
Engineering the nano-bio interface: challenges and opportunities for predicting the surface properties of monolayer-protected nanoparticles
Carlos A Huang-Zhu a and
Reid C. Van Lehn*ab
a and
Reid C. Van Lehn*ab
aDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: vanlehn@wisc.edu
bDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA
First published on 2nd June 2025
Abstract
The surface properties of biologically active nanoparticles (NPs) are often dictated by synthetic ligands that are grafted to the NP core to form a protecting monolayer. Ligand selection is thus critical in determining NP surface properties and corresponding interactions at the nano-bio interface, which are relevant to numerous applications including drug delivery and biosensing. However, chemically specific structure–property relationships for rationally selecting ligands to achieve desired biointeractions are largely lacking. In this Focus Article, we review the challenges associated with relating ligand chemical properties to monolayer-protected NP surface properties due to the interplay of ligand–ligand, ligand–solvent, and ligand–biomolecule interactions that are difficult to anticipate. In particular, we highlight unexpected spatially varying properties that emerge even for uniformly functionalized NPs due to the fluctuations of ligands at the nanoscale. We further review the capability of physics-based molecular simulations to reveal these unexpected behaviors, providing powerful computational methods to predict NP properties. Finally, we discuss the opportunity for such simulations to be combined with machine-learning methods to guide the computational design of monolayer-protected NPs prior to synthesis.
Introduction
Monolayer-protected nanoparticles (NPs) are versatile materials that have been utilized in diverse biologically relevant applications including drug delivery, biosensing, bioimaging, and photothermal therapy.1–8 These applications are facilitated by the modular nature of NP synthesis; NPs can be fabricated by choosing a core with a desired material composition, size, and shape, and then grafting organic ligands to the core at a high density such that they form a monolayer which dictates NP surface properties (Fig. 1).9–13 Numerous ligands have been synthesized and used to coat different core materials for diverse applications. As recent examples, silicon quantum dots have been coated with aromatic fluorophore ligands (phenanthrene, pyrene, and perylene) to enhance cancer cell bioimaging while retaining low cytotoxicity,14 iron oxide NPs have been coated with block copolymers incorporating poly(ionic liquids) for drug delivery applications,15 gold NPs have been functionalized with a combination of peptides to regulate gene transcription,16 and silver NPs have been coated with simple surfactants like sodium dodecyl sulfate to facilitate penetration of bacterial membranes.17,18 In all of these applications, ligand selection, and particularly the properties of solvent-exposed chemical groups, is critical to tuning intermolecular interactions at the nanoscale that drive behaviors such as aggregation,19 protein binding,20 and biomembrane adsorption.21 For example, ligands with charged groups will impact electrostatic interactions, ligands with solvent-exposed nonpolar surface area will influence hydrophobic interactions, and ligands with aromatic groups will promote π–π or cation–π interactions. Multiple ligands, or ligands with multiple functional groups, can also be incorporated into a single monolayer.6 This synthetic flexibility affords a large design space to modulate NP interactions with biosystems.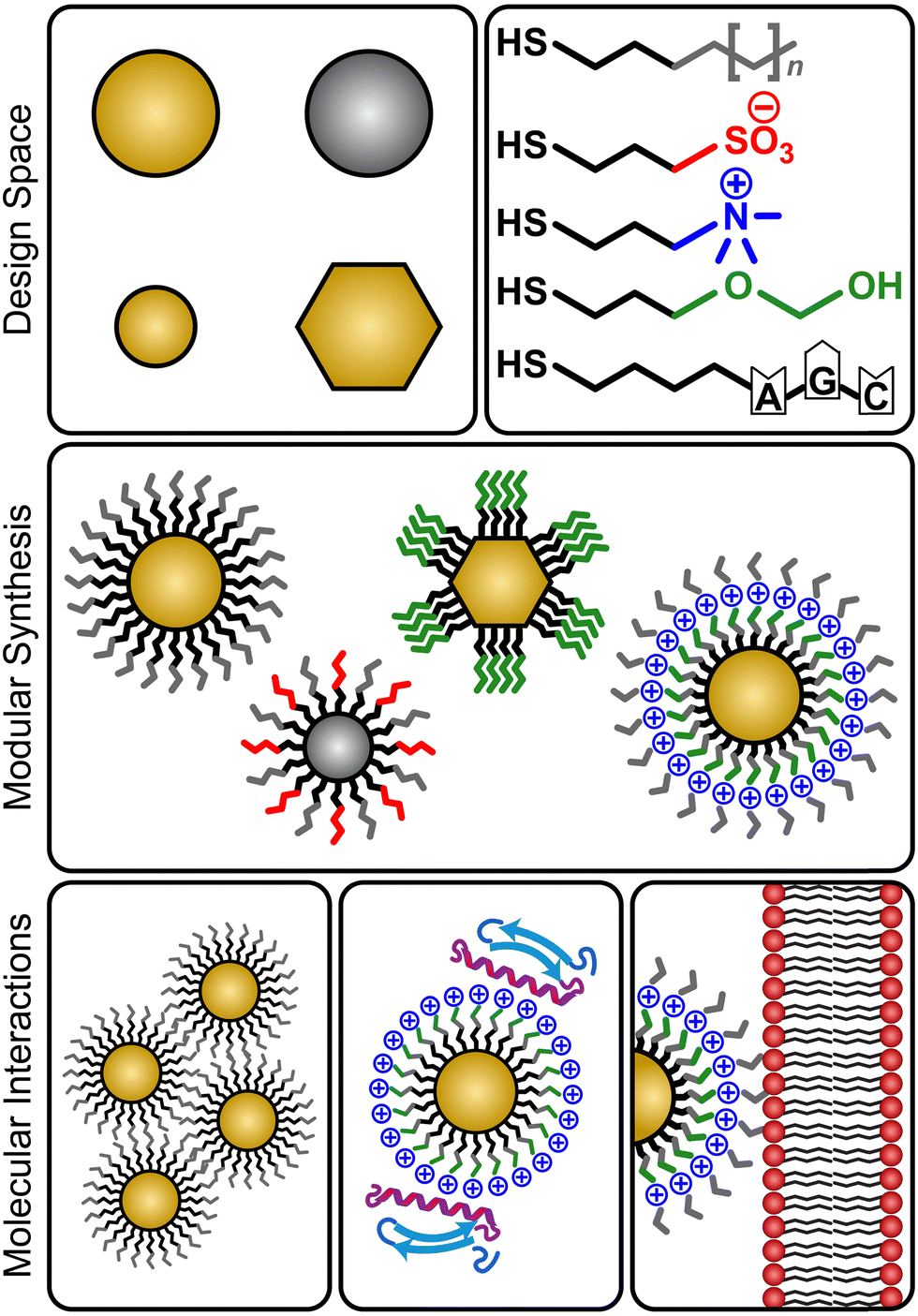 | ||
| Fig. 1 Schematic of design space for monolayer-protected nanomaterials. A wide variety of ligand structures can be grafted onto core materials of varying size, shape, and composition, which permits the tuning of NP surface properties and biointeractions. | ||
Anticipating how ligand selection affects biointeractions prior to NP synthesis remains challenging, however, inhibiting the rational design of monolayer-protected NPs. While numerous studies have revealed structure–property relationships that relate biointeractions to substantial variations in NP surface properties (e.g., the difference between net positively or negatively charged NPs),10,11 it is much more difficult to predict the effect of less significant variations in ligand chemistry, such as variations in the length of alkyl groups in alkanethiol ligands. This challenge is exacerbated by the inability of experimentally accessible macroscopic properties to resolve subtle differences in ligand chemical properties that manifest as large changes in NP behavior.22–24 For example, varying the positioning of charged moieties in zwitterionic alkanethiol ligands was found to influence cellular uptake pathways of small gold NPs even though the NPs exhibited similar hydrodynamic radii and zeta-potentials.23 The difficulty in predicting the effect of ligand selection on biointeractions, coupled with the large set of feasible ligand designs that is synthetically accessible, significantly slows the development of biomedically relevant NPs by requiring substantial experimental trial-and-error.
In this Focus Article, we discuss the effect of ligand selection on the physicochemical properties and interactions of NPs in biological environments, with a focus on small (<10 nm in diameter, a size commensurate with proteins, lipid membranes, and related biomolecules) alkanethiol monolayer-protected gold NPs as illustrated in Fig. 1. While NP properties are often anticipated based on ligand chemical structures alone, surface properties at the nanoscale emerge from a complex interplay of ligand–ligand and ligand–solvent interactions, which themselves can vary both with ligand chemistry and physical factors such as NP core size.25–27 These behaviors can lead to spatially varying properties at the nanoscale, even for NPs with uniform ligand coatings. We further discuss how molecular-scale simulations permit analysis of the interactions and fluctuations that dictate the local chemical environment at the nanoscale and can provide insight into unexpected ligand effects. Finally, we end by discussing opportunities to utilize molecular-scale descriptors of NP and ligand properties with machine-learning algorithms to guide the computational design of new NPs.
Experimental studies of ligand effects on biointeractions of gold nanoparticles
Gold NPs have been well-studied for biological applications in part because ligands with thiol groups can be self-assembled at high density onto the gold core to create a stable self-assembled monolayer (SAM). Ligands for gold NPs often have a modular alkanethiol structure consisting of a sulfur group that anchors to the gold surface, a nonpolar alkyl backbone of variable length, and an end group that can present a variety of possible functional groups to solvent.9–11 Independently tuning the length of the alkyl backbone and the number and type of functional groups present in the end group permits access to a large design space of possible ligands. Consequently, dozens of different ligands have been reported in the literature that encompass variations in alkyl chain length, saturation, and architecture, as well as end group charge, bulkiness, polarity, and related properties.28–34 Mixed SAMs comprising two (or more) ligand species have also been explored to further expand the potential design space.22,35,36The control over ligand chemistry, as well as control over gold core properties, have led to studies of gold NP libraries as useful models for ligand effects on NP behavior. For example, systematic variations in the properties of ligand end groups were used to explore the impact of NP surface properties and core size on the cellular uptake of small gold NPs, revealing distinct cellular uptake efficiencies and mechanisms depending upon the end group charge (cationic, anionic, or zwitterionic) as well as core size.23 Variations in the relative ratio of ligands functionalized with nonpolar or anionic end groups present in mixed SAMs, thereby impacting apparent NP hydrophobicity, were also shown to affect NP nonendocytic uptake, again depending upon the core size.22,37 Similar systematic variations in ligand structure were employed to study protein adsorption on planar mixed SAMs (representative of large NPs, with diameters >100 nm) to highlight the importance of hydrogen bond donors and acceptors on protein-resistant surfaces.38 Numerous other studies have assessed the effect of various ligand end groups on biointeractions as summarized in recent reviews.10,11,39
These prior studies highlight qualitative insights into structure–property relationships that can be obtained from experimental studies. Some studies have sought more quantitative relationships between NP biointeractions and computed descriptors of ligand chemical properties. For example, Rotello and coworkers pioneered a modular ligand design featuring a cationic quaternary ammonium end group which can present up to three additional functional groups.29,31,40 By varying these groups, gold NPs were fabricated with identical core sizes and functionalized with ligands with varying end group octanol–water partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P). LogP is a measure of a functional group's affinity for octanol compared to water and serves as a metric of ligand hydrophobicity. Variations in logP, and thus apparent NP hydrophobicity, were found to correlate with cellular uptake,23,28,29,41 antimicrobial activity,42,43 and protein adsorption,30,44 indicating the potential for simple descriptors of ligand chemical properties to predict complex biointeractions. However, ligands with similar end group logP values can still demonstrate different and unpredictable behaviors, such as distinct hemolytic activity (Fig. 2).30 Moreover, descriptors of ligand chemical structure may not capture properties that emerge due to interactions between or with ligands and are influenced by the NP core size; even nanometer variations in core size can impact NP biointeractions.37 Alternative approaches have sought to parameterize quantitative structure–activity relationship (QSAR) models utilizing multiple descriptors of NP properties, rather than descriptors of individual ligands.45–47 However, such approaches often rely upon experimentally measured macroscopic properties, such as the zeta potential, which limits predictions prior to synthesis. QSAR approaches may also lack physical interpretability.
P). LogP is a measure of a functional group's affinity for octanol compared to water and serves as a metric of ligand hydrophobicity. Variations in logP, and thus apparent NP hydrophobicity, were found to correlate with cellular uptake,23,28,29,41 antimicrobial activity,42,43 and protein adsorption,30,44 indicating the potential for simple descriptors of ligand chemical properties to predict complex biointeractions. However, ligands with similar end group logP values can still demonstrate different and unpredictable behaviors, such as distinct hemolytic activity (Fig. 2).30 Moreover, descriptors of ligand chemical structure may not capture properties that emerge due to interactions between or with ligands and are influenced by the NP core size; even nanometer variations in core size can impact NP biointeractions.37 Alternative approaches have sought to parameterize quantitative structure–activity relationship (QSAR) models utilizing multiple descriptors of NP properties, rather than descriptors of individual ligands.45–47 However, such approaches often rely upon experimentally measured macroscopic properties, such as the zeta potential, which limits predictions prior to synthesis. QSAR approaches may also lack physical interpretability.
 | ||
| Fig. 2 Ligand structure pioneered by Rotello and coworkers. Tuning the logP values of the R group was shown to correlate with the propensity for NPs to lyse red blood cells (quantified by % hemolysis). However, the correlation is imperfect, with dashed red circles showing ligand pairs with similar logP but different % hemolysis, indicating the challenge in utilizing single-ligand descriptors to predict biointeractions. Adapted from ref. 30. | ||
Experimental characterization of NP monolayer composition and structure
Beyond macroscopic characterization of NP properties, experimental techniques can characterize the composition and structural organization of ligands grafted to NPs. Typically, a series of spectroscopic and microscopic techniques are employed to characterize the physicochemical properties of synthesized NPs. To determine the composition of the ligand monolayer, Raman spectroscopy,48 nuclear magnetic resonance,49 matrix-assisted laser desorption/ionization mass spectrometry50 have been effective in characterizing mixed monolayers of alkanethiolates and aromatic thiols48 on gold NPs. Combining matrix-assisted laser desorption/ionization mass spectrometry with theoretical calculations have also enabled quantitative measurements of nanoscale phase separation for a variety of mixed monolayers (octanethiol, tiopronin, glutathione, 11-mercaptoundecanoic acid, and mercaptoundecyltetraethylene glycol).51 The binding mechanisms of ligands on inorganic surfaces can also be determined using experimental techniques that capture vibrational modes. For example, surface-enhanced Raman spectroscopy has shown that m-terphenyl isocyanide ligands bind differently to gold and silver nanospheres.52 In another study, the use of both Fourier transform infrared and X-ray photoelectron spectroscopy found that oleic acid binds covalently to cobalt NPs as a carboxylate with both oxygens strongly coordinated to the metal surface in a symmetric configuration.53Experimentally studying ligand fluctuations and monolayer structure is more challenging. For example, studies of ligand dynamics have used neutron scattering to observe ligand precession and rotation54,55 and solid-state nuclear magnetic resonance to quantify the dynamic volume of ligand chain segments.56 To visualize functionalized surfaces, microscopic techniques such as atomic force microscopy (AFM) and transmission electron microscopy (TEM) can be employed with some caveats. AFM can achieve atomic-level resolution to study electronic states, allowing one to even visualize resonant structures, but requires harsh operating conditions (ultra-high vacuum and near zero Kelvin temperatures) to achieve this resolution.57 Such operating conditions have been shown to be unsuitable for studying ligand dynamics at the molecular scale, as hydration plays a major role. For example, Bals and Liz-Marzán used aberration-corrected high-resolution TEM to capture different dynamics of cetyltrimethylammonium bromide and thiol-terminated polyethylene glycol ligands on the surface of gold nanorods at different degrees of hydration.58 By characterizing the contour of the ligand shell (by thickness), they demonstrated that hydration influences the organization of the ligands and the formation of micellar structures, and found their conclusions to be in agreement with molecular dynamics simulations reported elsewhere.59–61 Thus, while experiments have demonstrated the impact of ligand selection on different biointeractions, relating these interactions to the structure and surface properties of NPs is hindered by challenges associated with the limits of experimental measurements and their interpretability. These considerations motivate the development and application of computational methods to understand and predict how ligand selection influences NP surface structure, properties, and resulting biointeractions.
Simulations reveal NP surface properties and biointeractions at the molecular level
A common physics-based computational method to model biomolecular systems is molecular dynamics (MD) simulation (Fig. 3). An MD simulation models the time evolution of a system of particles according to Newton's equations of motion. Particles move based on forces computed at discrete timesteps to create a trajectory consisting of a sequence of system configurations (i.e., sets of particle positions and velocities). Forces are obtained from a potential energy function that includes bonded terms to model intramolecular bonds, angles, and dihedral angles and nonbonded terms to model intra- and intermolecular van der Waals and electrostatic interactions. The set of functional forms and parameters for a particular potential energy function is called the force field. Force field parameters are typically derived to reproduce more detailed quantum chemical calculations, although in classical MD simulations electron degrees of freedom are not explicitly modeled so that covalent bonds cannot be formed or broken during a simulation. | ||
| Fig. 3 Example all-atom and coarse-grained molecular dynamics simulations of monolayer-protected nanomaterials. All-atom simulations can access length scales suitable for modelling individual nanomaterials interacting in relatively simple biological environments, such as modelling the adsorption of a charged nanoparticle to a single-component lipid bilayer (simulation snapshot adapted from ref. 72). Coarse-grained simulations enable access to larger nanomaterial sizes and more complex system compositions, such as multicomponent lipid bilayers, albeit with reduced accuracy (simulation snapshot adapted from ref. 66). | ||
Biomolecular systems are often modeled using all-atom simulations in which all of the atoms in a set of biomolecules, solvent molecules, and ions are modeled as individual particles and assigned force field parameters which define their chemical identities and interactions. All-atom MD simulations can reach system sizes comprising up to millions of atoms over timescales of microseconds using timesteps on the order of a few femtoseconds, permitting a detailed molecular-scale view of the motion and interactions of biomolecules as well as analysis of behavior using tools from statistical mechanics. Advances in all-atom MD simulations of NPs, such as the development of force fields62,63 for gold or other metals and tools for modeling ligand grafting,64,65 permit simulations of NPs at realistic sizes (≥10 nm core diameter) with chemically explicit representations of ligands, solvent molecules, ions, and biomolecules. Alternative simulation methods can also be used to overcome computational limitations on the maximize system size and timescale accessible to all-atom MD simulations. For example, coarse-grained MD simulations treat groups of atoms as particles with force field parameters selected to model the effective interactions between these groups (Fig. 3). Reducing the number of atoms by grouping them together permits access to larger system sizes and timescales, which allows for more complex representations of biological systems (e.g., multicomponent lipid bilayers66–69) or more rapid screening of NP libraries.70,71
All-atom and coarse-grained MD simulations of NPs can complement experiments by revealing ligand-mediated biointeractions at the nanoscale. For example, all-atom simulations were used to model the adsorption of a library of small (2 nm core diameter) gold NPs with ligands of varying logP to single-component lipid bilayers. The simulations found that increasing ligand logP reduces kinetic barriers for the adsorption of NPs at lipid interfaces by promoting favorable ligand-lipid contact.72 This example shows how MD simulations can provide physical insight to interpret results from experimental screening of a NP library, thereby complementing the aforementioned QSAR methods. Building on these results, coarse-grained simulations were used to assess the impact of a wider variety of ligand structures – including variation in logP and variation in the degree of branching of ligand end groups at fixed logP – on the thermodynamics of lipid bilayer adsorption.73,74 These simulations revealed unexpected behaviors: for ligands with linear architectures, adsorption free energies were nonmonotonically related to logP with adsorption being most favorable for intermediate logP values; for ligands with similar end group logP values, adsorption was found to be more favorable for ligands with branched, rather than linear, architectures. These behaviors were related to the nanoscale organization of ligands at the NP surface, as further detailed below.
These examples highlight the ability of MD to provide detailed insight into how ligand selection can affect NP biointeractions, and in particular how MD simulations can reveal unexpected effects that would not be anticipated based on ligand chemical structure or single-ligand descriptors alone. Here, we do not seek to exhaustively review all applications of MD simulation to the study of NPs and direct the reader to recent reviews in this area.75–77 The following two sections will instead focus on how MD simulations can reveal spatially inhomogeneous structures and chemical properties at a NP surface that emerge from the interplay of ligand fluctuations, core size, and interactions between ligands and with the surrounding solvent, again using gold NPs as model systems.
Molecular simulations predict spatially inhomogeneous ligand structures and properties
Conventional schematic representations of monolayer-protected NPs commonly depict ligands as grafted to a NP core with ligands pointed radially outward and end groups presented to the solvent (as in Fig. 3 and labeled as ligand protrusions in Fig. 4). Such representations suggest that NP surfaces should be spherically symmetric with spatially homogeneous properties (assuming a single ligand component), and that ligand end groups should be available to interact with molecules in the surrounding solution. These considerations may drive ligand design by motivating the selection of specific functional groups to interact with species in solution. However, ligands can fluctuate due to atomic motions and rotations around bonds, leading to monolayer structures that reflect contributions from intramolecular energies associated with ligand conformations, intermolecular interactions between ligands within the monolayer, and the entropy gain associated with sampling multiple distinct ligand conformations. In particular, exposure of a ligand monolayer to an aqueous solution will lead to dynamic reorganization of ligand structures due to these fluctuations and due to interactions with the surrounding solvent environment.25,78–83 Such fluctuations are particularly pronounced for small NPs, where the curvature of the core leads to substantial free volume in the ligand monolayer through which ligands can move, and which is potentially accessible to solvent. Consequently, the structures obtained by individual ligands within a solvated ligand monolayer may substantially differ from schematic depictions.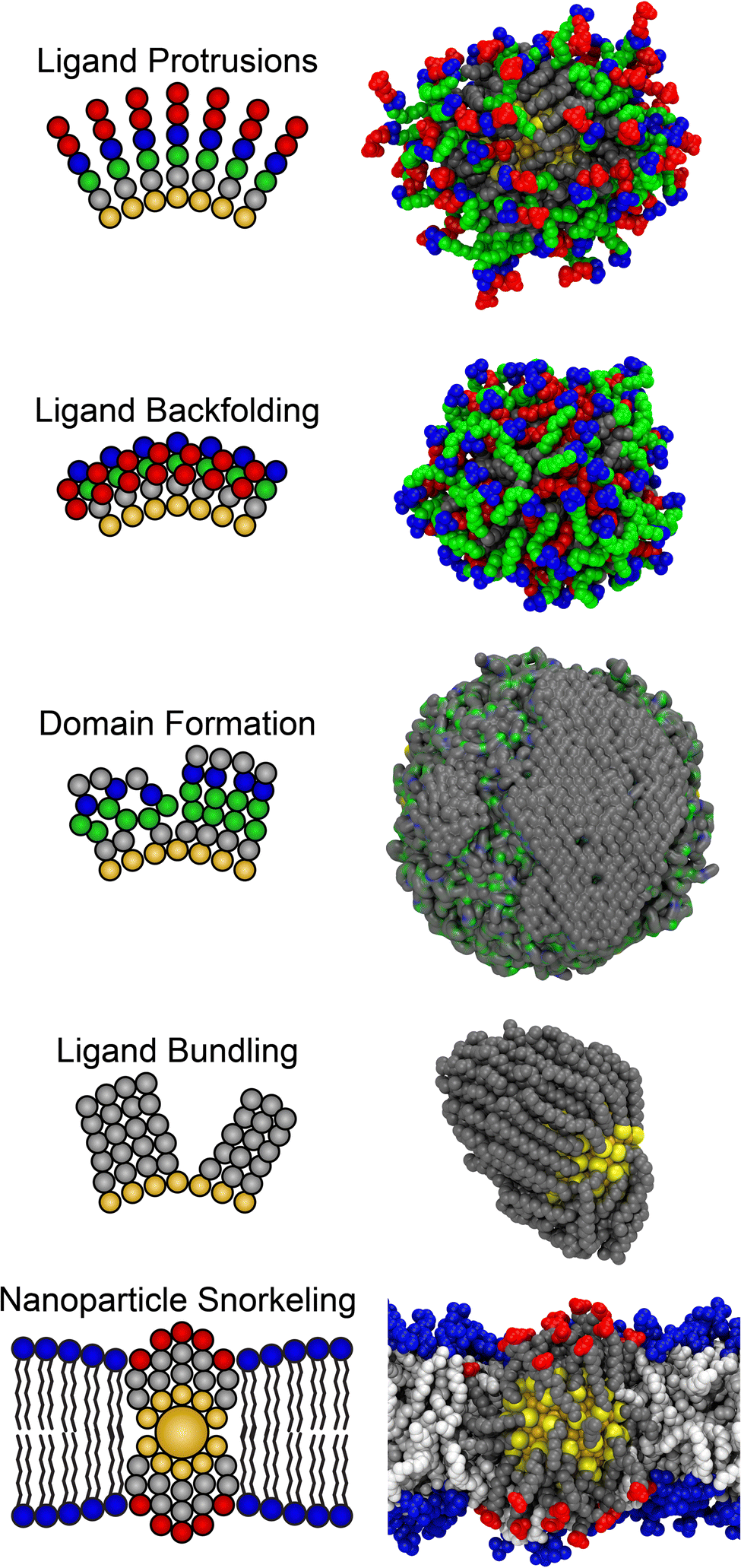 | ||
| Fig. 4 Example unusual ligand-mediated heterogeneous structures that emerge in water or in the presence of other biomolecules (e.g., lipids). The snapshot for domain formation was adapted from ref. 81. The snapshot for nanoparticle snorkeling was adapted from ref. 113. | ||
MD simulations have revealed several behaviors that emerge from ligand fluctuations and lead to changes in monolayer structure and apparent NP surface properties (Fig. 4). One such behavior is the packing of ligands against each other to maximize favorable interactions (typically van der Waals or hydrophobic interactions).25,26,78,82 Such packing leads to “bundles” of ligands that introduce anisotropic structures into even compositionally uniform NPs. Bundling is inherently a property that emerges from the interactions of multiple ligands and hence is difficult to predict; moreover, it depends upon backbone characteristics such as the degree of saturation or branching which may not be expected to directly affect NP surface properties. Similar anisotropic structures can be promoted by alternative ligand interactions, such as the formation of “ridges” of charged end groups due to electrostatic interactions.83
These structures can likewise introduce spatial variations in chemical properties by exposing ligand backbones, not just end groups, to solvent molecules. For example, ligands end-functionalized with polar groups that may be expected to produce uniformly polar NP coatings can instead lead to amphiphilic surface properties due to the exposure of nonpolar ligand backbones after bundle formation.25,78,79 Spatial variations in NP surface properties can also be introduced by the formation of ligand domains with distinct chemical properties, either due to the segregation of different ligand components in multicomponent multilayers (due to diffusion on the surface or conformational changes of specific components) or due to the interactions of different segments of complex ligand architectures.81 Similar spatially heterogeneous variations in surface properties on short (<1 nm) length scales can arise from stochastic ligand fluctuations, leading to distinct local chemical environments across the surface of a compositionally uniform NP which can have a substantial impact on interactions with biomolecules.
Ligand fluctuations can also drive behaviors in which functional groups are sequestered within the ligand monolayer to avoid exposure to solvent, even if a ligand's chemical structure suggests that they are solvent exposed. We refer to such a process as “backfolding”.72,73 For example, nonpolar terminal groups of functionalized ligands may backfold to maintain contact with nonpolar backbone groups of ligands rather than protruding into aqueous solution. Similar behavior can also be observed in mixed ligand monolayers; a long-chain nonpolar ligand may backfold to avoid exposure to water while permitting shorter-chain polar ligands to protrude into aqueous solution.79 Backfolding can thus influence NP surface properties by preventing end groups from being presented to the solvent environment, which has been shown to influence NP binding to lipid bilayers and proteins.72–74,84 This example also points to the challenge in designing ligand end groups: systematically varying logP, as in the example presented above, may not lead to commensurate changes in surface hydrophobicity if end groups backfold into the ligand monolayer to avoid surface exposure.
Finally, we note that the above discussion focuses on ligand fluctuations that affect the structure and surface properties of isolated NPs in solution. Perturbations to the structure of a ligand monolayer can also arise in response to interactions with other species in the environment. For example, NPs with charged ligand end groups can stably embed within lipid bilayers if the ligands deform to move charged groups into aqueous solution, rather than into contact with hydrophobic lipid tail groups, in a process referred to as “snorkeling”.85–87 Snorkeling thus produces anisotropic structures that arise dynamically due to a biointeraction and can lead to unintuitive behavior (i.e., the stable insertion of a charged object into the hydrophobic core of the bilayer).
NP surface properties emerge from complex interplay of ligand interactions
Another challenge with ligand selection is that critical NP surface properties depend on the local chemical context at the nanoscale, and thus emerge from the interplay of ligand–ligand and ligand–solvent interactions that is difficult to relate to single-ligand properties. Here, we focus on two such properties of a surface: hydrophobicity and electrostatic potential. Both properties are often estimated based on single-ligand descriptors. For example, hydrophobicity is typically estimated based on single-ligand descriptors like logP, or sometimes based on additive hydropathy scales adapted from calculations of protein hydrophobicity.88,89 The electrostatic potential can be computed using continuum methods that assume a surface charge density, which can itself be estimated based on the system pH and pKa of individual ligand functional groups. However, both of these properties will vary depending on the local chemical environment in ways that are difficult to predict but can be assessed using MD simulations (Fig. 5).25,27,90
 | ||
| Fig. 5 Example spatially inhomogeneous surface properties for NPs with single-component ligand monolayers. (A) Variations in interfacial hydrophobicity, quantified by the hydration free energy, μL, for a gold NP (GNP) with a uniformly nonpolar ligand monolayer. Adapted from ref. 25. (B) Variations in the electrostatic potential as indicated by the Voronoi tessellation (at right) due to the spatial distribution of ions and changes in protonation state of charged ligands. Reprinted (adapted) with permission from ref. 27. Copyright 2020, American Chemical Society. | ||
Hydrophobicity measures the thermodynamic affinity of water molecules for an interface.91 Materials can experience attractive, water-mediated hydrophobic interactions whose magnitude will depend upon their effective hydrophobicity; such interactions are critical in driving biointeractions with hydrophobic binding interfaces on proteins or with the hydrophobic core of the lipid bilayer. NP hydrophobicity can be difficult to predict in part because of ligand fluctuations that vary the exposure of nonpolar functional groups to water. However, hydrophobicity is also difficult to predict because it fundamentally emerges from the interfacial structure of water molecules, which reflects the spatial organization of water molecules near an interface due to their propensity for hydrogen bonding.
Nonpolar interfaces are generally considered to be hydrophobic because nonpolar groups cannot hydrogen bond with nearby water molecules, leading to thermodynamic penalties associated with hydration.92 Water structure can be also perturbed by local variations in chemical context that can substantially impact interfacial hydrophobicity. For example, introducing polar or charged functional groups near nonpolar regions of an interface can either increase or decrease the hydrophobicity of that nonpolar region depending on the chemical identity of the polar group.93 Similarly, varying the nanoscale order of ligands by changing backbone length or saturation (without varying exposure to water) or the local curvature of a uniformly nonpolar interface can likewise impact hydrophobic interactions.94 Hydrophobic interactions are also a strong function of temperature due to the effect of thermal motion on water structure. MD simulations can provide insight into these effects by quantifying thermodynamically well-defined descriptors of hydrophobicity, such as local water density fluctuations, to provide insight into how local chemical context influences hydrophobic interactions.94,95
The surface charge density, which influences the electrostatic potential, is also heavily influenced by the local chemical environment, particularly at highly charged interfaces. For example, charged NPs can have their surface charge density modulated by interactions with oppositely charged counterions, with the counterion density depending upon their size and valency.96,97 Highly charged NPs can introduce substantial counterion binding to largely neutralize the surface charge density in a process called counterion condensation.97 Spatial variations in counterion binding, along with the potential formation of anisotropic structures as noted in the preceding section, can introduce spatially varying electrostatic properties that would not be anticipated from single-ligand descriptors.27 Electrostatic interactions between charged ligands can also influence monolayer structure as noted above.27 Another challenge is predicting the ionization of weak acidic or basic groups, which will vary with both the system pH and variations in the local thermodynamic activity of protons near the NP–water interface.98 These factors will also all depend upon the electrolyte concentration in solution which will dictate the screening of electrostatic interactions.
MD simulations can explicitly model ligand–ion, ligand–solvent, ion–ion, and ion–solvent interactions to capture these complex electrostatic effects. While continuum electrostatics calculations, such as those based on Poisson–Boltzmann calculations, omit effects due to explicit ions, they can be utilized in conjunction with surface charge densities obtained from simulations to predict spatial variations in the electrostatic potential. Advanced simulation methods that permit alterations in the charge of functional groups based on the system pH (e.g., constant-pH simulations) have also been used to explore spatial variations in surface charge density.27,99,100 Finally, such simulations also account for changes in the local dielectric environment associated with perturbations to water structure (which, as noted above, also will impact hydrophobic interactions).93–95
Outlook: integration of simulation, experiment, and data science for predictive ligand design
The prior discussion highlights the power of MD simulation to reveal NP properties at the nanoscale. MD simulations still face substantial challenges due to their computational cost, however, which limits the compositional complexity of modeled systems. For example, most MD simulations of NP biointeractions utilize simplified representations of biomolecular systems, such as treating lipid bilayers with only a small number of lipid components and omitting proteins entirely. This limitation inhibits accurate modeling of NP–biosystems interactions with realistic chemical compositions. Nonetheless, the capability of MD simulations to compute quantitative descriptors of NP surface properties that account for ligand fluctuations and local chemical context suggests the potential for MD simulations to be integrated with machine learning methods and experimental studies of NP libraries to predict biointeractions a priori.101 Such simulations can also uniquely provide thermodynamic descriptors (e.g., adsorption free energies) of NP–biomolecule interactions as possible input for machine learning models.As steps in this direction, recent studies have generated virtual libraries of NPs then computed descriptors from static 3D structures of these NPs, including a ligand coating, as input to a QSAR model.102 Building upon these results, high-throughput MD simulations of a similar set of NPs were shown to produce descriptors of ligand fluctuations, ligand–ligand interactions, and ligand–solvent interactions that improved QSAR model accuracy when predicting cellular uptake (Fig. 6).101 Methods combining MD simulations of a small number of NPs with deep learning models (e.g., graph neural networks) have also recently been developed to predict NP–protein binding.103,104 We further envision opportunities to closely integrate experiment and computation through the development of feedback loops that use machine learning models to guide the selection of experiments at each iteration. Similar approaches have successfully been used in the design of related materials with targeted biointeractions, such as polymeric NPs,105 protein-binding polymers,106 and antifungal peptides.107
 | ||
| Fig. 6 Data-driven prediction of NP biointeractions using MD-derived descriptors. (A) Predicting NP–biosystem interactions from simulations of monolayer-protected gold NPs (AuNPs) in solution. MD simulations were used to compute descriptors that were input to regression models that show good linear correlation (values indicate Pearson's r) with experimental measurements. (B) Example AuNPs from 168-member library. Adapted from ref. 101. | ||
Conclusions
This Focus article has sought to highlight the difficulty in rationally designing monolayer-protected nanoparticles with desired biointeractions because of challenges with anticipating how ligand chemistry influences spatially varying surface properties at the nanoscale. Such properties emerge from the cooperative influence of ligand chemistry and NP size which makes it difficult to develop predictive structure–property models based on ligand chemistry alone. However, molecular simulations can both model these effects and quantify chemical properties, such as hydrophobicity and electrostatic potential, that depend upon the local chemical environment (including solvent and ions) and dictate critical interactions in biological environments. Such simulations can also provide molecular-scale descriptors of NP–biomolecule interactions, as opposed to descriptors of single-ligand properties or experimentally determined NP properties. Molecular simulations could thus provide valuable input, particularly when combined with machine learning models and analysis of experimental NP libraries, to guide ligand selection and, more broadly, predictive NP design.To further connect experimental characterization of ligand properties to simulation results, it is of great importance to continue developing ultra-high-resolution imaging and analytical techniques that can more clearly show interactions between individual molecules and can be employed under normal conditions (i.e., standard temperature and pressure, and liquid environments). Increasing the utility and accuracy of molecular simulation methods requires new coarse-graining methods that can reach longer time and length scales necessary to model NP–biomolecule interactions while still accurately capturing heterogeneous NP surface properties.76,108 Similarly, new enhanced sampling methods are needed to permit the calculation of complex free energy landscapes associated with nano-bio interactions,109,110 as well as methods to correctly capture the effect of pH on NP surface properties in biological environments.111,112 Technological advancements on these fronts would pave the way to fully bridge gaps between experiments and theory while fully synergizing both research areas to enable predictive models for future discovery and development of nanomaterials aimed at biomedical applications.
We note that while most of the examples in this Focus article involved gold NPs as model systems, the general observations and outlook for predictive methods should be equally appliable to other classes of NPs. For example, the importance of ligand fluctuations in dictating local changes to NP surface properties will be equally applicable to NPs with soft surface or surface coatings (e.g., polymerosomes or polymer-coated NPs) or lipid-based NPs with multiple lipids capable of diffusing on the surface (e.g., lipid NPs, vesicles). We thus expect the development of new simulation-informed methods for NP design to have a broad impact on engineering the nano-bio interface for diverse applications of biomedical relevance.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under grant No. DMR-2044997. C. A. H.-Z. and R. C. V. L. acknowledge the support provided by the Graduate Engineering Research Scholars–Advanced Opportunity Fellowship from the University of Wisconsin–Madison and the National Science Foundation through a Graduate Research Fellowship. C. A. H.-Z was supported in part by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number T32GM152341 (Chemistry–Biology Interface Training Program). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Gold Nanoparticles: Interesting Optical Properties and Recent Applications in Cancer Diagnostics and Therapy, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Engineered Nanoparticles for Drug Delivery in Cancer Therapy, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CrossRef CAS PubMed.
- D. P. O’Neal, L. R. Hirsch, N. J. Halas, J. D. Payne and J. L. West, Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles, Cancer Lett., 2004, 209, 171–176 CrossRef PubMed.
- R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Surface functionalization of nanoparticles for nanomedicine, Chem. Soc. Rev., 2012, 41, 2539–2544 RSC.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Gold Nanoparticles for Biology and Medicine, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Effect of Surface Properties on Nanoparticle–Cell Interactions, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- M. I. Anik, N. Mahmud, A. Al Masud and M. Hasan, Gold nanoparticles (GNPs) in biomedical and clinical applications: A review, Nano Sel., 2022, 3, 792–828 CrossRef CAS.
- M. F. Matus and H. Häkkinen, Understanding ligand-protected noble metal nanoclusters at work, Nat. Rev. Mater., 2023, 8, 372–389 CrossRef CAS.
- K. Bhattacharjee and B. L. V. Prasad, Surface functionalization of inorganic nanoparticles with ligands: a necessary step for their utility, Chem. Soc. Rev., 2023, 52, 2573–2595 RSC.
- M. D. Manning, A. L. Kwansa, T. Oweida, J. S. Peerless, A. Singh and Y. G. Yingling, Progress in ligand design for monolayer-protected nanoparticles for nanobio interfaces, Biointerphases, 2018, 13, 06D502 CrossRef CAS PubMed.
- A. Gupta, W. Ndugire, C.-M. Hirschbiegel, L. Grigely and V. M. Rotello, Interfacing Nanomaterials with Biology through Ligand Engineering, Acc. Chem. Res., 2023, 56, 2151–2169 CrossRef CAS PubMed.
- Y. Huang, T. A. Cohen, B. M. Sperry, H. Larson, H. A. Nguyen, M. K. Homer, F. Y. Dou, L. M. Jacoby, B. M. Cossairt, D. R. Gamelin and C. K. Luscombe, Organic building blocks at inorganic nanomaterial interfaces, Mater. Horiz., 2022, 9, 61–87 RSC.
- A. M. Alkilany, S. E. Lohse and C. J. Murphy, The Gold Standard: Gold Nanoparticle Libraries To Understand the Nano–Bio Interface, Acc. Chem. Res., 2013, 46, 650–661 CrossRef CAS PubMed.
- M. Abdelhameed, D. R. Martir, S. Chen, W. Z. Xu, O. O. Oyeneye, S. Chakrabarti, E. Zysman-Colman and P. A. Charpentier, Tuning the Optical Properties of Silicon Quantum Dots via Surface Functionalization with Conjugated Aromatic Fluorophores, Sci. Rep., 2018, 8, 3050 CrossRef PubMed.
- R. Li, C. Mbonu and P. Akcora, Structure-Dependent Ionic Conductivity in Poly(Ionic Liquid)-b-Poly(methyl methacrylate)-Grafted Nanoparticles, ACS Appl. Polym. Mater., 2025, 7, 3853–3862 CrossRef CAS.
- S. Patel, D. Jung, P. T. Yin, P. Carlton, M. Yamamoto, T. Bando, H. Sugiyama and K.-B. Lee, NanoScript: A Nanoparticle-Based Artificial Transcription Factor for Effective Gene Regulation, ACS Nano, 2014, 8, 8959–8967 CrossRef CAS PubMed.
- Y. Liu, N. Peng, Y. Yao, X. Zhang, X. Peng, L. Zhao, J. Wang, L. Peng, Z. Wang, K. Mochizuki, M. Yue and S. Yang, Breaking the nanoparticle's dispersible limit via rotatable surface ligands, Nat. Commun., 2022, 13, 3581 CrossRef CAS PubMed.
- X. Jin, N. Peng, A. Cui, Y. Liu, X. Peng, L. Huang, A. Ed-Dra, F. He, Y. Li, S. Yang and M. Yue, Sodium dodecyl sulfate-coated silver nanoparticles accelerate antimicrobial potentials by targeting amphiphilic membranes, mLife, 2024, 3, 551–564 CrossRef CAS PubMed.
- C. L. Bassani, G. van Anders, U. Banin, D. Baranov, Q. Chen, M. Dijkstra, M. S. Dimitriyev, E. Efrati, J. Faraudo, O. Gang, N. Gaston, R. Golestanian, G. I. Guerrero-Garcia, M. Gruenwald, A. Haji-Akbari, M. Ibáñez, M. Karg, T. Kraus, B. Lee, R. C. Van Lehn, R. J. Macfarlane, B. M. Mognetti, A. Nikoubashman, S. Osat, O. V. Prezhdo, G. M. Rotskoff, L. Saiz, A.-C. Shi, S. Skrabalak, I. I. Smalyukh, M. Tagliazucchi, D. V. Talapin, A. V. Tkachenko, S. Tretiak, D. Vaknin, A. Widmer-Cooper, G. C. L. Wong, X. Ye, S. Zhou, E. Rabani, M. Engel and A. Travesset, Nanocrystal Assemblies: Current Advances and Open Problems, ACS Nano, 2024, 18, 14791–14840 CrossRef CAS PubMed.
- C.-C. You, M. De and V. M. Rotello, Monolayer-protected nanoparticle–protein interactions, Curr. Opin. Chem. Biol., 2005, 9, 639–646 CrossRef CAS PubMed.
- E. Rascol, J.-M. Devoisselle and J. Chopineau, The relevance of membrane models to understand nanoparticles–cell membrane interactions, Nanoscale, 2016, 8, 4780–4798 RSC.
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H.-S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles, Nat. Mater., 2008, 7, 588–595 CrossRef CAS PubMed.
- Y. Jiang, S. Huo, T. Mizuhara, R. Das, Y.-W. Lee, S. Hou, D. F. Moyano, B. Duncan, X.-J. Liang and V. M. Rotello, The Interplay of Size and Surface Functionality on the Cellular Uptake of Sub-10 nm Gold Nanoparticles, ACS Nano, 2015, 9, 9986–9993 CrossRef CAS PubMed.
- Y. Zhang, N. V. Hudson-Smith, S. D. Frand, M. S. Cahill, L. S. Davis, Z. V. Feng, C. L. Haynes and R. J. Hamers, Influence of the Spatial Distribution of Cationic Functional Groups at Nanoparticle Surfaces on Bacterial Viability and Membrane Interactions, J. Am. Chem. Soc., 2020, 142, 10814–10823 CrossRef CAS PubMed.
- A. K. Chew, B. C. Dallin and R. C. Van Lehn, The Interplay of Ligand Properties and Core Size Dictates the Hydrophobicity of Monolayer-Protected Gold Nanoparticles, ACS Nano, 2021, 15, 4534–4545 CrossRef CAS PubMed.
- T. Kister, D. Monego, P. Mulvaney, A. Widmer-Cooper and T. Kraus, Colloidal Stability of Apolar Nanoparticles: The Role of Particle Size and Ligand Shell Structure, ACS Nano, 2018, 12, 5969–5977 CrossRef CAS PubMed.
- D. Liang, U. Dahal, M. Wu, C. J. Murphy and Q. Cui, Ligand Length and Surface Curvature Modulate Nanoparticle Surface Heterogeneity and Electrostatics, J. Phys. Chem. C, 2020, 124, 24513–24525 CrossRef CAS.
- Z.-J. Zhu, T. Posati, D. F. Moyano, R. Tang, B. Yan, R. W. Vachet and V. M. Rotello, The Interplay of Monolayer Structure and Serum Protein Interactions on the Cellular Uptake of Gold Nanoparticles, Small, 2012, 8, 2659–2663 CrossRef CAS PubMed.
- D. F. Moyano, K. Saha, G. Prakash, B. Yan, H. Kong, M. Yazdani and V. M. Rotello, Fabrication of Corona-Free Nanoparticles with Tunable Hydrophobicity, ACS Nano, 2014, 8, 6748–6755 CrossRef CAS PubMed.
- K. Saha, D. F. Moyano and V. M. Rotello, Protein coronas suppress the hemolytic activity of hydrophilic and hydrophobic nanoparticles, Mater. Horiz., 2013, 1, 102–105 RSC.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, Regulation of Macrophage Recognition through the Interplay of Nanoparticle Surface Functionality and Protein Corona, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- Y.-Q. Liu, Y.-C. Chao, S.-Q. Xu, Y.-R. Peng, J.-J. Syu, X.-H. Yang, Y.-K. Pan, P.-C. Lin, L.-L. Weng, I.-C. Chen and K.-T. Tan, Surface Functionalization of Gold Nanoparticles Using Alkyne Derivatives: Applications in Chemical Sensing, ACS Appl. Mater. Interfaces, 2024, 16, 58262–58273 CrossRef CAS PubMed.
- Y. Ishida, J. Suzuki, I. Akita and T. Yonezawa, Ultrarapid Cationization of Gold Nanoparticles via a Single-Step Ligand Exchange Reaction, Langmuir, 2018, 34, 10668–10672 CrossRef CAS PubMed.
- M. M. Paz, A. P. Veiga, T. Regueira, C. V. Vázquez and M. Arturo López Quintela, Facile generation of surface diversity in gold nanoparticles, J. Colloid Interface Sci., 2023, 641, 719–728 CrossRef CAS PubMed.
- A. K. Boal and V. M. Rotello, Radial Control of Recognition and Redox Processes with Multivalent Nanoparticle Hosts, J. Am. Chem. Soc., 2002, 124, 5019–5024 CrossRef CAS PubMed.
- O. Zeiri, Metallic-Nanoparticle-Based Sensing: Utilization of Mixed-Ligand Monolayers, ACS Sens., 2020, 5, 3806–3820 CrossRef CAS PubMed.
- R. C. Van Lehn, P. U. Atukorale, R. P. Carney, Y.-S. Yang, F. Stellacci, D. J. Irvine and A. Alexander-Katz, Effect of Particle Diameter and Surface Composition on the Spontaneous Fusion of Monolayer-Protected Gold Nanoparticles with Lipid Bilayers, Nano Lett., 2013, 13, 4060–4067 CrossRef CAS PubMed.
- E. Ostuni, B. A. Grzybowski, M. Mrksich, C. S. Roberts and G. M. Whitesides, Adsorption of Proteins to Hydrophobic Sites on Mixed Self-Assembled Monolayers, Langmuir, 2003, 19, 1861–1872 CrossRef CAS.
- R. Huang, D. C. Luther, X. Zhang, A. Gupta, S. A. Tufts and V. M. Rotello, Engineering the Interface between Inorganic Nanoparticles and Biological Systems through Ligand Design, Nanomaterials, 2021, 11, 1001 Search PubMed.
- K. K. Sandhu, C. M. McIntosh, J. M. Simard, S. W. Smith and V. M. Rotello, Gold Nanoparticle-Mediated Transfection of Mammalian Cells, Bioconjugate Chem., 2002, 13, 3–6 CrossRef CAS PubMed.
- K. Saha, S. T. Kim, B. Yan, O. R. Miranda, F. S. Alfonso, D. Shlosman and V. M. Rotello, Surface Functionality of Nanoparticles Determines Cellular Uptake Mechanisms in Mammalian Cells, Small, 2013, 9, 300–305 Search PubMed.
- X. Li, S. M. Robinson, A. Gupta, K. Saha, Z. Jiang, D. F. Moyano, A. Sahar, M. A. Riley and V. M. Rotello, Functional Gold Nanoparticles as Potent Antimicrobial Agents against Multi-Drug-Resistant Bacteria, ACS Nano, 2014, 8, 10682–10686 CrossRef CAS PubMed.
- A. Gupta, N. M. Saleh, R. Das, R. F. Landis, A. Bigdeli, K. Motamedchaboki, A. Rosa Campos, K. Pomeroy, M. Mahmoudi and V. M. Rotello, Synergistic antimicrobial therapy using nanoparticles and antibiotics for the treatment of multidrug-resistant bacterial infection, Nano Futures, 2017, 1, 015004 CrossRef.
- M. De, S. Rana, H. Akpinar, O. R. Miranda, R. R. Arvizo, U. H. F. Bunz and V. M. Rotello, Sensing of proteins in human serum using conjugates of nanoparticles and green fluorescent protein, Nat. Chem., 2009, 1, 461–465 CrossRef CAS PubMed.
- D. Fourches, D. Pu, C. Tassa, R. Weissleder, S. Y. Shaw, R. J. Mumper and A. Tropsha, Quantitative Nanostructure−Activity Relationship Modeling, ACS Nano, 2010, 4, 5703–5712 CrossRef CAS PubMed.
- W. Wang, A. Sedykh, H. Sun, L. Zhao, D. P. Russo, H. Zhou, B. Yan and H. Zhu, Predicting Nano–Bio Interactions by Integrating Nanoparticle Libraries and Quantitative Nanostructure Activity Relationship Modeling, ACS Nano, 2017, 11, 12641–12649 CrossRef CAS PubMed.
- N. Ouassil, R. L. Pinals, J. T. Del Bonis-O’Donnell, J. W. Wang and M. P. Landry, Supervised learning model predicts protein adsorption to carbon nanotubes, Sci. Adv., 2022, 8, eabm0898 CrossRef CAS PubMed.
- A. Cirri, A. Silakov, L. Jensen and B. J. Lear, Probing ligand-induced modulation of metallic states in small gold nanoparticles using conduction electron spin resonance, Phys. Chem. Chem. Phys., 2016, 18, 25443–25451 RSC.
- X. Liu, M. Yu, H. Kim, M. Mameli and F. Stellacci, Determination of monolayer-protected gold nanoparticle ligand–shell morphology using NMR, Nat. Commun., 2012, 3, 1182 Search PubMed.
- A. Dass, A. Stevenson, G. R. Dubay, J. B. Tracy and R. W. Murray, Nanoparticle MALDI-TOF Mass Spectrometry without Fragmentation: Au25(SCH2CH2Ph)18 and Mixed Monolayer Au25(SCH2CH2Ph)18−x(L)x, J. Am. Chem. Soc., 2008, 130, 5940–5946 CrossRef CAS PubMed.
- K. M. Harkness, A. Balinski, J. A. McLean and D. E. Cliffel, Nanoscale Phase Segregation of Mixed Thiolates on Gold Nanoparticles, Angew. Chem., Int. Ed., 2011, 50, 10554–10559 CrossRef CAS PubMed.
- L. Bi, Y. Wang, Z. Wang, A. Do, A. Fuqua, K. P. Balto, Y. Zhang, J. S. Figueroa, T. A. Pascal, A. R. Tao and S. Li, Molecular-Scale Insights into the Heterogeneous Interactions between an m-Terphenyl Isocyanide Ligand and Noble Metal Nanoparticles, Nano Lett., 2025, 25, 2027–2033 CrossRef CAS PubMed.
- N. Wu, L. Fu, M. Su, M. Aslam, K. C. Wong and V. P. Dravid, Interaction of Fatty Acid Monolayers with Cobalt Nanoparticles, Nano Lett., 2004, 4, 383–386 CrossRef CAS.
- N. Yazdani, M. Jansen, D. Bozyigit, W. M. M. Lin, S. Volk, O. Yarema, M. Yarema, F. Juranyi, S. D. Huber and V. Wood, Nanocrystal superlattices as phonon-engineered solids and acoustic metamaterials, Nat. Commun., 2019, 10, 4236 CrossRef PubMed.
- M. Jansen, F. Juranyi, O. Yarema, T. Seydel and V. Wood, Ligand Dynamics in Nanocrystal Solids Studied with Quasi-Elastic Neutron Scattering, ACS Nano, 2021, 15, 20517–20526 Search PubMed.
- W. Cao, Z. Pang, X. Zhou, Z. Cao, J. Li, Q. Wang, X. Peng and X. Kong, Calibrating ligand-ligand interaction on nanocrystals via the dynamic volume of chain segments, Cell Rep. Phys. Sci., 2023, 4, 101207 CrossRef CAS.
- Z. Ruan, J. Schramm, J. B. Bauer, T. Naumann, L. V. Müller, F. Sättele, H. F. Bettinger, R. Tonner-Zech and J. M. Gottfried, On-Surface Synthesis and Characterization of Pentadecacene and Its Gold Complexes, J. Am. Chem. Soc., 2025, 147, 4862–4870 Search PubMed.
- A. Pedrazo-Tardajos, N. Claes, D. Wang, A. Sánchez-Iglesias, P. Nandi, K. Jenkinson, R. De Meyer, L. M. Liz-Marzán and S. Bals, Direct visualization of ligands on gold nanoparticles in a liquid environment, Nat. Chem., 2024, 16, 1278–1285 CrossRef CAS PubMed.
- S. K. Meena and M. Sulpizi, Understanding the Microscopic Origin of Gold Nanoparticle Anisotropic Growth from Molecular Dynamics Simulations, Langmuir, 2013, 29, 14954–14961 Search PubMed.
- S. K. Meena and M. Sulpizi, From Gold Nanoseeds to Nanorods: The Microscopic Origin of the Anisotropic Growth, Angew. Chem., 2016, 128, 12139–12143 CrossRef.
- S. K. Meena, S. Celiksoy, P. Schäfer, A. Henkel, C. Sönnichsen and M. Sulpizi, The role of halide ions in the anisotropic growth of gold nanoparticles: a microscopic, atomistic perspective, Phys. Chem. Chem. Phys., 2016, 18, 13246–13254 Search PubMed.
- L. B. Wright, P. M. Rodger, S. Corni and T. R. Walsh, GolP-CHARMM: First-Principles Based Force Fields for the Interaction of Proteins with Au(111) and Au(100), J. Chem. Theory Comput., 2013, 9, 1616–1630 CrossRef CAS PubMed.
- H. Heinz, T.-J. Lin, R. Kishore Mishra and F. S. Emami, Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field, Langmuir, 2013, 29, 1754–1765 Search PubMed.
- S. Franco-Ulloa, L. Riccardi, F. Rimembrana, M. Pini and M. De Vivo, NanoModeler: A Webserver for Molecular Simulations and Engineering of Nanoparticles, J. Chem. Theory Comput., 2019, 15, 2022–2032 CrossRef CAS PubMed.
- Y. K. Choi, N. R. Kern, S. Kim, K. Kanhaiya, Y. Afshar, S. H. Jeon, S. Jo, B. R. Brooks, J. Lee, E. B. Tadmor, H. Heinz and W. Im, CHARMM-GUI Nanomaterial Modeler for Modeling and Simulation of Nanomaterial Systems, J. Chem. Theory Comput., 2022, 18, 479–493 CrossRef CAS PubMed.
- A. Marzouq, L. Morgenstein, C. A. Huang-Zhu, S. Yudovich, A. Atkins, A. Grupi, R. C. Van Lehn and S. Weiss, Long-Chain Lipids Facilitate Insertion of Large Nanoparticles into Membranes of Small Unilamellar Vesicles, Langmuir, 2024, 40, 10477–10485 CrossRef CAS PubMed.
- H. I. Ingólfsson, M. N. Melo, F. J. van Eerden, C. Arnarez, C. A. Lopez, T. A. Wassenaar, X. Periole, A. H. de Vries, D. P. Tieleman and S. J. Marrink, Lipid Organization of the Plasma Membrane, J. Am. Chem. Soc., 2014, 136, 14554–14559 Search PubMed.
- H. I. Ingólfsson, T. S. Carpenter, H. Bhatia, P.-T. Bremer, S. J. Marrink and F. C. Lightstone, Computational Lipidomics of the Neuronal Plasma Membrane, Biophys. J., 2017, 113, 2271–2280 Search PubMed.
- S. J. Marrink, L. Monticelli, M. N. Melo, R. Alessandri, D. P. Tieleman and P. C. T. Souza, Two decades of Martini: Better beads, broader scope, WIREs Comput. Mol. Sci., 2023, 13, e1620 Search PubMed.
- Y. Badhe, P. Sharma, R. Gupta and B. Rai, Elucidating collective translocation of nanoparticles across the skin lipid matrix: a molecular dynamics study, Nanoscale Adv., 2023, 5, 1978–1989 Search PubMed.
- S. Salassi, L. Caselli, J. Cardellini, E. Lavagna, C. Montis, D. Berti and G. Rossi, A Martini Coarse Grained Model of Citrate-Capped Gold Nanoparticles Interacting with Lipid Bilayers, J. Chem. Theory Comput., 2021, 17, 6597–6609 Search PubMed.
- C. A. Lochbaum, A. K. Chew, X. Zhang, V. Rotello, R. C. Van Lehn and J. A. Pedersen, Lipophilicity of Cationic Ligands Promotes Irreversible Adsorption of Nanoparticles to Lipid Bilayers, ACS Nano, 2021, 15, 6562–6572 Search PubMed.
- C. A. Huang-Zhu, J. K. Sheavly, A. K. Chew, S. J. Patel and R. C. Van Lehn, Ligand Lipophilicity Determines Molecular Mechanisms of Nanoparticle Adsorption to Lipid Bilayers, ACS Nano, 2024, 18, 6424–6437 Search PubMed.
- C. A. Huang-Zhu and R. C. Van Lehn, Influence of branched ligand architectures on nanoparticle interactions with lipid bilayers, Nanoscale, 2025, 17, 1659–1672 Search PubMed.
- S. Malola and H. Häkkinen, Prospects and challenges for computer simulations of monolayer-protected metal clusters, Nat. Commun., 2021, 12, 2197 Search PubMed.
- T. Vo, Theory and simulation of ligand functionalized nanoparticles – a pedagogical overview, Soft Matter, 2024, 20, 3554–3576 RSC.
- X. Zhang, G. Ma and W. Wei, Simulation of nanoparticles interacting with a cell membrane: probing the structural basis and potential biomedical application, NPG Asia Mater., 2021, 13, 1–18 CrossRef CAS.
- A. K. Chew and R. C. Van Lehn, Effect of Core Morphology on the Structural Asymmetry of Alkanethiol Monolayer-Protected Gold Nanoparticles, J. Phys. Chem. C, 2018, 122, 26288–26297 Search PubMed.
- R. C. Van Lehn and A. Alexander-Katz, Structure of Mixed-Monolayer-Protected Nanoparticles in Aqueous Salt Solution from Atomistic Molecular Dynamics Simulations, J. Phys. Chem. C, 2013, 117, 20104–20115 Search PubMed.
- R. C. Van Lehn and A. Alexander-Katz, Ligand-Mediated Short-Range Attraction Drives Aggregation of Charged Monolayer-Protected Gold Nanoparticles, Langmuir, 2013, 29, 8788–8798 CrossRef CAS PubMed.
- S. E. Hoff, D. Di Silvio, R. F. Ziolo, S. E. Moya and H. Heinz, Patterning of Self-Assembled Monolayers of Amphiphilic Multisegment Ligands on Nanoparticles and Design Parameters for Protein Interactions, ACS Nano, 2022, 16, 8766–8783 Search PubMed.
- C. Gabellini, M. Şologan, E. Pellizzoni, D. Marson, M. Daka, P. Franchi, L. Bignardi, S. Franchi, Z. Posel, A. Baraldi, P. Pengo, M. Lucarini, L. Pasquato and P. Posocco, Spotting Local Environments in Self-Assembled Monolayer-Protected Gold Nanoparticles, ACS Nano, 2022, 16, 20902–20914 CrossRef CAS PubMed.
- P. Guo, R. Sknepnek and M. Olvera de la Cruz, Electrostatic-Driven Ridge Formation on Nanoparticles Coated with Charged End-Group Ligands, J. Phys. Chem. C, 2011, 115, 6484–6490 CrossRef CAS.
- F. Simonelli, G. Rossi and L. Monticelli, Role of Ligand Conformation on Nanoparticle–Protein Interactions, J. Phys. Chem. B, 2019, 123, 1764–1769 Search PubMed.
- R. C. Van Lehn and A. Alexander-Katz, Fusion of Ligand-Coated Nanoparticles with Lipid Bilayers: Effect of Ligand Flexibility, J. Phys. Chem. A, 2014, 118, 5848–5856 Search PubMed.
- R. C. Van Lehn and A. Alexander-Katz, Membrane-Embedded Nanoparticles Induce Lipid Rearrangements Similar to Those Exhibited by Biological Membrane Proteins, J. Phys. Chem. B, 2014, 118, 12586–12598 CrossRef CAS PubMed.
- P. Gkeka, P. Angelikopoulos, L. Sarkisov and Z. Cournia, Membrane Partitioning of Anionic, Ligand-Coated Nanoparticles Is Accompanied by Ligand Snorkeling, Local Disordering, and Cholesterol Depletion, PLoS Comput. Biol., 2014, 10, e1003917 CrossRef PubMed.
- J. L. MacCallum and D. P. Tieleman, Hydrophobicity scales: a thermodynamic looking glass into lipid–protein interactions, Trends Biochem. Sci., 2011, 36, 653–662 CrossRef CAS PubMed.
- J. Ji, B. Carpentier, A. Chakraborty and S. Nangia, An Affordable Topography-Based Protocol for Assigning a Residue's Character on a Hydropathy (PARCH) Scale, J. Chem. Theory Comput., 2024, 20, 1656–1672 Search PubMed.
- C. A. Silvera Batista, R. G. Larson and N. A. Kotov, Nonadditivity of nanoparticle interactions, Science, 2015, 350, 1242477 Search PubMed.
- J. Monroe, M. Barry, A. DeStefano, P. A. Gokturk, S. Jiao, D. Robinson-Brown, T. Webber, E. J. Crumlin, S. Han and M. S. Shell, Water Structure and Properties at Hydrophilic and Hydrophobic Surfaces, Annu. Rev. Chem. Biomol. Eng., 2020, 11, 523–557 Search PubMed.
- D. Chandler, Interfaces and the driving force of hydrophobic assembly, Nature, 2005, 437, 640–647 Search PubMed.
- C. D. Ma, C. Wang, C. Acevedo-Vélez, S. H. Gellman and N. L. Abbott, Modulation of hydrophobic interactions by proximally immobilized ions, Nature, 2015, 517, 347–350 Search PubMed.
- A. S. Kelkar, B. C. Dallin and R. C. Van Lehn, Identifying nonadditive contributions to the hydrophobicity of chemically heterogeneous surfaces via dual-loop active learning, J. Chem. Phys., 2022, 156, 024701 Search PubMed.
- B. C. Dallin, A. S. Kelkar and R. C. Van Lehn, Structural features of interfacial water predict the hydrophobicity of chemically heterogeneous surfaces, Chem. Sci., 2023, 14, 1308–1319 RSC.
- R. J. E. Reinertsen, S. Kewalramani, F. Jiménez-Ángeles, S. J. Weigand, M. J. Bedzyk and M. Olvera, de la Cruz, Reexpansion of charged nanoparticle assemblies in concentrated electrolytes, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316537121 CrossRef CAS PubMed.
- Q. Tang and M. Rubinstein, Where in the world are condensed counterions ?, Soft Matter, 2022, 18, 1154–1173 RSC.
- F. Schulz, J. Hühn, M. Werner, D. Hühn, J. Kvelstad, U. Koert, N. Wutke, M. Klapper, M. Fröba, V. Baulin and W. J. Parak, Local Environments Created by the Ligand Coating of Nanoparticles and Their Implications for Sensing and Surface Reactions, Acc. Chem. Res., 2023, 56, 2278–2285 CrossRef CAS PubMed.
- M. V. Prud, A. Kyrychenko and O. N. Kalugin, pH-Controllable Coating of Silver Nanoparticles with PMMA-b-PDMAEMA Oligomers: A Molecular Dynamics Simulation Study, J. Phys. Chem. C, 2023, 127, 11748–11759 Search PubMed.
- N. Aho, P. Buslaev, A. Jansen, P. Bauer, G. Groenhof and B. Hess, Scalable Constant pH Molecular Dynamics in GROMACS, J. Chem. Theory Comput., 2022, 18, 6148–6160 Search PubMed.
- A. K. Chew, J. A. Pedersen and R. C. Van Lehn, Predicting the Physicochemical Properties and Biological Activities of Monolayer-Protected Gold Nanoparticles Using Simulation-Derived Descriptors, ACS Nano, 2022, 16, 6282–6292 Search PubMed.
- X. Yan, A. Sedykh, W. Wang, B. Yan and H. Zhu, Construction of a web-based nanomaterial database by big data curation and modeling friendly nanostructure annotations, Nat. Commun., 2020, 11, 2519 Search PubMed.
- A. Pihlajamäki, M. F. Matus, S. Malola and H. Häkkinen, GraphBNC: Machine Learning-Aided Prediction of Interactions Between Metal Nanoclusters and Blood Proteins, Adv. Mater., 2024, 36, 2407046 Search PubMed.
- J. C. Saldinger, M. Raymond, P. Elvati and A. Violi, Domain-agnostic predictions of nanoscale interactions in proteins and nanoparticles, Nat. Comput. Sci., 2023, 3, 393–402 CrossRef CAS PubMed.
- A. Ortiz-Perez, D. van Tilborg, R. van der Meel, F. Grisoni and L. Albertazzi, Machine learning-guided high throughput nanoparticle design, Digital Discovery, 2024, 3, 1280–1291 Search PubMed.
- M. J. Tamasi, R. A. Patel, C. H. Borca, S. Kosuri, H. Mugnier, R. Upadhya, N. S. Murthy, M. A. Webb and A. J. Gormley, Machine Learning on a Robotic Platform for the Design of Polymer–Protein Hybrids, Adv. Mater., 2022, 34, 2201809 Search PubMed.
- D. H. Chang, J. D. Richardson, M.-R. Lee, D. M. Lynn, S. P. Palecek and R. C. Van Lehn, Machine learning-driven discovery of highly selective antifungal peptides containing non-canonical β-amino acids, Chem. Sci., 2025, 16, 5579–5594 RSC.
- G. Giunta, G. Campos-Villalobos and M. Dijkstra, Coarse-Grained Many-Body Potentials of Ligand-Stabilized Nanoparticles from Machine-Learned Mean Forces, ACS Nano, 2023, 17, 23391–23404 Search PubMed.
- S. Mehdi, Z. Smith, L. Herron, Z. Zou and P. Tiwary, Enhanced Sampling with Machine Learning, Annu. Rev. Phys. Chem., 2024, 75, 347–370 Search PubMed.
- W. Shen, T. Zhou and X. Shi, Enhanced sampling in molecular dynamics simulations and their latest applications—A review, Nano Res., 2023, 16, 13474–13497 Search PubMed.
- N. Aho, P. Buslaev, A. Jansen, P. Bauer, G. Groenhof and B. Hess, Scalable Constant pH Molecular Dynamics in GROMACS, J. Chem. Theory Comput., 2022, 18, 6148–6160 Search PubMed.
- A. Jansen, N. Aho, G. Groenhof, P. Buslaev and B. Hess, phbuilder: A Tool for Efficiently Setting up Constant pH Molecular Dynamics Simulations in GROMACS, J. Chem. Inf. Model., 2024, 64, 567–574 CrossRef CAS PubMed.
- R. C. Van Lehn and A. Alexander-Katz, Grafting Charged Species to Membrane-Embedded Scaffolds Dramatically Increases the Rate of Bilayer Flipping, ACS Cent. Sci., 2017, 3, 186–195 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |