
Cu(I)/Pd(0) cooperative catalysis enabled regioselective C(sp2)-carboboration of 1,3-diynes†
Suman
Ghosh
,
Arnab
Rooj‡
,
Shreya
Deb‡
and
Venkataraman
Ganesh
 *
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur, West Bengal – 721 302, India. E-mail: ganesh.v@chem.iitkgp.ac.in
First published on 4th June 2025
Abstract
We present a regioselective C(sp2)-carboboration of 1,3-diynes using Cu(I)/Pd(0) cooperative catalysis, enabling access to a diverse range of conjugated aryl-substituted boryl enynes. Furthermore, sequential proto-/carbo-/di-boration strategies afforded highly conjugated multi-boron-substituted dienes. Cross-coupling of the boron end groups facilitated the modular synthesis of highly conjugated enynes and dienes with intriguing properties.
Aryl-decorated enynes and dienes' extended π-conjugation across the backbone imparts interesting photophysical properties, making them desirable candidates for optoelectronics and sensor applications. Multiarylated olefins such as tetraphenylethylene (TPE) and its derivatives show aggregation-induced emission (AIE), enabling their wide application as chemical and biosensing AIEgens (Scheme 1).1 There is a recent surge of interest in TPE-based soft materials possessing remarkable optical properties.2 Multi-arylated-1,3-butadienes (MAB), the higher homologs of TPE, show distinctive AIE with applications in multicolor switching display devices and in sensing toxic metal ions and explosives (Scheme 1). The AIE properties of MABs originate from the restricted intramolecular rotation mechanism.3 However, the perarylation of butadiene enhances its photo-lability to give hydronaphthalene.4
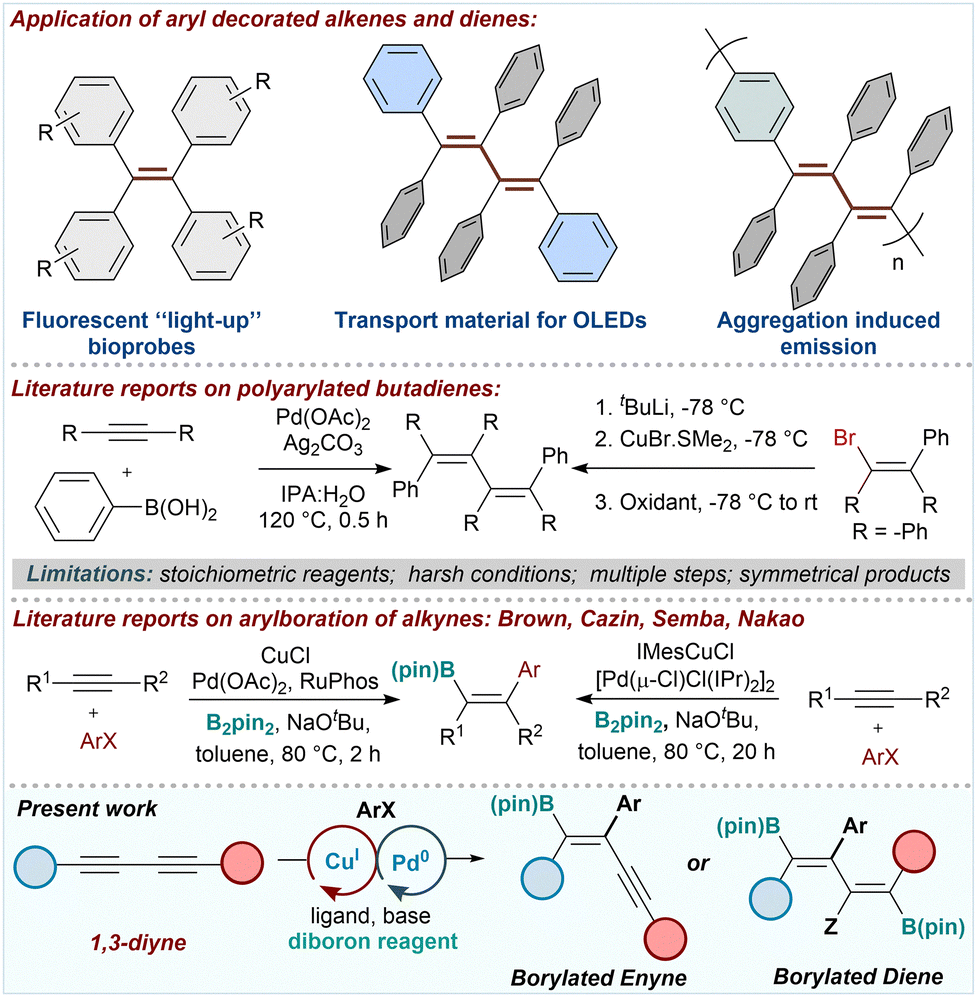 | ||
| Scheme 1 Prior art and present work on C(sp2)-carboboration. | ||
Miura pioneered the Pd0-catalyzed 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 coupling approach to MABs, employing terminal alkynes and boronic acids (Scheme 1).5 The Spring and Deng groups disclosed the reductive elimination of densely arylated divinyl metal intermediates mediated by oxidants to access MABs.6 The Brown group reported a CuI-catalyzed C(sp2)-carboboration of internal alkynes using ArI.7 Later, the Cazin, Semba, and Nakao group employed cooperative Cu(I)/Pd(0) catalysis for the C(sp2)-carboboration of internal alkynes and aryl halides (Scheme 1).8 C(sp2)-carboboration has been applied to alkyne or enynes to give 1,3-dienes.9 In alignment with our ongoing exploration of the regio- and stereoselective functionalization of 1,3-diynes,10 