 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurfactant-like peptide gels are based on cross-β amyloid fibrils†
Abhinaba Das a,
Ordy Gnewoua,
Xiaobing Zuob,
Fengbin Wang*c and
Vincent P. Conticello*a
a,
Ordy Gnewoua,
Xiaobing Zuob,
Fengbin Wang*c and
Vincent P. Conticello*a
aDepartment of Chemistry, Emory University, Atlanta, GA 30322, USA. E-mail: vcontic@emory.edu
bX-ray Science Division, Argonne National Laboratory, Lemont, IL 60439, USA
cBiochemistry and Molecular Genetics Department, University of Alabama at Birmingham, Birmingham, AL 35233, USA. E-mail: jerrywang@uab.edu
First published on 17th February 2025
Abstract
Surfactant-like peptides, in which hydrophilic and hydrophobic residues are encoded within different domains in the peptide sequence, undergo facile self-assembly in aqueous solution to form supramolecular hydrogels. These peptides have been explored extensively as substrates for the creation of functional materials since a wide variety of amphipathic sequences can be prepared from commonly available amino acid precursors. The self-assembly behavior of surfactant-like peptides has been compared to that observed for small molecule amphiphiles in which nanoscale phase separation of the hydrophobic domains drives the self-assembly of supramolecular structures. Here, we investigate the relationship between sequence and supramolecular structure for a pair of bola-amphiphilic peptides, Ac-KLIIIK-NH2 (L2) and Ac-KIIILK-NH2 (L5). Despite similar length, composition, and polar sequence pattern, L2 and L5 form morphologically distinct assemblies, nanosheets and nanotubes, respectively. Cryo-EM helical reconstruction was employed to determine the structure of the L5 nanotube at near-atomic resolution. Rather than displaying self-assembly behavior analogous to conventional amphiphiles, the packing arrangement of peptides in the L5 nanotube displayed steric zipper interfaces that resembled those observed in the structures of β-amyloid fibrils. Like amyloids, the supramolecular structures of the L2 and L5 assemblies were sensitive to conservative amino acid substitutions within an otherwise identical amphipathic sequence pattern. This study highlights the need to better understand the relationship between sequence and supramolecular structure to facilitate the development of functional peptide-based materials for biomaterials applications.
Introduction
Peptide-based materials have been the focus of significant research effort for over three decades.1–3 The intellectual driving force for these investigations has been the creation of functional nanomaterials, e.g., hydrogels, that mimic the level of structural control observed in native proteins and protein assemblies.4 Like protein folding, it has been postulated that the chemical information encoded within designed peptides can direct the formation of supramolecular structure through a hierarchical process that involves propagation of sequence information across length-scales. While sequence–structure correlations have been established for reliable prediction of native protein structure, most notably through the development of data-driven, deep learning algorithms such as AlphaFold and RoseTTAFold,5,6 a comparable degree of success in atomically accurate prediction of supramolecular structure,7 which is a prerequisite for the reliable design of peptide-based materials, has been difficult to achieve.8–12The macromolecular architectures of designed peptides, i.e., chain length, composition, and sequence, are much less complex than that of typical proteins, which, in theory, should facilitate the de novo design of peptide-based materials. Structural analyses of designed peptide filaments at near-atomic resolution revealed that the cohesive interactions between protomers in the assemblies often resulted from a combination of richly and sparsely designable interfaces.10,11 The presence of sparsely designable interfaces poses a significant challenge to the molecular design of filamentous peptide assemblies since they arise from the additive effect of relatively weak local interactions.10 These interactions may not be a priori predictable from statistical analyses of the frequency of occurrence of interfacial interactions within proteins in the PDB.13–15 In addition, the vast sequence space accessible to even short synthetic peptides is prohibitively difficult to systematically sample through experimentation, which has prompted the development of design rules that can simplify the process.16
The limited length of designed peptides necessitates that sufficient chemical information be encoded into relatively short sequences to specify the formation of a stable and unique supramolecular structure. These two considerations, i.e., thermodynamic stability and structural specificity, can be antagonistic in the context of peptide-based materials design. Thermodynamic stability is a prerequisite for the fabrication of peptide-based materials that can persist under a range of conditions such as those that would be required for applications as hydrogel biomaterials.2 Consequently, the sequences of peptide-based materials are designed to maximize the thermodynamic stability of a supramolecular structure. However, a strong thermodynamic driving force for self-assembly can lead to folding frustration,17 in which the observed structure of the assembly depends strongly on the initial conditions.11,18 Changes in assembly conditions can result in the formation of different structural polymorphs that may be difficult to reliably predict or reproducibly control. In contrast, structural specificity originates from the introduction of defined intra- and inter-molecular interactions into a peptide sequence to bias self-assembly toward a desired supramolecular structure through a combination of positive and negative design.16 However, sequence–structure correlations that would lead to predictable design are not well established for synthetic peptide-based materials, which can result in emergent self-assembly behaviour that results in the formation of unpredicted structures.10,11 Similar self-assembly behaviour is observed for amyloidogenic peptides, especially in vitro.18 The richly designable cross-β interface of β-amyloids is compatible with a potentially wide range of sparsely designable quaternary interactions, which can result in extensive structural polymorphism despite high thermodynamic stability.17,19–21
Here, we investigate the relationship between sequence and supramolecular structure for a pair of bola-amphiphilic peptides derived from semi-conservative mutagenesis of the designed sequence Ac-KIIIIK-NH2.22–24 The latter exemplifies the principles of surfactant-like peptide design, in which polar and apolar residues are encoded in different domains in the peptide sequence. Previous studies on this peptide and structurally related bola-like sequences have demonstrated that polar patterning promoted self-assembly into ordered supramolecular structures, e.g., nanosheets, nanotubes, nanoribbons, and nanofibers.25 The formation of different supramolecular structures was rationalized based on a surfactant-like packing model in which amino acid substitutions induced changes in lamellar curvature on the meso-scale. In this study, we propose an alternative explanation in which symmetry breaking in bola-like peptide sequences introduces localized changes in interfacial packing interactions within and between protofilaments composed of cross-β fibrils. Cryo-EM analysis26 of a nanotube derived from self-assembly of a bola-like peptide Ac-KIIILK-NH2 (L5) provided insight into the structures at near-atomic resolution. The structural arrangement of peptides in the assembly resembled the structured packing interfaces within β-amyloids. In addition, we demonstrate that alteration of the peptide sequence resulted in a morphological transition between filaments and lamellae despite conservation of peptide length, composition, and polar sequence pattern. These results suggest that the self-assembly behaviour of bola-like surfactant peptides is a surprisingly complex process in which the observed supramolecular structures would have been difficult to reliably predict a priori based on our current knowledge of peptide sequence–structure relationships.
Results and discussion
Peptide Ac-KIIIIK-NH2 displayed a typical bola-like peptide sequence pattern in which identically charged polar residues flank a longer apolar domain. In contrast to conventional bola-amphiphiles, bola-like peptide sequences have a net directionality due to the peptide backbone.27 The self-assembly behaviour of bola-peptides has been previously explored in different sequence contexts. For peptides having highly symmetric, quasi-palindromic sequences, e.g., Ac-KIIIIK-NH2, self-assembly resulted in the formation of lamellar sheets or wide-diameter nanotubes (Fig. 1).22–24,28–30 Previously reported spectroscopic evidence supported β-sheet formation within the assemblies of Ac-KIIIIK-NH2 and closely related derivatives.31–33 Since equal numbers of hydrophobic isoleucine residues were exposed at the surface of the filament, bifacial sheet stacking of cross-β fibrils should promote symmetric lamination with a concomitant reduction in sheet twisting.34–36 Small-angle neutron scattering (SANS) measurements performed on assemblies of Ac-KIIIIK-NH2 were fit to a 2D model in which the lamellar thickness corresponded to the length of the peptide.22 A monolayer model (Fig. 1) was proposed for the self-assembly of Ac-KIIIIK-NH2 in which nanoscale phase separation of the central hydrophobic domain drove formation of wide-diameter polymorphic nanotubes, which was confirmed by electron microscopy measurements. This process was hypothesized to mimic the self-assembly behaviour of more conventional bola-amphiphilic surfactants.37 | ||
| Fig. 1 Monolayer packing model for self-assembly of bola-like peptides Ac-KIIIXK-NH2 (X = I, L). The degree of curvature induced within the monolayer depends on the surface asymmetry of the cross-β fibril, which constitutes the fundamental structural subunit of the monolayer. | ||
Previously, Lu, Xu, and coworkers,32,33 reported the self-assembly behaviour of a series of peptides Ac-KIIIXK-NH2 in which different amino acid residues were substituted into the fifth position of the sequence. When leucine replaced isoleucine at the last site in the hydrophobic block, the resultant peptide, Ac-KIIILK-NH2 (L5), formed high aspect-ratio, narrow-diameter nanotubes.32 This difference in behaviour between the two peptides was rationalized in terms of the influence of the isoleucine to leucine substitution on the curvature of the resultant lamellae. Despite similar hydrophobicity, leucine residues display different stereochemical properties with respect to the core isoleucine residues. Nanotube formation was envisioned to arise from formation of asymmetrically substituted monolayer lamellae. This surface asymmetry was proposed to induce tighter coiling than observed for assemblies of the more symmetric parent peptide, which resulted in a narrow diameter tubular filament. Unbalanced surface stresses in assemblies of soft materials have been demonstrated to induce coiling or scrolling of lamellae into tubular structures.38 This effect has been described previously for collagen-mimetic peptide nanosheets that displayed surface asymmetry.39 To investigate the effect of these mutations on supramolecular structure, we have re-examined the self-assembly behaviour of the peptide L5. In addition, a related peptide, Ac-KLIIIK-NH2 (L2) was prepared as a retro-peptide analogue of L5 in which the peptide sequence was reversed. This pair of peptides was expected to display a similar degree of surface asymmetry within a putative cross-β monolayer. However, the unique Leu residue would be placed on opposite sides of the corresponding lamellae that resulted from β-sheet formation. We hypothesized that self-assembly of L2 and L5 would result in the formation of similar supramolecular structures, which would provide a test of the previously proposed monolayer model and provide further insight into the self-assembly behaviour of surfactant-like peptide-based materials.
