
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal decomposition of peroxyacrylic nitric anhydride (APAN)†
Amanda L.
Gomez
 ,
Kevin D.
Easterbrook
,
Nicole M.
Johnson
,
Shanu
Johnson
and
Hans D.
Osthoff
*
,
Kevin D.
Easterbrook
,
Nicole M.
Johnson
,
Shanu
Johnson
and
Hans D.
Osthoff
*
Department of Chemistry, University of Calgary, 2500 University Drive N.W., Calgary, T2N 1N4, Alberta, Canada. E-mail: hosthoff@ucalgary.ca
First published on 30th April 2025
Abstract
The peroxycarboxylic nitric anhydrides (PANs; RC(O)O2NO2 with R ≠ H) are important trace gas constituents of the troposphere. One of the lesser studied molecules of the PAN family is peroxyacrylic nitric anhydride (APAN; CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC(O)O2NO2) which is found in elevated concentration in biomass burning (BB) plumes and downwind from petrochemical plants. In this work, we conducted laboratory and field experiments to constrain the thermal decomposition (TD) rates of APAN in the atmosphere. The TD of APAN was studied in laboratory experiments using a Pyrex reaction coil at temperatures between 295.2 K and 320.7 K as a function of flow rate (i.e., residence time). Gas streams containing APAN were generated from a diffusion source containing a synthetic sample stored in tridecane at water-ice temperature. Nitric oxide (NO) was added to this gas stream to prevent recombination of the TD products. Concentrations of APAN were monitored by gas chromatography with electron capture detection (PAN-GC). The TD rate constant is best described by 10(17.88±0.80)
CHC(O)O2NO2) which is found in elevated concentration in biomass burning (BB) plumes and downwind from petrochemical plants. In this work, we conducted laboratory and field experiments to constrain the thermal decomposition (TD) rates of APAN in the atmosphere. The TD of APAN was studied in laboratory experiments using a Pyrex reaction coil at temperatures between 295.2 K and 320.7 K as a function of flow rate (i.e., residence time). Gas streams containing APAN were generated from a diffusion source containing a synthetic sample stored in tridecane at water-ice temperature. Nitric oxide (NO) was added to this gas stream to prevent recombination of the TD products. Concentrations of APAN were monitored by gas chromatography with electron capture detection (PAN-GC). The TD rate constant is best described by 10(17.88±0.80)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−(121.2±4.8) kJ mol−1/(RT) s−1, where R is the universal gas constant, and T is the temperature in kelvin. We report ambient air mixing ratios of peroxyacetic nitric anhydride (PAN), peroxypropionic nitric anhydride (PPN), and APAN measured by PAN-GC at the Calgary Central (Inglewood) air quality station from April 17 to May 31, 2023. From May 16 to May 21, the measurement location was blanketed by a BB plume as judged from co-located observations of fine particulate matter (PM2.5) and carbon monoxide (CO). During this time, mixing ratios as high as 3.4 ppbv (PAN), 455 pptv (PPN), and 220 pptv (APAN) were observed. After sunset, mixing ratios of the PANs decreased with pseudo-first order kinetics, rationalized by a combination of dry deposition and loss by TD.
e−(121.2±4.8) kJ mol−1/(RT) s−1, where R is the universal gas constant, and T is the temperature in kelvin. We report ambient air mixing ratios of peroxyacetic nitric anhydride (PAN), peroxypropionic nitric anhydride (PPN), and APAN measured by PAN-GC at the Calgary Central (Inglewood) air quality station from April 17 to May 31, 2023. From May 16 to May 21, the measurement location was blanketed by a BB plume as judged from co-located observations of fine particulate matter (PM2.5) and carbon monoxide (CO). During this time, mixing ratios as high as 3.4 ppbv (PAN), 455 pptv (PPN), and 220 pptv (APAN) were observed. After sunset, mixing ratios of the PANs decreased with pseudo-first order kinetics, rationalized by a combination of dry deposition and loss by TD.
Environmental significanceThe peroxyacyl nitrates (PANs) are important tropospheric trace gases acting as NOx reservoir species that are removed mainly by thermal decomposition (TD). Peroxyacrylic nitric anhydride (APAN; also called peroxyacryloyl nitrate or vinyl-PAN, CH2=CHC(O)O2NO2) is found in elevated concentration in motor vehicle exhaust, petrochemical, and biomass burning (BB) plumes. Grosjean et al. (1994) studied the TD of APAN but reported Arrhenius parameters at odds with other PAN-type compounds. Here, we report rate constants for APAN's TD measured in laboratory experiments and revised Arrhenius parameters which will allow for more accurate modeling of nitrogen oxides in BB plumes. We also report ambient air measurements of PANs (including APAN) during a BB episode in Calgary and investigated the removal of PANs after sunset. |
1. Introduction
Peroxycarboxylic nitric anhydrides (PANs; RC(O)O2NO2, R ≠ H), also referred to as peroxyacyl nitrates, are important trace gas constituents of the troposphere.1,2 The simplest and usually most abundant of the PANs is peroxyacetic nitric anhydride (PAN, R = –CH3), commonly referred to as peroxyacetyl nitrate. Its mixing ratio ranges from several parts per billion (10−9, ppbv) in polluted areas to a few parts per trillion (10−12, pptv) in remote regions.3 Although more than 45 different PANs are known,2 only a few have been quantified in ambient air. These include peroxypropionic (PPN, R = –C2H5), peroxyisobutyric (PiBN, R = –(CH3)2CH), peroxymethacrylic (MPAN, R = –CH2C(CH3)), and peroxyacrylic nitric anhydride (APAN, R = –CH2CH).4–7 Observations of APAN (on which this paper focuses) in ambient air have been relatively sparse (Table 1).
| Reference | Campaign | Platform | Major source type | Max. APAN (pptv) | APAN:PAN (%) |
|---|---|---|---|---|---|
| 8 | RISOTTO 1999–2000 | Ground | n/d | 65 | 4 |
| 9 | TexAQS 2000 | Ground | O&G | 502 | 30 |
| 10 | SENEX 2013 | Aircraft | BB | 26 | 5.3 ± 0.7 |
| 6 | OPECE 2018 | Ground | O&G | >400 | 1–4 |
| 7 | SUNVEx 2021 | Ground | BB | 80 | 3.7 ± 0.1 |
| 11 | KORUS-AQ 2016 | Aircraft | O&G | 850 | 14.2 |
| This work | FOOBAR 2023 | Ground | BB | 220 | 4–8 |
The PANs are primarily formed as by-products of photochemical ozone (O3) and smog production, i.e., from the photooxidation of volatile organic compounds (VOCs) in the presence of NOx (= NO + NO2), and are often associated with biomass burning (BB) plumes.7,12 Aldehydes are usually the most common VOC precursors of PANs. For example, PAN is mainly derived from the hydroxyl radical (OH) initiated oxidation of acetaldehyde, and the unsaturated APAN and MPAN are primarily derived from oxidation of acrolein and methacrolein, respectively, e.g.,
 | (R1) |
Subsequent reaction of the acrolein peroxyacyl (APA) radical with NO2 yields APAN:
| CH2CHC(O)O2 + NO2 → CH2CHC(O)O2NO2, k2 | (R2) |
Acrolein stems mainly from the photooxidation of 1,3-butadiene, though also has primary sources such as automobile exhaust;13 acrolein and 1,3-butadiene have also been associated with emissions from petrochemical industries.11 In contrast, methacrolein is mainly derived from isoprene, which is predominantly biogenic in origin. Consequently, the relative abundances of MPAN and APAN provide insight into whether anthropogenic or biogenic hydrocarbons dominated the O3 production upwind.4
The PANs are important molecules for two main reasons: for one, because their production consumes and their decomposition releases nitrogen dioxide (NO2), the PANs can act as reservoir species of NOx and odd oxygen (Ox = O3 + NO2) and can transport NOx and Ox from polluted urban air to remote regions where nitrogen oxides are scarce. In this way, the PANs contribute to the redistribution of NOx and, by extension, affect the rates of tropospheric O3 and secondary organic aerosol (SOA) production throughout the troposphere.14,15 The second reason why PANs are important molecules is that the PANs are noxious in high concentration. For instance, PAN has been shown to be phytotoxic,16 and PAN and PPN are known lachrymators.17 The toxicity of the other PANs has not been studied though may be inferred from the relative toxicity of their precursors in the atmosphere. For example, given acrolein's higher toxicity relative to that of acetaldehyde,18 APAN likely surpasses the potency of PAN (at least on a per-molecule basis).
