
In situ interfacial engineering of 1D Bi2S3/2D g-C3N4 heterostructures for antibiotics degradation in aqueous media via light mediated peroxymonosulfate activation†
Muhammad Mateen *ac,
Guanrong Chenab,
Na Guo*ad and
Wee Shong Chin*ab
*ac,
Guanrong Chenab,
Na Guo*ad and
Wee Shong Chin*ab
aAdvance Manufacturing and Material Center, National University of Singapore (Chongqing) Research Institute, Chongqing 400000, P. R. China. E-mail: mateenchem@hotmail.com; phyguon@u.nus.edu
bDepartment of Chemistry, Faculty of Science, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmcws@nus.edu.sg
cSchool of Chemistry and Chemical Engineering, Chongqing University, 400000, P. R. China
dDepartment of Physics, Faculty of Science, National University of Singapore, 3 Science Drive 3, 117543, Singapore
First published on 21st July 2025
Abstract
Interfacial engineering between metal sulfides (MS) and graphitic carbon nitride (g-C3N4) offers a promising strategy to design semiconductors for the efficient degradation of persistent water pollutants. However, conventional multi-step methods used to prepare MS/g-C3N4 heterostructures often result in weak interfacial interactions between the building blocks, thereby leading to inefficient charge separation and sub-optimal catalytic performance. To overcome this limitation, we present here a novel single-step strategy for the in situ preparation of 1D Bi2S3(n)/2D g-C3N4 heterostructures, producing intimate interactions between the 1D and 2D architectures as evidenced by experimental and theoretical findings. Remarkably, these robust interfacial interactions establish a strong internal electric field (IEF), favoring spatial separation of high charge flux at the 1D/2D interface via an S-scheme mechanism. Importantly, the lowered charge transfer barrier at the interface speeds up the activation kinetics of peroxymonosulfate (PMS) and O2, to achieve a high tetracycline degradation efficiency of 98.5% with a rate constant of 0.06 min−1. DFT calculation results reveal that the effective coupling between the 1D/2D counterparts induced a charge redistribution and electron density accumulation at the interface, facilitating cleavage of the O–O bond in PMS and O2. Furthermore, DFT calculations identified a unique PMS adsorption configuration on Bi sites and revealed the competence of S atoms in activating the peroxide bond in PMS. This work offers a cost-effective and environmentally friendly approach for the rational engineering of interfacial interactions in MS/g-C3N4 heterostructures, enabling highly efficient applications in energy and environmental remediation.
New conceptsThis communication introduces a new single-step in situ synthesis approach to forming 1D Bi2S3(n)/2D g-C3N4 S-scheme van der Waals (vdW) heterostructures. The in situ 1D/2D integration allowed the strong interfacial coupling, thereby enabling us to address the poor interfacial interactions faced by the traditional multi-step methods. Furthermore, in this work we clarified the underlying role of intimate interfacial interactions in constructing a robust internal electric field (IEF) for activating the S-scheme charge separation mechanism and decreasing the interfacial charge transfer barrier. Through DFT calculations we demonstrated that charge accumulation near the 1D Bi2S3/2D g-C3N4 facilitated PMS adsorption and lowered the energy barrier for O–O bond cleavage in PMS, while electronically modifying Bi and S sites served as pivotal catalytic centers. Beyond its superior catalytic activity, this work presents a scalable green approach to engineer metal sulfide/g-C3N4 interfaces, offering a universal design strategy for advanced nanomaterials via atomic-level interfacial control. Integrating material design (1D/2D coupling), mechanistic understanding (IEF-driven S-scheme), and practical application (PMS-based advanced oxidation processes), our findings provide significant advances in water purification technologies. |
1. Introduction
Rampantly increasing consumption of stubborn antibiotics as medicinal therapy against life-threatening microbial infections has triggered the emergence and transmission of multidrug resistant (MDR) bacterial strains in water reservoirs,1–3 thus imposing detrimental effects upon both human health and aquatic life.4 Considering the severity of the situation, the World Health Organization (WHO) has declared the sub-lethal accumulation and dissemination of antibiotic resistant organisms as an escalating threat to human life,5 forecasting a MDR bacterial associated mortality toll of 10 million by the year 2050.6,7 There is therefore a dire need for a globally collaborative approach from policymakers to mitigate the transmission of MDR strains and prevent waterborne infections. Up to now, adequate degradation and mineralization of refractory antibiotics by conventional wastewater treatment technologies have remained daunting challenges and demand urgent development of a highly efficient and economically feasible technology capable of effectively eliminating antibiotics to address the ongoing safe drinking water crisis.Advanced oxidation processes (AOPs),8,9 specifically persulfate based (PS-AOP) ones, have emerged as a cutting edge next generation water treatment technology to effectively degrade a wide spectrum of persistence micropollutants, including antibiotics.10,11 Compared to the conventional hydroxyl radicals process (˙OH), sulfate SO4˙− radicals possess higher redox potentials (2.5–3.1 eV), longer half-lives (30–40 μs) and wider pH (2–8) operative windows.12,13 PS-AOP could completely disintegrate antibiotics into CO2 and H2O rather than simply oxidizing them into non-toxic intermediates.14,15 In practice, strong oxidants such as peroxymonosulfate (PMS) and peroxydisulfate (PDS) with a half-life span of 3–4 × 10−5 s are employed to generate multiple reactive oxygen species (ROS) through appropriate activation means such as catalysts, light, heat, radiations, etc.16,17 In comparison with PDS, PMS based PS-AOP has gained more popularity due to the asymmetric geometry of the PMS molecule (H–O–O–SO3−), which renders it to serve as an electron acceptor as well as donor to activate superoxide bonds (O–O, 1.326 Å) for the non-selective generation of free radicals (SO4˙−, ˙OH, and O2˙−) and non-free radicals (1O2).18 In recent years, light mediated heterogeneous PMS activation has been demonstrated as an excellent strategy to dramatically improve antibiotics degradation efficiency.19 This triggers immense research focus because of the natural abundance of solar light, simple operation protocol and high efficiency. Nevertheless, rational design of a robust catalyst that can efficiently capture visible light to generate charge carriers is of tremendous importance for effective light-induced PMS activation.
Since 2009,20 metal-free conjugated polymeric g-C3N4 featuring an appealing electronic structure, suitable band gap and superior physiochemical stability has been the material of choice for a breadth of visible light driven catalysis including remediation of hazardous water pollutants.21–23 However, pristine g-C3N4 exhibits inferior PMS activation ability because of poor redox active sites,24 sluggish charge carrier separation,25 inadequate visible light absorption26 and low electric conductivity.27 Hitherto, elemental doping,28 constructing M–Nx atomic sites29,30 and heterostructure engineering with other suitable semiconductors has been explored in the pursuit of boosting PMS activation and degradation performance.31 Interfacial engineering of g-C3N4 with narrow bandgap metal oxides, metal chalcogenides, metal phosphides and metal carbides has been developed rapidly in recent years with the aim of constructing catalysts with good photocatalytic activity through simultaneously improved visible light utilization and amplified charges transportation.32,33 Amongst the semiconductors, metal sulfides,34 especially Bi2S3, an n-type narrow bandgap (1.3–1.7 eV) semiconductor, could be the best selection due to its distinct advantages such as low cost, non-toxicity and strong absorption capability in the visible and near-infrared region.35,36
Given the distinct advantages of g-C3N4 and Bi2S3 individually, constructing a Bi2S3/g-C3N4 heterostructure interface is a plausible strategy to significantly enhance catalytic performance.37,38 However, the synthesis of Bi2S3/g-C3N4 heterostructures has up until now relied on tedious multi-step methods, often requiring expensive precursors and post anchoring of Bi2S3 nanostructures onto the surface of pre-synthesized g-C3N4.39,40 Additionally, most reported Bi2S3/g-C3N4 heterojunctions suffer from inhomogeneous distribution of Bi2S3 and weak chemical or physical interfacial interactions, resulting in sub-optimal photocatalytic performance. Strong interfacial interactions are critical in regulating the electronic structure and charge transport properties. Such interactions could form an internal electric field (IEF) and enhance the adsorption behavior of PMS and antibiotics, thereby accelerating reaction kinetics of PMS-activated AOPs. Consequently, developing a strategy to engineer a robust Bi2S3/g-C3N4 heterojunction is highly desired to address the weak interfacial interaction challenges and to optimize the photocatalytic performance.
Herein, we present a facile single-step gas–solid reaction strategy for the in situ synthesis of a series of strongly attached 1D Bi2S3 chains on 2D g-C3N4 nanosheets with tunable Bi2S3 content. Our experimental evidences revealed that the interfacial interactions could be appropriately controlled by regulating the growth density of 1D Bi2S3 onto the 2D g-C3N4 sheets. The synthesized 1D/2D heterostructures exhibited optimal interfacial coupling, enhanced charge separation efficiency and appropriate number of active sites, thereby resulting in great antibiotic degradation performances in light mediated PMS-AOP processes. Density functional theory (DFT) simulations have unveiled the electronic charge redistribution on the Bi and S atoms at the 1D/2D interface. DFT calculations further demonstrated strong PMS adsorption on the electronically modified Bi and S atoms via distinct configurations, stimulating ROS generation and thereby enhancing antibiotic degradation activity.
2. Experimental section
2.1. Materials
Details of the chemicals and reagents employed in this study are provided in the ESI.†2.2. Synthesis protocol of 1D Bi2S3(n)/2D g-C3N4 heterostructures
Synthesis of 1D Bi2S3(n)/2D g-C3N4 heterostructures with varying 1D Bi2S3 content was accomplished via a single step strategy. In detail, bismuth acetate (1 mM, 2 mM and 3 mM) was dissolved in 20 mL DI water under stirring in a 50 mL centrifuge tube to make a yellow solution. Next, 3 g of thiourea and 2 g of urea was added into the above yellow solution and stirred at 40 °C until urea and thiourea completely dissolved. Then, the resulting solution was freeze-dried using liquid nitrogen and kept in a lyophilizer at −50 °C for three days. The obtained yellow solid was crashed into powder form and subjected to pyrolysis in a tube furnace at 560 °C under an inert atmosphere (Ar) at a heating rate of 4 °C minute−1 for 2 hours. After cooling to normal temperature, the samples were collected and designated as 1D Bi2S3(n)/2D g-C3N4, where n = 0.1, 0.2, and 0.3 represent the feed Bi concentration in mM. A control pristine 2D g-C3N4 sample was prepared by mixing 3 g thiourea and 2 g urea under the same conditions used to prepare the heterostructures. The procedure for synthesizing the pristine 1D Bi2S3 sample is given in the ESI.†2.3. Characterizations
Details of the instruments used for the characterization of the prepared samples and procedures for evaluating degradation activity are described in the ESI.†2.4. Density functional theory (DFT) calculations
Details of DFT calculations are provided in the ESI.†3. Results and discussion
3.1. Synthesis, morphological and structure analysis and DFT structure optimization
Fig. 1a schematically illustrates the preparation of 1D Bi2S3(n)/2D g-C3N4 heterostructures via the in situ solid–gas phase reaction. In this process, urea and thiourea were employed as the sources of carbon (C), nitrogen (N), and sulfur (S), while bismuth acetate served as the precursor for the bismuth (Bi) ions. Three representative heterostructures were synthesized and labeled as 1D Bi2S3(n)/2D g-C3N4, where n represents the feed concentration of Bi in millimoles (0.1, 0.2, and 0.3 mM). The formation mechanism of these heterostructures involves firstly, the polymerization of urea and thiourea into 2D g-C3N4 nanosheets along with the release of reactive CS2 gas;41 and the simultaneous decomposition of bismuth acetate into Bi2O3 under the pyrolysis conditions. Next, the in situ solid–gas reaction between Bi2O3 and CS2 resulted in the growth of 1D Bi2S3 nanostructures onto the 2D g-C3N4 surfaces to ultimately construct the 1D Bi2S3(n)/2D g-C3N4 heterostructures. | ||
| Fig. 1 (a) Schematic illustration of the synthesis process of 1D Bi2S3(n)/2D g-C3N4 heterostructures. Detailed analysis of the morphology of the 1D Bi2S3(0.2)/2D g-C3N4 sample: (b) SEM, (c) TEM, (d) HAADF-STEM, and (e)–(h) respective EDX elemental mapping of C, N, Bi and S, and (i) HR-TEM image showing the lattice spacing. DFT optimized models for: (j) 1D Bi2S3, (k) 2D g-C3N4, and (l) and (m) 1D Bi2S3/2D g-C3N4, respectively along the α and β directions. | ||
Field-emission scanning electron microscopy (FE-SEM) images of the as-obtained 1D Bi2S3(n)/2D g-C3N4 heterostructures (Fig. 1b and Fig. S1, ESI†) clearly revealed the presence of 1D Bi2S3 nanostructures uniformly distributed on the surface of 2D g-C3N4. As a comparison, the pristine g-C3N4 prepared under identical conditions gave a typical 2D sheet-like structure (Fig. S2a, ESI†), while the prepared pristine Bi2S3 gave a typical 1D morphology (Fig. S2b, ESI†). Notably, as the Bi3+ concentration was increased from 0.1 to 0.3 mM, a corresponding increase in the density of 1D Bi2S3 nanostructures could be observed on the 2D g-C3N4 surfaces (Fig. S1a–c, ESI†). We illustrate detailed morphological analysis of the 1D Bi2S3(0.2)/2D g-C3N4 sample in Fig. 1b-i, as this n = 0.2 mM feed ratio resulted in the highest catalytic performance in subsequent study. The SEM (Fig. 1b), TEM (Fig. 1c) and HAADF-STEM (Fig. 1d) images of the 1D Bi2S3(0.2)/2D g-C3N4 sample revealed good contact between the 1D Bi2S3 formed in situ onto the 2D g-C3N4 nanosheets. Energy-dispersive X-ray (EDX) mapping revealed decent spatial distribution of the Bi and S composition along C and N elements in the 1D Bi2S3(0.2)/2D g-C3N4 heterostructure (Fig. 1e–h).
In contrast, no such close contact was observed in the SEM (Fig. S3a, ESI†) and TEM (Fig. S3b, ESI†) images taken of a physically mixed 1D Bi2S3 and 2D g-C3N4 sample. The HR-TEM micrograph (Fig. 1i) clearly unveils the presence of distinct amorphous g-C3N4 and crystalline Bi2S3 phases, confirming the formation of an intimate heterostructure interface that is of outmost importance to set up IEF. The lattice fringes of 0.37 nm and 0.34 nm spacing correspond well to the d-spacing of the (130) planes of orthorhombic Bi2S3, further substantiating the successful formation of the 1D Bi2S3 structure.
Spin-polarized DFT simulations were performed to further investigate the structure synthesized. The heterostructure model was constructed with optimized structures of a 1 × 9 1D Bi2S3 molecular chain (Fig. 1j) and a 5 × 5 supercell of 2D g-C3N4 (Fig. 1k) with a lattice mismatch of less than 3%. As there exists two binding facets of 1D Bi2S3 (α and β, as indicated in Fig. S4a, ESI†), either of these facets could be oriented along the a, b or diagonal direction of the 2D g-C3N4 surface (Fig. S4b, ESI†). Among the models simulated, the α/diagonal configuration, designated as 1D Bi2S3/2D g-C3N4-α (Fig. 1l), was found to give stable van der Waals (vdW) interactions at an average interface space of about 3.38 Å and binding energy of −6.94 eV. In a similar manner, the β/diagonal configuration (1D Bi2S3/2D g-C3N4-β, Fig. 1m) was obtained with a binding energy of −7.04 eV. These energetically almost degenerate 1D Bi2S3/2D g-C3N4-α and 1D Bi2S3/2D g-C3N4-β configurations suggest that both heterostructures are stable and could be formed experimentally.
The crystal structure and phase composition of 1D Bi2S3(n)/2D g-C3N4 heterostructures were further confirmed by executing powder X-ray diffraction (XRD) analysis (Fig. 2a). The XRD pattern of 2D g-C3N4 exhibited two characteristic peaks at 13.1° and 27.6° for the graphitic layers of the (002) and (100) planes respectively. On the other hand, the pristine 1D Bi2S3 showed diffraction peaks that could be indexed to the crystal planes of the orthorhombic Bi2S3 phase (PDF no. 17-0320). All the 1D Bi2S3(n)/2D g-C3N4 heterostructures exhibited only diffraction peaks that corresponded to 1D Bi2S3 and 2D g-C3N4, confirming the successful fabrication with high phase purity. Notably, the diffraction peaks of the 1D Bi2S3 phase appeared sharper in the 1D Bi2S3(n)/2D g-C3N4 sample as compared to the pristine 1D Bi2S3 sample, indicating better crystallinity in the heterostructures. Furthermore, the peak intensity of the 1D Bi2S3 diffraction increased linearly with higher Bi3+ feed concentrations, while the 2D g-C3N4 peak intensity decreased steadily, as expected with increasing growth of 1D Bi2S3 on the surface of 2D g-C3N4.