Peptides L2 and L5 were prepared using solid-phase peptide synthesis (Fig. S1 and S2†) and were assembled from aqueous solution (32 mM) for two weeks using the preparative conditions previously described, which resulted in the formation of self-supporting hydrogels (Fig. S3†).32 Circular dichroism (CD) spectropolarimetric measurements indicated that the peptides adopted a β-sheet conformation (Fig. S4†), which was consistent with previous structural analyses of L5. In each case, negative-stain TEM imaging provided evidence for the formation of supramolecular assemblies (Fig. 2a). TEM images of L5 agreed with previously published reports in which narrow diameter nanotubes (∼8 nm) were observed to form upon incubation at ambient temperature.32 In contrast, TEM imaging of assemblies derived from peptide L2 revealed the presence of flat ribbon-like assemblies of variable diameter and length. The formation of morphologically similar nano-ribbons has been observed for surfactant-like peptide displaying different polar sequence patterns.40,41 Notably, in TEM images of the L2 assemblies (Fig. 2a), small filaments can be seen emerging from the ends of the nano-ribbons, which was consistent with the predictions of the monolayer model (Fig. 1), in which lamination of thin cross-β fibrils (∼2 nm thickness) resulted in formation of lamellae.
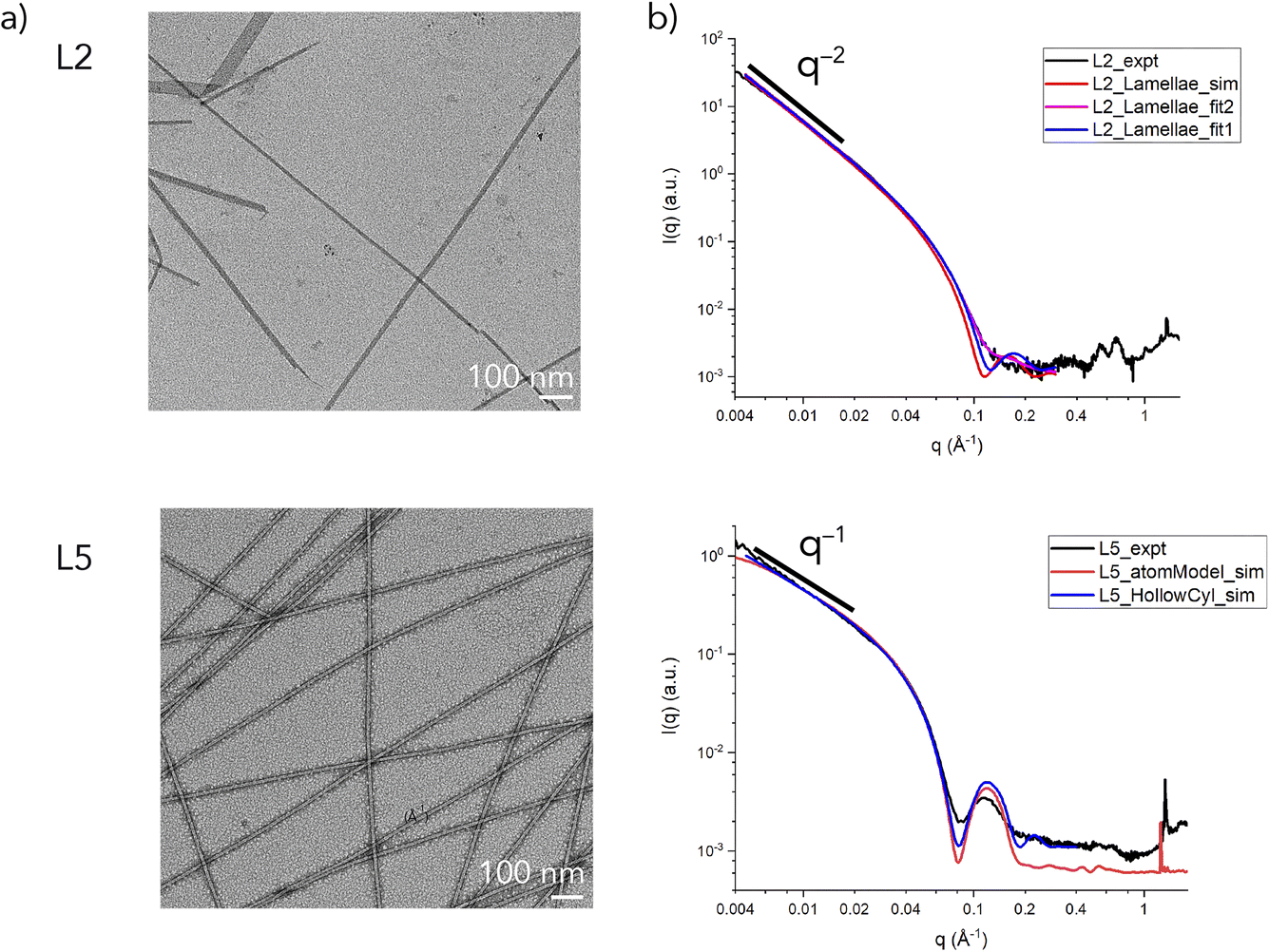 | ||
| Fig. 2 Representative negative stain TEM images (a) and synchrotron small-angle X-ray scattering (SAXS) measurements (b) of L2 and L5 assemblies. The L2 scattering curve was simulated or fit using a lamellar model (sim: t = 53.6 Å; fit1: t = 50 Å; fit2: t = 50.0 × (1.00 ± 0.20)). The L5 scattering curve was simulated using either a hollow cylinder model or an all-atom model derived from the cryo-EM structure. | ||
Synchrotron small-angle and wide-angle X-ray scattering (SAXS/WAXS) measurements on solutions of the peptide assemblies confirmed the results from TEM imaging (Fig. 2b). The SAXS intensity profiles for L5 indicated that the momentum transfer (q) decayed based on the relationship I ∝ q−1 (for q ≤ 0.02 Å−1), whereas, for L2, the observed decay in q could be fit to I ∝ q−2 (for q ≤ 0.02 Å−1). The difference in the respective power law exponents was consistent with the formation of filamentous assemblies for L5 and lamellar assemblies for L2. The scattering intensity of L5 was fit to a hollow cylinder model. The estimated values for the inner radius (∼20 Å) and wall thickness (∼18 Å) predicted an outer diameter of ∼76 Å, which agreed well with the corresponding value reported from TEM measurements and a previous SANS analysis.32
In contrast, the scattering intensity data in the Guinier region (qmaxRt ≤ 1.3) for the L2 assemblies could be fit to a model for sheet-like forms (Fig. S5†). An average value for the sheet thickness (t) was estimated to be ∼53.6 Å for the L2 assemblies. The estimated lamellar thickness for L2 was significantly larger than the projected length of the peptide in an extended β-sheet conformation (∼21 Å), which indicated that stacking of lamellae occurred. The latter result suggested that the peptide packing was more complex than that predicted from the monolayer packing model proposed from SANS analysis of Ac-KIIIIK-NH2 (Fig. 1), in which the peptide length defined the thickness of the lamellar monolayer. The SAXS data for L2 was fit to a lamellar model using the values of average sheet thickness derived from the Guinier analysis (Fig. S5†). A better fit of the experimental scattering data was obtained when a correction was applied for polydispersity in sheet thickness (Fig. 2b). For both peptide assemblies, a strong diffraction peak was observed at a q value of 1.3 Å−1, corresponding to d = 4.7 Å, which was associated with the hydrogen-bonding distance within a cross-β fibril structure (Fig. S6†). While different morphologies were observed for the L2 and L5 assemblies, the supramolecular structures of the two different peptides were based on a common core. AlphaFold-Multimer42 predicted a parallel cross-β fibril to be a common component of these peptide assemblies, which was consistent with the WAXS analysis. We propose that alterations in sheet packing could account for the differences in morphology that are observed between the L2 and L5 sequences.17,43
In the proposed monolayer model for peptide self-assembly (Fig. 1), surface asymmetry influences the degree of monolayer curvature since the variant residues are located only on one side of the parallel β-sheet. Greater curvature would result from increased asymmetry between the two lamellar surfaces. Tighter coiling of the supramolecular assemblies was postulated as the driving force for a structural transition from the lamellar structure of the symmetric Ac-KIIIIK-NH2 to a filamentous structure for the mono-substituted derivative L5. However, the TEM and SAXS analyses of L2 did not support this proposed model for peptide self-assembly, since a similar degree of surface asymmetry, albeit on the opposite sheet face, resulted in a significant difference in morphology between the L2 and L5.
