soc., 2006, 128, 14436 CrossRef CAS PubMed; (e) T. Schareina, A. Zapf and M. Beller, Chem. Commun., 2004, 1388 RSC; (f) M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 1661 CrossRef CAS PubMed; (g) K. Ouchaou, D. Georgin and F. Taran, Synlett, 2010, 2083 CAS; (h) N. Chatani and T. Hanafusa, J. Org. Chem., 1986, 51, 4714 CrossRef CAS; (i) M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 CrossRef CAS.
- (a) M. Chaitanya and P. Anbarasan, Org. Biomol. Chem., 2018, 16, 7084 RSC; (b) J. Cui, J. Song, Q. Liu, H. Liu and Y. Dong, Chem. – Asian J., 2018, 13, 482 CrossRef CAS PubMed.
- (a) T. V. Hughes and M. P. Cava, J. Org. Chem., 1999, 64, 313 CrossRef CAS PubMed; (b) P. Anbarasan, H. Neumann and M. Beller, Chem. – Eur. J., 2010, 16, 4725 CrossRef CAS PubMed; (c) P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519 CrossRef CAS PubMed; (d) Y. Cai, X. Qian, A. Rérat, A. Auffrant and C. Gosmini, Adv. Synth. Catal., 2015, 357, 3419 CrossRef CAS; (e) J. T. Reeves, C. A. Malapit, F. G. Buono, K. P. Sidhu, M. A. Marsini, C. A. Sader, K. R. Fandrick, C. A. Busacca and C. H. Senanayake, J. Am. Chem. Soc., 2015, 137, 9481 CrossRef CAS PubMed.
- (a) L. R. Mills, J. M. Graham, P. Patel and S. A. L. Rousseaux, J. Am. Chem. Soc., 2019, 141, 19257 CrossRef CAS PubMed; (b) J. Li, W. Xu, J. Ding and K.-H. Lee, Tetrahedron Lett., 2016, 57, 1205 CrossRef CAS; (c) M. S. Ahmad, Z. Shafiq and K. Meguellati, Synthesis, 2022, 3077 CrossRef CAS; (d) H. Li, S. Zhang, X. Yu, X. Feng, Y. Yamamoto and M. Bao, Chem. Commun., 2019, 55, 1209 RSC.
- (a) M. Chaitanya, D. Yadagiri and P. Anbarasan, Org. Lett., 2013, 15, 4960 CrossRef CAS PubMed; (b) M. Chaitanya and P. Anbarasan, Org. Lett., 2015, 17, 3766 CrossRef CAS PubMed; (c) M. Chaitanya and P. Anbarasan, J. Org. Chem., 2015, 80, 3695 CrossRef CAS PubMed.
- (a) J. K. Pierce, L. D. Hiatt, J. R. Howard, H. Hu, F. Qu and K. H. Shaughnessy, Organometallics, 2022, 41, 3861 CrossRef CAS; (b) Y. Coquerel, P. Brémond and J. Rodriguez, J. Organomet. Chem., 2007, 692, 4805 CrossRef CAS; (c) J. A. Molina de la Torre, P. Espinet and A. C. Albéniz, Organometallics, 2013, 32, 5428 CrossRef CAS.
- (a) R. Vedantham, V. P. R. Vetukuri, A. Boini, M. Khagga and R. Bandichhor, Org. Process Res. Dev., 2013, 17, 798 CrossRef CAS; (b) J. F. M. Hewitt, L. Williams, P. Aggarwal, C. D. Smith and D. J. France, Chem. Sci., 2013, 4, 3538 RSC.
- (a) S. B. Madasu, N. A. Vekariya, C. Koteswaramma, A. Islam, P. D. Sanasi and R. B. Korupolu, Org. Process Res. Dev., 2012, 16, 2025 CrossRef CAS; (b) G.-X. Wang, B.-P. Sun and C.-H. Peng, Org. Process Res. Dev., 2011, 15, 986 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02381e |
| This journal is © The Royal Society of Chemistry 2025 |