
Tannic acid-mediated surface engineering of CNTs for enhanced bifunctional oxygen electrocatalysis†
Xiangmin Tangab,
Luyao Zoub,
Xiaopeng Liab,
Zhipeng Xu c,
Huilin Fan*a,
Chao Lin*ab and
Jung-Ho Lee
*c
c,
Huilin Fan*a,
Chao Lin*ab and
Jung-Ho Lee
*c
aSchool of New Energy, Ningbo University of Technology, Ningbo, 315336, China. E-mail: hlfanjy@nbut.edu.cn
bState Key Laboratory of Advanced Fiber Materials, College of Materials Science and Engineering, Donghua University, Shanghai, 201620, P. R. China. E-mail: linc@dhu.edu.cn
cDepartment of Materials and Chemical Engineering, Hanyang University, Ansan, Gyeonggi-do 15588, Korea. E-mail: jungho@hanyang.ac.kr
First published on 24th June 2025
Abstract
Developing stable and efficient bifunctional electrocatalysts for oxygen reduction and evolution reactions is essential for rechargeable metal–air batteries. Here, we reported a facile surface modification strategy of carbon nanotubes (CNTs) as air electrocatalysts for zinc–air batteries (ZABs). By leveraging the versatile binding affinity of tannic acid with metal ions and carbon surfaces, the CNTs surface was coated with a uniform, thin layer of nitrogen-doped carbon featuring atomically dispersed cobalt. The atomic cobalt and rich nitrogen endow the material with high intrinsic activity in oxygen electrocatalysis. Moreover, the carbon layer optimizes the hydrophilicity of CNTs, and the interwoven CNTs network enables fast electron transfer and accelerated reactant diffusion. The assembled aqueous and solid-state ZABs deliver good rate performance (discharging current density in the liquid-state ZAB: 0–100 mA cm−2, solid-state ZAB: 0.5–10 mA cm−2) and nice cycling stability.
 Jung-Ho Lee | We are very pleased to have our paper published in the 10th Anniversary Collection of Nanoscale Horizons. Since 2019, we have published four papers in Nanoscale Horizons, which have helped us to further deepen our research on nanocatalysis and batteries. We hope to share cutting-edge research results in the field of nanomaterial-based secondary batteries through this journal. Nanoscale Horizons will continue to be an important forum for researchers to discuss key issues related to various nanomaterials. |
New conceptsThe widespread application of ZABs faces significant challenges due to high voltage polarization caused by slow oxygen reduction/evolution reactions. Transition metal/N-doped carbon bifunctional oxygen electrocatalysts, typically derived from pyrolyzed metal–organic compounds, have shown promise due to their high activity. However, their high cost and scalability limitations remain critical barriers to ZABs commercialization. Here, we introduce a coordination-driven approach using naturally abundant tannic acid to create ultrathin cobalt-chelated polyphenol coatings on CNTs. This polyphenol layer transforms into a carbon layer rich in Co–N–C and nitrogen species (Co–N/CNTs), which exhibit excellent bifunctional oxygen electrocatalytic activity. Additionally, the carbon layer improves the hydrophobicity of CNTs, while the interconnected CNTs network facilitates rapid electron transfer and efficient diffusion of reactants such as oxygen and ions. When the optimized electrocatalyst is integrated into the air electrode, both aqueous and solid-state ZABs demonstrate high power densities and remarkable cycling stability. This CNTs modification strategy provides a cost-effective method for developing efficient electrocatalysts, with potential applications extending to other sustainable energy conversion and storage technologies. |
Introduction
The energy crisis and environmental pollution caused by depleting fossil fuels pose a significant challenge to the sustainable development of human society. To address these issues, the economic transition from fossil fuels to clean energy must be undertaken. This shift is driving intense research into sustainable energy storage and conversion devices, among which ZABs have garnered significant attention due to their unique half-open configurations using oxygen as the energy source to minimize the mass and volume of batteries, leading to their theoretical energy density as high as 1084 W h kg−1.1,2 However, both the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) that occur during the charging and discharging of ZABs are multielectron reactions and require high overpotentials, resulting in low round-trip efficiency.3–6 Although conventional noble-metal-based electrocatalysts (e.g., Pt + RuO2) demonstrate high intrinsic activity for bifunctional oxygen electrocatalysis, their practical utilization is constrained by limited stability and prohibitively high costs.7–9 This underscores the critical imperative to develop efficient, durable, and earth-abundant alternatives to promote the application of ZABs.In recent decades, transition metal/nitrogen-doped carbon (M–N–C) materials have solidified their status as promising bifunctional oxygen electrocatalysts due to their adjustable active sites and tailorable electronic structures.10–12 Conventional synthetic methods, including pyrolysis of metal–organic frameworks (MOFs), covalent-organic frameworks (COFs), or hydrogen-bonded organic frameworks (HOFs), predominantly utilize high-temperature calcination (e.g., 900 °C) to engineer highly conductive framework architectures. This process intrinsically triggers metal agglomeration.13 As exemplified by Zeng et al.,14 an ORR electrocatalyst synthesized using a COF/ZIF-67 core–shell hybrid demonstrated enhanced ORR activity and stability over commercial Pt/C in 0.1 M KOH electrolyte. Notably, however, the coexistence of both Co nanoparticles and atomic Co–N4 configurations within the catalyst underscores a fundamental challenge: thermally induced metal aggregation preferentially produces metallic nanoparticles over atomically dispersed M–N–C sites, significantly undermining active-site availability and structural robustness.15–17 These unresolved ambiguities in active-site attribution, coupled with insufficient operational durability, accentuate the pressing necessity for new methodologies to attain exact atomic-level metal dispersion in carbon-based matrices.