we envisaged C(sp2)-carboboration of 1,3-diynes employing Cu(I)/Pd(0) cooperative catalysis as a streamlined route to diborylated MABs. The two boron end groups provide substantial versatility in incorporating various functional groups, including aryl groups of varying stereoelectronic properties (Scheme 1). The ability to precisely modulate the nature of the aryl groups will enable fine-tuning of the fluorophoric properties and the aggregation modes of MABs. Recently, Wang's group reported a Ni-catalyzed alkyne dimerization strategy to access MABs.11 The Song group reported C(sp)-carboboration of 1,3-diynes.12 However, there are no reports on the C(sp2)-carboboration of 1,3-diynes, providing the flexibility of multiple boron handles installed regioselectively. Here, we report the C(sp2)-carboboration of 1,3-diynes employing Cu(I)/Pd(0) cooperative catalysis to achieve boryl enynes and diboryldienes (Scheme 1).
:2 coupling approach to MABs, employing terminal alkynes and boronic acids (Scheme 1).5 The Spring and Deng groups disclosed the reductive elimination of densely arylated divinyl metal intermediates mediated by oxidants to access MABs.6 The Brown group reported a CuI-catalyzed C(sp2)-carboboration of internal alkynes using ArI.7 Later, the Cazin, Semba, and Nakao group employed cooperative Cu(I)/Pd(0) catalysis for the C(sp2)-carboboration of internal alkynes and aryl halides (Scheme 1).8 C(sp2)-carboboration has been applied to alkyne or enynes to give 1,3-dienes.9 In alignment with our ongoing exploration of the regio- and stereoselective functionalization of 1,3-diynes,10 we envisaged C(sp2)-carboboration of 1,3-diynes employing Cu(I)/Pd(0) cooperative catalysis as a streamlined route to diborylated MABs. The two boron end groups provide substantial versatility in incorporating various functional groups, including aryl groups of varying stereoelectronic properties (Scheme 1). The ability to precisely modulate the nature of the aryl groups will enable fine-tuning of the fluorophoric properties and the aggregation modes of MABs. Recently, Wang's group reported a Ni-catalyzed alkyne dimerization strategy to access MABs.11 The Song group reported C(sp)-carboboration of 1,3-diynes.12 However, there are no reports on the C(sp2)-carboboration of 1,3-diynes, providing the flexibility of multiple boron handles installed regioselectively. Here, we report the C(sp2)-carboboration of 1,3-diynes employing Cu(I)/Pd(0) cooperative catalysis to achieve boryl enynes and diboryldienes (Scheme 1).