 | ||
| Fig. 2 Comparison of 1D Bi2S3, 2D g-C3N4 and 1D Bi2S3(n)/2D g-C3N4 heterostructures: (a) XRD patterns, (b) and (c) FTIR spectra, XPS profiles of (d) C 1s, (e) N 1s and (f) Bi 4f peaks respectively. DFT simulated work function (WF, φ) for: (g) 2D g-C3N4, (h) 1D Bi2S3 and (i) 1D Bi2S3/2D g-C3N4 heterostructure respectively. Bader charge analysis for the optimized (j) 1D Bi2S3/2D g-C3N4-α, and (k) 1D Bi2S3/2D g-C3N4-β structures. | ||
The effect of in situ growth of 1D Bi2S3 on the chemical structure of 2D g-C3N4 was investigated by FTIR spectroscopy (Fig. 2b). In the case of pristine 1D Bi2S3, the absorption band observed in the range 510–640 cm−1 was assigned to the stretching mode of the Bi–S bond.42 The FTIR peaks of 2D g-C3N4 at ∼800 cm−1 and 1200–1700 cm−1 were respectively attributed to the bending vibrations of triazine/heptazine rings and the stretching modes of CN heterocycles within these rings.43 Specifically, the peaks at 1320 cm−1 and 1232 cm−1 correspond to the stretching vibrations of N–(C)3 and C–NH–C linking units within the tri-s-triazine structure.44 Broader peaks in the 2900–3400 cm−1 region could be associated to the stretching vibrations of terminal amine groups (–NH and –NH2) and hydroxyl (OH) groups of physically adsorbed water molecules.45 Notably, all the 1D Bi2S3(n)/2D g-C3N4 heterostructures retained the basic chemical skeleton of g-C3N4, as evidenced by the retention of characteristic absorption bands. However, as can be seen from the magnified FTIR spectra (Fig. 2c), the characteristic stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C heterocycles in the heterostructures exhibited a slight blue shift, suggesting the presence of interfacial interactions and electronic charge redistribution between the 1D Bi2S3 and 2D g-C3N4 counterparts.46
C and C–C heterocycles in the heterostructures exhibited a slight blue shift, suggesting the presence of interfacial interactions and electronic charge redistribution between the 1D Bi2S3 and 2D g-C3N4 counterparts.46
XPS analysis was conducted to ascertain the surface chemical structures and affirm the electronic interactions within the 1D Bi2S3(n)/2D g-C3N4 heterostructures. The XPS binding energy (B.E.) of all the peaks was calibrated with respect to C 1s peak at 284.8 eV. Apart from the dominant peaks of C 1s and N 1s, XPS survey scans of the 1D Bi2S3(n)/1Dg-C3N4 heterostructures (Fig. S5, ESI†) displayed minor peaks for Bi 4f (157.4 eV) and S 2s (225.01 eV), thus confirming the successful integration of 1D Bi2S3 on 2D g-C3N4 as observed by EDX mapping. In agreement with the XRD and SEM results, 1D Bi2S3(0.3)/2D g-C3N4 exhibited comparatively intense Bi 4f and S 2s peaks as compared to the 1D Bi2S3(0.1)/2D g-C3N4 and 1D Bi2S3(0.2)/2D g-C3N4 samples. The C 1s spectrum of 2D g-C3N4 (Fig. 2d) could be fitted into two well-resolved Gaussian–Lorentzian components, assignable respectively to the sp2 hybridized C–C (284.8 eV) in the graphitic structure and sp2 carbon of the C–NC (288.2 eV) bond in the s-triazine ring.47 Interestingly, a blue shift in the C 1s B.E. of the C–NC component was noted for the heterostructures, suggesting a modification in the electronic density around the carbon in the C–NC bond.
The N 1s spectrum of 2D g-C3N4 was deconvoluted into three component peaks (Fig. 2e). The peaks at 398.9 eV and 400.9 eV were respectively attributed to the sp2 hybridized N atom of the C–NC bond and tertiary nitrogen of N–(C)3,48 while the peak at 404.8 eV could be ascribed to π–π* excitations between the stacking 2D g-C3N4 interlayers or amino functional group (–NH2).49,50 Interestingly, the N 1s B.E. of the C–NC component peak red-shifted to varying degrees in the heterostructures, with the apparent shift (Δ) decreasing in the order: 1D Bi2S3(0.1)/2D g-C3N4 (Δ = 0.32 eV) > 1D Bi2S3(0.2)/2D g-C3N4 (Δ = 0.2 eV) > 1D Bi2S3(0.3)/2D g-C3N4 (Δ = 0.1 eV). This N 1s B.E. trend provides exclusive evidence for the existence of robust interfacial electronic interactions between 1D Bi2S3 and 2D g-C3N4, which triggered the electronic charge modification on the N atom of the C–NC bond and the nearby C atom.
As shown in Fig. 2f, the Bi 4f XPS profile of 1D Bi2S3 exhibited two peaks for the characteristic Bi 4f7/2 (158.5 eV) and Bi 4f5/2 (163.8 eV) with spin orbital splitting of 5.3 eV, indicating the existence of Bi3+.51 Notably, Bi 4f B.E. of the heterostructures exhibited a blue shift, suggesting a redistribution of electronic charge on Bi. The Bi 4f peaks shifted in the order of 1D Bi2S3(0.1)/2D g-C3N4 (Δ = 0.7 eV) > 1D Bi2S3(0.2)/2D g-C3N4 (Δ = 0.64 eV) > 1D Bi2S3(0.3)/2D g-C3N4 (Δ = 0.54 eV), implying a weakening of interfacial interactions with increasing population of 1D Bi2S3 in the heterostructures. From the XPS data, it could be inferred that the existence of strong interfacial interactions in 1D Bi2S3(n)/2D g-C3N4 heterostructures set up an IEF that directed the charge flow across the interface, similar to a previous report.52
In order to confirm the charge redistribution at the 1D Bi2S3/2D g-C3N4 interface, we performed the work function, charge differential and Bader charge analysis (Fig. 2g–k). According to band theory, the electron transfer and band alignment of the semiconductor closely relate to its work function (WF, φ). Therefore, we computed the WF from the equation: φ = Evac − EF, where EF and Evac are respectively the Fermi energy level and electrostatic vacuum level potentials. The values of WFs (φ) for the 2D g-C3N4, 1D Bi2S3 and 1D Bi2S3/2D g-C3N4 structures were respectively simulated to be 5.2 eV, 4.9 eV and 4.6 eV (Fig. 2g–i). On the basis of the mismatch of the Fermi energy levels of 2D g-C3N4 and 1D Bi2S3, it would be expected that the construction of the 1D Bi2S3/2D g-C3N4 interface would inevitably push electrons to flow from 2D g-C3N4 to 1D Bi2S3 until a unified Fermi energy level was achieved. This electronic charge redistribution caused an increased electron density at the interface near the 1D Bi2S3 side and electron depletion on the 2D g-C3N4 side, which eventually led to the formation of IEF directed from 2D g-C3N4 to 1D Bi2S3. IEF acted as the energizing force to drive the holes and electrons in opposite directions, thereby prolonging their lifetimes and improving catalytic performance. The charge density differences (CDD) analysis conducted by creating isosurfaces further verified the electronic charge accumulation (orange-purple color regions in Fig. 2j and k) around the Bi and S atoms with unsaturated coordination around the newly formed bonds at the 1D Bi2S3/2D g-C3N4 interface, while charge depletion (green color regions in Fig. 2j and k) on the 2D g-C3N4 surface. Additionally, Bader charge analysis revealed about 0.1 electronic charge (e−) transfer across the interface, thus fully corroborating the XPS results.