To provide insight into the mechanism of peptide assembly, cryo-EM analysis was employed to determine the structure of the L5 nanotube at near-atomic resolution (Fig. 3a). The reference-free 2D class averages indicated the presence of a population of peptide nanotubes of uniform diameter. Single-particle helical reconstruction generated a three-dimensional density map into which a reliable atomic model could be built (Fig. 3b).26,44,45 The reconstruction process involved testing all possible symmetries that could be indexed from the averaged power spectrum (Fig. S7†). A prominent layer-line was observed at 1/4.9 Å in the averaged power spectrum of the L5 nanotube assemblies, which was attributable to the packing of strands in a cross-β fibril (Fig. 3b).46–48 The correct solution yielded recognizable features in the density map that were consistent with the presence of the L5 peptide (Fig. 3b). Based on the map![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :map Fourier Shell Correlation (FSC) (Fig. S8†), the resolution of the reconstructed volume for the L5 nanotubes was estimated to be 2.7 Å. The structure of the L5 nanotube exhibited C5 rotational symmetry with a helical rise of 4.94 Å and a twist of −1.77° (Fig. 3b and c). The well-resolved peptide side-chain density and clear separation of the cross-β strands supported this symmetry assignment. The asymmetric unit (ASU) of the L5 nanotube was based on three peptides (Fig. 3d), which formed the structural repeat of three symmetry independent cross-β fibrils that defined five protofilaments within the structure. The five protofilaments propagated along a set of left-handed 5-start helices (Fig. 3c). The protofilament cross-sectional thickness at its widest point measured ∼27 Å, and the diameter of the lumen was estimated as ∼40 Å. The filament outer diameter of ∼81 Å compared well to estimates from fitting of the SAXS (∼76 Å) to a hollow-cylinder model (Fig. 2b). An all-atom model derived from the cryo-EM structure was employed to simulate the SAXS intensity profiles observed for the L5 nanotube and accurately captured the main oscillation peak at 0.12 Å−1 (Fig. 2b).
:map Fourier Shell Correlation (FSC) (Fig. S8†), the resolution of the reconstructed volume for the L5 nanotubes was estimated to be 2.7 Å. The structure of the L5 nanotube exhibited C5 rotational symmetry with a helical rise of 4.94 Å and a twist of −1.77° (Fig. 3b and c). The well-resolved peptide side-chain density and clear separation of the cross-β strands supported this symmetry assignment. The asymmetric unit (ASU) of the L5 nanotube was based on three peptides (Fig. 3d), which formed the structural repeat of three symmetry independent cross-β fibrils that defined five protofilaments within the structure. The five protofilaments propagated along a set of left-handed 5-start helices (Fig. 3c). The protofilament cross-sectional thickness at its widest point measured ∼27 Å, and the diameter of the lumen was estimated as ∼40 Å. The filament outer diameter of ∼81 Å compared well to estimates from fitting of the SAXS (∼76 Å) to a hollow-cylinder model (Fig. 2b). An all-atom model derived from the cryo-EM structure was employed to simulate the SAXS intensity profiles observed for the L5 nanotube and accurately captured the main oscillation peak at 0.12 Å−1 (Fig. 2b).
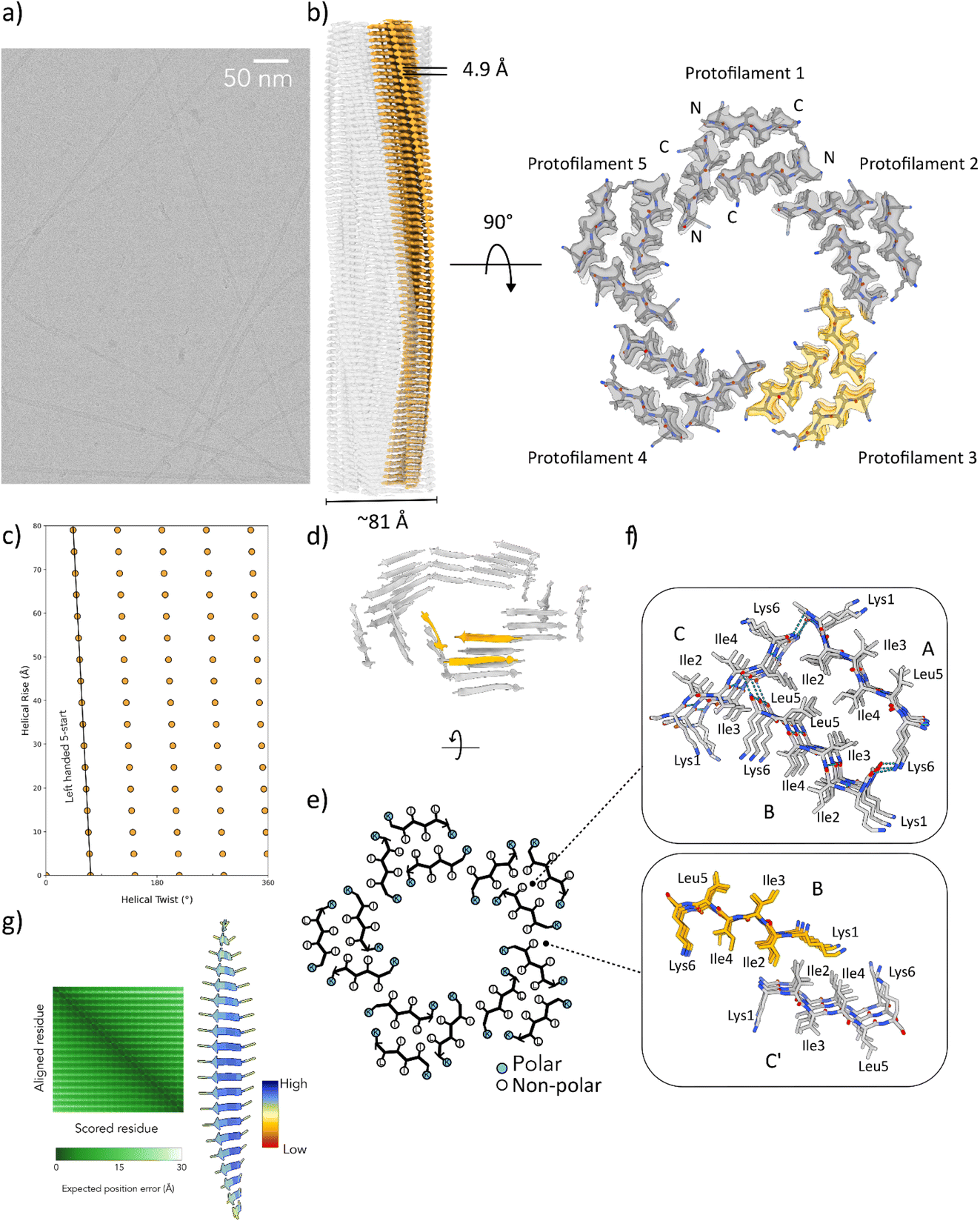 | ||
| Fig. 3 (a) Representative raw cryo-EM micrograph of L5 peptide nanotube assemblies. (b) Cryo-EM density map of the 3D reconstruction is shown on the left. The cross-sectional density map is shown on the right with the atomic model built into the map. In both cases, a single protofilament is highlighted in orange. (c) Helical net diagram for the L5 nanotube in which the unrolled surface lattice is viewed by convention from the outside of the filament. (d) A ribbon diagram of a cross-section corresponding to three layers of the L5 structure is on the upper left. The symmetry independent peptides in one ASU are highlighted. (e) A schematic view of a single layer of the structure is depicted below in which the positions of sidechains and the direction of the strands is indicated. (f) The packing of steric zipper interfaces is shown on the right for interfaces within and between ASUs. (g) AlphaFold-Multimer prediction of the structure of an oligomer of Ac-KIIILK-NH2 in which colouring is based on pLDDT. The graph depicts the predicted aligned error (PAE) on a per residue basis. | ||
In contrast to the predictions of the monolayer packing model (Fig. 1), the structure of the L5 nanotube closely resembled the reported structures of amyloid assemblies, in which cross-β fibrils interacted through the formation of steric zipper interfaces (Table 1).49 The slight left-handed twist of the 5-start (−1.77°) protofilaments in the L5 nanotube occurred within the typical range observed for cross-β protofilaments in amyloid fibrils.17 For comparison, a twist of −3.2° was observed for the 2-start protofilaments within a representative β-amyloid such as the Aβ1–42 type I structural polymorph (PDB: 7Q4B).50 While paired helical filaments are the most commonly observed morphology for amyloid-like fibrils, higher numbers of protofilaments have been observed in the structures of assemblies derived from native18,51–58 and designed10 peptide sequences. Several amyloid structures have been determined in which the protofilaments are composed of multiple peptides within the ASU. For example, the cryo-EM structure of a nanotube derived from self-assembly of an undecapeptide 247DLIIKGISVHI257 (PDB: 5W7V) from TDP-43 RRM2 contained three protofilaments based on an ASU containing nine symmetry independent peptides for a total of twenty-seven cross-β fibrils.54
| L5 | Aβ42 (type 1) | ||
|---|---|---|---|
| Shape complementarity | 0.72 | 0.75 | |
| Inter-strand distance | 4.9 Å | 4.88 Å | |
| Inter-sheet distance | 10.9 Å | 10.7 Å | |
| Buried surface area | Inter-sheet surface area |  |
 |
| Intra-sheet surface area |  |
 |
|
The cross-β fibrils in the protofilaments resulted from stacking of asymmetric units (ASUs) through axial hydrogen bonding interactions and were aligned in-register and parallel. The interdigitation of the side chains observed in the trimeric ASU of L5 was representative of an amyloid cross-β fold (Fig. 3e and f).17,49,59 Two chains (labelled chains A and B in Fig. 3f) were tightly mated through a dry, steric-zipper interaction involving residues Ile2(A), Ile4(A), Ile3(B), and Leu5(B). The distance between these two closely mated sheets, ∼11 Å, was typical for amyloid sheet stacking interfaces.60 The polar face-to-back sheet interface between chains A and B was characteristic of Class 4 amyloid steric zippers (using the nomenclature introduced in Sawaya, et al.).59 In contrast, chain C lay at angle to the binary interface between chains A and B in the ASU of L5. The interdigitated packing of the hydrophobic sidechains of residues Ile2(A), Leu5(B), and Leu5(C) mediated a ternary interaction between the three peptides in the ASU. This lateral association was further reinforced by an interaction between the sidechains of Lys6(B) and Ile3(C) on the opposite face of chain A. The arrangement of peptides within the ASU may be additionally stabilized through a network of hydrogen bonding interactions between polar groups including the N-terminal acetyl groups (A, B), the C-terminal amide groups (B, C) and the sidechain of Lys6(A) (Fig. 3f). A steric zipper-like motif was also observed between chain B and chain C′ at a protofilament interface. Interdigitation of sidechains of Lys1(B), Ile2(B), Ile2(C′), and Ile4(C′) resulted from a face-to-face sheet packing interaction corresponding to a Class 1 amyloid steric zipper. The two peptides were offset across the stacking interface presumably to alleviate a potential repulsive electrostatic interaction between Lys1(B) and Lys6(C′) that would have resulted from an in-register alignment. The chemical properties that governed the stability of the L5 nanotubes, such as buried surface area and shape complementarity between sheets, closely resembled those observed between peptide segments of similar length at cross-β interfaces within a representative amyloid structure (Table 1).
Since the cryo-EM structural analysis of the L5 nanotube indicated that the chain packing within the ASU was comparable to those observed in cross-sectional layers of β-amyloids, it stands to reason that the intra- and inter-molecular interactions that stabilize the packing arrangement within the nanotube should resemble those that stabilize the internal interfaces within cross-β amyloids. Analysis of hydrophobic and electrostatic surfaces of the L5 cross-sectional layers indicated that the apolar and polar residues segregated into spatially distinct domains that were stacked in register between adjacent rungs of the ladder-like structures (Fig. 4a and b). The bola-like polar sequence pattern should favour this spatial arrangement when the individual peptides are packed as parallel strands in minimally twisted cross-β fibrils. Similar packing arrangements are observed within amyloid structures, in which polar and apolar residues sequester into different spatial domains.17 Unlike many amyloid structures, in which polar residues can be localized in the hydrophobic core leading to charge frustration,17 the block-like sequence of the L5 peptide promoted nanoscale segregation between polar and apolar domains. The charged Lys sidechains were positioned at the solvent contacting the outer surface and inner lumen, while the stacking of hydrophobic residues occurred in the interior of the assembly. The solvent-accessible surface area (SASA) buried at the axial interfaces between rungs of the L5 nanotube (∼5000 Å2 or ∼1000 Å2 per protofilament) was comparable to segments of amyloid structures of comparable length (Fig. 4c and Table 1).
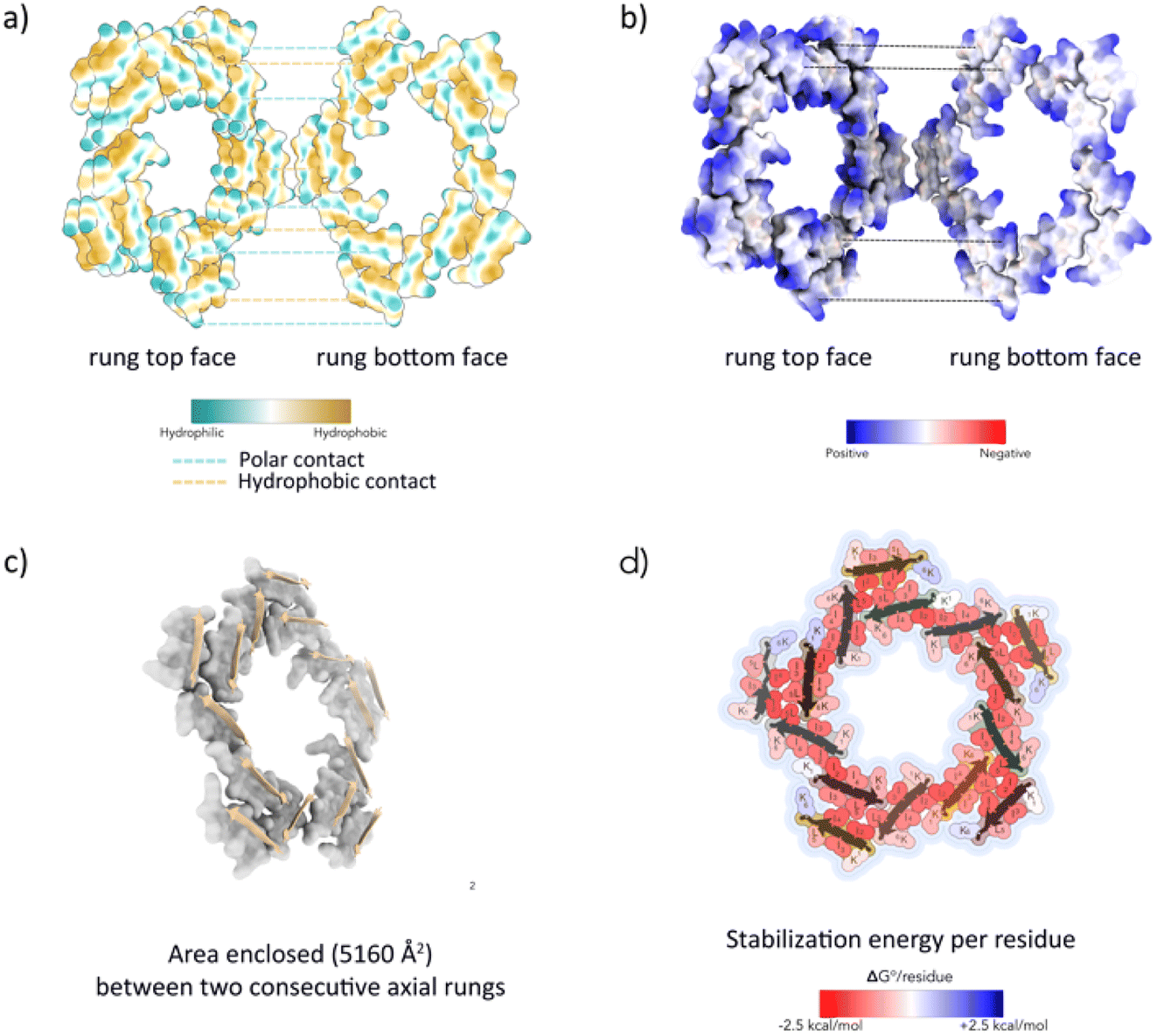 | ||
Fig. 4 Analysis of peptide interactions within cross-sectional layers of the L5 nanotube. (a) Hydrophobic surface representation of two aligned rungs of L5. (b) Electrostatic surface representation of two aligned rungs of L5. (c) Representation of buried axial surface area at cross-sectional layers of L5. (d) Stabilization energy distribution maps of cross-sectional layers of the L5 nanotube. Residues are coloured by calculated individual values of  . . | ||
The stabilization energy distribution maps17,61 (Fig. 4d) for the L5 cross-sectional layers confirmed that the sequestration of hydrophobic residues in the core domain of the filament provided the main energetic driving force for self-assembly. The mean stabilization free energy per residue  of −0.96 kcal mol−1 for L5 was more energetically favourable than the mean value of
of −0.96 kcal mol−1 for L5 was more energetically favourable than the mean value of  observed for amyloid fibrils,62 which is indicative of a strong per-residue driving force for self-assembly. However, the stabilization free energy per chain
observed for amyloid fibrils,62 which is indicative of a strong per-residue driving force for self-assembly. However, the stabilization free energy per chain  of −5.76 kcal mol−1 for L5 was significantly less stabilizing than the mean value of
of −5.76 kcal mol−1 for L5 was significantly less stabilizing than the mean value of 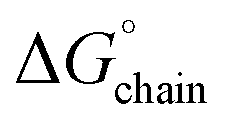 (ca. −24 kcal mol−1) determined for amyloids. The sequence of the L5 peptide is much shorter than those of most structurally characterized cross-β amyloids, which necessitates that the
(ca. −24 kcal mol−1) determined for amyloids. The sequence of the L5 peptide is much shorter than those of most structurally characterized cross-β amyloids, which necessitates that the  must be very energetically favourable to promote self-assembly of the nanotube.
must be very energetically favourable to promote self-assembly of the nanotube.
The  and
and 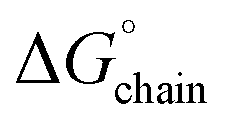 values for the L5 nanotube are comparable to the corresponding values determined for the structures of amyloid assemblies derived from relatively short peptides containing protofilaments composed of multiple peptides in the ASU, e.g., uperin-3.5
values for the L5 nanotube are comparable to the corresponding values determined for the structures of amyloid assemblies derived from relatively short peptides containing protofilaments composed of multiple peptides in the ASU, e.g., uperin-3.5  and TDP-43247–251
and TDP-43247–251  .54,55 The less favourable value of
.54,55 The less favourable value of  for the L5 nanotube suggested that self-assembly should be reversible. Upon incubation of a 10-fold dilution of the nanotube preparation, the filaments dis-assembled and could no longer be detected using TEM or SAXS measurements. We propose that these bola-like peptide sequences behave similarly to functional amyloid-like assemblies, in which reversible self-assembly behaviour is commonly observed. Thermodynamic analysis of functional amyloids indicated that free energy of stabilization of the fibrillar structures is much less than that for most pathogenic amyloids for which self-assembly is typically irreversible.17
for the L5 nanotube suggested that self-assembly should be reversible. Upon incubation of a 10-fold dilution of the nanotube preparation, the filaments dis-assembled and could no longer be detected using TEM or SAXS measurements. We propose that these bola-like peptide sequences behave similarly to functional amyloid-like assemblies, in which reversible self-assembly behaviour is commonly observed. Thermodynamic analysis of functional amyloids indicated that free energy of stabilization of the fibrillar structures is much less than that for most pathogenic amyloids for which self-assembly is typically irreversible.17
The accurate design of supramolecular structure constitutes a significant challenge to the development of peptide-based materials for applications. Most of the early research on peptide-based materials focused on the design and structural analysis of β-sheet filaments.63–69 These peptides are attractive substrates for materials design since the directional hydrogen bonding between β-strands provides a strongly stabilizing and designable interaction that can be encoded within short sequences that are amenable to preparative-scale synthesis. These research efforts also drew inspiration from concurrent structural analyses of amyloid fibrils,70,71 which display high thermodynamic stability that derives from a cross-β structure46 similar to that envisioned for designed β-sheet assemblies. Notably, like L2 and L5, many amyloidogenic peptide sequences form hydrogels upon fibrillation.72
Recent advances in cryo-EM, solid-state NMR, and microcrystal electron diffraction3 have enabled structural determination on amyloid assemblies at near-atomic resolution, which has resulted in a proliferation of high-resolution atomic models.73 These studies revealed that the amyloid structural landscape was far more complex than a conventional representation of the cross-β filament.43 Structural polymorphism is quite common within the lateral packing interfaces of the cross-β fold and amyloid structure was shown to depend strongly on the experimental conditions under which the peptides were assembled. In contrast, fewer structural analyses of designed β-sheet assemblies have been performed at near-atomic resolution.11,74–76 However, the results of these studies provided evidence for the presence of polymorphism in which the population of different species depended on experimental conditions. These analyses have demonstrated that the principles of peptide design are imperfectly understood at present with respect to the relationship between sequence and supramolecular structure for synthetic peptide-based materials.
Cryo-EM structural analysis of the L5 nanotube provided evidence for the similarity in self-assembly behaviour between surfactant-like peptide-based materials and amyloidogenic peptides. The cross-sectional layers of the L5 nanotube consisted of intra- and inter-protofilament interfaces based on different types of steric zipper interactions. These layers stack through hydrogen-bonding interactions to form an amyloid-like cross-β fibril. In this respect, the structure of the L5 nanotube can be understood in terms of the structural principles revealed from high resolution structural analyses of amyloid fibrils. In the latter, a richly designable and highly conserved cross-β interaction parallel to the fibril axis is combined with sparsely designable and structurally equivocal lateral packing interfaces within the cross-sectional layers.77 The well-known propensity for polymorphism in amyloid structures is a consequence of this potential ambiguity in lateral packing.
Remarkably, the cryo-EM structural analysis of L5 suggested that the peptide self-assembled into a homogeneous population of nanotubes despite the potential for packing polymorphism. The mean stabilization free energy per chain  calculated for the L5 nanotubes was more consistent with those calculated for functional amyloids, in which self-assembly behaviour was often observed to be reversible.17 The self-assembly of L5 was demonstrated to be reversible upon dilution, which was consistent with the calculated values of
calculated for the L5 nanotubes was more consistent with those calculated for functional amyloids, in which self-assembly behaviour was often observed to be reversible.17 The self-assembly of L5 was demonstrated to be reversible upon dilution, which was consistent with the calculated values of  for the L5 interface (Fig. 4d). Reversible self-assembly provides the opportunity for error correction and relaxation to the most thermodynamically favourable structure. The selective self-assembly of L5 into a monomorphic nanotube might be considered as an example of molecular frustration in which the intentional introduction of repulsive and attractive interactions into peptide sequences enables a greater degree of supramolecular structure control. This principle has been previously employed for the design of multi-domain peptides, in which the extent of self-assembly can be controlled through manipulation of the balance of attractive and repulsive forces through changes in peptide sequence and assembly conditions.78–81
for the L5 interface (Fig. 4d). Reversible self-assembly provides the opportunity for error correction and relaxation to the most thermodynamically favourable structure. The selective self-assembly of L5 into a monomorphic nanotube might be considered as an example of molecular frustration in which the intentional introduction of repulsive and attractive interactions into peptide sequences enables a greater degree of supramolecular structure control. This principle has been previously employed for the design of multi-domain peptides, in which the extent of self-assembly can be controlled through manipulation of the balance of attractive and repulsive forces through changes in peptide sequence and assembly conditions.78–81
Experimental
Materials and methods
Peptides L2 (Ac-KLI3K-NH2) and L5 (Ac-KI3LK-NH2) were obtained from GenScript USA, Inc. (Piscataway, NJ) at ≥95% purity. Stock solutions of peptides were prepared by solubilizing the purified, lyophilized peptides in aqueous solution at a concentration of 32 × 10−3 M. The pH value of the peptide solutions was measured as ∼4.0. The resulting solutions were incubated at ambient temperature for 2 weeks to achieve equilibrium. During this period, the initially clear solutions transformed into self-supporting hydrogels.Circular dichroism (CD) spectropolarimetry
CD measurements were performed on a Jasco J-1500 CD spectropolarimeter using 0.20 mm thick quartz plates from Hellma USA Inc (Plainview, NY). Three spectra were collected and averaged in a wavelength range from 190 to 260 nm at a scanning range of 100 nm min−1 with a bandwidth of 2 nm and a data pitch of 0.2 nm. Peptide concentration was determined spectrophotometrically at 214 nm using molar extinction coefficients calculated from the amino acid composition of the respective peptides.82 Samples were diluted to ∼25 mM prior to acquisition of CD data.Negative stain TEM analysis
TEM grids were prepared using 10–15-fold dilutions of peptide in aqueous solution at pH 4.0. Samples of the peptide solutions (4 μL) were deposited onto a 200-mesh carbon-coated copper grid from Electron Microscopy Services (Hatfield, PA). After 90 s of incubation on the grid, excess liquid was wicked away, leaving a thin film of sample. Uranyl acetate negative stain, 4 μL of a 1% solution, was deposited onto the thin film. After 1 min, the remaining moisture was wicked away, and the grid was dried overnight in a desiccator. Electron micrographs were captured on a JEOL JEM-1400 transmission electron microscope equipped with a LaB6 filament operating at an accelerating voltage of 80 kV.Synchrotron small-angle/wide-angle X-ray scattering (SAXS/WAXS) measurements
SAXS/WAXS measurements were performed at the 12-ID-B beamline of the Advanced Photon Source at Argonne National Laboratory. A SAXS/WAXS simultaneous setup was utilized, and the sample-to-detector distances were set such that the overall scattering momentum transfer (q) range was achieved from 0.003 to 2.4 Å−1, where q = 4πsin(θ)/λ, with 2θ denoting the scattering angle and λ denoting the X-ray wavelength. The wavelength was set at 0.9322 Å during the measurements. Scattered X-ray intensities were measured using a Pilatus 2M (DECTRIS) detector for SAXS and Pilatus 300K for WAXS. SAXS/WAXS measurements were performed on aqueous solutions of the peptide assemblies in quartz capillaries at concentrations of approximately 32 mM in sodium acetate buffer (10 mM, pH 4.0) at 25 °C. The 2D scattering images were converted to 1D SAXS curves through azimuthally averaging after solid angle correction and then normalizing with the intensity of the transmitted X-ray beam using the software package at beamline 12-ID-B. The 1D curves of the samples were averaged and subtracted with the background measured from the corresponding buffers. The choice of the model to fit the data was justified by negative-stain and cryo-EM observations. The simulated SAXS curves were calculated using the programs CRYSOL and Pepsi-SAXS with fitting to all-atom cryo-EM structural models. To reproduce the SAXS features for the all-atom model of the L5 nanotube, an atomic model was generated from the cryo-EM reconstruction in which the length:diameter aspect ratio was approximately 10:1.
Cryo-EM imaging and analysis
A sample of the L5 peptide assemblies was applied to glow-discharged lacey carbon grids and vitrified using a Leica plunge freezer. Grids were imaged on a Titan Krios (300 keV, Thermo Fisher) with a K3 camera (Gatan). A total number of 5292 micrographs were collected under electron counting mode at 1.08 Å per pixel, using a defocus range of 1–2 μm with ∼50 electrons per Å2 distributed into 40 fractions. Motion correction and CTF estimation were done in cryoSPARC.83 A few million particles were automatically picked using “Filament Tracer” with a shift of 11 pixels. Next, non-peptide junk particles and particles in low-resolution averages were removed by multiple rounds of reference-free 2D classifications. Particles having clear 2D average patterns were then selected. All possible helical symmetries were calculated from an averaged power spectrum of the raw particles and then were tested by trial and error in cryoSPARC until recognized peptide features, such as clear separation of β-sheets and good side chains densities, were observed.26 The final volumes were then sharpened using local sharpening and DeepEMhancer available in cryoSPARC. Data collection statistics are listed in Table S1.†Model building
The L5 nanotubes reached ∼2.7 Å resolution according to map:map FSC (Fig. S8†). Since the filaments are made of only β-sheets, the hand of the cryo-EM map cannot be determined directly, which is unlike cryo-EM maps that contain an α-helix, in which the hand is obvious when the resolution is 4.5 Å or better. In published cross-β structures, the parallel β-sheets typically have a left-handed twist.84 However, this observation may not be deducible to short peptides. Therefore, we performed model building for both hands of the map. First, the model was manually adjusted in Coot85 and then real-space refined in PHENIX.86 The model fits the map in the left-handed cross-β protofilament with better RSCC and better hydrogen bonds within β-sheets. Therefore, we suggest that the map has a left-handed twist in the 5-start helices (Fig. 3c). The refinement statistics of the L5 nanotubes are shown in Table S1.†
Structural prediction
The colabfold implementation87 of AlphaFold-Multimer v2 was employed for prediction of oligomeric structures of the peptides (16 copies). The following parameters were employed in the structural prediction: templates, none, MSAs, single sequence;88 recycles, 12; and seeds, 4. The highest ranked models, based on ipTM scores, were used to generate the atomic models of the oligomers. As controls, the following peptide sequences having different polar patterns were employed as controls: IIKKIL, KKIIIL, IIILKK, and IKIIKL. Except for the highest ranked model predicted for KKIIIL, no peptide sequences displayed an ordered cross-β structure with high pLDDT (≥0.8) or high ipTM (≥0.6) scores. ZipperDB89 was employed to calculate the energy values for formation of steric zipper interfaces for L2 and L5 (https://zipperdb.mbi.ucla.edu/). The calculated Rosetta energy unit scores for L2 (−28.12 kcal mol−1) and L5 (−27.13 kcal mol−1) were well below the energetic threshold for formation of stable steric zipper interfaces. The hydrophobic and electrostatic surfaces for the L5 nanotube cross-sections were calculated using ChimeraX.90 The values for the stabilization energies and the stabilization energy distribution maps were calculated using the method from https://people.mbi.ucla.edu/sawaya/amyloidatlas/.17Conclusions
Here, we examined the self-assembly behaviour of L2 and L5, a pair of bola-amphiphilic peptides. Polar sequence patterning was employed as a principle to guide the design of β-sheet forming peptides that were capable of nanoscale phase segregation. While polar patterning could drive peptide self-assembly through sequestration of the hydrophobic domains, we observed morphological differences between the L2 and L5 assemblies that could not be rationalized using a previously proposed model based on principles derived from surfactant self-assembly, e.g., Fig. 1. The experimental data supported the hypothesis that amyloid-like behaviour dominated the properties of these materials. Like amyloidogenic peptides, the self-assembly behaviour of L2 and L5 exhibited a significant sequence dependence despite identical length, composition, and polar sequence pattern. The sensitivity of supramolecular structure to sequence substitutions for these bola-like peptides was consistent with the known role of disease-related mutations on the self-assembly landscape of pathological amyloids.91 It has been well documented that mutagenesis or covalent modification of amyloidogenic peptides drives the self-assembly of alternative structural polymorphs. We recently described a similar phenomenon in the cryo-EM analysis of cross-α filaments, in which semi-conservative mutagenesis resulted in the formation of different supramolecular structures despite the presence of common core conformation.10 This behaviour differs from that observed for highly designable protein folds, in which native structure is resistant to disruption from semi-conservative mutagenesis.AlphaFold-Multimer42 and ZipperDB89 predicted that L2 and L5 sequences could adopt a parallel cross-β fold and form stable steric zipper interfaces. The experimental data also support a common core structure of a β-sheet filament for the L2 and L5 assemblies. Therefore, the formation of lamellae versus filaments for L2 versus L5 must result from subtle differences in local packing interfaces between β-sheets that are propagated hierarchically into distinct supramolecular structures. The geometries of these packing interactions are difficult to predict a priori as they only emerge at longer length scales where the cumulative effect of many such local interactions along the contour length of the filament mediates association into higher order structures. Unfortunately, the lamellar structure of the L2 assembly was not amenable to single particle reconstruction, which precluded a more detailed structural analysis that would have provided further insight into the factors that determined its preference for lamellae versus filaments.
Additionally, our results suggest that the plethora of structural information available from high-resolution analysis of amyloid fibrils can potentially inform the design of synthetic peptide-based materials that self-assemble through the formation of cross-β fibrils. However, structural determination at near-atomic resolution will be critical to develop an understanding of the relationship between sequence and supramolecular structure. These methods of structural analysis, particularly cryo-EM helical reconstruction, need to be more widely applied to the study of designed peptide filaments. The construction of a larger database of high-resolution structures of filamentous assemblies can provide a starting point from which more reliable principles can be formulated for peptide-based materials design.
Data availability
The reconstruction map of the L5 nanotube was deposited in the Electron Microscopy Data Bank with accession number EMD-46060. The corresponding filament model was deposited in the Protein Data Bank with accession number 9CZ3. Additional data that support the findings of this study are provided in the supplementary information† or are available from the lead author upon request.Author contributions
Conceptualization, A. D., O. G., F. W. and V. P. C.; methodology, A. D., X. Z., F. W., and V. P. C.; investigation, A. D., O. G., X. Z. and F. W.; supervision, F. W. and V. P. C.; validation, A. D. and F. W.; visualization, A. D., F. W., and V. P. C.; writing – original draft, A. D. and V. P. C.; writing – review and editing, A. D., X. Z., F. W., and V. P. C.; funding acquisition, F. W., and V. P. C.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The cryo-EM imaging of the L5 nanotubes was performed at the Molecular Electron Microscopy Core Facility at the University of Virginia, which is supported by the School of Medicine and built with NIH grant G20-RR31199. In addition, the Titan Krios (SIG S10-RR025067) and K3/GIF (U24-GM116790) were purchased, in part or in full, with the designated NIH grants. Research funding was provided by grants from the NSF (CHE-2108621) to V. P. C. and the NIH (GM138756) to F. W. The circular dichroism spectropolarimeter was acquired through funding from an NSF grant (DBI-1726544). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors thank Ed Egelman for useful discussions.Notes and references
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed.
- T. L. Lopez-Silva and J. P. Schneider, Curr. Opin. Chem. Biol., 2021, 64, 131–144 CrossRef CAS PubMed.
- J. G. Miller, S. A. Hughes, C. Modlin and V. P. Conticello, Q. Rev. Biophys., 2022, 55, e2, DOI:10.1017/S0033583522000014.
- J. Zhu, N. Avakyan, A. Kakkis, A. M. Hoffnagle, K. Han, Y. Li, Z. Zhang, T. S. Choi, Y. Na, C. J. Yu and F. A. Tezcan, Chem. Rev., 2021, 121, 13701–13796 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millan, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- P. Zhou, C. Yuan and X. Yan, Curr. Opin. Colloid Interface Sci., 2022, 62, 101645 CrossRef CAS.
- E. H. Egelman, C. Xu, F. DiMaio, E. Magnotti, C. Modlin, X. Yu, E. Wright, D. Baker and V. P. Conticello, Structure, 2015, 23, 280–289 CrossRef CAS PubMed.
- S. A. Hughes, F. Wang, S. Wang, M. A. B. Kreutzberger, T. Osinski, A. Orlova, J. S. Wall, X. Zuo, E. H. Egelman and V. P. Conticello, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14456–14464 CrossRef CAS PubMed.
- F. Wang, O. Gnewou, C. Modlin, L. C. Beltran, C. Xu, Z. Su, P. Juneja, G. Grigoryan, E. H. Egelman and V. P. Conticello, Nat. Commun., 2021, 12, 407 CrossRef CAS PubMed.
- F. Wang, O. Gnewou, S. Wang, T. Osinski, X. Zuo, E. H. Egelman and V. P. Conticello, Matter, 2021, 4, 3217–3231 CrossRef CAS PubMed.
- L. Pieri, F. Wang, A. A. Arteni, M. Vos, J. M. Winter, M. H. Le Du, F. Artzner, F. Gobeaux, P. Legrand, Y. Boulard, S. Bressanelli, E. H. Egelman and M. Paternostre, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120346119 CrossRef PubMed.
- C. O. Mackenzie, J. Zhou and G. Grigoryan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7438–E7447 CrossRef CAS PubMed.
- J. Zhou and G. Grigoryan, Protein Sci., 2015, 24, 508–524 CrossRef CAS PubMed.
- J. Zhou, A. E. Panaitiu and G. Grigoryan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1059–1068 CrossRef CAS PubMed.
- V. P. Conticello, Curr. Opin. Solid State Mater. Sci., 2023, 27, 101066 CrossRef CAS.
- M. R. Sawaya, M. P. Hughes, J. A. Rodriguez, R. Riek and D. S. Eisenberg, Cell, 2021, 184, 4857–4873 CrossRef CAS PubMed.
- S. Lovestam, F. A. Koh, B. van Knippenberg, A. Kotecha, A. G. Murzin, M. Goedert and S. H. W. Scheres, eLife, 2022, 11, e76494 CrossRef CAS PubMed.
- W. Close, M. Neumann, A. Schmidt, M. Hora, K. Annamalai, M. Schmidt, B. Reif, V. Schmidt, N. Grigorieff and M. Fandrich, Nat. Commun., 2018, 9, 699 CrossRef PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fandrich, Nat. Commun., 2019, 10, 4760 CrossRef PubMed.
- J. Adamcik and R. Mezzenga, Angew. Chem., Int. Ed., 2018, 57, 8370–8382 CrossRef CAS PubMed.
- Y. Zhao, J. Wang, L. Deng, P. Zhou, S. Wang, Y. Wang, H. Xu and J. R. Lu, Langmuir, 2013, 29, 13457–13464 CrossRef CAS PubMed.
- L. Deng, P. Zhou, Y. Zhao, Y. Wang and H. Xu, J. Phys. Chem. B, 2014, 118, 12501–12510 CrossRef CAS PubMed.
- Y. Zhao, L. Deng, J. Wang, H. Xu and J. R. Lu, Langmuir, 2015, 31, 12975–12983 CrossRef CAS PubMed.
- J. Li, J. Wang, Y. Zhao, P. Zhou, J. Carter, Z. Li, T. A. Waigh, J. R. Lu and H. Xu, Coord. Chem. Rev., 2020, 421, 213418 CrossRef CAS.
- F. Wang, O. Gnewou, A. Solemanifar, V. P. Conticello and E. H. Egelman, Chem. Rev., 2022, 122, 14055–14065, DOI:10.1021/acs.chemrev.1c00753.
- F. Qiu, Y. Chen, C. Tang, Q. Zhou, C. Wang, Y. K. Shi and X. Zhao, Macromol. Biosci., 2008, 8, 1053–1059 CrossRef CAS PubMed.
- E. R. da Silva, W. A. Alves, V. Castelletto, M. Reza, J. Ruokolainen, R. Hussain and I. W. Hamley, Chem. Commun., 2015, 51, 11634–11637 RSC.
- E. R. da Silva, M. N. Walter, M. Reza, V. Castelletto, J. Ruokolainen, C. J. Connon, W. A. Alves and I. W. Hamley, Biomacromolecules, 2015, 16, 3180–3190 CrossRef CAS PubMed.
- I. W. Hamley, S. Burholt, J. Hutchinson, V. Castelletto, E. R. da Silva, W. Alves, P. Gutfreund, L. Porcar, R. Dattani, D. Hermida-Merino, G. Newby, M. Reza, J. Ruokolainen and J. Stasiak, Biomacromolecules, 2017, 18, 141–149 CrossRef CAS PubMed.
- Y. Zhao, L. Deng, W. Yang, D. Wang, E. Pambou, Z. Lu, Z. Li, J. Wang, S. King, S. Rogers, H. Xu and J. R. Lu, Chem. – Eur. J., 2016, 22, 11394–11404 CrossRef CAS PubMed.
- Y. Zhao, W. Yang, D. Wang, J. Wang, Z. Li, X. Hu, S. King, S. Rogers, J. R. Lu and H. Xu, Small, 2018, 14, e1703216 CrossRef PubMed.
- Y. Zhao, X. Hu, L. Zhang, D. Wang, S. M. King, S. E. Rogers, J. Wang, J. R. Lu and H. Xu, J. Colloid Interface Sci., 2021, 583, 553–562 CrossRef CAS PubMed.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS PubMed.
- I. A. Nyrkova, A. N. Semenov, A. Aggeli and N. Boden, Eur. Phys. J. B, 2000, 17, 481–497 CrossRef CAS.
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, T. C. B. McLeish, I. Nyrkova, S. E. Radford and A. Semenov, J. Mater. Chem., 1997, 7, 1135–1145 RSC.
- N. Nuraje, H. Bai and K. Su, Prog. Polym. Sci., 2013, 38, 302–343 CrossRef CAS.
- B. Lotz and S. Z. D. Cheng, Polymer, 2005, 46, 577–610 CrossRef CAS.
- A. D. Merg, G. Touponse, E. v. Genderen, T. B. Blum, X. Zuo, A. Bazrafshan, H. M. H. Siaw, A. McCanna, R. Brian Dyer, K. Salaita, J. P. Abrahams and V. P. Conticello, J. Am. Chem. Soc., 2020, 142, 19956–19968, DOI:10.1021/jacs.0c08174.
- M. Wang, J. Wang, P. Zhou, J. Deng, Y. Zhao, Y. Sun, W. Yang, D. Wang, Z. Li, X. Hu, S. M. King, S. E. Rogers, H. Cox, T. A. Waigh, J. Yang, J. R. Lu and H. Xu, Nat. Commun., 2018, 9, 5118 CrossRef PubMed.
- Y. Hu, R. Lin, P. Zhang, J. Fern, A. G. Cheetham, K. Patel, R. Schulman, C. Kan and H. Cui, ACS Nano, 2016, 10, 880–888 CrossRef CAS PubMed.
- R. Evans, M. O’Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Žídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, bioRxiv, 2022, preprint, DOI:10.1101/2021.10.04.463034.
- R. Gallardo, N. A. Ranson and S. E. Radford, Curr. Opin. Struct. Biol., 2020, 60, 7–16 CrossRef CAS PubMed.
- E. H. Egelman, Ultramicroscopy, 2000, 85, 225–234 CrossRef CAS PubMed.
- E. H. Egelman, J. Struct. Biol., 2007, 157, 83–94 CrossRef CAS PubMed.
- E. D. Eanes and G. G. Glenner, J. Histochem. Cytochem., 1968, 16, 673–677 CrossRef CAS PubMed.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS PubMed.
- R. Diaz-Avalos, C. Long, E. Fontano, M. Balbirnie, R. Grothe, D. Eisenberg and D. L. Caspar, J. Mol. Biol., 2003, 330, 1165–1175 CrossRef CAS PubMed.
- D. S. Eisenberg and M. R. Sawaya, Annu. Rev. Biochem., 2017, 86, 69–95 CrossRef CAS PubMed.
- Y. Yang, D. Arseni, W. Zhang, M. Huang, S. Lövestam, M. Schweighauser, A. Kotecha, A. G. Murzin, S. Y. Peak-Chew, J. Macdonald, I. Lavenir, H. J. Garringer, E. Gelpi, K. L. Newell, G. G. Kovacs, R. Vidal, B. Ghetti, B. Ryskeldi-Falcon, S. H. W. Scheres and M. Goedert, Science, 2022, 375, 167–172 CrossRef CAS PubMed.
- A. K. Paravastu, R. D. Leapman, W. M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS PubMed.
- J. X. Lu, W. Qiang, W. M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell, 2013, 154, 1257–1268 CrossRef CAS PubMed.
- N. G. Sgourakis, W. M. Yau and W. Qiang, Structure, 2015, 23, 216–227 CrossRef CAS PubMed.
- E. L. Guenther, P. Ge, H. Trinh, M. R. Sawaya, D. Cascio, D. R. Boyer, T. Gonen, Z. H. Zhou and D. S. Eisenberg, Nat. Struct. Mol. Biol., 2018, 25, 311–319 CrossRef CAS PubMed.
- R. Bucker, C. Seuring, C. Cazey, K. Veith, M. Garcia-Alai, K. Grunewald and M. Landau, Nat. Commun., 2022, 13, 4356 CrossRef PubMed.
- E. H. Chen, H. W. Kao, C. H. Lee, J. Y. C. Huang, K. P. Wu and R. P. Chen, J. Am. Chem. Soc., 2022, 144, 13888–13894 CrossRef CAS PubMed.
- M. Wilkinson, R. U. Gallardo, R. M. Martinez, N. Guthertz, M. So, L. D. Aubrey, S. E. Radford and N. A. Ranson, Nat. Commun., 2023, 14, 1190 CrossRef CAS PubMed.
- Y. Yang, W. Zhang, A. G. Murzin, M. Schweighauser, M. Huang, S. Lovestam, S. Y. Peak-Chew, T. Saito, T. C. Saido, J. Macdonald, I. Lavenir, B. Ghetti, C. Graff, A. Kumar, A. Nordberg, M. Goedert and S. H. W. Scheres, Acta Neuropathol., 2023, 145, 325–333 CrossRef CAS PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. Ø. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- A. W. Fitzpatrick and H. R. Saibil, Curr. Opin. Struct. Biol., 2019, 58, 34–42 CrossRef CAS PubMed.
- D. Eisenberg and A. D. McLachlan, Nature, 1986, 319, 199–203 CrossRef CAS PubMed.
- B. A. Nguyen, V. Singh, S. Afrin, A. Yakubovska, L. Wang, Y. Ahmed, R. Pedretti, M. D. C. Fernandez-Ramirez, P. Singh, M. Pekala, L. O. Cabrera Hernandez, S. Kumar, A. Lemoff, R. Gonzalez-Prieto, M. R. Sawaya, D. S. Eisenberg, M. D. Benson and L. Saelices, Nat. Commun., 2024, 15, 581 CrossRef CAS PubMed.
- S. Zhang, C. Lockshin, R. Cook and A. Rich, Biopolymers, 1994, 34, 663–672 CrossRef CAS PubMed.
- M. R. Caplan, P. N. Moore, S. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomacromolecules, 2000, 1, 627–631 CrossRef CAS PubMed.
- D. M. Marini, W. Hwang, D. A. Lauffenburger, S. Zhang and R. D. Kamm, Nano Lett., 2002, 2, 295–299 CrossRef CAS.
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- K. Lu, J. Jacob, P. Thiyagarajan, V. P. Conticello and D. G. Lynn, J. Am. Chem. Soc., 2003, 125, 6391–6393 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS PubMed.
- T. P. Knowles and M. J. Buehler, Nat. Nanotechnol., 2011, 6, 469–479 CrossRef CAS PubMed.
- V. K. Belwal and N. Chaudhary, Soft Matter, 2020, 16, 10013–10028 RSC.
- M. R. Sawaya, Amyloid Atlas, 2024, https://people.mbi.ucla.edu/sawaya/amyloidatlas/.
- A. R. Cormier, X. Pang, M. I. Zimmerman, H. X. Zhou and A. K. Paravastu, ACS Nano, 2013, 7, 7562–7572 CrossRef CAS PubMed.
- M. Lee, T. Wang, O. V. Makhlynets, Y. Wu, N. F. Polizzi, H. Wu, P. M. Gosavi, J. Stohr, I. V. Korendovych, W. F. DeGrado and M. Hong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6191–6196 CrossRef CAS PubMed.
- K. Nagy-Smith, E. Moore, J. Schneider and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9816–9821 CrossRef CAS PubMed.
- R. van der Kant, N. Louros, J. Schymkowitz and F. Rousseau, Structure, 2022, 30, 1178–1189 CrossRef CAS PubMed.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS PubMed.
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS PubMed.
- E. L. Bakota, O. Sensoy, B. Ozgur, M. Sayar and J. D. Hartgerink, Biomacromolecules, 2013, 14, 1370–1378 CrossRef CAS PubMed.
- A. N. Moore and J. D. Hartgerink, Acc. Chem. Res., 2017, 50, 714–722 CrossRef CAS PubMed.
- B. J. Kuipers and H. Gruppen, J. Agric. Food Chem., 2007, 55, 5445–5451 CrossRef CAS PubMed.
- A. Punjani, J. L. Rubinstein, D. J. Fleet and M. A. Brubaker, Nat. Methods, 2017, 14, 290–296 CrossRef CAS PubMed.
- C. Chothia, J. Mol. Biol., 1973, 75, 295–302 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- P. V. Afonine, B. K. Poon, R. J. Read, O. V. Sobolev, T. C. Terwilliger, A. Urzhumtsev and P. D. Adams, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 531–544 CrossRef CAS PubMed.
- M. Mirdita, K. Schutze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- K. I. Albanese, R. Petrenas, F. Pirro, E. A. Naudin, U. Borucu, W. M. Dawson, D. A. Scott, G. J. Leggett, O. D. Weiner, T. A. A. Oliver and D. N. Woolfson, Nat. Chem. Biol., 2024, 20, 991–999, DOI:10.1038/s41589-024-01642-0.
- L. Goldschmidt, P. K. Teng, R. Riek and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3487–3492 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- D. Li and C. Liu, Structure, 2023, 31, 1335–1347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data and cryo-EM data collection and refinements statistics. See DOI: https://doi.org/10.1039/d4fd00190g |
| This journal is © The Royal Society of Chemistry 2025 |