We began our investigation by using dibutyl buta-1,3-diyne (1a) as the standard substrate with B2pin2 and 3-bromoanisole as the aryl electrophile. Various Pd0/phosphine and Cu(I)Ln combinations were studied to facilitate the desired arylboration of 1a (see ESI,† for details). The best results were obtained with SIPrCuCl/Pd2(dba)3·CHCl3/SPhos catalyst combination, operating cooperatively to deliver the desired product 3a in 81% NMR yield with rr 90:10 (isolated yield: 78%; entry 1, see ESI,† page S6). In the absence of either Pd0 or CuI catalyst, 1a remained unreacted, suggesting the cooperative involvement of both metal complexes in the catalytic cycle. Screening other Pd sources showed moderate reactivity (y 59–75%; entry 3). Bases like KOtBu and Cs2CO3 were incompatible for arylboration, delivering poor yields of 3a (0–30%; entry 4). A variety of phosphines like Johnphos, CyJohnphos, dppf, dppe, PCy2Ph, and P(p-anisyl)3 were studied; however, the desired product 3a was obtained only in moderate yields and regioselectivity (y 24–59%; rr up to 80:20, entry 5, see ESI,† page S6.
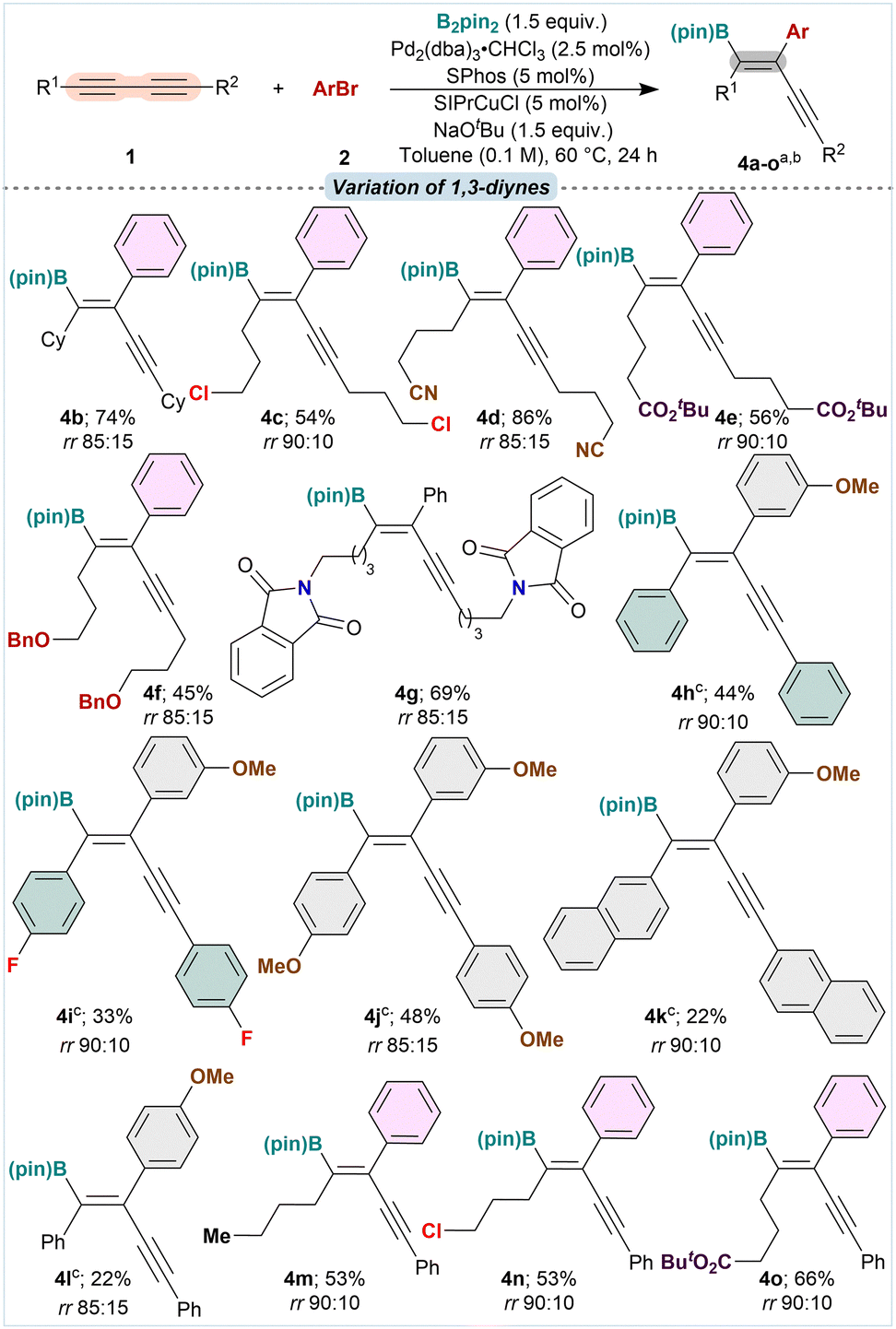
Using the optimized conditions, the scope of aryl halides was studied for the arylboration using 1a as the standard substrate. The effect of halides/pseudohalides on the arylboration was investigated by subjecting Ph–Cl, Ph–Br, Ph–I, and Ph–OTf under standard conditions. Among the aryl halides, bromobenzene provided the corresponding product 3b in 84% isolated yield with rr 85:15 (Table 1). Other electrophiles bearing –Cl, –I, and –OTf resulted in inferior yields of the arylboration products (y 39–59%; rr ranging from 80:20 to 90:10). Then, the substituent effects on bromobenzene were studied using 1a under standard conditions. Electron-rich aryl bromides containing functional groups like meta-OMe (2a), –Me (2c), para-OMe (2d), and para-(NTs)2 (2e) provided the corresponding products 3a, 3c–e in 66–78% yields with 90:10 regioselectivity, respectively. Under the standard conditions, aryl bromides bearing electron-withdrawing substituents like –F, –CF3, –CO2tBu, and –CN, all delivered the product 3f–3i in good yields (58–74%; rr between 85:15 and 90:10). With polycyclic aryl bromides like 2-naphthyl-(2j) and 9-anthracenyl bromide (2k) as partners, the reaction showed excellent yields of 3j and 3k with improved regioselectivity (78 and 68%; rr >95:5). The present conditions could smoothly accommodate heteroaryl bromides like 3-bromothiophene, 5-bromobenzothiophene, and tosyl-protected 5-bromoindole to result in the corresponding heteroaryl-substituted borylenynes 3l–3n in 50–56% yield, and rr ranging between 85:15 and 90:10. Estrone-derived aryl triflate (2o), which was easily prepared from triflation of the phenolic moiety of estrone, delivered the corresponding borylenyne 3o in 62% yield and rr >95:5. Under the standard conditions, with styryl iodide as a C(sp2)-electrophile, the corresponding alkynyl diene boronate 3p was obtained in moderate yield (25%; Table 1).

Then, we examined the scope of 1,3-diynes under standard conditions with bromobenzene as the C(sp2)-electrophiles. 1,3-Diynes bearing simple alkyl groups like –nBu (1a) and –Cy (1b) reacted smoothly with bromobenzene and B2pin2 under the cooperativity of Cu(I)/Pd(0) catalysts to provide the corresponding arylborated products 4a and 4b in 84 and 74% yields, respectively (rr 85:15). 1,3-Diynes with alkyl group bearing pendant functionalities like –Cl, –OBn, –CN, –ester, and –phthalimide groups demonstrated excellent functional group tolerance, delivering the arylboration products 4c–g in 45–86% yields and rr ranging between 85:15 to 90:10. Diaryl-1,3-diynes under the standard arylboration conditions resulted in a mixture of products due to uncontrolled side reactions with the boron moiety in the desired product. Only a trace amount of the products was observed with these substrates. A quick solvent and base screening, retaining the rest of the optimized conditions, identified DCM as a better choice of solvent for C(sp2)-carboboration of diaryl-1,3-diyne substrates. Diphenyl-1,3-diyne with m-bromoanisole as the C(sp2)-partner, under the modified conditions in DCM, provided the desired arylboration product 4h in 44% yield and rr 90:10. 1,3-Diynes with aryl groups bearing functionalities with diverse electronic properties like -naphthyl, p-fluorophenyl, and p-anisyl also provided the desired arylboration products 4i–k, albeit in low to moderate yields (22–48%) and rr ranging between 85:15–90:10. Using 4-bromoanisole as the C(sp2)-partner, 1h provided the desired product 4l in 22% yield and rr 85:15. Surprisingly, with unsymmetrically substituted 1,3-diynes bearing an alkyl group on one side and an aryl group on the other, viz., 1m smoothly reacted under the standard conditions in toluene to provide the product 4m in 53% yield and rr 90:10. The boryl moiety was transferred exclusively at the alkyl attached carbon on the alkyne functionality. The reaction showed a similar trend with substrates bearing alkyl groups with pendant –Cl and –ester groups providing the corresponding products 4n and 4o in 53 and 66% yields with rr 90:10, respectively (Table 2).