3.2. Optical and photo-electrical properties
As the light absorption capacity of semiconductors profoundly affects their catalytic efficiency, we characterized the 1D Bi2S3(n)/2D g-C3N4 heterostructures and control samples (pristine 2D g-C3N4 and 1D Bi2S3) by ultraviolet-visible diffused reflectance spectroscopy (UV-vis DRS) as depicted in Fig. 3a. The absorption edge of 2D g-C3N4 at ∼460 nm blue-shifted by the induction of 1D Bi2S3, with the 1D Bi2S3(0.3)/2D g-C3N4 heterostructure found to display the largest shift to ∼420 nm. Importantly, 1D Bi2S3 induction significantly extended the light absorption ability of 2D g-C3N4 across the 200–800 nm region as indicated by its dark color (Fig. 3c). To evaluate the bandgap energy (Eg), the Kubelka–Munk function, (αhv)n = k(hv − Eg), was applied to convert the UV-vis DRS spectral data to Tauc plots as shown in Fig. 3b. Compared to 2D g-C3N4 (Eg = 2.65 eV), 1D Bi2S3(n)/2D g-C3N4 samples exhibited narrowed Eg in the order: 1D Bi2S3(0.1)/2D g-C3N4 (Eg = 2.4 eV) > 1D Bi2S3(0.2)/2D g-C3N4 (Eg = 2.22 eV) > 1D Bi2S3(0.3)/2D g-C3N4 (Eg = 2.14 eV).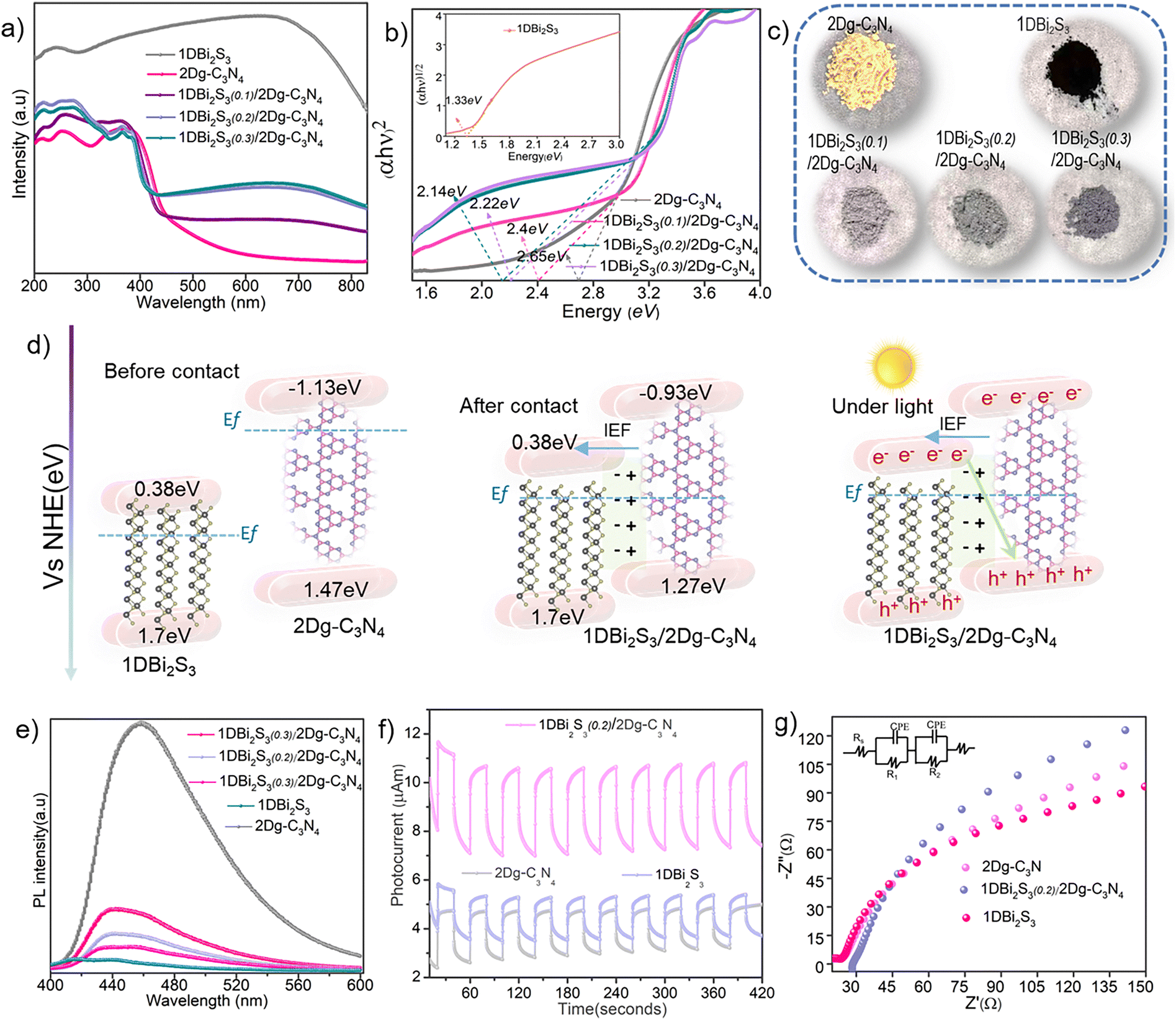 | ||
| Fig. 3 (a) and (b) UV-Visible DRS spectra and corresponding Tauc plots, and (c) digital photographs of 2D g-C3N4, 1D Bi2S3 and 1D Bi2S3(n)/2D g-C3N4 heterostructures. (d) Schematics showing the energy band alignment and charge transfer mechanism of the 1D Bi2S3/2D g-C3N4 heterostructure. (e) Comparison of PL spectra of 2D g-C3N4, 1D Bi2S3 and the various 1D Bi2S3(n)/2D g-C3N4 heterostructures. (f) and (g) Photocurrent response and Nyquist plots of the 1D Bi2S3, 2D g-C3N4 and 1D Bi2S3(0.2)/2D g-C3N4 heterostructures, respectively. | ||
The bandgap narrowing of 2D g-C3N4 upon coupling with 1D Bi2S3 was also validated by the DFT calculations (Fig. S6, ESI†). The valence band edge (EVB) potentials and conduction band edge (ECB) potentials of 2D g-C3N4 and 1D Bi2S3 were estimated using the empirical formulas: EVB = χ + 0.5Eg − E0 and ECB = EVB − Eg, where χ stands for absolute electronegativity, which is 4.73 eV and 5.27 eV for g-C3N4 and Bi2S3 respectively.53,54 E0 is the energy of free electrons on the hydrogen scale which is approximately ∼4.5 eV. Correspondingly, the EVB and ECB potentials of 2D g-C3N4 were respectively estimated to be 1.53 eV and −1.07 eV, while those for 1D Bi2S3 were estimated to be 1.7 eV and 0.38 eV. Notably, for the 1D Bi2S3(0.2)/2D g-C3N4 heterostructure, EVB and ECB edges of 2D g-C3N4 were lowered to 1.27 eV and −0.93 eV respectively, due to the formation of the intimate interface. According to the EVB and ECB potentials, band alignments of 1D Bi2S3(0.2)/2D g-C3N4 before contact, after contact and under light may be schematically represented as shown in Fig. 3d. Besides light absorption, the efficient separation of charge carriers (e−/h+) is also essential to activate surface-adsorbed molecules.
Photoluminescence (PL) spectroscopy offers an indirect means to investigate charge transfer characteristics, as the correlation between intensity and wavelength of the emitted photons produces a steady state PL spectrum that reflects the efficiency of charge separation. A significantly decreased PL spectral intensity of the 1D Bi2S3(n)/2D g-C3N4 heterostructures as compared to 2D g-C3N4 unveiled the suppression of e−/h+ recombination (Fig. 3e). Besides, the lowest PL intensity of 1D Bi2S3(0.2)/2D g-C3N4 among all the heterostructures implies the existence of strong interfacial interactions in 1D Bi2S3(0.2)/2D g-C3N4, constructing an IEF that steered up the directional charge transfer and hindered the recombination rate of e−/h+ pairs. Excessive surface coverage of 2D g-C3N4 by 1D Bi2S3 in the 1D Bi2S3(0.3)/2D g-C3N4 heterostructure has resulted in an increased PL intensity, which could be due to the weakening of interfacial interactions as also indicated by XPS analysis. Moreover, the higher transient photocurrent response of 1D Bi2S3(0.2)/2D g-C3N4 as compared to 2D g-C3N4 and 1D Bi2S3 (Fig. 3f) under visible light (λ > 420 nm) on–off cycling further indicated the reduced recombination rate of charge carriers and improved electron transport across the 1D Bi2S3(0.2)/2D g-C3N4 interface. In addition, electrochemical impedance spectroscopy (EIS) analysis was performed to probe the interface charge transfer resistance. Generally, a smaller arc radius of Nyquist plot obtained from EIS analysis signifies a better charge transfer tendency. The smaller arc radius observed for 1D Bi2S3(0.2)/2D g-C3N4 as compared to those of 2D g-C3N4 and 1D Bi2S3 indeed inferred the former has better charge transfer ability (Fig. 3g). On the basis of the above analysis, 1D Bi2S3(0.2)/2D g-C3N4 with abundant active sites and improved charge separation is expected to profoundly improve catalytic activity.
4. Catalytic evaluation
4.1. Catalytic performance of 1D Bi2S3(n)/2D g-C3N4 towards antibiotics degradation
Tetracycline (TC), due to its high concentration in underground and drinking water, extended half-life and poor metabolization, was selected as the model antibiotic to evaluate the degradation performance of our prepared 1D Bi2S3(n)/2D g-C3N4 heterostructures as shown in Fig. 4a. Prior to determining the contribution of visible light and PMS, the adsorption–desorption of TC in the dark was carried out for 30 minutes. Pristine 1D Bi2S3 exhibited better TC adsorption ability than 2D g-C3N4, which could be due to the interaction between surface functional groups on TC molecules and Bi+3 sites in the 1D Bi2S3 structure. Although both 2D g-C3N4 and 1D Bi2S3 are visible light responsive semiconductors, they failed to achieve adequate TC degradation efficiency due to the high e−/h+ recombination rate. Among the prepared heterostructures, 1D Bi2S3(0.2)/2D g-C3N4 exhibited the best TC degradation competency of 76% (Fig. S7, ESI†), followed by 1D Bi2S3(0.1)/2D g-C3N4 (65%) and 1D Bi2S3(0.3)/2D g-C3N4 (54%). This activity trend signifies that the optimal 1D Bi3S3 amount with strong contact with the 2D g-C3N4 surface is crucially important to optimize the degradation performance. Next, the PMS activation ability of 1D Bi2S3(0.2)/2D g-C3N4 was investigated. Under dark conditions, the 1D Bi2S3/PMS and 2D g-C3N4/PMS systems degraded 37% and 16% of TC respectively, while the 1D Bi2S3(0.2)/2D g-C3N4/PMS system exhibited significantly improved TC degradation of 61%. This may be attributed to the electronic charge accumulation at the 1D Bi2S3(0.2)/2D g-C3N4 interface that could be readily transferred to the adsorbed PMS for facile activation. | ||
| Fig. 4 (a) TC degradation performance of 1D Bi2S3, 2D g-C3N4 and 1D Bi2S3(0.2)/2D g-C3N4 under different conditions (with/without PMS/light). (b) Kinetics curves of the TC degradation of photocatalyst/PMS/light systems. (c) Comparison of TC degradation efficiency of this work with some literature reported Bi and g-C3N4 heterostructures. (d) TC degradation efficiency of the 1D Bi2S3(0.2)/2D g-C3N4/PMS/light system in various water matrices. (e) General applicability of the 1D Bi2S3(0.2)/2D g-C3N4/PMS/light system towards various antibiotics and dye degradation (inset: rate constant (k) values). (Reaction condition: [TC] = 20 mg L−1, [catalyst] = 30 mg, PMS = 2 mM and pH = 7). | ||
Next, we explored the light and PMS synergistic effect to ameliorate the TC degradation. Visible light alone degraded only 14% of TC, exhibiting weak PMS activation ability. Remarkably, under the simultaneous action of light and PMS, 1D Bi2S3(0.2)/2D g-C3N4 achieved optimized activity of 98.5% TC degradation, exceeding by 22% and 37% respectively when compared to 1D Bi2S3(0.2)/2D g-C3N4/light and 1D Bi2S3(0.2)/2D g-C3N4/PMS systems. Time-dependent UV-visible spectra of TC degradation was monitored for the 1D Bi2S3(0.2)/2D g-C3N4/light/PMS system (Fig. S8, ESI†) and the activities under different conditions are compared in Fig. 4a. The superiority of the 1D Bi2S3(0.2)/2D g-C3N4 system for the effective degradation of TC under the cooperative action of light and PMS could be credited to the enhanced light harvesting capacity, electronic charge modulation at the 1D/2D interface, and better charge separation via IEF. These thus collectively enabled 1D Bi2S3(0.2)/2D g-C3N4 to demonstrate impressive PMS activation under light as compared to its pristine 1D Bi2S3 and 2D g-C3N4 counterparts. In addition, adsorbed PMS itself may capture the photogenerated electrons to produce radicals or non-radical reactive oxygen species. The kinetic data of TC degradation could be described well by pseudo first-order reaction kinetics (Fig. 4b). The rate constant (k) value of TC degradation for the 1D Bi2S3(0.2)/2D g-C3N4/light/PMS system (0.06 min−1) was determined to be 7 and 4.6 times higher than the 2D g-C3N4/light/PMS (0.01 min−1) and 1D Bi2S3/light/PMS (0.015 min−1) systems, respectively. TC degradation comparison in Fig. 4c clearly illustrates the superiority of our 1D Bi2S3(0.2)/2D g-C3N4 system over most of the other literature reported Bi and g-C3N4 heterostructures.