In the lower troposphere, the removal of PANs is usually driven by thermal decomposition (TD; (R-2)), for example:
 | (R-2) |
To correctly model the abundances of PANs and their impact in the troposphere, accurate knowledge of their TD kinetics is needed. Table 2 summarizes measured Arrhenius parameters for the TD of PAN, PPN, MPAN, and APAN. While the TD of PAN has been the subject of numerous studies, there have only been two studies of MPAN and only a single study of APAN (prior to this work). Grosjean et al.31,36 investigated the TD of both compounds at T between 285 K and 300 K using samples generated in situ in a Teflon chamber. They reported activation energies that are quite different from the other PANs (in part due to a relatively narrow T range) and appear specious.9 Furthermore, the MPAN data are inconsistent with measurements at T > 302 K by Roberts and Bertman.27 Hence, corroboration of the TD parameters by Grosjean et al.31,36 is warranted, especially for APAN for which no data at T > 300 K exist.
| Molecule | A (s−1) | E a (kJ mol−1) | k −2 (298 K) (10−4 s−1) | Reference |
|---|---|---|---|---|
| PAN | — | — | 2.8 ± 0.8 | 23 |
| PAN | (0.8 ± 1.1) × 1015 | 104 ± 3 | 4.7 | 24 |
| PAN | (1.9 ± 2.7) × 1016 | 113 ± 4 | 3.7 ± 0.4 | 25 |
| PAN | 1018 | 121 ± 8 | 5.4 | 26 |
| PAN | 3.16 × 1016 | 113 | 4.9 | 27 |
| PAN | 2.52 × 1016 | 112.85 | 4.17 | 28 |
| PAN | 1.21 × 1017 | 101 ± 4 | 4.12 ± 0.25 | 29 |
| PAN | — | — | 3.1 | 30 |
| PAN | (2.5 ± 2.3) × 1017 | 119 ± 2 | 3.1 | 27 |
| PAN | (1.6 ± 5.8) × 1016 | 113 ± 9 | 3.0 | 31 |
| PAN | (0.5 ± 1.3) × 1016 | 109 ± 6 | 4.2 | 32 |
| PAN | — | — | 4.40 ± 0.13 | 33 |
| PPN | 2 × 1015 | 106 | 4.4 | 34 |
| PPN | (1.6 ± 4.7) × 1025 | 164 ± 7 | 3.4 | 31 |
| PPN | 7.2 × 1016 | 116 ± 2 | 3.5 | 35 |
| PPN | — | — | 3.67 ± 0.10 | 33 |
| Fur-PAN | (3.7 ± 0.2) × 1016 | 113.6 ± 4.2 | 4.6 | 7 |
| MPAN | (1.6 ± 2.6) × 1016 | 112 ± 4 | 3.5 | 27 |
| MPAN | (1.6 ± 6.7) × 1023 | 153 ± 10 | 2.3 | 36 |
| APAN | (0.3 ± 4.1) × 1020 | 131 ± 31 | 3.0 | 31 |
| APAN | (0.75 ± 1.39) × 1018 | 121.2 ± 4.8 | 4.3 | This work |
In this work, we studied the TD of APAN using a T-controlled flow reactor coupled to a gas chromatograph with an electron capture detector (PAN-GC) to monitor APAN concentrations. We report TD rate constants of APAN at T between 295.2 K and 320.7 K and at a pressure of (662 ± 11) torr and calculate Arrhenius parameters. To corroborate our results, we measured the TD of PAN at 313.2 K for which literature data are available.27
We also present ambient air measurements of PAN, PPN and APAN mixing ratios which were made at the Calgary Central (Inglewood) air quality station from April 17 to May 31, 2023, as part of the “Focus on nitrOgen diOxide at the Bird sAnctuaRy” (FOOBAR) campaign. From May 16 to May 21, elevated concentrations of carbon monoxide (CO), fine particulate matter (PM2.5), and PANs were observed, associated with BB emissions that originated from several out-of-control wildfires to the north-northwest (NNW) of the measurement location. On several occasions, mixing ratios of PAN, PPN, and APAN decreased exponentially after sunset, allowing for an assessment of APAN loss rates in ambient air with the revised TD rates obtained in this work.
2. Methods
2.1 Synthesis of PANs
All chemicals were obtained from Sigma-Aldrich and used as received. In preliminary experiments, we attempted to generate APAN dynamically from photolysis of acryloyl chloride in the presence of NOx.37 However, in spite of previous success generating PAN, PPN or PiBN in high yield with 254 nm or 285 nm light sources,38,39 we were unable to generate gas streams containing APAN in high purity by acryloyl chloride photolysis, as judged from the many, most likely chlorinated, byproducts that appeared in the chromatogram (not shown). Instead, APAN was synthesized and delivered from a diffusion source as described by Tokarek et al.40 Briefly, 4.0 mL of acryloyl chloride was placed in an ice-cold 100 mL round bottom flask (RBF) containing a magnetic stirring bar. Cold hydrogen peroxide (50%, 1.5 mL) was then added dropwise, followed by 20.0 mL of cold tridecane. The RBF was then placed in an ice-water bath, and the mixture was stirred. Caution needs to be exercised at this stage as we observed, on one occasion, rapid and sudden gas evolution after ∼1 h, resulting in some liquid splattering upwards and into the fume hood. After 2 hours, cold concentrated sulfuric (2.5 mL) and nitric acid (3.0 mL) were added dropwise, in turn. After 15 minutes, the organic and aqueous layers were separated in a pre-cooled 125 mL separatory funnel. The organic layer was washed three times with ∼50 mL of cold deionized water, dried over magnesium sulfate, and filtered through glass wool. Aliquots of the synthesized APAN in tridecane solution were stored in 2.0 mL polypropylene centrifuge tubes (VWR) in a freezer prior to use. Once thawed, the APAN samples were generally less stable than those of other PANs and needed to be replaced on an approximately weekly schedule. In addition to APAN, batches of PAN were synthesized from acetic anhydride as described earlier.40–42To corroborate the identity of APAN using relative elution times, gas streams containing PAN, PPN or PiBN were generated from photolysis at 285 nm of acetone, diethyl ketone, or diisopropyl ketone in the presence of NOx as described previously.39
2.2 Measurement of TD kinetics in laboratory experiments
The experimental setup for the kinetics experiments is based on that described by Roberts and Bertman27 and is shown in Fig. 1. A gas stream containing the PAN under investigation (i.e., APAN or PAN) was delivered with “zero” air (>99.999% purity, Air Liquide ALPHAGAZ 1) using a 100 standard-cubic-centimeter-per-minute (sccm; calibrated to a temperature of 273.15 K and a pressure of 1 bar) capacity mass flow controller (MFC, MKS) from a Pyrex diffusion source maintained at 273.15 K. The diffusion source output was combined with a flow of ∼15 sccm of 101 parts-per-million by volume (ppmv, 10−6) of nitric oxide (NO) in O2- and H2O-free N2 (Praxair) delivered via a 100 sccm capacity, all-metal MFC (MKS). The role of the NO is to rapidly titrate the APA or peroxyacetyl (PA) radicals to prevent the re-formation of PANs via(R2), for example:| CH2CHC(O)O2 + NO → CH2CHC(O)O + NO2, k3 | (R3) |
 | ||
| Fig. 1 Schematic of the experimental set-up (not to scale). MFC = mass flow controller. | ||
The value of k3 has not been experimentally determined for the reaction of NO with the APA radical but is likely of similar magnitude as that for NO + PA. Using the rate coefficient reported by Villalta and Howard43 for reaction of the PA radical and NO, we calculate a PA and APA radical lifetime of <1 ms with respect to (R3) under our experimental conditions, i.e., the re-formation of PANs via(R2) is calculated to be negligible.
The radical generated in (R3) dissociates to carbon dioxide (CO2) and vinyl radical (CH2CH), which will react with O2 to form formaldehyde (HCHO) and formyl radical (HCO),44 which in turn reacts with O2 to form carbon monoxide (CO) and hydroperoxyl radical (HO2).45 Under our experimental conditions, HO2 is expected to be titrated by NO to form OH, which in turn is expected to react with NO or the inner walls of reaction vessel (rather than adding to the double bond of APAN and speeding up its decomposition).
Kinetic measurements were conducted using a reaction vessel constructed from ∼1 cm outer diameter (o.d.) and ∼0.8 cm inner diameter (i.d.) Pyrex glass that was double-coiled (12 revolutions downwards in an inner coil and another 12 revolutions on the outside back to the top) and submerged in a T-controlled water bath (Lauda Proline RP 1290, ±0.01 °C). The internal volume of this reaction vessel was (234 ± 2) mL, determined by measuring the volume of water needed to fill it. When not in use (e.g., overnight), the reaction vessel and connecting tubing were continuously purged by a flow of ∼5 sccm of zero air. Thermal decomposition in the connecting tubing outside and downstream of the heated vessel (internal volume ∼5 cm3) was estimated at ∼0.1% and hence negligible.
The vessel effluent was combined with an always-on flow of 50 to 55 sccm of dry N2 and analyzed by PAN-GC. The line pressure was monitored using a transducer (MKS Baratron 722A) mounted on the overflow vent line (not shown) and ranged from 650 torr to 671 torr (855 to 883 hPa).
Concentrations of PANs were monitored using a customized Hewlett-Packard (HP) 5890 PAN-GC described previously.42 This GC was equipped with a 50 μL stainless steel sample loop and a 15 m long megabore analytical column (Restek RTX-200) with a film thickness of 1 μm. The GC was operated with N2 carrier and make-up gas delivered from the “blow-off” of a liquid N2 dewar. Injections were automated (and usually every 5 min). At a column flow rate of 15 mL min−1, PAN, PPN and PiBN eluted at 110 s, 181 s, and 296 s, respectively, whereas APAN eluted at 163 s (Fig. S1A†). These relative elution order and times are consistent with those reported by Roberts et al.9 who also used an RTX-200 column and with values by Tokarek et al.40 who utilized a marginally more polar RTX-1701 column. Chromatographic peak areas were determined by fitting the parameters of a Gaussian expression to the observed peaks as described by Tokarek et al.40
Concentrations of APAN (or PAN) entering (C0) and exiting (Ct) the reaction vessel were measured by manually bypassing the gas flow with the aid of 2-way valves (Swagelok PFA-43S4). The residence time (tres), calculated by dividing the reactor volume by the total volumetric flow rate, was then systemically varied (Table S1†). In a typical experiment (Fig. 2), the reaction vessel was initially bypassed, and the PAN under investigation was eluted from the diffusion source. Once a stable (or only slowly changing) peak area (i.e., concentration, C0) was observed by the PAN-GC, the reaction coil was switched in-line to measure Ct and bypassed again after consecutive GC injections showed stable peak areas. Values of Ct/C0 were calculated by dividing the peak areas observed with the reactor in-line (shown as red squares in Fig. 2) by a linear interpolation of the peak areas when the reactor was bypassed (shown as a blue line in Fig. 2). In the example shown in Fig. 2, Ct/C0 equaled 0.544 ± 0.014, where the error is 1σ precision. The apparent loss rate constant (k′) in the reaction coil was then obtained by linear regression from plots of 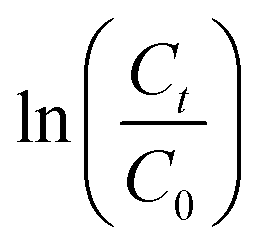 versus tres.
versus tres.
 | ||
| Fig. 2 Sample data collected in a representative experiment. In this example, the reaction coil was maintained at a temperature of (310.65 ± 0.01) K. The total volumetric flow rate through the coil was 64.4 mL min−1, from which a residence time of 218 s was calculated. The circles (shown in blue colour) and squares (shown in red colour) show the APAN peak areas when the reaction coil was bypassed and inline, respectively. Data obtained during the transition between bypass and inline flows are shown as open symbols (□) were not considered in the analysis. The insert shows the corresponding chromatograms. The column flow rate was 15.0 mL min−1. | ||
Pyrex surfaces can destroy PANs, particularly at low humidity,27,46 which adds to k′:
| k′ = k−2 + kPyrex. | (1) |
For experiments at T < 300 K, TD rates were slow, necessitating long run times that made it challenging to maintain stable diffusion source outputs. In those cases, k′ was calculated by dividing  by tres for individual experiments and averaging the results at each T. For experiments at T > 300 K, k′ values were calculated from the slopes of plots of
by tres for individual experiments and averaging the results at each T. For experiments at T > 300 K, k′ values were calculated from the slopes of plots of  versus tres. At higher T, both approaches gave equivalent results (Table 3), with the second method being more precise.
versus tres. At higher T, both approaches gave equivalent results (Table 3), with the second method being more precise.
 versus tres (Fig. 3). Stated uncertainties are at the 1σ precision level. n/d = not determined
versus tres (Fig. 3). Stated uncertainties are at the 1σ precision level. n/d = not determined
| Reference | T (K) | n | k′a (10−5 s−1) | k′b (10−5 s−1) | r | k −2 (10−5 s−1) |
|---|---|---|---|---|---|---|
a Average and standard deviation of k−2 values calculated in individual experiments (Table S1).
b Derived from linear fits of  versus tres (Fig. 3).
c
k
Pyrex = 2.8 × 10−4 s−1 subtracted (see text).
d Experiments conducted with humidified zero air and Teflon-coated linear flow reactor. versus tres (Fig. 3).
c
k
Pyrex = 2.8 × 10−4 s−1 subtracted (see text).
d Experiments conducted with humidified zero air and Teflon-coated linear flow reactor.
|
||||||
| This work | 320.7 | 6 | 1336 ± 20 | 1334 ± 9 | 99.98% | 1306 ± 9 |
| This work | 318.2 | 6 | 968 ± 53 | 958 ± 17 | 99.8% | 930 ± 17 |
| This work | 315.2 | 6 | 674 ± 67 | 680 ± 20 | 99.6% | 652 ± 20 |
| This work | 313.2 | 7 | 534 ± 50 | 527 ± 14 | 99.6% | 499 ± 14 |
| This work | 310.7 | 8 | 364 ± 58 | 352 ± 20 | 97.7% | 324 ± 20 |
| This work | 308.2 | 8 | 230 ± 37 | 228 ± 13 | 97.7% | 200 ± 13 |
| This work | 303.2 | 6 | 137 ± 30 | 130 ± 15 | 94.8% | 102 ± 15 |
| This work | 295.2 | 2 | 63 ± 2 | n/d | n/d | n/d |
| This workd | 295.2 | 4 | 51 ± 4 | 51 ± 2 | 99.6% | n/d |
| Grosjean et al.31 | 299.1 | 1 | 67.0 ± 12.9 | |||
| Grosjean et al.31 | 297.8 | 1 | 29.9 ± 2.5 | |||
| Grosjean et al.31 | 293.2 | 1 | 8.3 ± 0.2 | |||
| Grosjean et al.31 | 293.2 | 1 | 8.3 ± 0.2 | |||
| Grosjean et al.31 | 290.2 | 1 | 6.5 ± 0.5 | |||
| Grosjean et al.31 | 290.2 | 1 | 7.7 ± 0.4 | |||
To corroborate the results with APAN, we measured the TD rate of PAN at T = 313.2 K and compared to literature values. Complete lists of experiments conducted for APAN and PAN are shown in Tables S1 and S2,† respectively.
Rates for TD reactions are commonly shown in the form of an Arrhenius plot, i.e., of ln(k−2) versus 1/T. In this work, the k′ data were fit to the parameters of a rearranged Arrhenius expression:
 | (2) |
2.3 Observations of APAN in ambient air
Ambient air mixing ratios of PAN, PPN, and APAN were measured by PAN-GC as part of the “Focus on nitrOgen diOxide at the Bird sAnctuaRy” (FOOBAR) campaign, which took place in Calgary from March 20 to May 31, 2023. The PAN-GC was a Varian 3380 CP modified and operated in a similar fashion as described by Tokarek et al.40 This instrument was equipped with a 500 μL stainless steel sample loop and a 30 m long megabore column (Restek RTX-1701; film thickness 1 μm) maintained at 298.2 K. It was operated with high purity (99.999%) helium carrier gas at a flow rate of 19.1 mL min−1 (increased to 37.4 mL min−1 on May 8), N2 make-up gas at a flow of 13.2 mL min−1, an electron capture detector T of 100 °C, and the “N2 high” sensitivity setting. The instrument response factors for PAN and PPN were determined using photochemically generated gas mixtures calibrated against blue diode thermal dissociation cavity ring-down spectroscopy (TD-CRDS) as described by Rider et al.39 The response factor for APAN was calculated from the relative APAN:PAN response factor measured by Tokarek et al.40 A sample ambient air chromatogram is shown in Fig. S1B.† The instrument reported 10 min data from April 17 to May 31, 2023.Auxiliary measurements included mixing ratios of NO2 and ΣPAN by TD-CRDS,47 NO, total odd nitrogen (NOy) and O3 using commercial NO–O3 chemiluminescence and absorption analyzers (Thermo Scientific 42i-Y and 49i), and meteorological data collected using a commercial sensor (Vaisala WXT520). All instruments sampled from the same height, ∼4.5 m above ground.
The instruments were housed in a mobile laboratory parked adjacent to the Calgary Central (Inglewood) air quality station located at 51° 1′ 47.7840′′ North Latitude and 114° 0′ 29.1996′′ West Longitude and operated under the guidance of the Calgary Region Airshed Zone (CRAZ), a non-profit association with members from government agencies (federal, provincial, and municipal), non-government organizations, industry, and the public. The Inglewood station houses several continuously operated instruments, including a commercial carbon monoxide (CO) infrared analyzer (Thermo Scientific 48i) and a real-time particulate monitor (Thermo Scientific 5030 SHARP) quantifying sub-2.5 μm aerosol mass (PM2.5).
3. Results
3.1 Laboratory studies
An overview of the experiments conducted is given in Tables S1 (for experiments with APAN) and S2 (for experiments with PAN).† For each experiment, a value for k−2 was calculated by dividing by tres (Tables S1 and S2†). Averages and standard deviations of these values for APAN at each T are summarized in Table 3 under the column heading k′a. Alternatively, values of k−2 were calculated, by linear regression with forced “zero” intercept, from the slopes of plots of
by tres (Tables S1 and S2†). Averages and standard deviations of these values for APAN at each T are summarized in Table 3 under the column heading k′a. Alternatively, values of k−2 were calculated, by linear regression with forced “zero” intercept, from the slopes of plots of  against tres at each T (Fig. 3); these results are summarized in Table 3 under the column heading k′b and were consistent with the first method though more precise, with a relative standard deviation ranging from 0.7% at 320.7 K to 11.7% at 303.2 K. The absence of curvature in the plots shown in Fig. 3, judged from their large Pearson correlation coefficients (r) of typically >99% (Table 3) indicates that the APAN decay rate was not affected by secondary chemistry arising from CH2CHC(O)O that is generated by (R3).
against tres at each T (Fig. 3); these results are summarized in Table 3 under the column heading k′b and were consistent with the first method though more precise, with a relative standard deviation ranging from 0.7% at 320.7 K to 11.7% at 303.2 K. The absence of curvature in the plots shown in Fig. 3, judged from their large Pearson correlation coefficients (r) of typically >99% (Table 3) indicates that the APAN decay rate was not affected by secondary chemistry arising from CH2CHC(O)O that is generated by (R3).
 | ||
| Fig. 3 Plots of ln(Ct/C0) for APAN versus tres (i.e., reaction time) as a function of reactor temperature. Error bars are at the 1σ precision level. | ||
To probe whether the Pyrex walls contributed to the apparent APAN loss in our experiments, experiments were conducted at 295.2 K using humidified flows and with an internally Teflon-coated flow reactor. These experiments yielded lower k′ values than those obtained with the reaction coil and the dry zero air, (51 ± 4) × 10−5 s−1versus (63 ± 2) × 10−5 s−1 (Table 3), confirming that APAN indeed is partially scrubbed on the inner walls of the Pyrex reactor. A lower limit for kPyrex of ≥1.2 × 10−4 s−1 was calculated from the difference of the value with and without Teflon. This value is a lower limit as humidification and the Teflon coating may not fully eliminate wall losses.
The reactor wall loss contributes to all experiments but will have a lesser impact at the higher T where it is not as fast as TD and hence TD dominates. With the assumption that kPyrex is not T-dependent, we estimated its magnitude from r values of plots of ln(k′ − kPyrex) versus in the 303.2–320.7 K T range as a function of kPyrex (not shown). A maximum r value of 0.996 was observed at kPyrex = 2.8 × 10−4 s−1. This value was subtracted from all k′ values to calculate k−2; the results are summarized in Table 3 under the column heading k−2c. These values are plotted in Fig. 4 and were fitted to eqn (1), with the results of the fit shown in Table 2. By extrapolation, a loss rate constant of k−2 = 4.3 × 10−4 s−1 at T = 298 K was calculated, in excellent agreement with k−2 (298 K) of other PANs (Table 2). In contrast, the same analysis with assumed kPyrex = 1.2 × 10−4 s−1 gave k−2 (298 K) of 5.1 × 10−4 s−1, which would be inconsistent with other PANs.
in the 303.2–320.7 K T range as a function of kPyrex (not shown). A maximum r value of 0.996 was observed at kPyrex = 2.8 × 10−4 s−1. This value was subtracted from all k′ values to calculate k−2; the results are summarized in Table 3 under the column heading k−2c. These values are plotted in Fig. 4 and were fitted to eqn (1), with the results of the fit shown in Table 2. By extrapolation, a loss rate constant of k−2 = 4.3 × 10−4 s−1 at T = 298 K was calculated, in excellent agreement with k−2 (298 K) of other PANs (Table 2). In contrast, the same analysis with assumed kPyrex = 1.2 × 10−4 s−1 gave k−2 (298 K) of 5.1 × 10−4 s−1, which would be inconsistent with other PANs.
 | ||
| Fig. 4 Arrhenius plot of k−2 (blue circles) versus 1/T for APAN. Error bars for the laboratory data shown are ±1.4 × 10−4 s−1 (½ of the magnitude of kPyrex). Data from Grosjean et al.31 and pseudo-first order loss rate constants derived from ambient air data are shown as red squares and green triangles, respectively. The grey bar indicates the range of dry deposition loss rates for PAN reported by Roberts.2 | ||
For PAN, we measured k′ = (3.3 ± 0.1) × 10−3 s−1 at T = 313.2 K (Table S2 and Fig. S2†). Subtracting kPyrex = 2.8 × 10−4 s−1 yields k−2 = (3.0 ± 0.1) × 10−3 s−1, in excellent agreement with the value of (2.9 ± 0.5) × 10−3 s−1 reported by Roberts27 at that T.
3.2 Ambient air observations of PAN, PPN and APAN in BB plumes
A time series of ambient air observations from May 16, midnight, to May 21, noon, local or mountain daylight time (MDT), 2023, is shown in Fig. 5. On the morning of May 16, the city of Calgary was blanketed by a BB plume, as indicated by a sudden increase of CO and PM2.5 abundance, which peaked at 2.9 ppmv and 450 μg m−3, respectively. The smoke originated from several out-of-control wildfires burning to the NNW of the measurement location (Fig. S3†), including the Grizzly Complex wildfire south of Slave Lake which forced an evacuation of the town of Swan Hills in Northern AB on May 16. Media reports described the air quality in Calgary as “among the worst in the world”, “while the thick haze gave off an acrid smell, glowed orange and reduced visibility to half a mile”.48 Downwind, the wildfire emissions generated record-breaking O3 anomalies across the U.S. upper Midwest,49 and measurable health effects were reported as far away as New York City.50 | ||
| Fig. 5 Partial time series of quantities measured during FOOBAR. (A) PAN (left axis) and PPN and APAN (right axis). (B) Carbon monoxide (CO; left axis) and PM2.5 (right axis). (C) Nitric oxide (NO), nitrogen dioxide (NO2) and total odd nitrogen (NOy; left axis), and ozone (O3) and odd oxygen (Ox = O3 + NO2; right axis). (D) Ambient air temperature. The background shading indicates night (light gray) and day (light yellow). Regions shaded in white colour were selected for further analysis (see text). | ||
Mixing ratios of PAN, PPN, and APAN were elevated during this event, peaking at 3.4 ppbv, 455 pptv, and 220 pptv, respectively. These data are amongst the highest mixing ratios of BB-generated APAN reported at a surface site to date (Table 1) and are the largest mixing ratios measured by this research group.40 Prior to (and after) the BB event, APAN mixing ratios were <10 pptv. In the plume, the APAN:PAN ratio ranged from 4% to 8% (Fig. S4†), which is a larger ratio than reported in other BB studies though smaller than APAN:PAN ratios associated with industrial sources (Table 1). Average ratios of PPN to PAN and APAN to PAN mixing ratios were 12.5% and 6.0%, respectively (Fig. S4†).
In addition to the period shown in Fig. 5, BB smoke was observed on the morning of May 22 as well as from May 24 to May 28, 2023 (data not shown). During those times, PAN, PPN, and APAN mixing ratios peaked at 1.15 ppbv, 200 pptv, and 40 pptv, respectively, and CO and PM2.5 peaked at 602 ppbv and 47 μg m−3, respectively.
3.3 APAN loss rate constants derived from ambient air data
Mixing ratios of the PANs generally increased during daytime, when PANs are photochemically produced, and decreased starting in the late afternoon and into the night when the photochemical production of PA radicals (e.g., from reaction of OH and aldehydes) diminishes (Fig. 5). The time periods after sunset thus provided an opportunity to probe PAN loss kinetics. In addition to chemical production and loss, ambient air mixing ratios may have also increased or decreased due to air mass shifts, i.e., changes in BB plume intensity. Using seemingly stable concentrations of CO and PM2.5 as an initial guide, nocturnal time periods showing pseudo-first order exponential decay in the APAN mixing ratio were selected for further analysis (Table 4); three of these periods are indicated with a white background in Fig. 5. Concentrations of CO and PM2.5 concentrations were nearly constant during those selected times, suggesting that loss of PAN, PPN, and APAN was likely not due to transport, i.e., not a result of shifting air masses. In all cases, NO mixing ratios were below our ability to quantify (i.e., less than the limit of quantification if ∼50 pptv). Further, NO2 and O3 mixing ratios were sufficiently large to sustain a nitrate radical (NO3) production rate, calculated from k4[NO2][O3],51 of between 0.35 and 0.77 ppb h−1.| NO2 + O3 → NO3 + O2, k4 | (R4) |
 for PANs in ambient air. LOQ = limit of quantification (∼50 pptv for NO). Times are mountain daylight time (MDT). Errors denote ±1σ precision
for PANs in ambient air. LOQ = limit of quantification (∼50 pptv for NO). Times are mountain daylight time (MDT). Errors denote ±1σ precision
| Quantity\Period | May 17 22:00–midnight | May 18 01:40–02:40 | May 21 01:00–04:00 | May 25–May 26 23:30–02:50 |
|---|---|---|---|---|
| a Calculated based on values by Kabir et al.33 b Based on laboratory experiments in this work. | ||||
| PAN0 (pptv) | 815 | 511 | 1222 | 762 |
| PPN0 (pptv) | 106 | 69 | 174 | 113 |
| APAN0 (pptv) | 47 | 24 | 89 | 22 |
| NO (pptv) | <LOQ | <LOQ | <LOQ | <LOQ |
| NO2 (ppbv) | 14.1 ± 3.5 | 7.0 ± 0.5 | 10.4 ± 1.4 | 16.8 ± 3.8 |
| NOy (ppbv) | 16.5 ± 3.5 | 9.1 ± 0.5 | 16.1 ± 1.5 | 18.9 ± 3.8 |
| O3 (ppbv) | 31.3 ± 8.0 | 29.5 ± 4.5 | 20.4 ± 5.6 | 18.3 ± 5.9 |
| P(NO3) (ppb h−1) | 0.77 ± 0.10 | 0.35 ± 0.06 | 0.41 ± 0.13 | 0.47 ± 0.15 |
| RH (%) | 44 ± 10 | 52 ± 5 | 64 ± 8 | 75 ± 6 |
| CO (ppbv) | 436 ± 36 | 256 ± 5 | 943 ± 26 | 409 ± 18 |
| PM2.5 (μg m−3) | 33.4 ± 2.3 | 21.0 ± 2.6 | 164.3 ± 3.7 | 23.2 ± 1.3 |
| T (°C) | 12.0 ± 2.0 | 8.0 ± 1.1 | 13.2 ± 1.3 | 9.1 ± 1.2 |
| k −2 (PAN)a | 5.6 × 10−5 s−1 | 2.9 × 10−5 s−1 | 6.9 × 10−5 s−1 | 3.5 × 10−5 s−1 |
| k −2 (PPN)a | 4.7 × 10−5 s−1 | 2.4 × 10−5 s−1 | 5.7 × 10−5 s−1 | 2.9 × 10−5 s−1 |
| k −2 (APAN)b | 4.7 × 10−5 s−1 | 2.3 × 10−5 s−1 | 5.9 × 10−5 s−1 | 2.8 × 10−5 s−1 |
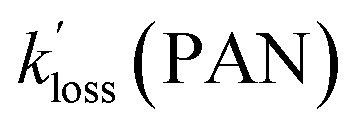
|
(3.5 ± 0.4) × 10−5 s−1 | (6.7 ± 1.0) × 10−5 s−1 | (4.6 ± 0.9) × 10−5 s−1 | (3.5 ± 0.5) × 10−5 s−1 |
| r | 0.948 | 0.968 | 0.785 | 0.853 |

|
(3.6 ± 0.4) × 10−5 s−1 | (7.0 ± 0.2) × 10−5 s−1 | (3.8 ± 0.8) × 10−5 s−1 | (3.2 ± 0.5) × 10−5 s−1 |
| r | 0.939 | 0.9991 | 0.780 | 0.862 |
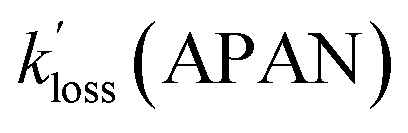
|
(3.7 ± 0.7) × 10−5 s−1 | (6.2 ± 0.7) × 10−5 s−1 | (4.1 ± 0.9) × 10−5 s−1 | (3.3 ± 1.7) × 10−5 s−1 |
| r | 0.862 | 0.981 | 0.767 | 0.485 |
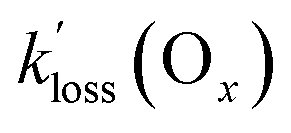
|
(4.8 ± 0.2) × 10−5 s−1 | (9.8 ± 0.6) × 10−5 s−1 | (4.3 ± 0.3) × 10−5 s−1 | (4.2 ± 0.2) × 10−5 s−1 |
For the selected cases, plots of ln(C0/Ct) versus time were linear as judged from their r values (Fig. S5† and Table 4). Further, PAN, PPN, and APAN decayed in unison (Fig. 5) with statistically identical values of their apparent loss rate coefficients 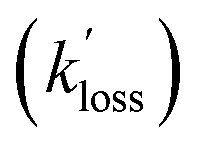 during each episode (Table 4).
during each episode (Table 4).
The 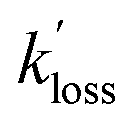 values for PAN and PPN were compared to k−2 values calculated using the parameterization by Kabir et al.33 The
values for PAN and PPN were compared to k−2 values calculated using the parameterization by Kabir et al.33 The 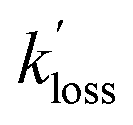 values for APAN are superimposed in Fig. 4 for comparison with k−2 predicted from the laboratory parameterization determined in this work. In all cases, the
values for APAN are superimposed in Fig. 4 for comparison with k−2 predicted from the laboratory parameterization determined in this work. In all cases, the  values were of the same order of magnitude as k−2. The apparent loss rates were ∼1/3 slower than the predicted k−2 on May 17 and May 21 and equal to the k−2 on May 26. For the May 18 data, however, the rate constants derived from the scatter plot shown in Fig. S5† were three times as large as the predicted k−2 value.
values were of the same order of magnitude as k−2. The apparent loss rates were ∼1/3 slower than the predicted k−2 on May 17 and May 21 and equal to the k−2 on May 26. For the May 18 data, however, the rate constants derived from the scatter plot shown in Fig. S5† were three times as large as the predicted k−2 value.
Mixing ratios of odd oxygen (Ox = O3 + NO2) also decreased with first-order exponential kinetics and rate constants between 4.2 × 10−5 and 9.8 × 10−5 (Table 4), factors between 0.9 and 1.5 larger than those determined for the PANs. Mixing ratios of Ox were chosen (as opposed to O3) to account for titration of O3 by NO to NO2.
4. Discussion
4.1 Laboratory measurements
This work constitutes only the second investigation of the TD kinetics of APAN reported in the literature. The first study was that by Grosjean et al.31 who reported k−2 values in the range 290 K ≤ T ≤ 299 K, whereas this study covered the 303.2 K ≤ T ≤ 320.7 K range (Table 3). As is evident from Fig. 4, our data leads to a revised Arrhenius expression with an activation barrier that is lower than that reported by Grosjean et al.,31 (121 ± 5) kJ mol−1versus (139 ± 6) kJ mol−1, and is more in line with values reported for other PANs (Table 2). We believe our data to be more accurate than the values by Grosjean et al.31 as our results are corroborated by the good agreement of our measured k′ and k−2 values for PAN with literature (Fig. S2†). It is also worth noting that the study by Grosjean et al.31 encompassed only four data points over a narrow T range, a limitation noted by Grosjean et al.31 themselves; it is hence not surprising that their Arrhenius parameters would differ from one derived from data collected over a wider T range (Table 2). Furthermore, our data are more linear (r = 0.992) than the data reported by Grosjean et al.31 which appear more scattered (Fig. 4). Finally, the discrepancy echoes what had occurred for MPAN, for which Grosjean et al.36 reported an Ea value of (153 ± 10) kJ mol−1 in contradiction with Roberts and Bertman27 who reported (112 ± 4) kJ mol−1.However, there is a fundamental difference between the two studies: the experimental approach. Grosjean et al.31 utilized a Teflon smog chamber and generated APAN in situ using sunlight, acrolein, and NO, whereas we utilized a glass reaction coil and an APAN diffusion source. Following APAN generation, their chamber would have contained a considerable amount of NO2, which increases the effective lifetime of APAN (eqn (3)) as the TD fragments would have had a reasonable chance to recombine rather than react with NO. The apparent PAN loss rate constant  under these conditions is
under these conditions is
 | (3) |
In contrast to the smog chamber studies by Grosjean et al.,31,36 the study by Roberts and Bertman27 and this work utilized a Pyrex reactor to dissociate PANs. These experimental setups suffer from a potential bias: We observed that APAN decomposes faster on dry Pyrex surfaces than on either humidified or Teflon-coated Pyrex, necessitating a correction by kPyrex = 2.8 × 10−4 s−1, assumed to be T-independent. An outstanding question is why the decomposition of APAN is faster on dry than on humidified Pyrex as has been reported for other PANs.27,46 Theoretical studies, e.g., by density functional theory,52 are needed to probe and rationalize the role of water in the TD of PANs on Pyrex surfaces.
The choice of kPyrex limits the accuracy of our study: For example, if we assume a value of kPyrex = 1.2 × 10−4 s−1 (the minimum value at T = 295.2 K), we would have obtained k−2 = 10(16.9±0.8)e−(115.1±4.6) kJ mol−1/(RT) s−1 and k−2 (298 K) = 5.1 × 10−4 s−1 which are quite different from the values calculated with kPyrex = 2.8 × 10−4 s−1 that are shown in Table 2. Even so, our kinetic data are corroborated by observations made with heated instrument inlets. For example, Veres and Roberts37 reported APAN to have TD kinetics of similar magnitude as those of the other PANs. Overall, a consistent picture emerges: The TD parameters for all PANs fall in the ranges of 106 kJ mol−1 < Ea < 122 kJ mol−1 and 2.8 × 10−4 s−1 < k−2 (298 K) < 4.6 × 10−4 s−1 (if studies published before the year 1980 are omitted). This makes sense chemically, as the TD rate is mainly determined by the strength of the –O–NO2 bond, and the nature of the side chain would be of lesser importance. Only the values reported by Grosjean et al.31,36 fall outside those ranges (Table 2).
4.2 Field measurements and atmospheric significance
The large PAN, PPN and APAN mixing ratios observed in this study highlights the importance of this class of compounds as the abundances of these lachrymators is set to increase with global climate change, which increases the frequency and severity of BB events. The data presented in this manuscript inform about APAN emission factors from BB plumes via APAN:PAN and PPN:PAN ratios (Fig. S4†) as well as ratios with respect to NOy (Table 4).As such, the ambient air observations provided a fortuitous opportunity to evaluate the revised parameterization for TD of APAN from this paper's laboratory experiments under “real-world” conditions. During the periods selected, the mixing ratios of PAN, PPN and APAN decreased with identical pseudo-first order rate constants,  . Further, the observed
. Further, the observed 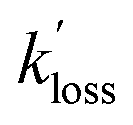 values appear to agree (within error limits) with k−2 values predicted using laboratory parameterizations (Table 4 and Fig. 4). For APAN, this parameterization was extrapolated from the T range of our lab experiments (295.2–320.7 K) to that of the ambient air data (280–287 K).
values appear to agree (within error limits) with k−2 values predicted using laboratory parameterizations (Table 4 and Fig. 4). For APAN, this parameterization was extrapolated from the T range of our lab experiments (295.2–320.7 K) to that of the ambient air data (280–287 K).
At first glance, this seems like an excellent result. However, because of these relatively cool temperatures, the lifetimes of the PANs with respect to TD was relatively long, making it possible for other loss pathways to dominate. Further, for TD to dominate the atmospheric removal loss of PANs, the generated PA radicals need to be removed, and quickly. For the four ambient air periods selected, the NO mixing ratios were below our ability to quantify (i.e., <50 pptv), in contrast to the laboratory experiments (Section 3.1), where kloss (PA) was fast because NO was deliberately added in high concentration. It is unclear which reaction(s), if any, other than (R3) would have removed PA radicals and to what extent. Potential candidates for PA radical removal include aerosol uptake,53 and reaction with NO3 (ref. 54) or with organic peroxy radicals, XO2.22,55 Villalta et al.53 reported a relatively large uptake coefficient (γ) of 0.0043 for uptake of CH3C(O)O2 radicals on deionized water at 274 K, and the aerosol surfaces in the BB plume, while not quantified, were likely large. Thus, aerosol uptake of PA radicals was likely their largest sink. Still, one would expect that reaction of PA radicals with NO2(R2) would have increased the apparent lifetime of the PANs (eqn (3)), i.e., the apparent loss rate constitutes a lower limit to k−2. Perhaps not surprisingly then, the field data (shown as green triangles in Fig. 4) tend to fall below the linear fit of the TD rate extrapolated from higher T data.
Another assumption of this analysis is that nocturnal sources of PANs such as reactions of NO3 with aldehydes54 were negligible. Neither mixing ratios of NO3 nor those of VOCs (from which NO3 abundances could be constrained) were quantified during FOOBAR. Nevertheless, the instantaneous NO3 production rate, P(NO3), was large during the selected periods, 0.35 to 0.77 ppbv h−1 (Table 4), which is within the range of 0.1 to 1.5 ppbv h−1 reported by Decker et al.56 who observed nocturnal BB plumes from an aircraft platform. Thus, all of the selected periods were conducive to NO3 formation. However, Decker et al.56 also noted exceptionally large total NO3 reactivity toward BB-VOCs of between 17 and 70 s−1 that only gradually decreased during the several hours transport time between the emission source and observation point. Large NO3 sinks were likely also present during the BB events in this data set, implying that NO3 mixing ratios would have been small during those times. How much chemistry went through the NO3 + aldehyde channel (which can yield PANs) during FOOBAR is unclear.
Finally, the analysis assumes that direct sinks of PANs (other than TD followed by removal of the PA radicals) such as aerosol uptake57 and dry deposition20–22 were also negligibly small. Lifetimes of PAN with respect to dry deposition have been estimated to be in the 5–10 h range,2 which translate to a loss rate constant of between 5.6 × 10−5 s−1 to 2.8 × 10−5 s−1, which is on par with the 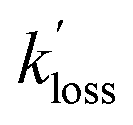 values determined in this work (Table 4). In this context, the Ox loss constants,
values determined in this work (Table 4). In this context, the Ox loss constants,  , are informative since deposition velocities (vd) for O3 are of similar magnitude (with a range of 0.1 to 0.7 cm s−1)58 as those of PAN, for which vd values between 0.13 and 0.54 cm s−1 have been reported.2 The values of
, are informative since deposition velocities (vd) for O3 are of similar magnitude (with a range of 0.1 to 0.7 cm s−1)58 as those of PAN, for which vd values between 0.13 and 0.54 cm s−1 have been reported.2 The values of 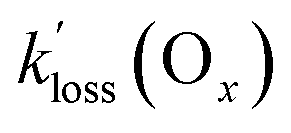 are slightly larger than
are slightly larger than  of the PANs, which supports the notion that both O3 and PANs are removed mainly by dry deposition. However, Decker et al.56 reported a total O3 reactivity toward BB-VOCs of up to ∼6 × 10−4 s−1 which rapidly decreased with plume age to ∼2 × 10−4 s−1, about a factor of two to four larger than
of the PANs, which supports the notion that both O3 and PANs are removed mainly by dry deposition. However, Decker et al.56 reported a total O3 reactivity toward BB-VOCs of up to ∼6 × 10−4 s−1 which rapidly decreased with plume age to ∼2 × 10−4 s−1, about a factor of two to four larger than  in this work. In other words, the observed Ox loss could have entirely been due to chemical reactions as opposed to dry deposition; in the absence of speciated VOC, it is not possible to tell. For APAN, the implication is that the nocturnal losses are explained through a combination of dry deposition and TD.
in this work. In other words, the observed Ox loss could have entirely been due to chemical reactions as opposed to dry deposition; in the absence of speciated VOC, it is not possible to tell. For APAN, the implication is that the nocturnal losses are explained through a combination of dry deposition and TD.
5. Conclusions
The laboratory experiments have yielded revised and more accurate parameterization of the TD kinetics for APAN than previously reported by Grosjean et al.31 Large APAN mixing ratios were observed in ambient air during a spring BB episode. The ambient air temperatures were ultimately too cold to allow for additional constraints of the TD kinetics of APAN as other sinks (e.g., dry deposition) were competitive. Overall, analysis of field data should not be construed as a substitute for properly conducted laboratory studies.Data availability
Data supporting the laboratory experiments described in this article have been included as part of the ESI.† Data supporting the field experiments are available at https://doi.org/10.5683/SP3/VXLT6V (after acceptance). During review, please use the following URL for anonymous access: Private URL: https://borealisdata.ca/privateurl.xhtml?token=eab128af-bdc4-4ad7-8905-6a01bbb9b077.Author contributions
NMJ and KDE synthesized the APAN samples. NMJ, ALG, and KDE carried out the experiments and reduced the data. ALG, KDE, and SJ acquired ambient air data. HDO conceptualized the experiments and drafted the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was made possible by the financial support of the Natural Sciences and Engineering Research Council of Canada (NSERC) in the form of a Discovery grant to HDO (RGPIN-2022-03128). KDE acknowledges an NSERC undergraduate student research award (USRA). Acquisition of ambient air data was supported by Alberta Environment and Protected Areas. The authors thank the Calgary Region Airshed Zone (CRAZ) for providing access to and power at the Calgary Central (Inglewood) AQ station and time-resolved CO and PM2.5 data. Partial funding for the FOOBAR study was provided by a grant from Alberta Environment and Protected Areas (23GRRSD63).References
- J. M. Roberts, The atmospheric chemistry of organic nitrates, Atmos. Environ., Part A, 1990, 24, 243–287 CrossRef.
- J. M. Roberts, in Volatile Organic Compounds in the Atmosphere, ed. R. Koppmann, Blackwell Publishing, Oxford, UK, 2007, pp. 221–268 Search PubMed.
- J. S. Gaffney and N. A. Marley, The Impacts of Peroxyacetyl Nitrate in the Atmosphere of Megacities and Large Urban Areas: A Historical Perspective, ACS Earth Space Chem., 2021, 5, 1829–1841 CrossRef CAS.
- J. M. Roberts, B. T. Jobson, W. Kuster, P. Goldan, P. Murphy, E. Williams, G. Frost, D. Riemer, E. Apel, C. Stroud, C. Wiedinmyer and F. Fehsenfeld, An examination of the chemistry of peroxycarboxylic nitric anhydrides and related volatile organic compounds during Texas Air Quality Study 2000 using ground-based measurements, J. Geophys. Res., 2003, 108, 4495 Search PubMed.
- W. Zheng, F. M. Flocke, G. S. Tyndall, A. Swanson, J. J. Orlando, J. M. Roberts, L. G. Huey and D. J. Tanner, Characterization of a thermal decomposition chemical ionization mass spectrometer for the measurement of peroxy acyl nitrates (PANs) in the atmosphere, Atmos. Chem. Phys., 2011, 11, 6529–6547 CrossRef CAS.
- Y. Lee, L. G. Huey, Y. Wang, H. Qu, R. Zhang, Y. Ji, D. J. Tanner, X. Wang, J. Tang, W. Song, W. Hu and Y. Zhang, Photochemistry of Volatile Organic Compounds in the Yellow River Delta, China: Formation of O3 and Peroxyacyl Nitrates, J. Geophys. Res.: Atmos., 2021, 126, e2021JD035296 CrossRef CAS.
- J. M. Roberts, J. A. Neuman, S. S. Brown, P. R. Veres, M. M. Coggon, C. E. Stockwell, C. Warneke, J. Peischl and M. A. Robinson, Furoyl peroxynitrate (fur-PAN), a product of VOC–NOx photochemistry from biomass burning emissions: photochemical synthesis, calibration, chemical characterization, and first atmospheric observations, Environ. Sci.: Atmos., 2022, 2, 1087–1100 CAS.
- H. Tanimoto and H. Akimoto, A new peroxycarboxylic nitric anhydride identified in the atmosphere: CH2=CHC(O)OONO2 (APAN), Geophys. Res. Lett., 2001, 28, 2831–2834 CrossRef CAS.
- J. M. Roberts, F. Flocke, A. Weinheimer, H. Tanimoto, B. J. Jobson, D. Riemer, E. Apel, E. Atlas, S. Donnelly, V. Stroud, K. Johnson, R. Weaver and F. C. Fehsenfeld, Observations of APAN during TexAQS 2000, Geophys. Res. Lett., 2001, 28, 4195–4198 CrossRef CAS.
- Z. C. J. Decker, K. J. Zarzana, M. Coggon, K.-E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dubé, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti and S. S. Brown, Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations, Environ. Sci. Technol., 2019, 53, 2529–2538 CrossRef CAS PubMed.
- Y. R. Lee, L. G. Huey, D. J. Tanner, M. Takeuchi, H. Qu, X. X. Liu, N. L. Ng, J. H. Crawford, A. Fried, D. Richter, I. J. Simpson, D. R. Blake, N. J. Blake, S. Meinardi, S. Kim, G. S. Diskin, J. P. Digangi, Y. Choi, S. E. Pusede, P. O. Wennberg, M. J. Kim, J. D. Crounse, A. P. Teng, R. C. Cohen, P. S. Romer, W. Brune, A. Wisthaler, T. Mikoviny, J. L. Jimenez, P. Campuzano-Jost, B. A. Nault, A. Weinheimer, S. R. Hall and K. Ullmann, An investigation of petrochemical emissions during KORUS-AQ: Ozone production, reactive nitrogen evolution, and aerosol production, Elem. Sci. Anth., 2022, 10, 00079 CrossRef.
- E. Aruffo, F. Biancofiore, P. Di Carlo, M. Busilacchio, M. Verdecchia, B. Tomassetti, C. Dari-Salisburgo, F. Giammaria, S. Bauguitte, J. Lee, S. Moller, J. Hopkins, S. Punjabi, S. J. Andrews, A. C. Lewis, P. I. Palmer, E. Hyer, M. Le Breton and C. Percival, Impact of biomass burning emission on total peroxy nitrates: fire plume identification during the BORTAS campaign, Atmos. Meas. Tech., 2016, 9, 5591–5606 CrossRef.
- M. M. Roy, HPLC analysis of aldehydes in automobile exhaust gas: Comparison of exhaust odor and irritation in different types of gasoline and diesel engines, Energy Convers. Manage., 2008, 49, 1111–1118 CrossRef CAS.
- H. B. Singh and P. L. Hanst, Peroxyacetyl nitrate (PAN) in the unpolluted atmosphere – an important reservoir for nitrogen oxides, Geophys. Res. Lett., 1981, 8, 941–944 CrossRef CAS.
- E. V. Fischer, D. J. Jacob, R. M. Yantosca, M. P. Sulprizio, D. B. Millet, J. Mao, F. Paulot, H. B. Singh, A. Roiger, L. Ries, R. W. Talbot, K. Dzepina and S. Pandey Deolal, Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution, Atmos. Chem. Phys., 2014, 14, 2679–2698 CrossRef CAS PubMed.
- O. C. Taylor, Importance of Peroxyacetyl Nitrate (PAN) as a Phytotoxic Air Pollutant, J. Air Pollut. Control Assoc., 1969, 19, 347–351 CrossRef CAS PubMed.
- A. P. Altshuller, Assessment of the Contribution of Chemical Species to The Eye Irritation Potential of Photochemical Smog, J. Air Pollut. Control Assoc., 1978, 28, 594–598 CrossRef CAS PubMed.
- R. M. LoPachin and T. Gavin, Molecular Mechanisms of Aldehyde Toxicity: A Chemical Perspective, Chem. Res. Toxicol., 2014, 27, 1081–1091 Search PubMed.
- R. K. Talukdar, J. B. Burkholder, A. M. Schmoltner, J. M. Roberts, R. R. Wilson and A. R. Ravishankara, Investigation of the loss processess for peroxyacetyl nitrate in the atmosphere – UV Photolysis and reaction with OH, J. Geophys. Res., 1995, 100, 14163–14173 CrossRef.
- W. Schrimpf, K. Lienaerts, K. P. Muller, J. Rudolph, R. Neubert, W. Schussler and I. Levin, Dry deposition of peroxyacetyl nitrate (PAN): Determination of its deposition velocity at night from measurements of the atmospheric PAN and 222Radon concentration gradient, Geophys. Res. Lett., 1996, 23, 3599–3602 CrossRef CAS.
- A. A. Turnipseed, L. G. Huey, E. Nemitz, R. Stickel, J. Higgs, D. J. Tanner, D. L. Slusher, J. P. Sparks, F. Flocke and A. Guenther, Eddy covariance fluxes of peroxyacetyl nitrates (PANs) and NOy to a coniferous forest, J. Geophys. Res., 2006, 111, D09304 CrossRef.
- G. M. Wolfe, J. A. Thornton, R. L. N. Yatavelli, M. McKay, A. H. Goldstein, B. LaFranchi, K. E. Min and R. C. Cohen, Eddy covariance fluxes of acyl peroxy nitrates (PAN, PPN and MPAN) above a Ponderosa pine forest, Atmos. Chem. Phys., 2009, 9, 615–634 CrossRef CAS.
- C. T. Pate, R. Atkinson and J. N. Pitts, Rate constants for the gas phase reaction of peroxyacetyl nitrate with selected atmospheric constituents, J. Environ. Sci. Health, Part A, 1976, 11, 19–31 CrossRef.
- R. A. Cox and M. J. Roffey, Thermal decomposition of peroxyacetylnitrate in the presence of nitric oxide, Environ. Sci. Technol., 1977, 11, 900–906 CrossRef CAS.
- D. G. Hendry and R. A. Kenley, Generation of peroxy radicals from peroxy nitrates (RO2NO2). Decomposition of peroxyacyl nitrates, J. Am. Chem. Soc., 1977, 99, 3198–3199 CrossRef CAS.
- R. Guicherit, Photochemical Smogformation in the Netherlands, TNO, Commissie Milieuprojecten, Nederlandse Organisatie voor Toegepast Natuurwetenschappelijk Onderzoek, 1978 Search PubMed.
- J. M. Roberts and S. B. Bertman, The thermal decomposition of peroxyacetic nitric anhydride (PAN) and peroxymethacrylic nitric anhydride (MPAN), Int. J. Chem. Kinet., 1992, 24, 297–307 CrossRef CAS.
- E. C. Tuazon, W. P. L. Carter and R. Atkinson, Thermal decomposition of peroxyacetyl nitrate and reactions of acetyl peroxy radicals with nitric oxide and nitrogen dioxide over the temperature range 283–313 K, J. Phys. Chem., 1991, 95, 2434–2437 CrossRef CAS.
- I. Bridier, F. Caralp, H. Loirat, R. Lesclaux, B. Veyret, K. H. Becker, A. Reimer and F. Zabel, Kinetic and Theoretical Studies of the reactions CH3C(O)O2+NO2+M <-> CH3C(O)O2NO2+M between 248 K and 393 K and between 30 Torr and 760 Torr, J. Phys. Chem., 1991, 95, 3594–3600 CrossRef CAS.
- N. Roumelis and S. Glavas, Thermal decomposition of peroxyacetyl nitrate in the presence of O2, NO2 and NO, Monatsh. Chem., 1992, 123, 63–72 CrossRef CAS.
- D. Grosjean, E. Grosjean and E. L. Williams, Thermal decomposition of PAN, PPN and vinyl-PAN, J. Air Waste Manage. Assoc., 1994, 44, 391–396 CrossRef CAS.
- J. Sehested, L. K. Christensen, T. Mogelberg, O. J. Nielsen, T. J. Wallington, A. Guschin, J. J. Orlando and G. S. Tyndall, Absolute and relative rate constants for the reactions CH3C(O)O2+NO and CH3C(O)O2+NO2 and thermal stability of CH3C(O)O2NO2, J. Phys. Chem. A, 1998, 102, 1779–1789 CrossRef CAS.
- M. Kabir, S. Jagiella and F. Zabel, Thermal Stability of n-Acyl Peroxynitrates, Int. J. Chem. Kinet., 2014, 46, 462–469 CrossRef CAS.
- G. Mineshos and S. Glavas, Thermal decomposition of peroxypropionyl nitrate: Kinetics of the formation of nitrogenous products, React. Kinet. Catal. Lett., 1991, 45, 305–312 CrossRef CAS.
- F. Kirchner, A. Mayer-Figge, F. Zabel and K. H. Becker, Thermal stability of Peroxynitrates, Int. J. Chem. Kinet., 1999, 31, 127–144 CrossRef CAS.
- D. Grosjean, E. Grosjean and E. L. Williams, Thermal-Decomposition of C3-Substituted Peroxyacyl Nitrates, Res. Chem. Intermed., 1994, 20, 447–461 CrossRef CAS.
- P. R. Veres and J. M. Roberts, Development of a photochemical source for the production and calibration of acyl peroxynitrate compounds, Atmos. Meas. Tech., 2015, 8, 2225–2231 CrossRef CAS.
- A. Furgeson, L. H. Mielke, D. Paul and H. D. Osthoff, A photochemical source of peroxypropionic and peroxyisobutanoic nitric anhydride, Atmos. Environ., 2011, 45, 5025–5032 CrossRef CAS.
- N. D. Rider, Y. M. Taha, C. A. Odame-Ankrah, J. A. Huo, T. W. Tokarek, E. Cairns, S. G. Moussa, J. Liggio and H. D. Osthoff, Efficient photochemical generation of peroxycarboxylic nitric anhydrides with ultraviolet light-emitting diodes, Atmos. Meas. Tech., 2015, 8, 2737–2748 CrossRef CAS.
- T. W. Tokarek, J. A. Huo, C. A. Odame-Ankrah, D. Hammoud, Y. M. Taha and H. D. Osthoff, A gas chromatograph for quantification of peroxycarboxylic nitric anhydrides calibrated by thermal dissociation cavity ring-down spectroscopy, Atmos. Meas. Tech., 2014, 7, 3263–3283 CrossRef.
- L. H. Mielke and H. D. Osthoff, On quantitative measurements of peroxycarboxylic nitric anhydride mixing ratios by thermal dissociation chemical ionization mass spectrometry, Int. J. Mass Spectrom., 2012, 310, 1–9 CrossRef CAS.
- K. D. Easterbrook, M. A. Vona, K. Nayebi-Astaneh, A. M. Miller and H. D. Osthoff, Measurement of Henry's law and liquid-phase loss rate constants of peroxypropionic nitric anhydride (PPN) in deionized water and in n-octanol, Atmos. Chem. Phys., 2023, 23, 311–322 CrossRef CAS.
- P. W. Villalta and C. J. Howard, Direct kinetics study of the CH3C(O)O2+NO reaction using chemical ionization mass spectrometry, J. Phys. Chem., 1996, 100, 13624–13628 CrossRef CAS.
- A. H. Laufer and A. Fahr, Reactions and Kinetics of Unsaturated C2 Hydrocarbon Radicals, Chem. Rev., 2004, 104, 2813–2832 CrossRef CAS PubMed.
- M. Martínez-Ávila, J. Peiró-García, V. c. M. Ramírez-Ramírez and I. Nebot-Gil, Ab initio study on the mechanism of the HCO+O2→HO2+CO reaction, Chem. Phys. Lett., 2003, 370, 313–318 CrossRef.
- H. B. Singh and L. J. Salas, Methodology for the analysis of Peroxyacetyl nitrate (PAN) in the unpolluted atmosphere, Atmos. Environ., 1983,(17), 1507–1516 CrossRef CAS.
- D. Paul and H. D. Osthoff, Absolute Measurements of Total Peroxy Nitrate Mixing Ratios by Thermal Dissociation Blue Diode Laser Cavity Ring-Down Spectroscopy, Anal. Chem., 2010, 82, 6695–6703 CrossRef CAS PubMed.
- I. Livingston, Calgary smothered in wildfire smoke, as plumes surge into Lower 48 states, The Washington Post, 2023, retrieved online at https://www.washingtonpost.com/weather/2023/05/17/calgary-wildfire-smoke-alberta-united-states/ Search PubMed.
- O. R. Cooper, K.-L. Chang, K. Bates, S. S. Brown, W. S. Chace, M. M. Coggon, A. M. Gorchov Negron, A. M. Middlebrook, J. Peischl, A. Piasecki, N. Schafer, C. E. Stockwell, S. Wang, C. Warneke, K. Zuraski, K. Miyazaki, V. H. Payne, E. A. Pennington, J. R. Worden, K. W. Bowman and B. C. McDonald, Early Season 2023 Wildfires Generated Record-Breaking Surface Ozone Anomalies Across the U.S. Upper Midwest, Geophys. Res. Lett., 2024, 51, e2024GL111481 CrossRef.
- T. Joo, M. J. Rogers, C. Soong, T. Hass-Mitchell, S. Heo, M. L. Bell, N. L. Ng and D. R. Gentner, Aged and Obscured Wildfire Smoke Associated with Downwind Health Risks, Environ. Sci. Technol. Lett., 2024, 11, 1340–1347 CrossRef CAS PubMed.
- S. S. Brown and J. Stutz, Nighttime radical observations and chemistry, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- X. X. Zhao and F. L. Liu, Mechanism for the Gas-Phase Hydrogen Fluoride-Mediated Decomposition of Peroxyacetyl Nitrate (PAN) Studied by DFT Method, Int. J. Quantum Chem., 2010, 110, 1214–1223 CrossRef CAS.
- P. W. Villalta, E. R. Lovejoy and D. R. Hanson, Reaction probability of peroxyacetyl radical on aqueous surfaces, Geophys. Res. Lett., 1996, 23, 1765–1768 CrossRef CAS.
- J. F. Doussin, B. Picquet-Varrault, R. Durand-Jolibois, H. Loirat and P. Carlier, A visible and FTIR spectrometric study of the nighttime chemistry of acetaldehyde and PAN under simulated atmospheric conditions, J. Photochem. Photobiol., A, 2003, 157, 283–293 CrossRef CAS.
- P. B. Shepson, D. R. Hastie, K. W. So and H. I. Schiff, Relationships between PAN, PPN and O3 at urban and rural sites in Ontario, Atmos. Environ., Part A, 1992, 26, 1259–1270 CrossRef.
- Z. C. J. Decker, M. A. Robinson, K. C. Barsanti, I. Bourgeois, M. M. Coggon, J. P. DiGangi, G. S. Diskin, F. M. Flocke, A. Franchin, C. D. Fredrickson, G. I. Gkatzelis, S. R. Hall, H. Halliday, C. D. Holmes, L. G. Huey, Y. R. Lee, J. Lindaas, A. M. Middlebrook, D. D. Montzka, R. Moore, J. A. Neuman, J. B. Nowak, B. B. Palm, J. Peischl, F. Piel, P. S. Rickly, A. W. Rollins, T. B. Ryerson, R. H. Schwantes, K. Sekimoto, L. Thornhill, J. A. Thornton, G. S. Tyndall, K. Ullmann, P. Van Rooy, P. R. Veres, C. Warneke, R. A. Washenfelder, A. J. Weinheimer, E. Wiggins, E. Winstead, A. Wisthaler, C. Womack and S. S. Brown, Nighttime and daytime dark oxidation chemistry in wildfire plumes: an observation and model analysis of FIREX-AQ aircraft data, Atmos. Chem. Phys., 2021, 21, 16293–16317 CrossRef CAS.
- M. Sun, Y. Zhou, Y. Wang, X. Qiao, J. Wang and J. Zhang, Heterogeneous Reaction of Peroxyacetyl Nitrate on Real-World PM2.5 Aerosols: Kinetics, Influencing Factors, and Atmospheric Implications, Environ. Sci. Technol., 2022, 56, 9325–9334 CrossRef CAS PubMed.
- M. L. Wesely and B. B. Hicks, A review of the current status of knowledge on dry deposition, Atmos. Environ., 2000, 34, 2261–2282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ea00032g |
| This journal is © The Royal Society of Chemistry 2025 |