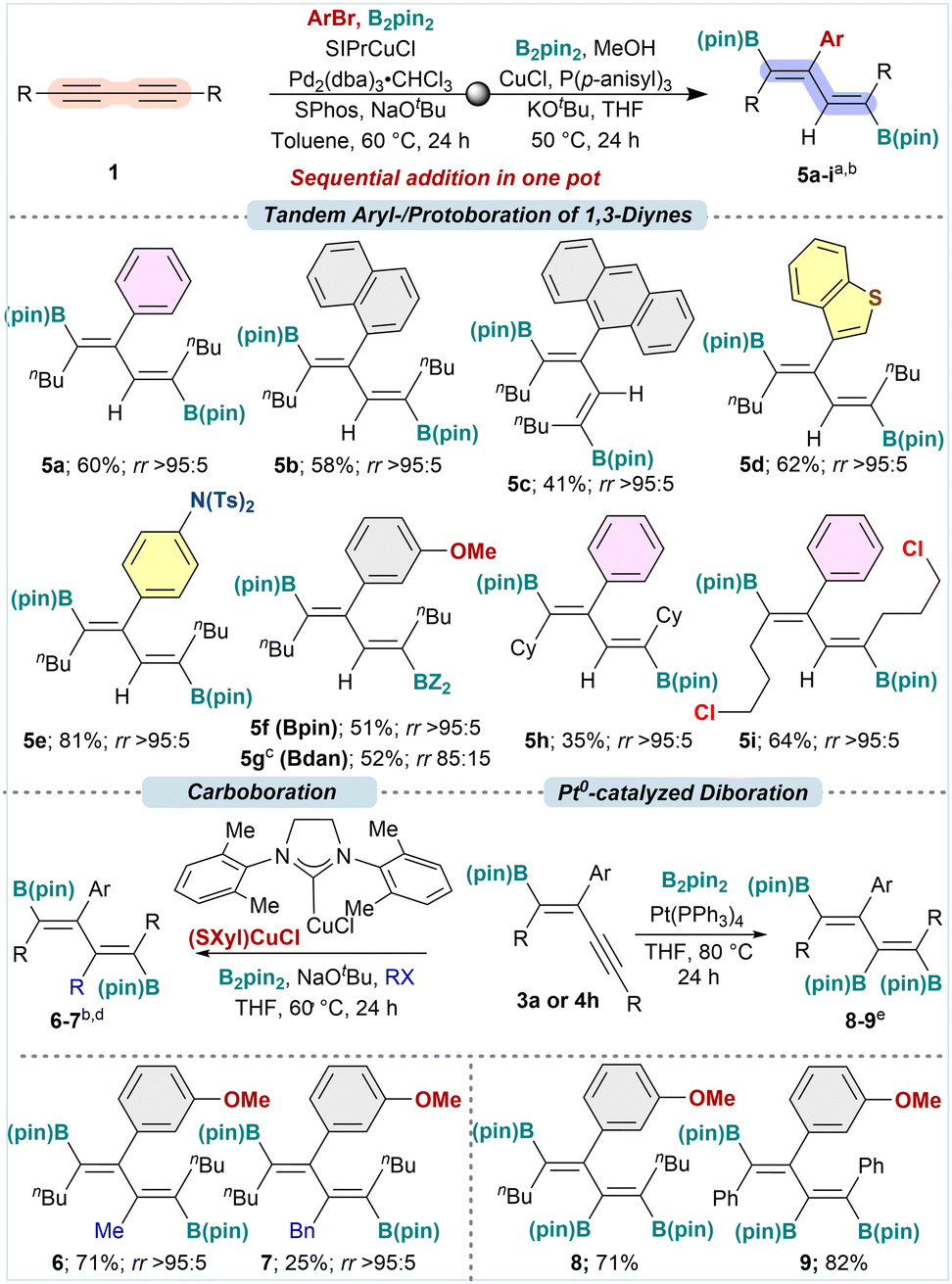
After achieving arylboration of 1,3-diynes to obtain boryl enynes 3 and 4, we aimed to synthesize aryl-decorated 1,4-diboryl-1,3-dienes directly via a second proto-/carboboration. Based on our previous studies on Cu(I)-catalyzed proto- and carboboration of boryl enynes, we explored a tandem arylboration/protoboration cascade using the developed Cu(I)/Pd(0) cooperative catalysis. The first arylboration would require catalyst cooperativity, whereas the second protoboration step could proceed with the help of Cu(I)-catalyst. Our initial attempts with 1a failed, yielding only arylborated product 3b. Further investigation showed that SIPrCuCl was ineffective for protoboration, necessitating a Cu(I)/phosphine catalyst system in THF (see ESI†). The tandem reactivity was achieved by converting 1a to 3b under our arylboration conditions with SIPrCuCl, then adding CuCl/P(p-anisyl)3, B2pin2, KOtBu, MeOH, and THF, yielding 5f in 51% with rr >95:5 (Table 3). The minor regioisomers were undetectable due to further side reactions generating multi-arylated dienes in the presence of ArX and Pd0. These tandem conditions were tested using higher aromatic homologs of ArBr, like naphthyl-, anthracenyl-, and benzothiophenyl bromides. In all cases, the reaction provided the expected penta-substituted 1,4-diboryl-1,3-dienes 5b–d in 41–62% yields, respectively, with excellent regioselectivity. Other functional groups, such as para-N(Ts)2 and meta-OMe on the aryl partner, conveniently transformed to the desired diboryl diene 5e–f in 81 and 51% yields with rr >95:5. Further, we envisaged the introduction of an orthogonally protected boron moiety by sequentially adding B2pin2 for arylboration, followed by a mixed diboron reagent, pinB-Bdan, for the protoboration. Following the above sequence of addition of the diboron reagent, the desired product 5g was obtained in 51% yield with rr 85:15. 