To circumvent these limitations, decoupling the carbonization process of conductive substrates from the construction of atomic-level active sites presents a viable strategy to inhibit high-temperature-induced metal agglomeration. This approach enables atomic dispersion of metal sites through low-temperature coordination-driven thermal decomposition of metal–organic ligand complexes anchored on highly conductive carbon frameworks.18 Critical to this methodology is the judicious selection of organic ligands with low decomposition thresholds and conductive carbon substrates. Tannic acid (TA), a naturally abundant and scalable biomaterial, has emerged as an ideal candidate for this purpose. TA can self-polymerize under specific conditions to form porous scaffolds that immobilize and coordinate metal ions via its phenolic groups, serving as a versatile precursor for metal–organic compounds.19–21 Furthermore, TA's hydrophobic aromatic rings and hydrophilic hydroxyl groups facilitate robust adhesion to diverse materials through covalent and non-covalent interactions (e.g., π–π stacking and hydrogen bonding).22,23 Concurrently, CNTs, distinguished by their high conductivity, large surface area, chemical stability, mechanical flexibility, and commercial viability, represent optimal graphitic carbon substrates.24,25 Integrating TA with CNTs as a structural scaffold synergistically leverages their complementary advantages, positioning the hybrid system as an exemplary precursor for advanced oxygen electrocatalysts.
Therefore, we engineered a cobalt-chelated polyphenol layer on CNTs through a facile one-step TA polymerization strategy. Subsequent carbonization transformed this polymeric layer into a nitrogen-enriched carbon matrix hosting Co–N–C active sites (denoted as Co–N/CNTs), optimized for bifunctional oxygen electrocatalysis. The derived carbon coating simultaneously tailored the hydrophobicity of CNTs while preserving their intrinsic conductivity. When integrated into air electrodes, the Co–N/CNTs self-assembled into an interpenetrating conductive network, establishing hierarchical pore structures that facilitate rapid oxygen and ion diffusion and minimize charge-transfer resistance. This structural synergy enabled both aqueous and solid-state ZABs to deliver high power densities (liquid-state: 224.5 mW cm−2; solid-state: 86.3 mW cm−2) alongside nice cycling durability (>500 cycles for liquid-electrolyte ZABs; >270 cycles for solid-state configurations).
Results and discussion
Synthesis and characterization of Co–N/CNTs
The fabrication of Co–N/CNTs is illustrated in Fig. 1. In the initial phase, TA and formaldehyde were thoroughly mixed in a water–ethanol solution under basic conditions, which initiated a condensation reaction to form TA–formaldehyde (TAF) oligomers. CNTs were then introduced into the solution, and the TAF oligomers with the abundant hydrophobic aromatic rings could be adsorbed onto their surface. Subsequently, the TAF can be further polymerized, forming a crosslinked network. Meanwhile, the Co ions can be chelated by combining with the rich phenolic groups in the crosslinked network. Note that formaldehyde plays a key role in forming methylene bridges between TA molecules, which randomly link the TA units, promoting isotropic and uniform growth of the crosslinked TAF layer. The TAF layer was then converted into a cobalt and nitrogen co-doped carbon layer on the CNTs surface (denoted as Co–N/CNTs) upon pyrolysis in the pure ammonia atmosphere, thereby modifying the surface chemistry of CNTs. In addition, by adjusting the Co doping concentration, three samples were prepared: Co–N/CNTs-x (x = 0.04, 0.06, and 0.08, where x represents the mass ratio of doped Co ions relative to a constant 25 mg of CNTs). | ||
| Fig. 1 Schematic illustration of the preparation process of Co–N/CNTs. | ||
The phase composition and crystalline nature of the samples were confirmed by powder X-ray diffraction (XRD). As shown in Fig. 2a, the XRD patterns of the samples doped with varying Co content all exhibited broad peaks similar to those of CNTs. The strong diffraction peaks observed at Bragg angles of 26° and 44° corresponded to the (002) and (101) lattice planes of carbon, respectively. The absence of Co-related peaks is indicative of the high dispersion of cobalt species.
 | ||
| Fig. 2 (a) XRD patterns of CNTs, Co–N/CNTs-0.04, Co–N/CNTs-0.06, and Co–N/CNTs-0.08. (b) SEM image, (c) TEM image, and (d) AC-STEM image of Co–N/CNTs-0.06. (e) The corresponding elemental mapping images of Co–N/CNTs-0.06. (f) Contact angle of CNTs, Co–N/CNTs-0.06 and 20% Pt/C. (g) Co 2p and (h) N 1s XPS spectra of Co–N/CNTs-0.06. (i) The relative proportion of different types of N species on the surface of Co–N/CNTs-0.04, Co–N/CNTs-0.06, and Co–N/CNTs-0.08. | ||
The morphology and structure of Co–N/CNTs-0.06 were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). It can be found that the Co–N/CNTs-0.06 maintained the morphology of CNTs with a diameter of 20–30 nm (Fig. 2b), suggesting that the introduction of N and Co species in CNTs during calcination did not compromise the primary structure of the CNTs, illustrating their structural stability. In addition, the CNTs were interwoven together, forming a porous structure, which is beneficial for the transport of reactant molecules. The HR-TEM analysis of Co–N/CNTs-0.06 (Fig. 2c and Fig. S1, ESI†) revealed distinct lattice fringes exhibiting a characteristic interplanar spacing of 0.34 nm, which corresponds to the (002) crystallographic planes of graphitic carbon. The observed amorphous carbon layers encapsulating the CNT surfaces are attributed to carbon-based active species derived from cobalt-chelated polyphenol precursors during the thermal decomposition process. Aberration-corrected scanning transmission electron microscopy (AC-STEM) images of Co–N/CNTs-0.06 (Fig. 2d) demonstrate the uniform distribution of isolated cobalt atoms across the carbon nanotube matrix, with no observable metallic clusters or nanoparticles. This atomic dispersion aligns with the XRD analysis (Fig. 2a), where only the characteristic peaks of graphitic carbon are observed, confirming the absence of crystalline cobalt phases. The combined STEM and XRD results validate the successful formation of atomically dispersed Co–Nx sites through our synthesis strategy. The elemental mapping images (Fig. 2e) confirm the successful coating of the carbon layer on the CNTs, indicating the presence of atomically dispersed N and Co species on the CNTs. The porous architecture arising from the interwoven CNT network in Co–N/CNTs-0.06 is validated by Brunauer–Emmett–Teller (BET) analysis. As evidenced in Fig. S2 (ESI†), both pristine CNTs and Co–N/CNTs-0.06 display comparable multiscale porosity profiles, confirming that the surface functionalization process preserves the intrinsic interconnected pore structure of the CNT framework. This retention of inherent porosity ensures unaltered mass transport pathways while introducing catalytically active sites. As illustrated in Fig. S3 (ESI†), the Raman spectra reveal distinct vibrational modes at 1342 cm−1 (D-band, associated with sp3 hybridized defects) and 1579 cm−1 (G-band, corresponding to sp2 graphitic domains, E2g mode).26 The calculated ID/IG ratios of 1.4 for CNTs and 1.36 for Co–N/CNTs-0.06 demonstrate that the 600 °C pyrolysis process employed for Co/N doping induces minimal alterations in graphitic ordering. This structural preservation correlates well with the maintained hierarchical porosity revealed by our BET analysis, confirming the stability of the carbon matrix during the modification process. Moreover, inductively coupled plasma mass spectrometry (ICP-MS) was employed to determine the exact cobalt content in the Co–N/CNTs. As shown in Table S1 (ESI†), the measured cobalt content for Co–N/CNTs-0.06 is 0.86 wt%. Contact angles were measured to evaluate the changes in the surface chemistry of CNTs (Fig. 2f). The hydrophobicity of modified Co–N/CNTs-0.06 was enhanced, and its contact angle (CA) was 47.7°, which was larger than that of original CNTs (CA = 35.3°) and 20% Pt/C (CA = 28.7°). This improvement can facilitate oxygen diffusion across the electrode coated with the Co–N/CNTs.
X-ray photoelectron spectroscopy (XPS) further probed the surface chemistry of Co–N/CNTs-0.06. The survey XPS spectrum displayed C, N, O, and Co species (Fig. S4, ESI†). The Co 2p spectrum (Fig. 2g) exhibited dual chemical states: Co2+ (780.3 eV for Co 2p3/2 and 795.3 eV for Co 2p1/2) and Co–N coordination bonds (783.8 eV and 798.3 eV), confirming Co–N–C formation.27–29 The Co–N–C structures are known to be efficient active centers for the ORR and the OER.30–33 As shown in Fig. 2h, the N 1s spectrum of Co–N/CNTs-0.06 can be deconvoluted into five nitrogen species: pyridinic-N (398.3 eV), coordinated N with Co (399.2 eV), pyrrolic-N (400.1 eV), graphitic-N (401.4 eV), and oxidized-N (403.3 eV).29,34,35 For other samples, the chemical states (binding energy positions) of surface N and Co species were similar to those of Co–N/CNTs-0.06 (Fig. S5, ESI†). XPS semi-quantitative analysis revealed a positive correlation between surface Co and N concentrations (Table S2, ESI†). As the Co doping level increased, the N content also increased. Analysis of the N 1s XPS spectrum enabled quantification of surface N species proportions. As shown in Fig. 2i, optimal Co doping (Co–N/CNTs-0.06) maximized pyridinic-N (45.4%) and Co–N content (16.3%), whereas excessive doping (Co–N/CNTs-0.08) shifted the equilibrium toward pyrrolic-N (34.7%) with reduced pyridinic-N (35.8%). It is worth noting that, at the atomic scale, the effect of each N species on the local electronic structure of adjacent carbon atoms varies depending on the C–N bond configuration. Specifically, the formation of pyridinic-N leads to the carbon atoms adjacent to the nitrogen species becoming active sites with Lewis basicity, which is favorable for oxygen electrocatalysis.36–38
Electrochemical ORR and OER performances of Co–N/CNTs
To evaluate the ORR catalytic performance of Co–N/CNTs-0.06, a three-electrode electrochemical setup equipped with a rotating disk electrode (RDE) was established in an alkaline electrolyte (0.1 M KOH) saturated with O2. The cyclic voltammetry (CV) curves (Fig. 3a) of Co–N/CNTs-0.06 measured in a nitrogen-saturated 0.1 M KOH electrolyte exhibited no discernible reduction peaks. However, upon introducing oxygen into the electrolyte to saturation, a prominent reduction peak appeared at 0.8 V vs. RHE, indicating a significant redox catalytic activity for Co–N/CNTs-0.06. According to the polarization curves shown in Fig. 3b and c, Co–N/CNTs-0.06 exhibits a high half-wave potential (E1/2 = 0.83 V vs. RHE) and onset potential (Eonset = 1.09 V vs. RHE, the potential required to achieve a current density of 0.1 mA cm−2). In addition, the ORR performances of CNTs, Co–N/CNTs-0.04, Co–N/CNTs-0.08, and commercial 20% Pt/C were also assessed for comparison. It can be found that the Co–N/CNTs-0.06 delivers the highest activity, outperforming those of pristine CNTs (Eonset = 0.81 V vs. RHE, E1/2 = 0.71 V vs. RHE), Co–N/CNTs-0.04 (Eonset = 0.92 V vs. RHE, E1/2 = 0.83 V vs. RHE), and Co–N/CNTs-0.08 (Eonset = 0.97 V vs. RHE, E1/2 = 0.79 V vs. RHE) and comparable to the benchmark 20% Pt/C (Eonset = 0.91 V vs. RHE, E1/2 = 0.84 V vs. RHE). Meanwhile, Co–N/CNTs-0.06 also exhibited a much higher diffusion limited current (jL = 6.19 mA cm−2) than Co–N/CNTs-0.08 (jL = 5.91 mA cm−2), Co–N/CNTs-0.04 (jL = 4.07 mA cm−2), CNTs (jL = 3.09 mA cm−2) and 20% Pt/C (jL = 4.65 mA cm−2). Compared to recently reported precious metal-free electrocatalysts, Co–N/CNTs-0.06 shows decent electrocatalytic activities, as shown in Table S3 (ESI†). Fig. S6 (ESI†) shows the kinetic current density (Jk) derived from the K–L equation. Co–N/CNTs-0.06 showed an activity of 12.8 mA cm−2 at 0.8 V, which is higher than that of Co–N/CNTs-0.04 (11.6 mA cm−2 at 0.8 V) and Co–N/CNTs-0.08 (4.7 mA cm−2 at 0.8 V) and close to that of 20% Pt/C (14.5 mA cm−2 at 0.8 V). | ||
| Fig. 3 (a) CV curves of Co–N/CNTs-0.06 in N2- and O2-saturated 0.1 M KOH at the scan rate of 10 mV s−1. (b) The linear sweep voltammetry (LSV) curves, (c) the half-wave potential and onset potential, and (d) Tafel plots of the ORR on CNTs, Co–N/CNTs-0.04, Co–N/CNTs-0.06, Co–N/CNTs-0.08, and 20% Pt/C in 0.1 M KOH solution at 1600 rpm. (e) EIS pattern of Co–N/CNTs-0.06 and 20% Pt/C recorded at 1.03 V vs. RHE. (f) Calculated n and determined H2O2% at various potentials based on the RRDE. | ||
The Tafel slope of various materials was calculated to measure their reaction kinetics. As shown in Fig. 3d, the Tafel slope of Co–N/CNTs-0.06 is 99.3 mV dec−1, lower than that of Co–N/CNTs-0.08 (181.6 mV dec−1), Co–N/CNTs-0.04 (100.4 mV dec−1), bare CNTs (103.2 mV dec−1), and 20% Pt/C (138.2 mV dec−1), indicating its fast ORR kinetics. As depicted in Fig. 3e, Co–N/CNTs-0.06 demonstrated a lower series resistance (Rs) than 20% Pt/C in high-frequency regions, owing to the ultra-conductive 1D CNT networks that minimize inter-nanotube contact resistance compared to particle-based commercial catalysts. In the low-frequency region, the EIS curve approximates a semicircle, where the diameter represents the charge transfer resistance (Rct) at the electrode/electrolyte interface. Clearly, the charge transfer rate on the surface of Co–N/CNTs-0.06 is significantly faster than that of 20% Pt/C. The reduced Rct observed in Co–N/CNTs-0.06 can be attributed to the formation of a 3D network formed by interwoven CNTs, which facilitates mass transfer more efficiently than catalysts with conventional particle structures that are closely packed. Moreover, the enhanced hydrophobicity of this 3D structure significantly promotes oxygen transport, thereby improving the rate of charge transfer. Operational stability stands as a critical performance metric for ORR electrocatalysts. The Co–N/CNTs-0.06 catalyst exhibits excellent long-term stability in 0.1 M KOH solution, maintaining stable current density for 10 hours without significant degradation (Fig. S7, ESI†). The ORR selectivity of Co–N/CNTs-0.06 and pristine CNTs is quantitatively delineated in Fig. 3f and Fig. S8 (ESI†) through comparative analysis of electron transfer numbers (n) and peroxide yields (H2O2%). Across the 0.2–0.8 V vs. RHE potential window, Co–N/CNTs-0.06 demonstrates an average n value of 3.89–3.32, corresponding to a suppressed H2O2 yield of 5.4–34.3%. These metrics confirm the catalyst's predominant adherence to a concerted four-electron transfer pathway in alkaline media. In contrast, unmodified CNTs exhibit substantially higher parasitic two-electron activity, generating H2O2 at 11.8–68.5% yields under identical conditions.
The OER activity of Co–N/CNTs-0.06 was also evaluated in 1 M KOH. As shown in Fig. S9a (ESI†), the catalyst demonstrated a low overpotential of 250 mV to reach the current density of 10 mA cm−2 (EJ=10), lower than that of RuO2 (1.50 V vs. RHE). Further kinetic analysis revealed a Tafel slope of 91.6 mV dec−1 for Co–N/CNTs-0.06 (Fig. S9b, ESI†), the smallest value among all tested catalysts, confirming its fast OER kinetics. Chronoamperometry was employed to evaluate the OER stability of the catalyst. Co–N/CNTs-0.06 exhibited exceptional stability, retaining 91.5% of its initial current density after 30 hours of continuous operation (Fig. S10, ESI†). The above results suggest that Co–N/CNTs-0.06 is an ideal bifunctional electrocatalyst for rechargeable ZABs.
Zinc–air battery performance analysis
Motivated by the superior ORR and OER bifunctional activity of Co–N/CNTs-0.06, the homemade liquid rechargeable ZAB (Fig. 4a) was assembled to assess its practical applications in energy devices, and ZABs based on Co–N/CNTs-0.04, Co–N/CNTs-0.08, and 20 wt% Pt/C were also investigated for comparison. The battery configuration includes a zinc plate anode, a cathode composed of Co–N/CNTs-0.06 loaded on carbon fiber paper, and an electrolyte composed of 6.0 M KOH containing 0.2 M Zn(OAc)2. The open circuit voltage (OCV) of the battery assembled with Co–N/CNTs-0.06 was 1.45 V (Fig. 4b), which was higher than that of the 20% Pt/C (1.38 V), Co–N/CNTs-0.04 (1.32 V), and Co–N/CNTs-0.08 (1.33 V) based ZABs. The charge–discharge polarization curves indicate that the Co–N/CNTs-0.06-based ZAB exhibited a smaller charge–discharge potential difference compared to the other samples, suggesting superior rechargeability (Fig. 4c). Fig. 4d shows the discharge polarization curves and corresponding power density plots of ZABs assembled with different catalysts. Notably, the ZAB incorporating the Co–N/CNTs-0.06 catalyst exhibited a slower voltage decay with increasing current density compared to those utilizing other electrodes, suggesting effective mitigation of electrode polarization, which well corresponds to the ORR activity. The power density of the Co–N/CNTs-0.06-based ZAB reached 224.5 mW cm−2, which was also higher than that of Co–N/CNTs-0.04 (150.1 mW cm−2), Co–N/CNTs-0.08 (175.2 mW cm−2) and the commercial 20% Pt/C (131.1 mW cm−2) based ZABs. Furthermore, the Co–N/CNTs-0.06-based ZAB demonstrated stable rate capability without significant decay after discharging across a wide range of current densities from 5 to 100 mA cm−2 (Fig. 4e). Even as the upper limit of current density increased to 200 mA cm−2 or 300 mA cm−2, the Co–N/CNTs-0.06-based ZAB still demonstrated stable rate performance (Fig. S11, ESI†). It is noted that, after a discharge cycle test in large current density ranges (5 to 200 mA cm−2 and 5 to 300 mA cm−2), the discharge platform was restored to the initial values when the current density was reduced to 10 mA cm−2. The rechargeability and long-term cycling stability of the Co–N/CNTs-0.06-based ZAB were evaluated using galvanostatic charge/discharge cycle measurements (20 min per cycle) at a current density of 5 mA cm−2. As shown in Fig. 4f, the homemade battery with Co–N/CNTs-0.06 maintained a stable charge and discharge potential exceeding 160 hours (>500 cycles). In contrast, the ZAB with 20% Pt/C experienced a substantial decline in efficiency over time, likely caused by Pt aggregation and carbon corrosion. These results further confirm the exceptional stability of Co–N/CNTs-0.06 applied in the ZABs.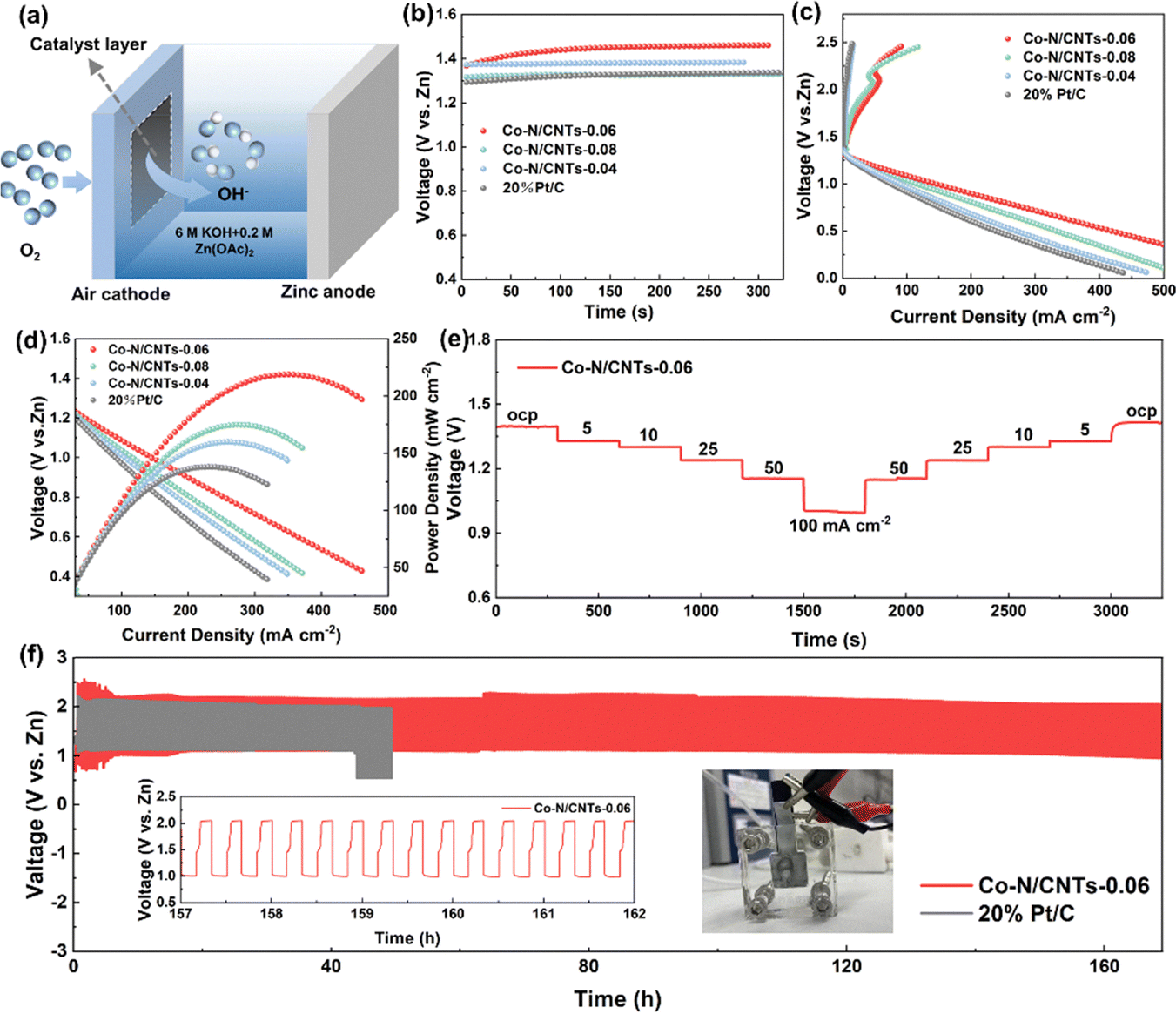 | ||
| Fig. 4 (a) Schematic of the basic configuration of a rechargeable ZAB. (b) Open circuit voltage and (c) discharge–charge polarization curves of rechargeable ZABs with Co–N/CNTs-0.04, Co–N/CNTs-0.06, Co–N/CNTs-0.08 and 20% Pt/C as the air cathodic catalysts. (d) Discharge polarization curves and corresponding power density plots. (e) Galvanostatic discharge curves at various current densities, including OCP, 5, 10, 20, 50, and 100 mA cm−2. (f) Discharge/charge cycling curves of ZABs using Co–N/CNTs-0.06 and 20% Pt/C as the air electrodes at a current density of 5 mA cm−2 (the inset on the left shows cycling curves from the 157th to the 162nd cycle; the inset on the right shows the image of a homemade liquid ZAB). | ||
The superior performance of the Co–N/CNTs-0.06-based ZAB can be attributed to the abundant active sites exposed on the catalyst surface. These active sites primarily consist of carbon atoms adjacent to pyridinic-N configurations and Co–N–C coordination structures, which collectively serve as dual-active centers for both the ORR and the OER. Apart from its well-maintained ORR activity in a strong alkaline environment, the excellent performance of the Co–N/CNTs-0.06-based ZAB also arises from its enhanced mass transfer properties. The hydroxyl ion diffusion coefficient (DOH−) can be derived from the Z′ vs. ω−1/2 plot of the EIS data. As shown in Fig. S12 (ESI†), the DOH− of the Co–N/CNTs-0.06-based ZAB is 38.2 × 10−11 cm2 s−1, which is significantly higher than that of 20% Pt/C (8.73 × 10−11 cm2 s−1). This suggests that the interwoven structure of the porous CNTs significantly enhances hydroxyl ion transport in the electrode. The enhanced mass transport of hydroxyl ions between the solid and liquid interface is consistent with the excellent limiting diffusion current density of Co–N/CNTs-0.06 in the ORR, as well as the discharge polarization curve analysis in ZABs. In addition, Co–N/CNTs-0.06 shows the advantage of mass transfer of oxygen between the gas and liquid interface. The enhanced hydrophobicity of Co–N/CNTs-0.06 facilitated the diffusion of gas on the electrode surface and efficiently removed water generated during the reaction. As a result, the liquid/gas/solid three-phase interface was maximized and stabilized, which not only facilitated the ORR but also improved the performance of the ZAB.
Flexible all-solid zinc–air battery performance analysis
Flexible all-solid zinc–air batteries (ASS–ZABs) were constructed to further explore the potential application of the Co–N/CNTs-0.06 electrocatalyst, which was loaded on carbon paper as the air cathode. The anode and solid electrolytes adopted a flexible zinc foil and copolymers of acrylic acid (AA) and methacryloyloxyethyl trimethyl ammonium chloride (DMC), respectively (Fig. 5a). It was found that the open-circuit voltage of the Co–N/CNTs-0.06-based ASS–ZAB was 1.57 V (Fig. 5b), which was higher than that of the Pt-based battery (1.44 V), further highlighting the superior intrinsic ORR activity of the Co–N/CNTs-0.06 catalyst. As shown in Fig. 5c, the charge–discharge curves of the Co–N/CNTs-0.06-based ASS–ZAB show a significantly smaller voltage gap compared to the Pt-based battery. Importantly, the peak power density of the Co–N/CNTs-0.06-based ASS–ZAB reached 83.6 mW cm−2, surpassing that of the Pt/C + RuO2-based battery (62.9 mW cm−2) (Fig. 5d). The specific capacity of an ASS–ZAB based on bioinspired Co–N/CNTs-0.06 reached up to 757.13 mA h g−1 (Fig. 5e), outperforming the batteries based on Pt/C + RuO2 (417.72 mA h g−1). In addition, the discharge rate capability (0.5–10 mA cm−2) of the Co–N/CNTs-0.06-based ASS–ZAB is significantly superior to those of the Pt/C + RuO2-based ASS–ZAB (Fig. 5f). Furthermore, the Co–N/CNTs-0.06-based ASS–ZAB exhibited excellent cycling stability (Fig. 5g). The battery operated smoothly at a current density of 5 mA cm−2 for nearly 90 h (>270 cycles), which is much longer than that of Pt/C + RuO2-based ASS–ZAB (≈10 h). | ||
| Fig. 5 (a) Schematic illustration of an ASS–ZAB. (b) Open circuit voltage, (c) charge and discharge polarization curves, (d) power density curves, and (e) discharging capacity plots at the current density of 2 mA cm−2 of Co–N/CNTs-0.06 and Pt/C + RuO2. (f) Discharging plateaus at different current densities. (g) Galvanostatic discharge–charge curves of a flexible ASS–ZAB at the current density of 2 mA cm−2 and a duration of 20 min per cycle. | ||
The enhanced bifunctional oxygen electrocatalytic performance of Co–N/CNTs stems from the concerted interplay of four structurally engineered advantages. Firstly, the atomic dispersion of cobalt and nitrogen doping into the carbon matrix creates abundant Co–N–C coordination structures and pyridinic-N species, which serve as active centers for both the ORR and the OER. These configurations optimize the local electronic structure of adjacent carbon atoms, enhancing their intrinsic catalytic activity. Secondly, the conformal carbon overlayer derived from tannic acid pyrolysis not only enhances CNTs' hydrophobicity to stabilize the triple-phase boundary but also promotes efficient oxygen permeation (Fig. 2f), as quantitatively evidenced by the superior limiting current density of Co–N/CNTs-0.06 (6.19 mA cm−2 vs. 3.09 mA cm−2 for CNTs and 4.65 mA cm−2 for Pt/C) in ORR polarization profiles (Fig. 3b). Thirdly, the three-dimensionally interpenetrating CNT network establishes express electron highways and streamlined ion diffusion channels, validated by its reduced charge-transfer resistance (Fig. 3e) and elevated OH− diffusion coefficient (Fig. S12b, ESI†) relative to Pt/C. Lastly, the porous architecture arising from the polyphenol-derived carbon coating maximizes accessible active sites while maintaining structural integrity during cycling. These synergistic structural and electronic modifications collectively enable record-breaking activity and stability metrics in zinc–air battery applications.
Conclusions
In summary, a Co, N co-doped CNT material, enriched with pyridinic-N and an atomically dispersed Co–N–C structure (Co–N/CNTs-0.06), was successfully synthesized by the combination of the organic ligand polymerization and ammonia pyrolysis strategies. Co–N/CNTs-0.06 delivers excellent ORR and OER performances under alkaline conditions. At the same time, its effective application in other sustainable energy conversion and storage devices has been confirmed through the self-assembled liquid rechargeable ZABs and all-solid electrolyte ZABs. The excellent electrochemical performance of Co–N/CNTs-0.06 mainly benefits from the following aspects: (I) the surface of Co–N/CNTs-0.06 is rich in active sites, including ORR and OER active centers formed by pyridinic-N and Co–N–C structures. (II) The CNTs in the catalyst possess high electrical conductivity and structural stability. (III) The 3D structure formed by the interwoven CNTs shortens the diffusion distance of ions in the electrolyte, promotes an efficient electron/ion transport pathway, and ensures adequate oxygen supply at the active site.Experimental section
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the Basic Science Research Program (NRF-2023R1A2C3003788) through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT. C. L. thanks the support from the National Natural Science Foundation of China (52402231) and the start-up Funding offered by Donghua University.Notes and references
- X. Bi, Y. Jiang, R. Chen, Y. Du, Y. Zheng, R. Yang, R. Wang, J. Wang, X. Wang and Z. Chen, Adv. Energy Mater., 2024, 14, 2302388 CrossRef.
- X.-W. Lv, Z. Wang, Z. Lai, Y. Liu, T. Ma, J. Geng and Z.-Y. Yuan, Small, 2024, 20, 2306396 CrossRef PubMed.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 Search PubMed.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef.
- J. Stacy, Y. N. Regmi, B. Leonard and M. Fan, Renewable Sustainable Energy Rev., 2017, 69, 401–414 CrossRef.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef PubMed.
- S. D. Bhoyate, J. Kim, F. M. de Souza, J. Lin, E. Lee, A. Kumar and R. K. Gupta, Coord. Chem. Rev., 2023, 474, 214854 CrossRef.
- T. Liu, C. Chen, Z. Pu, Q. Huang, X. Zhang, A. M. Al-Enizi, A. Nafady, S. Huang, D. Chen and S. Mu, Small, 2024, 20, 2405399 CrossRef PubMed.
- M. Gong, A. Mehmood, B. Ali, K.-W. Nam and A. Kucernak, ACS Catal., 2023, 13, 6661–6674 CrossRef PubMed.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, Y. Li, L. Xiao, X. Ren, J. Zhang and M. An, J. Mater. Chem. A, 2020, 8, 23187–23201 RSC.
- W. Shao, R. Yan, M. Zhou, L. Ma, C. Roth, T. Ma, S. Cao, C. Cheng, B. Yin and S. Li, Electrochem. Energy Rev., 2023, 6, 11 CrossRef.
- Y. Wang, L. Chen, Z. Mao, L. Peng, R. Xiang, X. Tang, J. Deng, Z. Wei and Q. Liao, Sci. Bull., 2019, 64, 1095–1102 CrossRef CAS PubMed.
- Z. Zhang, J. Dong, C. Huang, K. Wan, Z. Feng, B. Li, N. Zhao, L. Dai, L. Wang and Z. He, Chem. Eng. J., 2025, 506, 159958 CrossRef CAS.
- M. Liu, Q. Xu, Q. Miao, S. Yang, P. Wu, G. Liu, J. He, C. Yu and G. Zeng, J. Mater. Chem. A, 2022, 10, 228–233 RSC.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604–9633 CrossRef CAS PubMed.
- W.-J. Niu, J.-Z. He, B.-N. Gu, M.-C. Liu and Y.-L. Chueh, Adv. Funct. Mater., 2021, 31, 2103558 CrossRef CAS.
- H. Yin, H. Xia, S. Zhao, K. Li, J. Zhang and S. Mu, Energy Environ. Mater., 2021, 4, 5–18 CrossRef CAS.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- H. Huang, J. Zhao and R. Liu, J. Colloid Interface Sci., 2021, 582, 396–404 CrossRef CAS PubMed.
- M. A. Rahim, M. Björnmalm, N. Bertleff-Zieschang, Q. Besford, S. Mettu, T. Suma, M. Faria and F. Caruso, Adv. Mater., 2017, 29, 1606717 CrossRef PubMed.
- H. Li, K. Du, C. Xiang, P. An, X. Shu, Y. Dang, C. Wu, J. Wang, W. Du, J. Zhang, S. Li, H. Tian, S. Wang and H. Xia, J. Mater. Chem. A, 2020, 8, 17136–17149 RSC.
- J. Zhou, Z. Lin, Y. Ju, M. A. Rahim, J. J. Richardson and F. Caruso, Acc. Chem. Res., 2020, 53, 1269–1278 CrossRef CAS PubMed.
- X.-L. Liu, H.-C. Wang, T. Yang, X.-Z. Yue and S.-S. Yi, Chem. Commun., 2023, 59, 13690–13702 RSC.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M.-J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS.
- Q. He, S. Qiao, Y. Zhou, R. Vajtai, D. Li, P. M. Ajayan, L. Ci and L. Song, Adv. Funct. Mater., 2022, 32, 2106684 CrossRef CAS.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- D. Wang, M. Yuan, J. Xu, Y. Li, K. Shi, H. Yang, H. Li and G. Sun, ACS Sustainable Chem. Eng., 2021, 9, 16956–16964 CrossRef CAS.
- X. Wu, G. Han, H. Wen, Y. Liu, L. Han, X. Cui, J. Kou, B. Li and J. Jiang, Energy Environ. Mater., 2022, 5, 935–943 CrossRef CAS.
- C. Zhang, N. Huang, Z. Zhai, L. Liu, B. Chen, B. Yang, X. Jiang and N. Yang, Adv. Energy Mater., 2023, 13, 2301749 CrossRef CAS.
- Y. Ha, B. Fei, X. Yan, H. Xu, Z. Chen, L. Shi, M. Fu, W. Xu and R. Wu, Adv. Energy Mater., 2020, 10, 2002592 CrossRef CAS.
- J. Zhang, Y. Mou, W. Suo, S. Yang, J. Shen, H. Xu, Z. Zeng, R. Zhang, Z. Liang, Y. Wang, H. Zheng, J. Cao and R. Cao, Adv. Funct. Mater., 2025, 35, 2417621 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- X.-X. Ma, X.-Q. He and T. Asefa, Electrochim. Acta, 2017, 257, 40–48 CrossRef CAS.
- B. Li, Y. Ren, G. Zhang, C. Lv, L. Li, X. Yang, Z. Lu, X. Zhang and X. Yu, Appl. Surf. Sci., 2025, 679, 161212 CrossRef CAS.
- Z. Lei, Y. Tan, Z. Zhang, W. Wu, N. Cheng, R. Chen, S. Mu and X. Sun, Nano Res., 2021, 14, 868–878 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- T. Kondo, S. Casolo, T. Suzuki, T. Shikano, M. Sakurai, Y. Harada, M. Saito, M. Oshima, M. I. Trioni, G. F. Tantardini and J. Nakamura, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035436 CrossRef.
- Y. Li, C. Li, Q. Zhou and Y. Lian, Appl. Surf. Sci., 2025, 689, 162444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00179j |
| This journal is © The Royal Society of Chemistry 2025 |