The actual implication of the 1D Bi2S3(0.2)/2D g-C3N4/light/PMS system in real-world scenarios was further investigated by testing TC degradation in Yangtze river, rain, tap, and lake water matrices (Fig. 4d). Interestingly for tap water, 1D Bi2S3(0.2)/2D g-C3N4 exhibited comparable TC degradation (92%) with that in pure water. In more complex water matrices such as Yangtze river, lake, and rainwater samples, 1D Bi2S3(0.2)/2D g-C3N4 still degraded 89%, 88%, and 86% of TC, respectively, proving its enormous potential for PMS-AOP. Additionally, the 1D Bi2S3(0.2)/2D g-C3N4 heterostructure exhibited excellent to moderate degradation ability towards various other organic molecules (Fig. 4e), including levofloxacin, ofloxacin, sulfamethoxazole, ciprofloxacin, rhodamine B and methylene blue, establishing its higher and universal utility.
4.2. Effects of reaction parameters
The effect of various reaction parameters such as: 1D Bi2S3 content, PMS dosage, TC concentration, pH value and ion interference were investigated as presented in Fig. 5. First, the TC degradation activity of 1D Bi2S3(n)/2D g-C3N4 was optimized by varying the 1D Bi2S3 to 2D g-C3N4 ratio as illustrated in Fig. 5a. Initially, the increase in Bi concentration from 0.1 mM to 0.2 mM produced a positive impact as indicated by enhanced TC degradation from 60% to 98.5%. However, at 0.3 mM Bi, TC degradation sharply declined to 83%. This was due to the over-abundance of 1D Bi2S3, which provided an ample number of reaction sites for species quenching55 and thus lowered the overall degradation efficiency. These results signify that an appropriate amount of the 1D/2D intimate contact is crucial for effective TC removal. To prove this argument, we physically mixed 1D Bi2S3 and 2D g-C3N4 to fabricate a composite catalyst. This composite was found to exhibit only slight shifts in its XPS N 1s and Bi 4f B.E. relative to those of the 1D Bi2S3(n)/2D g-C3N4 heterostructure (Fig. S9, ESI†), confirming its weaker interfacial interactions. Expectedly, the physically mixed sample degraded merely 79% of TC (Fig. S10, ESI†) under the same conditions, thus reaffirming the argument that strong interfacial interactions are essential to promote electron–hole separation and PMS activation towards efficient TC degradation. | ||
| Fig. 5 Effect of various reaction parameters on the TC degradation: (a) 1D Bi2S3 concentrations, (b) PMS concentration, (c) influence of pH, (d) TC concentration, (e) effect of common ions/molecules, and (f) recycling experiment. | ||
TC degradation increased linearly with PMS concentration due to a proportionate increase in the number of active species until it reached 98.5% (k = 0.06 min−1) at a concentration of 3 mM L−1, then started to decline (Fig. 5b). The observed activity decline could be caused by the reverse reaction between SO4˙− and ˙OH radicals and excess HSO5− (eqn (1)–(4)), leading to the formation of radicals with lower oxidation potential such as HO2˙ and SO5˙−.56 In addition, degradation efficiency could be hampered by the self-quenching of SO4˙− radicals (eqn (5)) prior to attacking the antibiotics.56
| HSO5− + SO4˙− → SO42− + SO5˙− + H+ | (1) |
| HSO5− + ˙OH → SO5˙− + H2O | (2) |
| HSO5− + ˙OH → HO2˙ + SO42− + H+ | (3) |
| HSO5− + SO4˙− + H2O → HO2˙ + 2SO42− + H+ | (4) |
| SO4˙− + SO4˙− → S2O82− | (5) |
Next, it is of practical importance to investigate the pH tolerance, for the reason that TC can exist as cationic species (pH < 3.3), zwitterionic species (3.3 < pH < 7.7) or anionic species (pH > 7.7).57 Consequently, water pH can alter the adsorption behavior of TC onto the surface of the photocatalyst and also influence the selectivity of ROS.58 Under strong acidic conditions (pH = 1), TC degradation was found to drop to 69% (Fig. 5c), since the excessive H+ ions not only inhibit the generation of ROS by making HSO5− more stable, but also effectively react with SO4˙− or ˙OH,59 resulting in a deficiency of ROS. As the initial pH was changed progressively to neutral conditions, TC degradation increased gradually and 1D Bi2S3(0.2)/2D g-C3N4 achieved 86% and 93% TC degradation, respectively, at pH = 4 and pH = 6. More interestingly, 1D Bi2S3(0.2)/2D g-C3N4 exhibited an exceptional 99.9% TC degradation in alkaline conditions (pH = 11). The plausible reason for this trend is that the alkaline environment promotes TC deprotonation, rendering TC with increased electron density around the ring system, thus making it more vulnerable to oxidation by ˙OH and especially SO4˙− radical.60,61 Furthermore, alkaline conditions accelerate PMS activation to generate more ROS, thus enhancing TC degradation.62 Also, at pH = 11, 1D Bi2S3(0.2)/2D g-C3N4 exhibited negligible TC absorption, leaving more active sites available for PMS adsorption and activation. The influence of initial TC concentrations (10–50 mg L−1) revealed the highest TC degradation efficiency of 99.3% at 10 mg L−1 (Fig. 5d). However, the degradation of TC declined stepwise from 99.3 to 66.7%, when the initial concentration increased from 10–50 mg L−1. The reason is that the PMS concentration is fixed and more ROS are required to react with the increased number of TC molecules and its intermediate fragments.
Natural water matrices contain a mixture of inorganic anions and organic species that can potentially compete with ROS or adhere to the photocatalyst surface, resulting in severely compromised antibiotic degradation activity. It is therefore highly critical to study the inhibitory effect of various ions (Cl−, NO3−, HPO42−, CO32− and HCO3−) and organic molecules on the catalytic performance. As can be seen in Fig. 5e, the addition of 2 mM Cl− ions did not result in any noticeable interference. In contrast, the addition of 5 mM and 10 mM Cl− ions significantly enhanced the degradation of TC by 98.5% and 99.3% respectively. This could be due to the formation of chlorine species from the reaction between Cl− ion and PMS, ˙OH, and SO4˙, according to eqn (6)–(10).63 Although the chlorine radicals are less reactive than ˙OH and SO4˙ radicals, they possess a very strong affinity to attack the electron-rich regions of TC molecules, i.e., OH and NH2 groups and conjugated double bonds of the aromatic rings, thus leading to a boosted TC degradation as previously reported.63,64
| Cl− + HSO5− → SO42− + HOCl | (6) |
| 2Cl− + HSO5− + H+ → SO42− + Cl2 + H2O | (7) |
| ˙OH + Cl− → ClOH˙− | (8) |
| SO4˙− + Cl− → SO42− + Cl˙ | (9) |
| Cl˙ + Cl− → Cl2˙− | (10) |
The existence of CO32− and HCO3− ions is expected to have a negative effect on the AOP process because they can act as scavengers for SO4˙− and ˙OH radicals.65 However, in our experiment, a contradictory effect was observed because the existence of 10 mM CO32− and HCO3− ions significantly accelerated the TC degradation. There are a number of reasons that could be credited to the promotional effect of CO32− and HCO3− ions. First, the added CO32− and HCO3− ions could react with ˙OH, and SO4˙ radicals to produce CO3˙− and HCO3˙ radicals (eqn (11)–(15)) of lower reactivity but higher selectivity towards electron rich groups of TC such as phenol and dimethylammonium.66 Second, the nucleophilic character of both CO32− and HCO3− ions promoted the activation of asymmetric PMS molecules to produce more reactive species,67 thus resulted in higher degradation efficiency. Third, the presence of CO32−/HCO3− ions turned the reaction system alkaline, which promoted TC degradation as explained under the pH effect.66
| CO32− + H2O → OH− + HCO3− | (11) |
| ˙OH + HCO3− → CO3˙− + H2O | (12) |
| ˙OH + CO32− → CO3˙− + OH− | (13) |
| SO4˙− + HCO3− → HCO3˙ + SO42− | (14) |
| SO4˙− + CO32− → CO3˙− + SO42− | (15) |
Low concentrations of HPO4− did not caused any interference, but high concentrations improved TC degradation slightly because of the capability of HPO4− to break the O–O bond of PMS to yield SO4˙− radicals. In contrast to Cl−, CO32−, HCO3− and HPO42− ions, NO3− moderately inhibited the TC degradation via the scavenging of ˙OH, and SO4˙− radicals by NO3− ions to form less reactive nitrate radicals (eqn (16) and (17)).68
| NO3− + ˙OH → NO3˙ + OH− | (16) |
| NO3− + SO4˙− → NO3˙ + SO42− | (17) |
The TC degradation efficiency of 1D Bi2S3(0.2)/2D g-C3N4 reduced significantly from 98 to 57.7% with the addition of 10 mM of sodium salt of humic acid (HA). A possible reason could be due to the fact that HA not only competes for the active sites on the catalyst surface but also scavenges ROS more effectively because the functional groups on HA are more prone to attack from the SO4˙− and ˙OH radicals as previously described.69,70
4.3. Identification of reactive species and reaction mechanism
In order to identify the radical and non-radical reactive species formed during the entire TC degradation process, competitive scavenging experiments were carried out by adding tert-butyl alcohol (TBA), ethanol (EtOH), triethanolamine (TEO), L-histidine (L-His), and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL) as a specific scavenger for ˙OH, ˙OH and SO4˙−, h+, 1O2 and O2˙−, respectively (Fig. 6a). Quenching experiments revealed a moderate contribution of ˙OH radicals to the degradation of TC since the addition of TBA retarded the removal efficiency to 71%. The hydroxyl group of EtOH is expected to exhibit a strong ability to trap both ˙OH and SO4˙− radicals at high rates (k = (1.2–2.8) × 109 M−1 s−1 and k = (1.6–7.7) × 107 M−1 s−1, respectively). However, the addition of EtOH only showed a slightly stronger inhibitory effect compared to TBA in our experiment, indicating the minor role of SO4˙− radicals and their transfer to the other ROS. | ||
| Fig. 6 (a) Radical and non-radical quenching effect on TC degradation for the 1D Bi2S3(0.2)/2D g-C3N4 photocatalyst. Spin-trapping ESR spectra of 1D Bi2S3(0.2)/2D g-C3N4 for (b) DMPO–˙OH and DMPO–SO4˙, (c) DMPO–O2˙−, (d) TEMP–1O2, and (e) TEMP–h+. (f) Comparison of the Bi 4f XPS profile for fresh and spent 1D Bi2S3(0.2)/2D g-C3N4 samples. (g) Proposed TC degradation mechanism on the 1D Bi2S3(0.2)/2D g-C3N4 heterostructure under the synergistic action of light and PMS. | ||
Consistently, an electron paramagnetic resonance (EPR) spectra with a DMPO spin trapping agent revealed the characteristic peaks of DMPO˙–OH and DMPO˙–SO4 (Fig. 6b). This therefore provided direct evidence for the generation of ˙OH and SO4˙− radicals. Notably, relatively enhanced DMPO˙–OH signal intensity for the 1D Bi2S3(0.2)/2D g-C3N4/PMS/light system further confirmed the transfer of SO4˙− radicals to ˙OH radicals according to eqn (18). The O2˙− and 1O2 quenching effect was more obvious because the addition of 2 mM TEMPOL and 2 mM L-His suppressed the TC degradation to 66% and 57%, respectively. The appearance of intense characteristic peaks of DMPO–O2˙− (Fig. 6c) and TEMP–1O2 (Fig. 6d) in the EPR spectrum of the 1D Bi2S3(0.2)/2D g-C3N4/PMS/light system proved the formation of O2˙− and 1O2. The increase in TEMP–1O2 EPR signals of the 1D Bi2S3(0.2)/2D g-C3N4/PMS/light system, as compared to the 1D Bi2S3(0.2)/2D g-C3N4/light system, suggests that the activation of PMS generated additional 1O2 species. Hole (h+) quenching by a TEAO scavenger restricted the TC degradation to 76%, which implies their direct contribution by reacting with TC molecules or indirect contribution by reacting with HSO5, and O2˙− to form 1O2 species according to eqn (19) and (20). To our surprise, the intensity of the TEMP–h+ signal was significantly reduced when PMS was present (Fig. 6e), indicating part of the h+ was consumed by PMS to produce 1O2 species, which is consistent with the increased intensity of TEMP–1O2 signals. Quenching all the ROS (˙OH, SO4˙−, O2˙− and 1O2) with NaN3 retarded the TC degradation to 21%. Consequently, radical and non-radical species quenching together with EPR results conclusively proved that 1D Bi2S3(0.2)/2D g-C3N4 would efficiently trigger the activation of O2 and PMS to produce ROS; and further evidenced that effective TC degradation was achieved via the synergistic action of various reactive species as shown in Fig. 6g.
| SO4˙− + H2O → SO42− + ˙OH + H+ | (18) |
| HSO5 + h+ → 1/21O2 + ˙SO4 + H− | (19) |
| O2˙− + h+ → 1O2 | (20) |
4.4. DFT investigation on O2 and PMS activation
To further elucidate the activation mechanism, we simulated the adsorption behavior of O2 and PMS molecules on 2D g-C3N4, 1D Bi2S3, and separately on the α and β configurations of the 1D Bi2S3/2D g-C3N4 heterostructure. As the 1D Bi2S3(0.2)/2D g-C3N4/light system exhibited significant TC degradation under aerobic conditions, we first investigated the adsorption and activation of O2. The O2 molecule showed no adsorption on pristine g-C3N4 but strong adsorption on 1D Bi2S3 (ΔEad(O2) = −1.94 eV, Fig. S11a, ESI†), which stretched and eventually ruptured the O–O bond. In the 1D Bi2S3/2D g-C3N4-α and 1D Bi2S3/2D g-C3N4-β configurations, O2 adsorbed weakly on the 2D g-C3N4 sites, with ΔEad(O2) values of −0.06 eV and −0.07 eV, respectively (Fig. S11b and c, ESI†). For the 1D Bi2S3/2D g-C3N4-α model, two distinct O2 adsorption sites were identified: one through the unsaturated S atom (ΔEad(O2) = −0.93 eV, Fig. S11d, ESI†) and the other sandwiched between S and Bi atoms (ΔEad(O2) = −0.42 eV, Fig. S11e, ESI†). In contrast, for the case of the 1D Bi2S3/2D g-C3N4-β model, O2 adsorption solely occurred at the unsaturated S sites (ΔEad(O2) = −1.82 eV, Fig. S11f, ESI†). The observed slight increase in ΔEad(O2) at the 2D g-C3N4 sites of the heterostructure relative to its pristine 2D structure, and slight decrease at the 1D Bi2S3 sites relative to its pristine 1D structure could be attributed to the electronic charge redistribution as evidenced by XPS and CDD analysis. These findings confirm the crucial role of 1D Bi2S3/2D g-C3N4 heterostructures in modulating electronic properties, facilitating O2 adsorption and activation process.Next, we examined the possible adsorption configurations of the PMS molecule on the pristine 2D g-C3N4, 1D Bi2S3, and the 1D Bi2S3/2D g-C3N4 heterostructure. Simulations revealed weak interactions with 2D g-C3N4 (Fig. S12a, ESI†) but robust adsorption on 1D Bi2S3 (ΔEad = −3.98 eV, Fig. S12b, ESI†) through both Bi and S atoms. These findings confirm the high capability of 1D Bi2S3 towards PMS activation and are in good agreement with the observed TC degradation performance. To understand the PMS (HO–OSO3−) activation mechanism, we simulated the adsorption energy of the peroxide O–O bond in PMS on the 1D Bi2S3/2D g-C3N4-α model. Previous work has revealed that activation of PMS to generate SO4˙− and ˙OH radicals is accomplished through the adsorption of the peroxide O–O bond, either via the HO– or the –SO4 side.71 Our simulations disclosed that the peroxide O–O bond adsorbed strongly on both Bi and S sites on the 1D Bi2S3/2D g-C3N4-α heterostructure (Fig. 7). Notably on the Bi site (Fig. 7a), the O–O bond exhibited a unique adsorption mode with both the O atoms interacting effectively with the Bi atom (ΔEad(HSO5−) = −1.72 eV, system a(II) in Fig. 7a). During adsorption, the O–O bond was instantaneously stretched from 1.33 Å (gas phase) to 1.49 Å and the system energy decreased to −4.00 eV (system a(III) in Fig. 7a), which signifies barrier-less O–O bond breaking. The CDD analysis, as shown in the inset of Fig. 7a, revealed a significant charge depletion around the Bi atom (indicated by green isosurfaces) and charge accumulation around the O atoms of the adsorbed PMS (indicated by orange-purple isosurfaces). Bader charge analysis endorsed these findings and affirmed the transfer of 1.82e− from the Bi site to the adsorbed PMS molecule. This observation thus emphasizes that the Bi atom played a central role in facilitating the activation of PMS. The charge redistribution around the Bi site was also confirmed by performing XPS analysis of the spent catalyst. The B.E. of the Bi 4f5/2 and Bi 4f7/2 XPS peaks shifted towards higher values by 0.8 eV (Fig. 6f), indicating a reduction in electron density around the Bi atom, consistent with the charge transfer mechanism predicted theoretically.
 | ||
| Fig. 7 Schematic illustration of DFT simulated free energy changes against the reaction coordinate for PMS adsorption: (a) on the Bi site, and (b) on the S site, for the simulated 1D Bi2S3/2D g-C3N4-α model. Insets of (a) and (b) are the CDD analysis. | ||
Similarly, DFT simulations predicted strong adsorption of the peroxide O–O bond in PMS on the S site (Fig. 7b) through the O atom of –SO4 (ΔEad(HSO5−) = −3.18 eV, system b(II) in Fig. 7b). The robust adsorption resulted in the stretching of the O–O bond length to 2.54 Å, causing splitting of PMS into ˙OH and SO4˙− radicals. These radicals adsorbed on the same S atom, with adsorption energies of ΔEad(˙OH) = −0.44 eV and ΔEad(SO4˙−) = −2.74 eV respectively (systems b(III) and b(IV) in Fig. 7b). CDD analysis (inset in Fig. 7b) and Bader charge transfer calculations revealed the transfer of 1.86e− charge from the S site to PMS, supporting the cleavage of the O–O bond on the S site. The relatively mild adsorption energy of ˙OH radicals on Bi and S sites suggests that they can easily desorb and participate in the TC degradation process. This agrees with the increased intensity of EPR signals for ˙OH radicals measured in the presence of PMS. On the other hand, the stronger affinity of SO4˙− towards Bi and S sites indicates that it may react with water molecules (eqn (18)) to generate additional reactive oxygen species (ROS), as certified by the radical trapping experiments. These DFT findings together with experiment results predicted that the formation of the 1D Bi2S3/2D g-C3N4 heterostructure is beneficial for the efficient PMS activation and subsequent radical generation processes.
In light of radical trapping experiments, EPR analysis and DFT calculations, the plausible mechanism of TC degradation by the 1D Bi2S3(n)/2D g-C3N4 heterostructure in a light-assisted PMS-AOP can be rationally put forward as illustrated in Fig. 6g. First, the loading of 1D Bi2S3 nanostructures on the 2D g-C3N4 surface facilitated the light absorption, separation of charge carriers and electronic tuning of surface-active sites to collectively improve the activation of PMS activation and antibiotic degradation process. Second, optimized growth of 1D Bi2S3 effectively adjusted the interfacial interactions, creating a strong IEF that enabled rapid electron transfer from the 2D g-C3N4 substrate to the 1D Bi2S3 active sites, as supported by both XPS and DFT results. Third, the increased electron density on the Bi and S sites enabled strong binding with PMS molecules to form a stable transition state structure. As a result, compared to standalone 2D g-C3N4 and 1D Bi2S3, the in situ engineered 1D Bi2S3(n)/2D g-C3N4 heterostructure exhibited robust homolytic cleavage of the peroxide O–O bond to yield ˙OH and SO4˙− radicals at significantly lowered energy barriers as predicted by DFT calculations. Ultimately, the abundance of ROS produced by the simultaneous activation of O2 and PMS on the DBi2S3(0.2)/2D g-C3N4/light system collectively reacted with the pollutants, leading to their degradation and mineralization in aqueous media.
4.5. Stability and recyclability of photocatalysts
The structural stability and recyclability of photocatalysts is crucial for practical catalytic implementation. Thus, the stability of 1D Bi2S3(0.2)/2D g-C3N4 was investigated by carrying out TC degradation experiments for four consecutive cycles. After each cycle, the spent 1D Bi2S3(0.2)/2D g-C3N4 catalyst was recovered and washed with deionized water and ethanol to remove the adsorb organic and inorganic species and then dried in an oven at 80 °C. As shown in Fig. 5f, the TC degradation efficiency of recycled 1D Bi2S3(0.2)/2D g-C3N4 declined approximately to 92%, i.e. ∼6% lower than that of first cycle. The apparent loss in activity could be attributed to the loss of catalyst upon filtration and/or re-oxidation of Bi as evident by the post XPS results. The XRD diffraction patterns of the fresh and recycled samples exhibited the characteristic peaks of 1D Bi2S3 (Fig. S13a, ESI†), suggesting promising structural stability. Additionally, SEM and TEM images of the recycled catalysts revealed no visible structural changes (Fig. S13b–d, ESI†), indicating the high structural stability of the prepared 1D Bi2S3(0.2)/2D g-C3N4 heterostructure.Conclusion
In summary, we have designed a novel single-step in situ solid–gas reaction strategy to fabricate 1D Bi2S3(n)/2D g-C3N4 (n = 0.1, 0.2 and 0.3) heterostructures with adjustable densities of 1D Bi2S3 architectures. XPS and DFT studies verified the presence of strong interfacial interactions, which efficiently modified the electronic structure, especially for the Bi and S atoms at the interface. Notably, the optimized 1D Bi2S3(0.2)/2D g-C3N4 heterostructure exhibited exceptional performance, and degraded 98.5% and 99.9% of tetracycline (TC) and rhodamine B (RhB), respectively, exceeding most of the documented g-C3N4-based systems. DFT calculations unveiled that electronic modification of Bi and S sites at the 1D Bi2S3/2D g-C3N4 interface promoted the PMS adsorption and decreased the energy barrier for the cleavage of the peroxide O–O bond, thereby accelerating the ROS generation and pollutant degradation efficiency. This in situ strategy is general and could be used for the rational design of metal sulfide/g-C3N4 heterostructures to explore their potential for real-world energy and environmental remediation applications.Conflicts of interest
There is no conflict of interest to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
M. Mateen, N. Guo and W. S. Chin contributed equally to this work. The authors appreciate the financial support from the National University of Singapore (Chongqing) Research Institute and Chongqing Liangjiang New Area Municipal Government.References
- L. Zhao, W. Zhou, M. Wen, Q. Wu, W. Li, Y. Fu, Q. Zhu, S. Chen and J. Ran, Energy Environ. Mater., 2023, 6, e12299 CrossRef CAS.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- H. Long, S. F. Miller, C. Strauss, C. Zhao, L. Cheng, Z. Ye, K. Griffin, R. Te, H. Lee, C.-C. Chen and M. Lynch, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2498–E2505 CrossRef CAS.
- X. Zhang, B. Xu, S. Wang, X. Li, C. Wang, Y. Xu, R. Zhou, Y. Yu, H. Zheng, P. Yu and Y. Sun, Appl. Catal., B, 2022, 306, 121119 CrossRef CAS.
- M. Z. Akbari, Y. Xu, Z. Lu and L. Peng, Environ. Adv., 2021, 5, 100111 CrossRef CAS.
- E. Gómez, R. Cestaro, L. Philippe and A. Serrà, Appl. Catal., B, 2022, 317, 121703 CrossRef.
- H. Kim, S. Lee, H. W. Seo, B. Kang, J. Moon, K. G. Lee, D. Yong, H. Kang, J. Jung, E.-K. Lim, J. Jeong, H. G. Park, C.-M. Ryu and T. Kang, ACS Nano, 2020, 14(12), 17241–17253 CrossRef CAS PubMed.
- B. C. Hodges, E. L. Cates and J.-H. Kim, Nat. Nanotechnol., 2018, 13, 642–650 CrossRef CAS PubMed.
- S. Zhang, H. Zheng and P. G. Tratnyek, Nat. Water, 2023, 1, 666–681 CrossRef CAS.
- R. Guo, B. Xi, C. Guo, W. Liu, N. Lv and J. Xu, Environ. Funct. Mater., 2022, 1, 239–252 Search PubMed.
- T. Yu, H. Chen, T. Hu, J. Feng, W. Xing, L. Tang and W. Tang, Appl. Catal., B, 2024, 342, 123401 CrossRef CAS.
- Q.-Y. Wu, Z. W. Yang, Z. W. Wang and W. L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(16), e2219923120 CrossRef CAS PubMed.
- X. Yang, X. Xie, S. Li, W. Zhang, X. Zhang, H. Chai and Y. Huang, J. Hazard. Mater., 2021, 419, 126360 CrossRef CAS PubMed.
- U. Ushani, X. Lua, J. Wang, Z. Zhang, J. Dai, Y. Tan, S. Wang, W. Li, C. Niu, T. Cai, N. Wang and G. Zhen, Chem. Eng. J., 2020, 402, 126232 CrossRef CAS.
- J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54(6), 3064–3081 CrossRef CAS PubMed.
- N. Zrinyi and A. L.-T. Pham, Water Res., 2017, 120, 43–51 CrossRef CAS PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- Q. Zhou, C. Song, P. Wang, Z. Zhao, Y. Li and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2016, 120(13), e2300085120 CrossRef PubMed.
- R. Su, Y. Zhu, B. Gao and Q. Li, Water Res., 2024, 251, 121119 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116(12), 7159–7329 CrossRef CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27(13), 2150–2176 CrossRef CAS PubMed.
- G. F. S. R. Roch, M. A. R. da Silva, A. Rogolino, G. A. A. Diab, L. F. G. Noleto, M. Antonietti and I. F. Teixeira, Chem. Soc. Rev., 2023, 52, 4878–4932 RSC.
- W.-D. Oh, V. W. C. Chang, Z.-T. Hu, R. Goei and T.-T. Lim, Chem. Eng. J., 2017, 323, 260–269 CrossRef CAS.
- L. Li, H. Zeng, R. Tang, Z. Zhou, S. Xiong, W. Li, Y. Huang and Y. Deng, Appl. Catal., B, 2024, 345, 345123693 Search PubMed.
- H. Shi, Y. He, Y. Li and P. Luo, ACS Catal., 2023, 13(13), 8973–8986 CrossRef CAS.
- C. Lu, Y. Yang and X. Chen, Nano Lett., 2019, 19(16), 4103–4111 CrossRef CAS PubMed.
- W. Zhang, D. Xu, F. Wang and M. Chen, Nanoscale Adv., 2021, 3, 4370–4387 RSC.
- J. Miao, J. Song, J. Lang, Y. Zhu, J. Dai, Y. Wei, M. Long, Z. Shao, B. Zhou, P. J. J. Alvarez, P. J. J. Alvarez and L. Z. Zhang, Environ. Sci. Technol., 2023, 57(10), 4266–4275 CrossRef CAS PubMed.
- C. Zhai, Y. Chen, X. Huang, A. B. Isaev and M. Zhu, Environ. Funct. Mater., 2022, 1, 219–229 Search PubMed.
- V. Hasija, V.-H. Nguyen, A. Kumar, P. Raizada, V. Krishnan, A. A. P. Khan, P. Singh, E. Lichtfouse, C. Wang and P. T. Huong, J. Hazard. Mater., 2021, 413, 125324 CrossRef CAS PubMed.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8(3), 1701503 CrossRef.
- A. Zhu, L. Qiao, P. Tan and J. Pan, Inorg. Chem. Front., 2020, 7, 4754 RSC.
- Y. Ren, D. Zeng and W.-J. Ong, Chin. J. Catal., 2019, 40(3), 289–319 CrossRef CAS.
- G. Manna, R. Bose and N. Pradhan, Angew. Chem., Int. Ed., 2014, 53, 6743–6746 CrossRef CAS PubMed.
- J. Li, Z. Li, X. Liu, C. Li, Y. Zheng, K. Wai, K. Yeung, Z. Cui, Y. Liang, S. Zhu, W. Hu, Y. Qi, T. Zhang, X. Wang and S. Wu, Nat. Commun., 2022, 12, 1224 CrossRef PubMed.
- J. Wang, S. Lin, N. Tian, T. Ma, Y. Zhang and H. Huang, Adv. Funct. Mater., 2021, 31(9), 2008008 CrossRef CAS.
- H. Zhang, Z. Wang, J. Zhang and K. Dai, Chin. J. Catal., 2023, 49, 42–67 CrossRef CAS.
- D. Chen, J. Fang, S. Lu, G. Y. Zhou, W. Feng, F. Yang, Y. Chen and Z. Q. Fang, Appl. Surf. Sci., 2017, 426, 427–436 CrossRef CAS.
- B. Zhanga, H. Shia, Y. Yana, C. Liua, X. Hub, E. Liua and J. Fan, Colloids Surf., A, 2021, 608, 125598 CrossRef.
- A. Balakrishnan, J. D. Groeneveld, S. Pokhrel and L. Mädler, Chem. – Eur. J., 2021, 27, 6390–6406 CrossRef CAS PubMed.
- B. Shao, X. Liu, Z. Liu, G. Zeng, Q. Liang, C. Liang, Y. Cheng, W. Zhang, Y. Liu and S. Gong, Chem. Eng. J., 2019, 368, 730–745 CrossRef CAS.
- J. Fan, H. Qin and S. Jiang, Chem. Eng. J., 2019, 359, 723–732 CrossRef CAS.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7(30), 16850–16856 CrossRef CAS PubMed.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 RSC.
- F. Zhang, J. Zhang, J. Li, X. Jin, Y. Li, M. Wu, X. Kang, T. Hu, X. Wang, W. Ren and G. Zhang, J. Mater. Chem. A, 2019, 7, 6939–6945 RSC.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- R. Shen, J. Xie, X. Lu, X. Chen and X. Li, ACS Sustainable Chem. Eng., 2018, 6(3), 4026–4036 CrossRef CAS.
- N. Tian, Y. Zhang, X. Li, K. Xiao, X. Du, F. Dong, G. I. N. Waterhouse, T. Zhang and H. Huang, Nano Energy, 2017, 38, 3772–3781 CrossRef.
- C. Li, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, M. Kraft, G. Chen and R. Xu, Funct. Mater., 2017, 27, 1604328 CrossRef.
- P. Li, X. Zhang, C. Hou, Y. Chen and T. He, Appl. Catal., B, 2018, 238, 656–663 CrossRef CAS.
- Q. Hao, C. Xie, Y. Huang, D. Chen, Y. Liu, W. Wei and B.-J. Ni, Chin. J. Catal., 2020, 4, 249–258 CrossRef.
- J. Zhang, Y. Hu, X. Jiang, S. Chen, S. Meng and X. Fu, J. Hazard. Mater., 2014, 280, 713–722 CrossRef CAS PubMed.
- Y. Yan, Z. Zhou, W. Li, Y. Zhu, Y. Cheng, F. Zhao and J. Zhou, RSC Adv., 2014, 4, 38558–38567 RSC.
- S. M. Abdel-Moniem, M. A. El-Liethy, H. S. Ibrahim and M. E. M. Ali, Ecotoxicol. Environ. Saf., 2021, 226, 112808 CrossRef CAS PubMed.
- Z. Li, M. Wang, C. Jin, J. Kang, J. Liu, H. Yang, Y. Zhang, Q. Pu, Y. Zhao, M. You and Z. Wu, Chem. Eng. J., 2020, 392, 123789 CrossRef CAS.
- M. Li, X. Liu, Z. Xie, C. Du and Y. Su, Environ. Sci.:Adv., 2024, 3, 290–303 CAS.
- Y.-H. Guan, J. Ma, X.-C. Li, J.-Y. Fang and L.-W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- H. Li, H. Wang, Q. Gao, B. Han, K. Xia and C. Zhou, J. Mater. Chem. A, 2020, 8, 20953–20962 RSC.
- Q. Wang, Y. Shi, S. Lv, Y. Liang and P. Xiao, RSC Adv., 2021, 11, 18525–18538 RSC.
- X. Ao, W. Sun, S. Li, C. Yang, C. Li and Z. Lu, Chem. Eng. J., 2018, 361, 1053–1062 CrossRef.
- Y. Wu, G. Liang, W.-B. Li, X.-F. Zhong, Y.-Y. Zhang, J.-W. Ye, T. Yang, Z.-W. Mo and X.-M. Chen, Chem. Sci., 2024, 15, 9733–9741 RSC.
- L. R. Bennedsen, J. Muff and E. G. Søgaard, Chemosphere, 2012, 86, 1092–1097 CrossRef CAS PubMed.
- P. Wang, Y.-L. He and C.-H. Huang, Water Res., 2011, 45, 1838–1846 CrossRef CAS PubMed.
- R. Zhang, P. Sun, T. H. Boyer, L. Zhao and C.-H. Huang, Environ. Sci. Technol., 2015, 49(5), 3056–3066 CrossRef CAS PubMed.
- Y. Liu, X. He, X. Duan, Y. Fu, D. Fatta-Kassinos and D. D. Dionysiou, Water Res., 2016, 95, 195–204 CrossRef CAS PubMed.
- J. Iqbal, N. S. Shah, J. A. Khan, M. Naushad, G. Boczkaj, F. Jamil, S. Khan, L. Li, B. Murtaza and C. Han, Sep. Purif. Technol., 2024, 347, 127458 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS.
- B. Wu, Z. Wang, Y. Jia, N. Xu, L. Liao, C. Zhang, Z. Wang, Y. Shan, W. Feng and H. Xue, Environ. Technol. Innov., 2024, 35, 103736 CrossRef CAS.
- S. Zhang, Z. Shao and D. Wu, Arab. J. Chem., 2024, 17, 105483 CrossRef CAS.
- J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nh00265f |
| This journal is © The Royal Society of Chemistry 2025 |