5g would be a versatile intermediate for the orthogonal functionalization on the 1,3-diene via iterative cross-coupling. Other representative diynes substrates like 1b and 1c also underwent tandem aryl-/protoboration with B2pin2 smoothly to provide 5h and 5i in 35 and 64% yields with rr >95:5, respectively.
| a Standard conditions: (1) standard conditions from Table 1; (2) CuCl (5 mol%), P(p-anisyl)3 (12.5 mol%); B2Pin2 (2 equiv.), KOtBu (50 mol%), MeOH (4 equiv.), THF (1 mL for 0.15 mmol of 1), 50 °C, 18 h. b rr was determined using 1H NMR. c KOtBu (1 equiv.) was used. d Enyne 3a or 4h (1 equiv.), SXylCuCl (10 mol%), B2pin2 (2 equiv.), RX (1.5–6 equiv.), NaOtBu (1.5 equiv.), THF (0.15 M), 60 °C, 48 h. e Enyne 3a or 4h (1 equiv.), Pt(PPh3)4 (2 mol%), B2pin2 (1.2 equiv.), THF (0.1 M), 80 °C, 24 h. |
|---|
|
Then, we attempted a double arylboration and aryl-/alkylboration in one pot by simply doubling the reagent amounts. However, these reactions stopped after the first arylboration, giving the arylborated enynes. Even the stepwise diarylboration failed due to steric strain in achieving hexa-substituted diene. Pleasingly, the arylborated enynes 3a and 4h could undergo another round of alkylboration and diboration to provide hexasubstituted dienes. Applying the previously established conditions,10d with Me–I and Bn–Br as C(sp3)-electrophiles, 3a was converted to the desired hexa-substituted 1,4-diboryl-1,3-diene 6 and 7 in 71 and 25% yields with rr >95:5. Further, a Pt(0)-catalyzed diboration10d of 3a and 4h provided 1,3,4-triboryl-1,3-dienes 8 and 9 in 71 and 82% yields.
Having demonstrated the aryl boration of 1,3-diynes, the synthetic utility of the resultant arylated borylenyne 4h and 4j was demonstrated. Compound 4l was successfully coupled with 4-iodobenzonitrile to get the desired donor–acceptor-based tetrasubstituted enyne 10 in 99% yield. Similarly, using 1,4-diiodobenzene as a coupling partner, 4h delivered a highly conjugated arylated enyne 11 in 68% yield. Compounds 10 and 11 exhibited solid-state fluorescence under UV light (356 nm). The fluorescence emission maxima for 10 and 11 in chloroform appeared at 404 nm and 387 nm. Then, with compound 10, protoboration and diboration reactions delivered the desired products 12 and 13 in 27 and 75% yields, highlighting the versatility of the enynes and dienes (Scheme 2).
In conclusion, aryl-decorated enynes and dienes hold an illustrious position due to their applications in biological systems and cutting-edge technology. The present approach provides access to aryl-decorated enynes and dienes boronates. These compounds serve as direct synthons for MABs, providing complete control over the selection and introduction of functional groups. The ability to functionalize the enyne backbone with donor–acceptor aryl substituents using our methodology facilitated the solid-state fluorescence properties. The work establishes a modular synthetic platform for the design of functionally diverse boron-containing molecules.
 | ||
| Scheme 2 Synthetic utility of 4h and 4l (1) 4h (2.2 equiv.), Pd(OAc)2 (10 mol%), SPhos (20 mol%), 1,4-diiodobenzene (1 equiv.), KOH (6 equiv.), THF, 70 °C, 24 h. (2) 4l (1 equiv.), Pd(OAc)2 (5 mol%), SPhos (10 mol%), 4-iodobenzonitrile (1.2 equiv.), KOH (3 equiv.), THF, 70 °C, 24 h. | ||
A. R. thanks MHRD for PMRF (File No. 2403435). V. G. thanks SERB for the Core Research Grant (CRG/2022/003322). The authors acknowledge IIT Kharagpur for the infrastructure.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Qayyum, et al. , ACS Omega, 2021, 6, 25447–25460 CrossRef CAS PubMed; (b) J. H. Wang, et al. , Chem. Commun., 2014, 50, 11407–11410 RSC.
- (a) Z. Chi, et al. , Chem. Soc. Rev., 2012, 41, 3878–3896 RSC; (b) N. Sun, et al. , Macromolecules, 2019, 52, 5131–5139 CrossRef CAS.
- Y. Zhang, et al. , Chem. – Eur. J., 2018, 24, 15965–15977 CrossRef CAS PubMed ; and references therein.
- J. Freudenberg, J. Org. Chem., 2014, 79, 11787–11791 CrossRef CAS PubMed.
- (a) T. Satoh, Angew. Chem., Int. Ed., 2004, 43, 5063–5065 CrossRef CAS PubMed; (b) H. Horiguchi, et al. , Adv. Synth. Catal., 2008, 350, 509–514 Search PubMed.
- (a) S. J. Aves, et al. , Synlett, 2012, 298–300 CAS; (b) Y. Liu, et al. , Organomet, 2015, 34, 4401–4407 CrossRef CAS.
- Y. Zhou, et al. , Angew. Chem., Int. Ed., 2014, 53, 3475–3479 CrossRef CAS PubMed.
- (a) M. Lesieur, et al. , Chemcatchem, 2015, 7, 2108–2112 CrossRef CAS; (b) K. Semba, et al. , BCS J., 2017, 90, 1340–1343 CrossRef CAS.
- (a) W. Li, et al. , ACS Catal., 2024, 14, 11318–11331 CrossRef CAS; (b) N. Vázquez-Galiñanes and M. Fañanás-Mastral, ChemCatChem, 2018, 10, 4817–4820 CrossRef.
- (a) S. Ghosh, et al. , ACS Catal., 2022, 12, 11660–11666 CrossRef CAS; (b) S. Ghosh, et al. , Org. Lett., 2024, 26, 6574–6579 CrossRef PubMed; (c) R. Chakrabortty, et al. , Org. Lett., 2024, 26, 792–797 CrossRef CAS PubMed; (d) S. Ghosh, et al. , Adv. Synth. Catal., 2025, 367, e202401188 CrossRef CAS.
- K. Chen, et al. , Nat. Commun., 2025, 16, 3077 CrossRef CAS PubMed.
- J. Xie, et al. , J. Am. Chem. Soc., 2024, 146, 10167–10176 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2444264. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02545a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |