
Synthesis planning for atomically precise metal nanoclusters
Jingkuan Lyu
 ab,
Jing Qian
ab,
Zhucheng Yang
ab and
Jianping Xie
*ab
ab,
Jing Qian
ab,
Zhucheng Yang
ab and
Jianping Xie
*ab
aJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Fuzhou 350207, P. R. China. E-mail: chexiej@nus.edu.sg
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore
First published on 3rd July 2025
Abstract
The rational design and synthesis of materials with tailored properties remains a long-standing goal in advanced materials science. Metal nanoclusters (MNCs), distinguished by their atomic precision and molecule-like properties—including discrete energy levels, strong photoluminescence, and high property tunability—represent promising platforms for applications spanning catalysis to biomedicine. This perspective presents a comprehensive synthesis planning framework comprising three critical stages, i.e., target design, route development, and condition optimization, systematically addressing MNC rational design and synthesis with special emphasis on thiolate-protected gold nanoclusters as exemplary systems. We first discuss design considerations for core and ligand shell engineering based on their profound influence on overall material properties. Subsequently, we examine methods and synthetic mechanisms for atomic-level tailoring of core and ligand shells to achieve target MNC synthesis. We then elucidate condition parameter tuning considerations based on their deterministic roles in reaction outcomes. While this structured approach provides a systematic methodology for MNC development, significant challenges persist owing to the high structural and synthetic complexity of MNCs. We then discuss the opportunities brought by recent advances in machine learning and high-throughput experimentation, which have demonstrated potential in addressing these challenges based on their superior computational and data analytical capabilities. We advocate for systematic adoption of this synthesis planning approach enhanced by data-driven methods, addressing inherent limitations in future development to better exploit these integrated approaches for accelerating rational MNC design and synthesis.
 Jingkuan Lyu | Jingkuan Lyu is a PhD student at the National University of Singapore under the supervision of Prof. Jianping Xie. He obtained his BS degree in chemical engineering from the same university. His research interest focuses on the automated synthesis of gold nanoclusters, integrating in situ and real-time characterization, robotic synthesis and machine learning. |
 Jing Qian | Jing Qian obtained his BS (2021) from the National University of Singapore. He is currently a PhD student under the supervision of Prof. Jianping Xie at NUS. His current research interests are centered on the structure prediction of metal nanoclusters with the assistance of machine learning algorithms. |
 Zhucheng Yang | Zhucheng Yang received his BS degree (2020) from the University of Chinese Academy of Sciences. He is currently pursuing his PhD degree under the supervision of Prof. Jianping Xie at the National University of Singapore. He is interested in the smart synthesis and ligand-driven programming of metal nanoclusters. |
 Jianping Xie | Dr Jianping Xie is a professor at the Department of Chemical & Biomolecular Engineering, National University of Singapore. He received his BS and MS from Tsinghua University and his PhD from the Singapore-MIT Alliance. Since joining NUS in 2010, he has led research on water-soluble noble metal nanoclusters for biomedical and catalytic applications. With 280+ publications, 37 |
1. Introduction
Rational design and synthesis represents the strategic process by which scientists systematically design and execute the efficient synthesis of desired compounds with specific properties. In contrast to traditional trial-and-error approaches that rely on serendipitous discovery, this methodology begins with identifying target materials possessing specific desired characteristics, guided by established structure–property relationships. Synthetic mechanistic insights are then leveraged to develop viable pathways between available precursors and the target products—exemplified by retrosynthetic analysis in organic chemistry. The field of drug discovery has adopted the design-make-test-analyse (DMTA) framework to implement this systematic approach, embracing the iterative nature of rational design through continuous validation and refinement of design decisions via experimental feedback.1 Crucially, data generated throughout these cycles not only inform immediate synthetic objectives but also enrich the broader knowledge base, creating a cumulative foundation for increasingly sophisticated materials development strategies.Atomically precise metal nanoclusters (MNCs) represent a unique class of nanomaterials distinguished by their monodispersity and well-defined molecular structures. These ultrasmall entities consist of a metallic core, typically below 3 nm in diameter, stabilized by a shell of metal–ligand motifs. Single-crystal X-ray diffraction (SCXRD) and mass spectrometry characterization enable precise determination of their structure and composition, often expressed in the form [Mn(L)m]q, where n and m denote the number of metal atoms (M) and ligands (L) respectively, and q is the net charge. Owing to their quantum confinement effects—arising when cluster dimensions become comparable to the electron de Broglie wavelength—MNCs exhibit molecule-like behaviours such as electron transition between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO),2 strong photoluminescence (PL),3–7 and intrinsic chirality,8,9 rendering them promising in diverse applications including catalysis,10–12 biomedicine,13–15 and photosensitizers.16
The molecule-like character renders MNC properties highly reliant on the cluster's size, structure, and composition, all engineerable via atomic-level manipulation of the metallic core and ligand shell.17–22 For instance, the growth of the bi-icosahedral metal core in [Au25(PR3)10(SR)10Cl2]+ to a tri-icosahedral structure in [Au37(PR3)10(SR)10Cl2]+ (where PR3 represents phosphine ligands and SR represents thiolate ligands) significantly reduces the optical energy gap from 1.73 eV to 0.83 eV.23 In another example, the peroxidase mimic catalytic activity of Au15(SR)13 was enhanced by incorporating ligands with higher electron-withdrawing capabilities.24 Such tunability not only enables fundamental structure–property relationship development,25–27 but also inspires advances in synthetic methods and growth mechanisms toward stepwise synthesis and control of MNCs,28,29 as exemplified by the conceptualization of “total synthesis” of MNCs.30 Understanding MNC formation processes has revealed opportunities for customized MNC synthesis through adjusting synthetic condition parameters,31–33 such as pH tuning to promote less stable species like highly luminescent Au22(SR)18,34 or exploiting additive–ligand interactions to control reaction kinetics.35 However, the chemical complexity, coupled with intricate parameter interdependencies, poses significant challenges to advancing rational design and synthesis towards higher precision. The limited availability of high-quality structural data further exacerbates these difficulties. Recent advances in data-driven methodologies—integrating machine learning (ML) algorithms with high-throughput experimentation (HTE)—have demonstrated promising capabilities in modelling complex relationships for rational design and synthesis across diverse materials.36–43 Autonomous laboratories exemplify fully integrated systems enabling data-guided material discovery, as demonstrated by Ceder and co-workers, who successfully identified 41 novel compounds.44 These developments provide timely inspiration for adopting data-centric strategies in MNC research.45–47
Targeting rational design and synthesis of MNC, this perspective presents a comprehensive synthesis planning framework, which comprises three critical stages: target design (designing the MNC core and ligand shell for desired functionality), route development (mapping the synthetic route from available precursors to targets), and condition optimization (fine-tuning reaction condition parameters for precise synthesis control) (Fig. 1). We emphasize thiolate-protected Au nanoclusters as exemplary systems given their extensive characterization and established relevance. We highlight the challenges faced and discuss how emerging data-driven approaches can address these barriers. Finally, we conclude with our perspectives on how to better exploit this integrated approach in future development. While existing reviews have focused on individual aspects of MNC design and synthesis,7,19,30,48 no comprehensive framework exists that systematically integrates target design, route development, and condition optimization into a unified synthesis planning approach. Although a recent perspective presents opportunities in MNC synthesis enabled by the integration of automation, data and advanced algorithms,45 it lacks in-depth discussion of theoretical considerations regarding the synthetic and structural complexities of MNCs. Moreover, previous reviews on data-driven nanomaterial discovery, such as those focusing on nanoparticles, do not address the atomic-precision considerations and molecular-like property dependencies essential for MNCs.49,50 By establishing this integrated framework which systematically decomposes MNC rational design and synthesis into tractable relationship modelling problems amenable to data-driven methodologies, this perspective aims to catalyse a paradigm shift toward predictive MNC synthesis that bridges the gap between the precision of molecular design and the complexity of nanomaterial engineering, ultimately enabling more efficient discovery of atomically precise MNCs with tailored properties.
 | ||
| Fig. 1 Schematic overview of the synthesis planning framework of MNCs, including three main processes, namely, target design, route development and condition optimization, and the opportunities endowed by data-driven approach adoption, in terms of structure prediction acceleration, global condition optimization modelling, synthesis–property relationship modelling, and model generalization. Colour label: yellow, core Au or Au cation; orange, motif Au; pink, S. The detailed flowchart below illustrates the individual steps during the three critical stages of synthesis planning for MNCs. After each successful synthesis, all relevant data are updated to the data libraries for future MNC developments, forming an iterative cycle. | ||
2. Target MNC design
Target design represents the critical first step in MNC synthesis planning, wherein researchers define the structural features needed to achieve specific functional properties prior to synthesis. Different from conventional trial-and-error approaches, rational target design leverages established structure–property relationships to anticipate how microscopic structural features will determine macroscopic characteristics.For atomically precise MNCs, design efforts focus on two interdependent structural domains: the metallic core and the surrounding ligand shell. Each component plays distinct yet complementary roles in determining the MNC's fundamental properties. Understanding these relationships enables researchers to design MNCs with tailored functionality for specific applications.
The metallic core—defined by its size, structure, and composition—predominantly governs the electronic structure of MNCs.2,51 Due to quantum confinement effects at the nanoscale, MNCs exhibit discrete electronic energy levels rather than continuous bands, resulting in molecule-like optical absorption profiles which are strongly affected by the metallic core.52 For instance, Au42(SCH2Ph)32 (SCH2Ph represents benzyl mercaptan) with its distinctive rod-like core structure demonstrates strong near-infrared absorption at 808 nm, yielding superior photothermal conversion efficiency compared to more spherical MNC species.53 The core can also significantly influence catalytic activity via size effects, following predictable trends that inform design strategies. Zheng et al. demonstrated that catalytic performance in resazurin reduction systematically increases as Au nanocluster size decreases from Au25 to Au18 to Au15.54 Similarly, Li et al. observed enhanced electrochemical CO2 reduction activity with decreasing Au nanocluster dimensions.55 Moreover, variations in core composition can tune MNC properties by modulating the HOMO–LUMO gap, further expanding the design landscape.56
While the core determines electronic structure, the protective ligand shell offers additional opportunities for property modulation. In addition to stabilizing the metal core, ligands influence solubility, aggregation behaviour, and interfacial interactions, which can markedly affect properties such as PL, particularly through solvent-mediated effects.4 Furthermore, the ligand can also alter MNC properties by exerting electronic effects. For example, replacing electron-donating 3-mercapto-2-methylpropanoic acid (MMPA) with N-acetylcysteine (NAC) in Au15(SR)13 nanoclusters enhances the peroxidase-like activity, illustrating the potential for ligand-directed catalytic tuning.24
Thus, rational design of MNCs demands integrated consideration of both core architecture and ligand shell composition to achieve desired property profiles. In the following section, we elaborate on specific principles for templated core design and ligand shell engineering that enable predictive synthesis of MNCs with targeted properties. We also address current challenges in translating these design principles into synthetic strategies that reliably produce the intended structures.
2.1. Templated core design
Stability is one of the fundamental considerations in the design of MNCs. From the valence electronic structure point of view, most of the Au nanoclusters are found with even number of valence electrons. The valence electron count can be calculated by N* = n − m − q (where n, m, and q share the same meaning with those in [Mn(L)m]q). These valence electrons can be considered to be contributed by stable basic building blocks, including Au4 tetrahedra,57 Au3 triangles58 and Au13 icosahedra,23 which constitute the core structures of Au nanoclusters. Both Au4 tetrahedra and Au4 triangles contribute 2 valence electrons (2e−) while the Au13 icosahedron contributes 8 valence electrons (8e−). These building blocks can fuse and stack with each other, resulting in numerous evolution pathways. These pathways function as templates that guide the prediction, design and synthesis of MNCs with desired sizes and structures. Subsequent composition adjustments of these cores allow further property tailoring for better PL or catalytic performance.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)18 (tBuPhCC represents 4-(tert-butyl)phenylacetylene).62 These possible arrangements result in numerous evolution pathways of MNCs.
C)18 (tBuPhCC represents 4-(tert-butyl)phenylacetylene).62 These possible arrangements result in numerous evolution pathways of MNCs.A representative face-centred cubic (fcc) evolution pathway begins with Au20(SR)16,63 progressing through Au21(SR)15 to [Au23(SR)16]−,64,65 where the core expands via symmetric Au3 additions.60 As illustrated in Fig. 2a, the Au10 core in Au21(SR)15 and subsequently the Au13 core in [Au23(SR)16]− arise from successive additions to a central Au7 bitetrahedral backbone. A similar template exists featuring double Au7 in the backbone which underlies Au28(SR)20 (8e−)66 and Au30(SR)18 (12e−)67 (Fig. 2b). Based on these priorly obtained structures, Xiong et al. predicted that the 10e− species Au29(SR)19, which was previously detected in mass spectroscopy (MS) experiments by Dass and colleagues,68 would exhibit an intermediate core structure comprising 2 Au7 and 1 additional Au3.69 Li et al. later confirmed the hypothesis via SCXRD, which exemplifies the predictive power of templated core design.58 Consistently, the HOMO–LUMO gap narrows as the core size of the species in the template increases (∼1.7 eV in Au28(SR)20 to 1.25 eV in Au30(SR)18).58,66 This evolution mode is further supplemented by the discovery of Au36(SR)22, which features an additional Au3 unit.70 It would be delightful to see the discovery of more species that behave according to this evolution template.
 | ||
| Fig. 2 Illustrations of various core growth templates of Au nanoclusters depicting the Au core structural evolution with stepwise addition of (a) the Au3 unit onto the Au7 backbone and (b) the Au3 unit onto the Au14 backbone. (c) Template with stepwise addition of 2 Au4 units forming a double-helix superstructure, and (e) their respective UV-Vis spectra. The mass formula with Au core size and valence electron number N* is indicated below each structure. Reprinted with permission from ref. 57. Copyright 2016, American Chemical Society. (d) Template with stepwise addition of Au13, and (f) UV-Vis-NIR spectra of Au25(I) and Au13(II) nanoclusters. Insets: Spectra on the photon energy scale: Au25(III) and Au13(IV). The Au core sizes are indicated below each structure. (g) UV-Vis-NIR spectra of the Au37 nanocluster. Inset: Spectrum on the photon energy scale. Reprinted with permission from ref. 23. Copyright 2015, American Chemical Society. | ||
An alternative uniform anisotropic growth was discovered starting from Au28(TBBT)20 (TBBT stands for 4-tert-butyl-benzenethiolate),71 which grows in 4e− steps through the addition of two Au4 units per stage. Unlike the parallel Au7 units in the earlier Au28(SR)20 isomer protected by S-c-C6H11 (cyclohexanethiolate), this isomer features crossed Au7 units forming a distinct architecture. Following the successful synthesis of Au28(TBBT)20, Jin and colleagues extended this template to Au36(TBBT)24,28 Au44(TBBT)28,57 Au52(TBBT)32,61 revealing a uniform core expansion via stacking of 2 Au4 units into a double-helix superstructure (Fig. 2c). Optical gaps decrease progressively from Au28, Au36, Au44, to Au52 (Eg = 1.77, 1.76, 1.51, and 1.39 eV, respectively) as reflected in red-shifted UV-Vis absorption peaks (absorption peaks at 702, 704, 820, and 890 nm, respectively) (Fig. 2e). Notably, all species in the template exhibit chirality with quasi-D2 symmetry except Au36, whose cubic geometry bestows it higher symmetry with quasi-D2d Au–S framework with no chirality. This template inspired theoretical design of new analogues, such as Au60(SR)36, Au68(SR)40, and Au84(SR)48.72,73 Au76(SR)44 synthesized by Takano et al.74 was proposed to follow the same trend, exhibiting 9 Au4 layers.75 This evolution template also inspired Liu et al., who demonstrated rational synthesis of a Au36(SR)24 isomer based on de novo design.76 The core structure features a 2-dimensional growth pattern proposed based on theoretical calculations. It would be valuable to observe further studies that provide verification for this hypothesized template.
Other less-explored pathways include ring-like tetrahedral growth around a Au7 backbone, observed in species like Au22(tBuPhCC)18, Au34(S-c-C6H11)22,77 and Au40(o-MBT)24 (o-MBT represents 2-methylbenzenethiolate).61 However, a structural gap remains between Au22 and Au34—potentially bridged by a nanocluster comprising a Au7 backbone encircled by an incomplete ring of two vertex-sharing tetrahedra—suggesting the possible existence of a novel structural isomer within the Au28(SR)20 family.
A vertex-sharing template is evident in mono-, bi-, and tri-icosahedral species such as [Au13(dppe)5Cl2]3+,82 [Au25(PPh3)10(SR)5Cl2]2+,80 and [Au37(PPh3)10(SC2H4Ph)10Cl2]+ (where dppe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 1,2-bis(diphenylphosphino)ethane, PPh3 = triphenylphosphine, and SC2H4Ph = phenylethylthiolate),23 respectively (Fig. 2d). Interactions among Au13 units lead to distinct electronic transitions while preserving the electronic features arising from a single Au13 unit. This is exemplified by the UV-Vis absorption peaks at 1230 and 795 nm resulting from the interacting tri-icosahedron in Au37 and the 670 nm peak from bi-icosahedral interaction in Au25 (Fig. 2f and g). As expected, the optical energy gap decreases with increasing core size (Eg ∼ 1.96 eV, 1.73 eV, and 0.83 eV respectively). As the core evolves further, a pentameric Au60 ring is formed.83 However, variations in the Au13 unit arrangement significantly alter the electronic transition behaviour. The absorption peak arising from the interaction within the pentamer structure is observed at 850 nm, different from the double-peak behaviour in Au37. This suggests that further studies—especially on tetra-icosahedral species—could clarify the evolution of optical properties. In thiolate-protected Au nanoclusters, while mono-icosahedral [Au25(SR)18]− and bi-icosahedral Au38SR24 have been discovered,2,81 larger analogues have not yet been reported. Given the distinct chiral behaviours of [Au25(SR)18]− and Au38(SR)24,9 uncovering structures of larger species in the template could yield new insights into chirality trends.
= 1,2-bis(diphenylphosphino)ethane, PPh3 = triphenylphosphine, and SC2H4Ph = phenylethylthiolate),23 respectively (Fig. 2d). Interactions among Au13 units lead to distinct electronic transitions while preserving the electronic features arising from a single Au13 unit. This is exemplified by the UV-Vis absorption peaks at 1230 and 795 nm resulting from the interacting tri-icosahedron in Au37 and the 670 nm peak from bi-icosahedral interaction in Au25 (Fig. 2f and g). As expected, the optical energy gap decreases with increasing core size (Eg ∼ 1.96 eV, 1.73 eV, and 0.83 eV respectively). As the core evolves further, a pentameric Au60 ring is formed.83 However, variations in the Au13 unit arrangement significantly alter the electronic transition behaviour. The absorption peak arising from the interaction within the pentamer structure is observed at 850 nm, different from the double-peak behaviour in Au37. This suggests that further studies—especially on tetra-icosahedral species—could clarify the evolution of optical properties. In thiolate-protected Au nanoclusters, while mono-icosahedral [Au25(SR)18]− and bi-icosahedral Au38SR24 have been discovered,2,81 larger analogues have not yet been reported. Given the distinct chiral behaviours of [Au25(SR)18]− and Au38(SR)24,9 uncovering structures of larger species in the template could yield new insights into chirality trends.
In addition to the abovementioned templates, additional pathways have been proposed but await verification from successful synthesis. For example, a bi-tetrahedral growth template is inferred from the evolution from Au20(SR)16 to the Au28(SR)20 isomer in the first template. Moreover, Au11 units are recurring in species like Au11(PPh3)7Cl3 and [Au20(PPhpy2)10Cl4]2+ (PPhpy2 = bis(2-pyridyl)-phenylphosphine) where 2 edge-sharing Au11 units are observed.84,85
 | ||
| Fig. 3 (a) Illustrations of two-step spherical jellium potentials for dopant Au13 doped with an atom of lower (left) or higher (right) valent element. Reprinted with permission from ref. 88. Copyright 2021, American Chemical Society. (b) Illustrations of the electronic energy levels of undoped [Au25]− and doped [PtAu24]0. α, β and γ denote different optical transitions which can be observed in their UV-Vis spectra. Reprinted with permission from ref. 87. Copyright 2017, Springer Nature Limited. | ||
Doping induced HOMO–LUMO gap variation also affects catalytic performance. When a single Pt atom is doped at the centre of the Au13 icosahedral core in [Au25(SC6H13)18]− (SC6H13 represents 1-hexanethiol), the 8e− core is changed to 6e− causing a Jahn–Teller-like distortion of the PtAu12 core accompanied by 1P orbital splitting (Fig. 3b).87 The resultant reduction potential matches well with the reduction potential of a proton, significantly enhancing its electrocatalytic performance in hydrogen evolution reaction. Moreover, due to the difference in electronegativity, the dopant could change the electron density of the MNC, thereby altering the adsorption behaviour during catalysis. Ag25 centrally doped by Au in the Ag13 icosahedral core exhibits significant electron donation from Ag to Au which creates more positively charged surface Ag facilitating the adsorption of electron-rich alkynes in the carboxylation of CO2 with terminal alkynes.91
2.2. Functionalization via ligand shell design
Ligand shell design plays a pivotal role in MNC functionalization for various applications including PL,92 catalysis,93 and biomedicine.13 A wide range of ligand types—thiolates,2 phosphines,84 α-alkynyls,62 sulfido,94 halide,80 N-heterocyclic carbene (NHC),95 multiple N donor ligand (MND),96 and carboranes97—have been employed in MNC diversification. The primary role of the ligand shell in MNCs is to stabilize the metal core, preventing aggregation and further growth. Taking the thiolate-protected Au nanocluster as an example, the strong Au–SR covalent bonds confer greater stability than the weaker Au–ligand interactions present in Au nanoparticles.98 Using ligands with distinct Au–ligand bonds99 or various ligand–ligand interactions100,101 can effectively alter MNC stability and enable the formation of different stable sizes.102 While comprehensive reviews on ligand design strategies exist,20,27 this section focuses on three key considerations on target nanocluster design for desired properties integrating recent developments: synergy with the metal core, intra-shell interactions, and interactions with external molecules.C)28 (PhCC represents ethynylbenzene) and Au44(TBBT)28 (Fig. 4b),103 due to the more active participation of the PhCC ligand in the frontier orbitals endowed by its different binding mode, according to theoretical studies.104 Furthermore, ligand's contribution to the electronic structure gives rise to ligand-to-metal charge transfer (LMCT) through the Au–ligand bond, which influences MNC PL performance. In [Au25(SR)18]−, replacing 1-hexanethiol with more electron-donating ligands such as 1-dodecanethiol and 2-phenylethanethiol (PET) increases fluorescence QY by 2.5 and 5 times respectively.92 Besides, a delocalized electron from the electron-rich ligand body of glutathione (GSH) can be directly donated to the metal core of [Au25(SR)18]−, boosting QY by ∼100 times relative to 1-hexanethiol.
 | ||
| Fig. 4 (a) UV-Vis absorption spectra of Au25 nanoclusters protected by various thiolates or selenolate. Reprinted with permission from ref. 17. Copyright 2021, Wiley-VCH. (b) UV-Vis spectra of Au44 nanoclusters in CH2Cl2. Reprinted with permission from ref. 103. Copyright 2017, Wiley-VCH. (c) Illustration of the core structures of Au24 nanoclusters. The mass formula with Au core size and valence electron number N* is indicated below each structure. | ||
Secondly, the choice of ligand determines the geometric structure of the metal core. For example, Au24(SCH2Ph-tBu)20 possesses a bi-tetrahedral Au8 core with anti-prismatic face-joint Au4 units,105 whereas Au24(SeC6H5)20 shares the same core size but different geometry. Its 2 cross-joint tetrahedral Au4 units have the same orientation in space (Fig. 4c).106 The same effects are observed in Au21 nanoclusters where the Au10 core structure differs between S-Adm (adamantanethiolate) and StBu (tert-butylthiol)-protected nanoclusters.107
CAr)18]− (Ar = 3,5-bis(trifluoromethyl)phenyl) whose 3 of the 12 Au atoms in staple motifs are twisted about 60° compared to its thiolate-protected counterpart, giving rise to its intrinsic chirality with a D3 symmetry (Fig. 5a and b).108 Ligand rigidity can also influence the thermodynamically stable surface structures of MNCs. In the case of Au28(SR)20, S-c-C6H11 stabilizes more protruded staple motifs compared to TBBT, which enhances carbon monoxide (CO) adsorption at the motif Au atoms during catalytic CO oxidation reaction.66
 | ||
| Fig. 5 Illustration of the different arrangements of three V-shaped surface motif staples in (a) alkynyl-protected and (b) thiolate-protected Au25 nanoclusters. Colour label: orange, blue and green spheres, Au; yellow spheres, sulfur; grey spheres, carbon. (c). Reprinted with permission from ref. 108. Copyright 2018, Wiley-VCH. Simulated structures of S-c-C6H11-protected and SG-protected Au18(SR)14 in the gas or solution phase. Reprinted with permission from ref. 100. Copyright 2018, American Chemical Society. | ||
Ligand–ligand interactions can also be utilized to rigidify the ligand shell for PL enhancement effects. Stronger π–π stacking interactions between the adjacent aromatic ligands, in addition to the bulkier ligand body, contribute to the more rigidified structure which suppresses the high-frequency optical phonons in [Au25(SR)18]− protected by aromatic ligands, leading to enhanced PL compared to [Au25(SR)18]− protected by non-aromatic ligands.109 Likewise, the rigid surface and strong internal π–π stacking interactions in NHC-stabilized Au13 result in superior QYs of 16%, much higher than those of other nanoclusters with the same Au13 core.110 Ligand–ligand hydrogen bonds can also provide surface structural rigidity. The extensive intermolecular hydrogen bonds between the H-donors (–OH and –NH) and the H-acceptor (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) functional groups between adjacent ligands led to smaller structural changes between the solid and solution phases for glutathione-protected Au18(SR)14 as compared to the S-c-C6H11-protected nanocluster (Fig. 5c), and superior PL performance.100
O) functional groups between adjacent ligands led to smaller structural changes between the solid and solution phases for glutathione-protected Au18(SR)14 as compared to the S-c-C6H11-protected nanocluster (Fig. 5c), and superior PL performance.100
MNC properties can be modulated through carefully designed electrostatic interactions. For instance, ligand's electrostatic interactions with cationic surfactant CTA+ (cetyltrimethylammonium) induced [Au25(p-MBA)18]− (p-MBA stands for para-mercaptobenzoic acid) isomerization.113 By means of the synergistic effect of electrostatic interactions (between the deprotonated carboxylic groups and the positively charged ammonium headgroup) and CH⋯π interactions (between the aromatic ring of p-MBA ligands and the small carbon tails at the ammonium headgroup), the CTA+ ions adsorbed onto the nanocluster surface, forming a double layer structure (Fig. 6a). The resultant surface rigidification effect stretches and rotates the inner metal core, forming a new isomer with distinct optical properties (Fig. 6b). Likewise, multi-layer ligand engineering was applied to ATT (6-Aza-2-thiothymine)-stabilized Au10 through hydrogen bonding with ARG (L-arginine), which is further ion-paired with TOA+ (tetraoctylammonium), effectively suppressing kernel vibrations.114
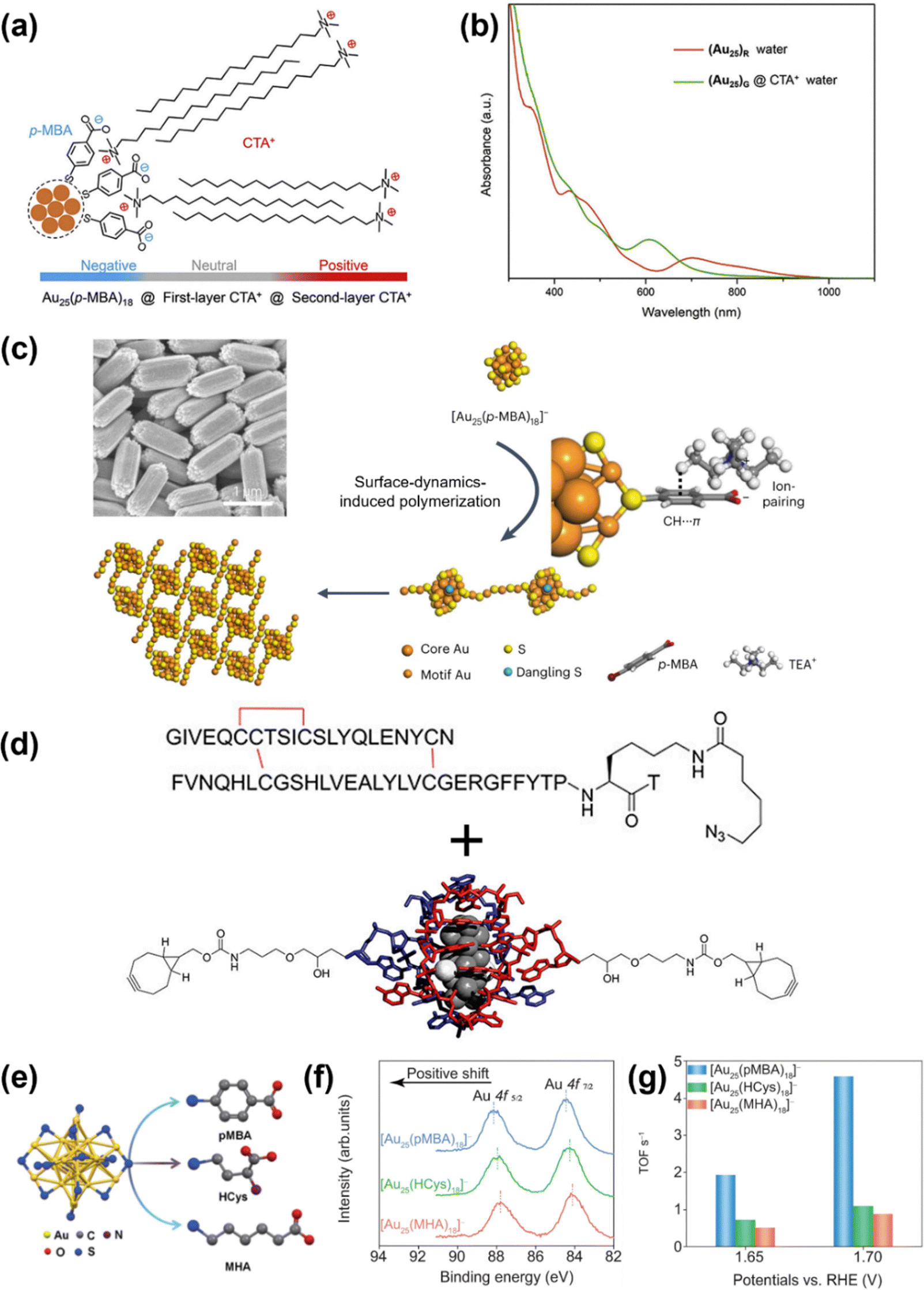 | ||
| Fig. 6 (a) Schematic illustration of the adsorption of CTA+ ions onto the [Au25(p-MBA)18]− nanocluster surface. (b) UV-Vis absorption spectra of [Au25(p-MBA)18]− and [Au25(p-MBA)18]− after isomerization. Reprinted with permission from ref. 113. Copyright 2021, Elsevier. (c) Field-emission scanning electron microscopy (FESEM) image of hexagonal rod-like supercrystals formed by [Au25(p-MBA)18]− and the schematic illustration of the surface rearrangement and crystallization processes induced by CH⋯π and ion-paring interactions between the ligand at the apex position and TEA+. Reprinted with permission from ref. 116. Copyright 2023, Springer Nature Limited. (d) Schematic illustration of the conjugation between human insulin and the bicyclononyne-terminated DNA–Ag16 nanocluster. Reprinted with permission from ref. 130. Copyright 2023, American Chemical Society. (e) Schematic illustration of [Au25(SR)18]− nanoclusters protected by various ligands, (f) their high-resolution Au 4f XPS (X-ray photoelectron spectroscopy) spectra, and (g) their turn-over frequency values at 1.65 and 1.7 V. Reprinted with permission from ref. 131. Copyright 2023, Springer Nature Limited. | ||
Intercluster interactions are pivotal to MNC self-assembly behaviour and supercrystal formation.115 [Au25(p-MBA)18]− crystallization was facilitated by combined CH⋯π and ion-paring interactions between p-MBA ligands at the apex position and tetraethylammonium cations (TEA+) (Fig. 6c).116 These interactions achieved an intricate balance, detaching the SR–[Au(I)–SR]2 motifs from the surface of [Au25(p-MBA)18]− to form SR–[Au(I)–SR]4 linkers that connected adjacent distorted monomers into an orderly three-dimensional architecture. Similarly, controlled van der Waals interactions between the surface ligands of Au29(S-Adm)19 enabled helical assembly of the MNC monomers.58 These ordered superstructures serve dual functions: facilitating structural determination for structure–property relationship construction and inducing synergistic properties distinct from those of individual MNC monomers. The dual ligand system in Au4Ag13(dppm)3(SR)9 (dppm = bis(diphenylphosphino)methane and SR specifically denotes 2,5-dimethylbenzenethiolate in this case) exemplifies this principle, where six pairs of intercluster CH⋯π interactions between dppm aromatic rings and SR aromatic hydrogens promoted supercrystal formation while compact packing significantly enhanced radiative transitions through intramolecular vibration/rotation restriction.117 Interestingly, the supercrystal formed by CH⋯π and π⋯π interactions between dppp (1,3-bis-diphenylphosphine propane) ligands in [Pt1Ag18(S-Adm)2(dppp)6Cl6]2+ not only exhibited crystallization-induced PL enhancement but also demonstrated promising optical waveguide performance with low optical loss and polarized emission, attributed to its distinct packing mode.118
Another crucial consideration is ligands’ interaction with the solvent environment, especially for bio-related applications. Ligands such as peptides, DNA, and proteins contribute to biocompatibility necessary in applications such as biosensing, bioimaging and photothermal therapy.119–122 For inherently hydrophobic ligands such as NHC, biocompatibility can be achieved through functionalization with polar water-soluble functional groups such as triethylene glycol monomethyl ether.123 Bioconjugation represents another effective strategy to achieve biocompatibility. Zhang et al. demonstrated stoichiometric conjugation between [Au25(SR)18]− and BSA (bovine serum albumin) via electrostatic interactions and hydrogen bonds which simultaneously enhanced NIR-II emission and potential theranostic applicability.124 Host–guest chemistry offers additional opportunities, as demonstrated by complex formation between β-cyclodextrin (CD) and the 4-(tert-butyl)benzyl mercaptan protected Au25 nanocluster.125
Exploiting the chemical reactivity of ligands also serves as a powerful tool for property enhancement and functional diversification. Deng et al. demonstrated that intracluster cross-linking between GSH ligands in Au22(SG)18 via Bis-Schiff base linkage formation enhanced the PL QY over 11-fold.126 Similarly, [Au11(PNHP)4Br2]+ (PNHP stands for [PPh2(CH2)2]2NH where PPh2 represents diphenylphosphine) was functionalized through amidation with acyl chlorides to simultaneously introduce chirality and desired functional groups.127
Recent advances include click chemistry-compatible MNCs, such as [Au25(SR)18]− protected by azide-functionalized thiolate (SCH2CH2-p-C6H4-N3).128 Its reactivity in strain-promoted azido–alkyne cycloaddition (SPAAC) was found to be affected by ligand regioisomerism with the para isomer exhibiting the highest reaction rates while the ortho isomer failed to stabilize the nanocluster during reaction.129 In a complementary approach, reactivity in SPAAC is incorporated into the DNA–Ag16 nanocluster by attaching ring-strained alkyne bicyclononyne to the DNA ligands (Fig. 6d).130 When conjugated with azido-modified human insulin, the nanocluster maintained its original photophysical properties while enabling specific staining of the Chinese hamster ovary membranes with promising stability.
Interactions in terms of molecular adsorption are crucial in designing MNCs with desired catalytic performance. Liu et al. reported enhanced OH− adsorption during oxygen evolution reaction in alkaline medium by ligands with stronger electron-withdrawing capability.131 Compared to [Au25(SR)18]− protected by MHA (6-mercaptohexanoic acid) and H-cys (homocysteine) (Fig. 6e), p-MBA in [Au25(p-MBA)18]− induced more positively charged Au(I) active sites in the surface motifs (Fig. 6f), rendering nearly 4 times catalytic performance enhancement (Fig. 6g). Through Tafel slope analysis and in situ Raman spectroscopy, the authors determined that the difference in charge density around the active site altered the rate determining step: decomposition of Au–O–OH for [Au25(p-MBA)18]− versus deprotonation of Au–OH for the other nanoclusters.
3. Route development
Route development, the second critical step in synthesis planning, involves designing the reaction pathway from commercially available starting materials to the desired target material. In organic chemistry, this step typically employs retrosynthetic analysis—working backward from the target molecule to identify viable precursors based on established reaction libraries. Multiple possible routes are then evaluated using criteria such as cost-effectiveness, safety, and synthetic feasibility.41,132 The synthesis of ketoprofen, a nonsteroidal anti-inflammatory drug, illustrates this approach. To avoid regulated substances, a viable retrosynthetic pathway begins with commercially available α,β-dibromopropanoate and benzophenone. These precursors initially undergo dehydrohalogenation and iodination, respectively, followed by a coupling reaction. The synthesized intermediate is subsequently hydrolysed and hydrogenated to yield the desired pharmaceutical product.133MNC synthesis planning adopts a similar conceptual framework but requires specialized approaches due to fundamental differences in reaction mechanisms. Unlike organic synthesis with its discrete covalent bond transformations, MNC formation involves complex formation processes with numerous concurrent reactions and intermediates. While organic chemistry can draw upon extensive libraries of well-characterized reactions, the high complexity and rapid kinetics of nanocluster formation have hindered the development of analogous elementary reaction libraries for MNCs.
Despite these challenges, researchers have developed qualitative “reaction maps” that guide MNC synthesis by correlating starting materials, intermediates, and conditions with structural outcomes. A typical MNC synthesis protocol adapted from the Brust–Schiffrin method involves metal–ligand complex formation through ligand exchange with metal salts (e.g., HAuCl4 with thiols), followed by reduction (often with NaBH4) to generate core–shell nanoclusters.134
Several distinct synthetic strategies have emerged for controlling specific MNC structural features. Direct synthesis through one-step reduction produces numerous well-defined nanoclusters like [Au25(SR)18]− and [Ag44(SR)30]4− through reduction-growth processes.29,135,136 “Size-focusing” approaches convert polydisperse mixtures into monodisperse products by leveraging thermodynamic stability differences.137 Seeded growth reactions use existing monodisperse MNCs as templates for larger structures, while oxidative etching provides routes to reduce core size.138 Surface-induced transformations offer pathways to core structure modification through physical means or chemical approaches like ligand exchange.113,139 Core composition can be manipulated via co-reduction,158 metal exchange with preformed MNCs,140 or intercluster reactions,141 each offering specific advantages for dopant control.
In this section, we systematically examine these synthetic routes for controlling four critical aspects of MNC structure: core size, core structure, core composition, and ligand shell structure. For each approach, we discuss the mechanistic principles and the criteria for selecting optimal routes based on target design requirements.
3.1. Synthesizing the desired core size
MNC synthesis typically employs the reduction of metal–ligand (M–L) complex precursors following adaptations of the Brust–Schiffrin method. During this process, a rich library of species is formed whose N*s are found to follow an evolution path of 0e− → 2e− → 4e− → 6e− → 8e− → 10e− via a stepwise 2e− hopping mechanism based on electrospray ionization mass spectrometry (ESI-MS) and UV-Vis absorption spectroscopy results.29,142 While reduction-growth reactions promote formation of larger species from smaller ones, concurrent reactions like isoelectronic addition, disproportionation, and comproportionation are also taking place (Fig. 7a and b). These reactions drive the formation of the thermodynamically favourable product in a “size-focusing” manner,137 as exemplified by the synthesis of [Au25(m-MBA)18]− (m-MBA represents meta-mercaptobenzoic acid) (Fig. 7c).29 Due to the complexity of the reduction process, reaction intermediates can be stabilized via kinetic control methods by carefully adjusting reaction condition parameters (detailed in Section 4). Following synthesis, various separation and purification techniques—including solvent fractionation,143 solvent extraction,144 thin-layer chromatography (TLC),145 high-performance liquid chromatography (HPLC),146 and electrophoresis135—can be applied to remove impurities and obtain product monodispersity. This one-step direct reduction approach has successfully yielded MNCs of varied sizes, compositions, and ligands, such as thiolate-stabilized Au15(SR)13,33 Au18(SR)14,33 Au20(SR)16,35 [Au27(SR)13]4+,35 [Au25(SR)18]−,29,135 Au36(SR)22,70 [Ag44(SR)30]4−,136 DNA-stabilized Ag16,147 Ag11,148 and amidinate-protected Cu11,149 just to name a few.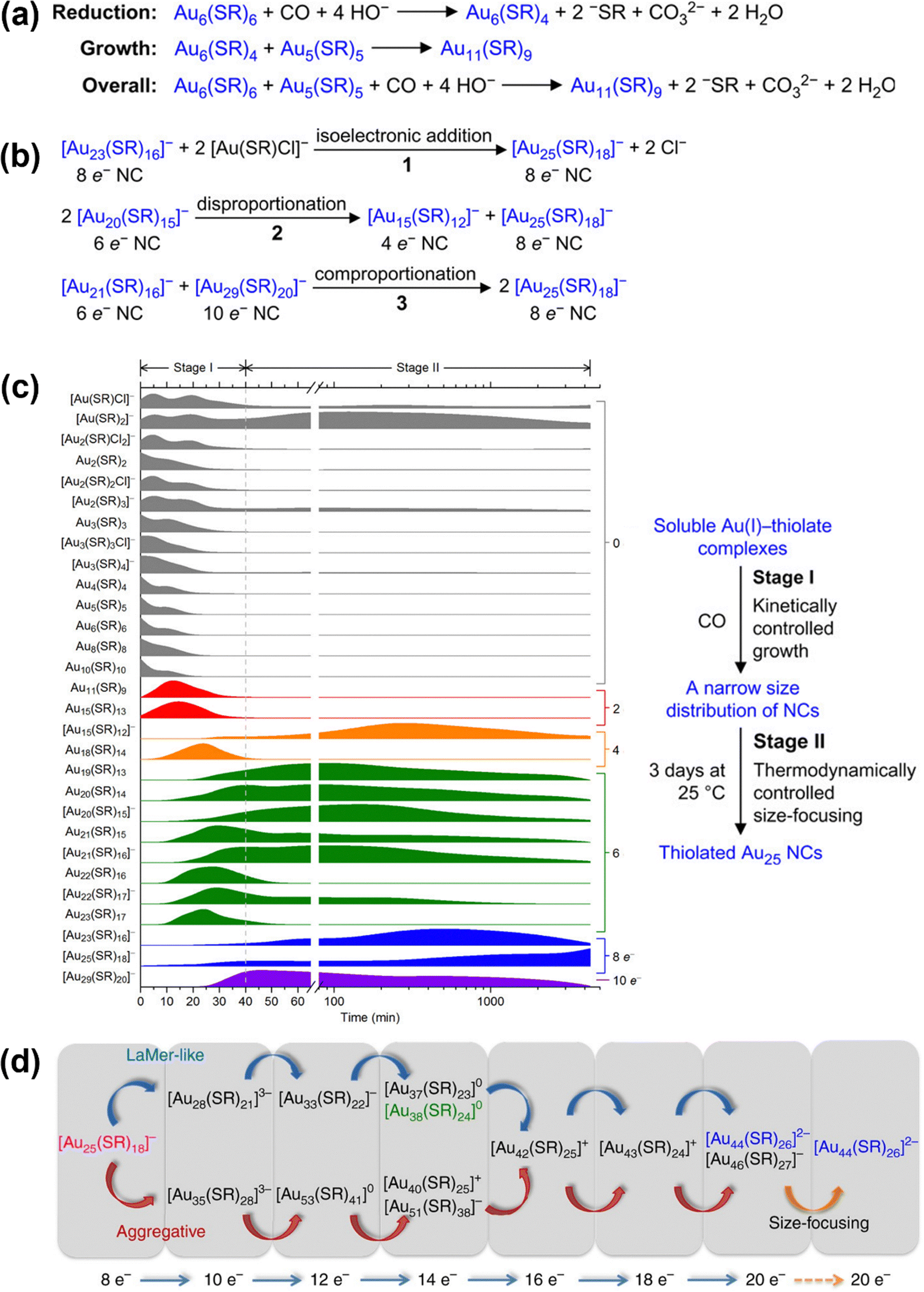 | ||
| Fig. 7 Proposed reaction schemes of (a) reduction-growth formation of Au11(SR)9 and (b) possible isoelectronic addition, disproportionation, and comproportionation reactions that occur during the size-focusing stage. (c) ESI-MS spectral profiles with normalization of the intensity of the complex precursors and nanocluster species throughout the synthesis (left) and the overall reaction scheme (right). Reprinted with permission from ref. 29. Copyright 2014, American Chemical Society. (d) Schematic illustration of the growth pathways from [Au25(SR)18]− to [Au44(SR)26]2− featuring two growth patterns. Reprinted with permission from ref. 138. Copyright 2017, Springer Nature Limited. | ||
As an alternative to direct synthesis, a two-step “size-focusing” route can be employed.137 This approach begins with generating a mixture of species through reduction-growth, followed by an additional step that adjusts reaction kinetics to favour more thermodynamically stable products.150 For example, introduction of an excess ligand can accelerate the etching reaction and promote the “size-focusing” process. Size-mixed Aux(TBBT)y obtained from NaBH4 reduction of Au–TBBT complexes can be reacted with excess TBBT thiol at elevated temperature to form Au52(TBBT)32.61 Similar approaches have enabled the synthesis of Au36(DMBT)24 (DMBT = 3,5-dimethylbenzenethiol).76 Alternatively, introducing different ligands can initiate simultaneous etching and ligand exchange, as demonstrated by the formation of Au38(SC12H25)24151 and Au21(StBu)15.60
Monodisperse MNCs can serve as seeds for continued reduction-growth reactions to yield larger core sizes. Yao et al. successfully synthesized Au38(SR)24 and [Au44(SR)26]2− by adding [Au25(SR)18]− as seed to the Au(I)–SR complex precursor followed by CO-mediated reduction.138 [Au25(SR)18]− played a duel role in this process—it reacts with the Au(I)–SR complexes or nanocluster species while also adsorbing CO molecules, making CO more susceptible to oxidation as evidenced by the successful detection of [Au25(SR)18CO]− via ESI-MS. Monitoring reaction intermediates revealed two parallel size growth patterns: LaMer-like monotonic size growth and volcano-shaped aggregative growth, both following the 2e− hopping mechanism (Fig. 7d). Fine-tuning of the reaction kinetics allowed optimization toward Au38(SR)24 as the major product. This seeded growth approach has also been successfully applied to silver nanoclusters, producing Ag50(dppm)6(SR)30 from [Ag44(SR)30]4−.152
Conversely, oxidative etching provides an effective method for reducing MNC core size, generating MNCs with higher ligand-to-metal ratios. In thiol-mediated etching of larger Au nanoparticles, oxygen molecules initiate the process by radicalizing thiol molecules to form thiyl and peroxy radicals. These radicals cleave surface motifs and oxidize Au(0) to form surface-exposed Au(I), creating new Au(I)–S bonds (Fig. 8a).153 A similar radical-induced mechanism takes place during the oxidative etching of [Au25(SR)18]− using excessive thiols.154 Mechanistic investigations reveal that the reaction proceeds through two reaction stages: decomposition and recombination (Fig. 8b). The initial decomposition stage follows a reverse 2e− hopping mechanism compared to the reduction-growth process. Subsequently, decomposition products recombine with Au(I)–SR complexes to form isoelectronic Au nanoclusters with identical N* values but higher SR:Au ratio—species not observed in simple reduction-growth processes (Fig. 8c). This allows the successful synthesis of Au25(SR)19 from [Au25(SR)18]−,155 which is predicted to possess a smaller core, consisting of one Au4 unit alongside a bitetrahedral Au7.156
 | ||
| Fig. 8 (a) Proposed mechanism of Au nanoparticle etching by thiol. Reprinted with permission from ref. 153. Copyright 2015, Wiley-VCH. (b) Schematic illustration of the etching process of [Au25(SR)18]− which depicts two reaction stages: decomposition and recombination. (c) Comparison of the distinct nanocluster species formed during etching and reduction-growth processes of [Au25(SR)18]−. The black dashed line is a guideline for reference. Reprinted with permission from ref. 154. Copyright 2021, Springer Nature Limited. (d) ESI-MS spectra (left) and the corresponding UV-Vis spectra (right) characterized at various points of time during the transformation reaction. Three grey shadows highlight three groups of peaks: Au36(TBBT)m(PET)24−m, Au38(TBBT)m(PET)24−m, and Au40(TBBT)m(PET)24−m, from left to right, respectively. The number of TBBT ligands exchanged onto the cluster (m) is highlighted on top of the mass peaks. (e) Schematic reaction pathway showing conversion from Au38(PET)24 to Au36(TBBT)24 which exhibits four stages: ligand exchange, structure distortion, disproportionation, and size-focusing. Reprinted with permission from ref. 139. Copyright 2013, American Chemical Society. | ||
Intercluster reaction offers another approach to modulate MNC core size. When [Au25(SR)18]− is oxidized to the neutrally charged Au25(SR)18, it loses the stability endowed by the shell-closing 8e− valence electron count.157 An elevation in temperature then initiates the intercluster fusion reaction forming Au38(SR)24 from two Au25(SR)18 nanoclusters.158 Remarkably, the Au13 icosahedral cores of individual Au25(SR)18 integrate to form Au38(SR)24 with a face-fused Au23 biicosahedral core.
Beyond redox reactions and intercluster reactions, the synergy between surface ligands and metal cores can induce size modification during ligand exchange processes. For example, treating Au38(PET)24 with TBBT in large excess results in complete ligand exchange to produce Au36(TBBT)24.139 Time-dependent ESI-MS and UV-Vis characterization revealed four distinct reaction stages in this transformation (Fig. 8d and e):
(1) Initial ligand exchange forming Au38(TBBT)m(PET)24−m (m < 12).
(2) Progressive structural distortion of Au38(TBBT)m(PET)24−m likely triggered by steric interactions from the bulky TBBT ligands.
(3) Disproportionation of structurally distorted Au38*(SR)24 to form Au36(SR)24 and Au40(SR)26 through internal reconstruction.
(4) Simultaneous ligand exchange and size-focusing, yielding Au36(TBBT)24 with approximately 90% yield.
This methodology has been extended to synthesize Au21(S-Adm)15 and Au16(S-Adm)12 from Au18(S-c-C6H11)14 and Au15(SG)13, respectively, utilizing the bulky HS-Adm ligand.107,159 Notably, unlike the Au36(TBBT)24 case, these products exhibit larger core sizes than their predecessors, indicating that the product core size is highly ligand-dependent.
3.2. Tuning for the desired core structure
Once the desired core size is achieved, further refinement of the core structure—with minor core size adjustments that preserve the valence electron count—can be accomplished through various surface-induced transformation methods. These approaches can be broadly categorized into physical measures and chemical approaches.Firstly, physical measures can effectively modify core structures without changing chemical composition. As discussed previously, pairing anionic surface ligands with bulky cations promotes electrostatic and CH⋯π interactions that rigidify the nanocluster surface, inducing core rotation in [Au25(SR)18]−.113 Similarly, modulating solvent pH can regulate inter-ligand hydrogen bonds to trigger isoelectronic transformations. For example, by adjusting the solvent pH from 5.5 to either 2.5 or 8, Au22(SG)18 is readily converted to isoelectronic Au24(SG)20 or Au18(SG)14 respectively.160 Although the exact structures of these glutathione-protected species have not been fully resolved, their UV-Vis absorption profiles—reliable indicators of their core size and structure—closely resemble those of structurally characterized analogues with different ligands. This suggests that they likely possess similar core structures: bitetrahedral Au8 in Au24, bitetrahedral Au7 in Au22, and bioctahedral Au9 in Au18 (Fig. 9a).105,161–163
 | ||
| Fig. 9 (a) Illustration of the core structures of Au24(SR)20, Au22(CCR)18, and Au18(SR)14. The mass formula with Au core size and valence electron number N* is indicated below each structure. (b) Proposed reaction pathway of conversion of the Au core from [Au23(S-c-C6H11)16]− to Au28(TBBT)20. Reprinted with permission from ref. 165. Copyright 2020, American Chemical Society. | ||
Surface dynamics can be strategically altered through ligand exchange reactions to induce core structure modifications. Reacting Au30(StBu)18 with excessive HSPhX (X = –H or –tBu) induces inter-template core structure conversion, forming monodisperse Au36(SPhX)24.164 This transformation fundamentally reconfigures the 12e− Au20 in Au30(StBu)18 (Fig. 2b)—which can be regarded as two Au3 units attached to two bitetrahedral Au7 units—into 2 crossing vertex-fused tritetrahedral Au10 in Au36(SPhX)24 (Fig. 2c). Notably, this process is reversible—the original Au30(StBu)18 can be regenerated by exchanging the surface ligand back to tert-butylthiol. Similarly, the 8e− Au13 core in [Au23(S-c-C6H11)16]− can transform to the double bitetrahedral Au14 core of Au28(TBBT)20 (Fig. 9b).165 The authors propose that progressive replacement of S-c-C6H11 with TBBT alters surface dynamics, inducing core distortion that ultimately results in core transformation once a threshold concentration of TBBT on the surface is reached.
In addition to ligand exchange, surface motif exchange provides another effective approach for tuning the core structure through surface dynamics modification. Reacting Au(I)–SR complexes with [Au23(SR)16]− can transform the cuboctahedral Au13 core into an icosahedron, forming [Au25(SR)18]−.166 Through hetero-ligand experiments, the authors proposed that the transformation process is initiated by the association of 2 SR–[Au(I)–SR]2 motifs. The process involves disruption of the original longer SR–[Au(I)–SR]3 motifs and ejection of SR–Au(I)–SR motifs, giving rise to an enhanced degree of freedom in the metal core which facilitates the core conversion.
3.3. Synthesizing the desired core composition
Controlling core composition through heteroatom doping is achievable via three primary routes: one-step co-reduction, two-step metal exchange with monodisperse MNCs, and intercluster reactions. These approaches have successfully incorporated various heteroatoms—including Au,167 Ag,168 Cu,169 Pt,118 Pd,170 Cd,171 Hg,172 Ru,86 Rh,86 Ir,86 and H173,174—into MNCs. Detailed structural analysis revealed that different dopants have distinct preferential sites. In the case of [MAu24(SR)18]− (where M represents a dopant), Pt and Pd preferentially replace the central atom in the icosahedral core, Ag, Cd and Hg tend to occupy positions on the icosahedral shell, and Cu typically localizes in the staple motifs.171,175,176 Such site preference is observed in Au16Cu6(tBuPhCC)18, where all 6 Cu atoms are located in the surface motifs as compared to its isostructural counterpart Au22(tBuPhCC)18.62
The one-step co-reduction approach involves mixing precursors of both host and dopant metals—either as metal salts or as metal–ligand complexes—to undergo simultaneous reduction. This facile method enables single-atom doping in various systems, including the Au13 icosahedral core of [Au25(SR)18]− with Pt and Pd,51,177 and phosphine-protected Au13 MNCs with Ru, Rh and Ir.86 If necessary, ligand shell modification can be thereafter conducted.178 When doping heteroatoms that are homologous to the host metal, such as Ag and Au, multi-doped MNC mixtures often form, such as [AgxAu25−x(SR)18]− and AgxAu38−x(SR)24.168,179 HPLC can then be employed to isolate species with different dopant number from the mixture.180,181 Notably, heteroatom doping can lead to surface reconstruction of the host MNC. Li et al. demonstrated that adding Cd(II)–(SR)2 to the Au precursors yields [Au23-xCdx(SR)16]− (x ≈ 2) which exhibits a structure similar to [Au23(SR)16]− but with 2 surface Au atoms replaced by Cd.171 Increasing the Cd(II)–(SR)2 precursor concentration results in [Au19Cd2(SR)16]−, where each metallic Cd replaces 2 surface Au atoms—preserving the Au13 core while reconstructing the surface motif configurations (Fig. 10a).
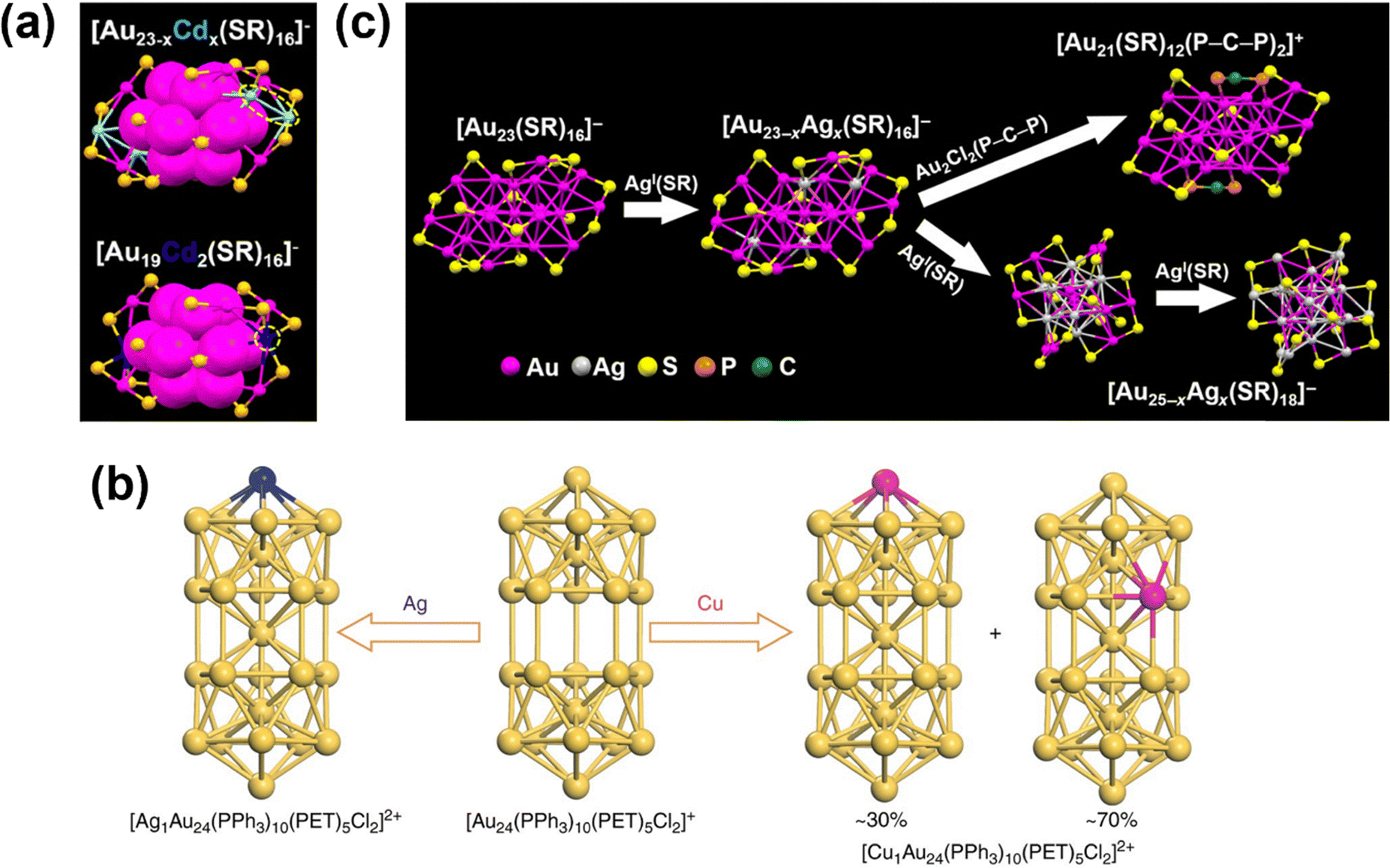 | ||
| Fig. 10 (a) Structures of [Au23−xCdx(SR)16]− and [Au19Cd2(SR)16]−. Colour label: magenta, Au; yellow, S; blue, Cd; light blue, partial occupancy of Cd/Au. Reprinted with permission from ref. 171. Copyright 2017, American Chemical Society. (b) Reaction pathway showing core structures of Au24 and the products of single-atom doping with Ag and Cu. Colour label: yellow, Au; blue, Ag; magenta, Cu. Reprinted with permission from ref. 140. Copyright 2017, Springer Nature Limited. (c) Reaction pathway of [Au23(SR)16]− with light doping forming [Au23−xAgx(SR)16]− (x = 1 to 2) and thereafter forming [Au21(SR)12(P–C–P)2]+ and heavy doping forming [Au25−xAgx(SR)18]− upon exchange reaction with Ag(I)–SR. Reprinted with permission from ref. 186. Copyright 2017, The American Association for the Advancement of Science. | ||
The second approach employs two-step metal exchange with preformed monodisperse MNCs. After synthesizing [Au24(PPh3)10(SC2H4Ph)5Cl2]+, addition of MCl salt (M = Ag/Cu) gives rise to single-atom doping where the dopant occupies the vertex position in the core, displacing one Au atom to the centre (Fig. 10b).140 Likewise, CdAu24(SR)18 and HgAu24(SR)18 can be obtained by reacting Cd2+ and Hg2+ ions with [Au25(SR)18]−.182 It is intriguing that the less noble Hg is reduced by Au in Au25, seemingly contradicting the galvanic sequence. This “anti-galvanic” behaviour is attributed to the decreased oxidation potential of these MNCs falling below the reduction potential of the dopant ions.183 Metal–ligand complexes can also serve as effective dopant sources. Bakr and colleagues demonstrated that reacting [Ag25(SR)18]− with Au complexes replaces the central Ag yielding [Ag24Au(SR)18]− and achieving single-atom doping in Au/Ag bimetallic nanoclusters.167 Mechanistic studies by Zheng et al. using real-time monitoring in hydrophilic systems suggest that the doped Au atom initially replaces an atom in the surface motif; subsequently, it diffuses dynamically to the icosahedral shell, and ultimately occupies the energetically favourable central position.184 However, exchange with complexes can also lead to surface motif replacement, as demonstrated when appropriate amounts of Au(I)–SR substitute all the surface motifs of [Ag44(p-MBA)30]4− forming [Ag32Au12SR30]4−.185
Precursor concentration is critical in metal exchange reactions. In the exchange reaction between [Au23(SR)16]− and Ag(I)–SR, light doping allows the formation of [Au23−xAgx(SR)16]− (x = 1 to 2) with Ag occupying core vertex positions, which can subsequently form [Au21(SR)12(P–C–P)2]+ with Au2Cl2(P–C–P) (P–C–P stands for bis(diphenylphosphino)methane). Conversely, heavy doping leads to structural conversion, forming [Au25−xAgx(SR)18]− (x ranging from ∼4 to ∼19).186,187 In this process, Ag dopants preferentially occupy icosahedral core vertices before filling surface motif positions (Fig. 10c). It should also be noted that the choice of dopant precursor form (ions versus complexes) can impact the synthesis outcome. While CdAu24(SR)18 can be synthesized from both Cd2+ ion and complex precursors,182,188 these precursors yield different products when reacted with [Au23(SR)16]−.189 Such nanocluster dependency was demonstrated by Zhu et al., who showed that Cd2+ ions induce conversion from [Au23(SR)16]− to Au28(SR)20 while Cd(II)–(SR)2 results in Au20Cd4(SH)(SR)29.
Composition adjustment can also be realized via intercluster reactions.141 By reacting [Au25(SR)18]− with [Ag25(SR)18]− in different molar ratios, the entire range of alloy composition [Ag25−xAux(SR)18]− (x = 1–24) becomes attainable.190 Researchers successfully captured the formation of [Ag25Au25(SR)36]2− dianionic adducts using ESI-MS, suggesting a reaction mechanism that begins with adduct formation, proceeds through metal atom exchange in transient dimers, and concludes with dimer dissociation into monomers.191 Of note, intercluster reactions typically generate polydisperse product mixtures requiring subsequent separation efforts.
3.4. Synthesizing the desired ligand shell
The most straightforward way to synthesize MNCs of desired ligand shell—whether comprising single or multiple ligands—is via one-step reduction-growth of the metal–ligand complex containing the target ligand(s).192–194 When target ligands are not commercially available, in-house synthesis provides researchers greater flexibility in ligand design, enabling the incorporation of specific functionalities or reactivities prior to complex formation with metal salts.123,127,130For fine-tuning the ligand shell of monodisperse MNCs, mild ligand exchange offers a versatile approach. Different from excessive ligand change, adding controlled amounts of foreign ligands can result in a multi-ligand surface with precisely regulated foreign ligand incorporation. SCXRD analysis of [Au25(SR)18]− with two exchanged foreign ligands revealed that these foreign ligands are symmetrically bonded to the most solvent-exposed Au atoms on the surface, replacing the host thiolate ligands at the core site (Fig. 11a).195 This observation is consistent with an associative mechanism where the ligand exchange is initiated by the association of the foreign ligand with the accessible Au atom. The site preference in ligand exchange reactions is also supported by HPLC results.196 Adopting this approach, Liu et al. successfully introduced chirality to the ligand shell of [Au23(S-c-C6H11)16]− via ligand exchange with chiral phosphoramidite ligands.8 To accommodate these bulky ligands, two short SR–Au(I)–SR staple motifs on the host nanocluster surface were replaced by one long SR–[Au(I)–SR]3 motif while the Au13 core structure was largely preserved despite slight twisting (Fig. 11b). In certain cases, complete ligand exchange can alter the structure of the ligand shell while maintaining core integrity, as exemplified by the reversible isomerism reactions between Au28(S-c-C6H11)20 and Au28(TBBT)20.66
 | ||
| Fig. 11 (a) Structure of Au25(PET)18(p-BBT)2 (p-BBT = 4-bromobenzenethiol) resolved by SCXRD. Colour label: orange, Au; yellow, S; grey, C; red, Br. Red arrows indicate the locations of exchanged p-BBT ligands and blue arrows indicate the Au atoms on the surface motif that bond to the exchanged p-BBT ligands. Reprinted with permission from ref. 195. Copyright 2014, American Chemical Society. (b) The total structures of [Au23(S-c-C6H11)16]− after (R configuration) and before the exchange reaction with phosphoramidite. Colour label: pint, core Au; blue, motif Au; yellow, S; green, P; orange, N; light blue, O; grey, C. Reprinted with permission from ref. 8. Copyright 2023, Springer Nature Limited. (c) Schematic illustration of the reaction process of the seeded growth of [Au25(SR)18]−. Reprinted with permission from ref. 197. Copyright 2021, Wiley-VCH. | ||
Seeded growth provides another effective route for ligand shell synthesis. Lei et al. developed a two-step measure for high yield synthesis of [Au25(SR)18]− by first preparing [Au13(dppp)4Cl4]+, which possesses an identical Au13 core structure and can be synthesized with high yield (Fig. 11c).197 In the second step, this Au13 precursor is reacted with Au(I)–SR complexes in the presence of NaBH4 as a reducing agent to form [Au25(SR)18]− with yields approaching 100%. The authors observed that the ligand body can significantly affect synthesis results. When aromatic thiols are replaced with alkanethiols such as PET and S-c-C6H11, [Au25(SR)18]− formation was prohibited, instead producing a mixture of Au20(SR)13 and Au33(SR)20. However, the detailed reaction mechanism and the role played by the ligand during the process remain to be fully elucidated.
After establishing the desired ligand shell architecture, further functionalization of ligand bodies based on their physical or chemical reactivities can be performed. As these reactions constitute crucial considerations during ligand shell design, they have been discussed extensively in Section 2.2.3 and are not repeated here.
4. Condition optimization
Optimization of reaction conditions represents the third step in synthesis planning, where parameters are systematically adjusted to achieve optimal reaction objectives. Based on the fundamental understanding of reaction mechanisms and kinetics, scientists fine-tune influential factors that affect reaction rates and product selectivity to determine the most effective parameter combinations. Common objectives in this optimization process include maximizing reaction yield and product purity. In many cases, scientists encounter multi-objective optimization scenarios where all desired outcomes cannot be simultaneously achieved, necessitating careful trade-off decisions to balance competing priorities while maintaining overall synthetic efficiency.The synthesis of MNCs with well-designed core and ligand shell architectures necessitates navigation through a complex reaction landscape involving numerous intermediates and competing pathways. The selection of specific ligand and metal precursors not only determines the thermodynamically stable structures achievable but also profoundly influences the reaction kinetics through their inherent reactivity, binding affinities, and steric properties as discussed in previous sections. These precursor effects operate synergistically with other reaction condition parameters,33,192,198 creating an optimization space of high dimensionality where strategic manipulation of conditions can control product yield and purity. This section examines the critical condition parameters that significantly impact synthesis outcomes, starting with physical parameters including reaction time, temperature and stirring condition, and thereafter focusing on chemical parameters: metal/ligand (M/L) ratio, solvent, reducing agent, pH, and use of additives (such as surfactant cations), elucidating how these factors interact with precursor chemistry to collectively determine synthesis outcomes and reviewing the optimization strategies commonly employed by researchers throughout the field.
4.1. Condition parameters at play
The synthesis of MNCs involves numerous intermediates whose evolution can be controlled through kinetic means. Condition parameters, both physical and chemical, play crucial roles during this process. In terms of physical parameters, reaction time critically influences the composition of product mixtures, thereby dictating outcomes in subsequent reaction steps. For instance, under identical conditions, a 7-day metal–ligand complex formation reaction time produced high-purity [Au25(MHA)18]− upon subsequent NaBH4 reduction reaction, while a 15-second reaction yielded the same product contaminated with larger nanoclusters as impurities.199 Similarly, in the two-step synthesis of Au22(SG)18, a 20-minute CO incubation time was found to yield maximum purity.200Temperature represents another fundamental physical parameter governing reaction kinetics. Through temperature variation analysis, Chen et al. revealed the endothermic nature of the reductive formation of the thermodynamically favourable [Au25(MHA)18]− using NaBH4, while noting that elevated temperature accelerates its decomposition.201 The optimal condition was identified at 40 °C, which provided approximately 95% yield with favourable kinetics. In another example, Zhu et al. enhanced [Au25(PET)18]− synthesis yield by lowering the solution temperature from room temperature to 0 °C before thiol addition, thereby modulating the kinetics of the Au(I)–SR complex formation.202 Temperature control in both complex formation and “size-focusing” thiol etching reactions enabled Zeng et al. to selectively obtain the smaller Au44(TBBT)28 rather than Au52(TBBT)32.57,61
Moreover, stirring conditions, which regulate mass transfer kinetics, can significantly influence synthesis outcomes. Reducing the stirring speed during NaBH4 reduction of Au(I)–SR complexes from 400 rpm to 100 rpm altered the reaction kinetics sufficiently to shift the major product from a mixture of 10e− Au39(SR)29 and Au40(SR)30 to 4e− Au24(SR)20.203 Further modification combining lower stirring speed, reduced NaBH4 addition rate, and shortened reaction time yielded Au20(SR)16. Beyond homogenization of the reaction mixture, stirring conditions were shown to affect silver nanocluster synthesis by influencing gas–liquid mass transfer rates, particularly oxygen uptake which led to desired product formation.204
:1 to 3:1) produce predominantly Au chloride or hydroxide complexes such as [AuCl2]− and [H4AuO4]− in the NaBH4 reduction precursor solution, with thiols primarily consumed in reducing Au(III) to Au(I) and forming disulfides (Fig. 12a).205 This insufficient protection from thiolate ligands results in the formation of larger Au nanoparticles which exhibit surface plasmon resonance. Intermediate M/L ratios (1:1 and 1:2) yield predominantly cyclic oligomeric and polymeric Au(I)–SR complexes like Au10(SR)10, alongside shorter chain-like complexes such as [Au2(SR)3]−, which successfully produce Au nanoclusters upon reduction. In low ratio scenarios (1:3 to 1:8), cyclic complexes are replaced by shorter species including [Au(SR)2]− and [Au2(SR)3]−, which provide enhanced protection to Au atoms and consequently exhibit decreased reactivity toward NaBH4 reduction. Despite the better protection, these short complexes arguably form short surface motifs that can only stabilize larger nanoparticles with smaller surface curvature compared to nanoclusters, ultimately leading to nanoparticle formation after sufficient reaction time. Yao et al. leveraged these smaller complexes formed at lower M/L ratios to suppress parallel aggregative growth pathways during the seeded growth of [Au44(SR)26]2−, facilitating the successful synthesis of Au38(SR)24.138
 | ||
| Fig. 12 (a) ESI-MS spectra of precursors formed with different SR:Au ratio and the corresponding UV-Vis absorption spectra. The species identified and labelled in ESI-MS spectra are tabulated on the right. Reproduced with permission from ref. 205. Copyright 2016, Royal Society of Chemistry. (b) Schematic illustration of the water–toluene two-phase synthesis method developed for the high-purity synthesis of Au15(SG)13 and Au18(SG)14. Reprinted with permission from ref. 209. Copyright 2013, Wiley-VCH. (c) UV-Vis absorption spectra and ESI-MS mass spectra of Au nanoclusters synthesized with different electron:Au ratios, where each mole of NaBH4 can supply 8 electrons to each mole of Au. Reprinted with permission from ref. 142. Copyright 2018, American Chemical Society. (d) The relative abundance of species possessing different N* identified by ESI-MS after each reduction step. (e) Time-dependent ESI-MS spectra showing normalized abundance of the complex and nanocluster species detected during sub-stoichiometric reduction, with the duration of each step being 30 minutes. Reprinted with permission from ref. 211. Copyright 2020, Elsevier. | ||
Additionally, ligands such as thiols function as etchants, as mentioned above, with their concentration significantly influencing etching kinetics and playing a critical role in the “size-focusing” process. Demonstrating this principle, Liao et al. successfully synthesized the larger Au49(SR)27 species by introducing reduced thiol quantities to the Aux(SR)y etching precursor and employing shorter etching duration, in contrast to the Au44(SR)26 nanocluster produced when following the original protocol.206,207
Beyond solubility effects, the solvent environment determines the reactivity of reducing agents and the degree of thiol deprotonation. In Au144(PET)60 synthesis, replacing toluene with methanol precipitates Aux(SR)y intermediates, preventing further growth and creating a mixture of nanoclusters with relatively narrow size distribution amenable to “size-focusing”.32 Simultaneously, methanol promotes both NaBH4 reduction and thiolate etching, synergistically modifying kinetics in both reduction-growth and “size-focusing” stages to yield high-purity Au144(PET)60. Similar solvent-dependent outcomes were observed with [Au37(PPh3)10(SC2H4Ph)10Cl2]+ formation in water, compared to [Au25(PPh3)10(SC2H4Ph)5Cl2]2+ in ethanol.23 Additionally, the use of THF as solvent significantly improved the purity and yield of Au25(SG)18 compared to the previously reported protocol using methanol.210
Beyond concentration manipulation, the selection of reducing agents with different reduction potentials offers another dimension for kinetic control that works synergistically with metal and ligand precursor chemistry. Wu et al. substituted NaBH4 with milder TBAB, observing a gradual colour evolution from yellow to orange to black over approximately 15 minutes—a marked contrast to the rapid blackening within seconds with NaBH4—resulting in the formation of 6e− Au19(PET)13 rather than the thermodynamically favoured [Au25(PET)18]−.212 Carbon monoxide represents another mild reducing agent successfully implemented by Xie and colleagues, leading to the discovery of Au22(SG)18,34 and high-purity synthesis of water-soluble nanoclusters of a wide size range: Au15(SR)13,33 Au18(SR)14,33 Au20(SR)16,35 [Au25(SR)18]−,33 [Au27(SR)13]4+,35 upon synergistic tinkering of other condition parameters such as solvent pH, solvent polarity, and use of assistant additives. Other weak reducing agents including trimethylamine borane ((CH3)3N·BH3) and sodium cyanoborohydride (NaBH3CN) have been effectively employed in nanocluster synthesis, each interacting uniquely with specific metal–ligand precursor combinations.213,214
Yuan et al. leveraged these mechanistic insights by adding NaOH prior to NaBH4 introduction, achieving high-purity synthesis of mono- and multi-thiolate protected [Au25(SR)18]− through precise balancing of reduction and etching kinetics.192 Conversely, Wu and colleagues employed acidic conditions, via nitric acid addition, to promote reduction while retarding thiolate etching, leading to successful synthesis of novel Au52(PET)32 nanoclusters that are isomeric to previously discovered Au52(TBBT)32.215 Notably, as acid introduction preceded thiol addition in their protocol, complex formation kinetics were altered as well, contributing to the formation of Au52(PET)32. Acid can be strategically introduced at different stages of the synthesis process to modulate specific reaction steps. Wu's group demonstrated this versatility by adding acid before NaBH4 during Au42(TBBT)26 synthesis,216 while Shichibu et al. incorporated acid at the beginning of the “size-focusing” step when working with AuN(dppe)xCly precursors to produce phosphine-protected [Au13(dppe)5Cl2]3+.82 When considering CO as the reducing agent, reaction kinetics are promoted by hydroxide ions, resulting in the formation of larger nanoclusters at higher pH values (Fig. 13a).33 These observations collectively demonstrate that pH serves as a powerful parameter for tuning nanocluster synthesis through its multifaceted effects on ligand behaviour and reducing agent performance, allowing researchers to navigate complex reaction landscapes with greater precision.
 | ||
| Fig. 13 (a) Schematic illustration of the facile synthesis of GSH-protected Au nanoclusters of various sizes by adjusting the pH value through the CO-reduction approach. Reprinted with permission from ref. 33. Copyright 2013, American Chemical Society. (b) Schematic illustration of the tailoring of Au nanocluster sizes via the introduction of duel electrostatic π–π stacking interactions between ionic liquid cations such as OMIm+ and deprotonated p-MBA. Reprinted with permission from ref. 35. Copyright 2021, Wiley-VCH. | ||
Extending this approach, Zhu et al. introduced ionic liquid cations such as 1-octyl-3-methylimidazolium (OMIm+) to interact with the deprotonated p-MBA ligands through duel electrostatic π–π stacking interactions (Fig. 13b).35 As a result, Au20(SR)16 is stabilized by the altered reaction kinetics. By systematically varying the side chain length of the cation while simultaneously adjusting other parameters (cation concentration, pH, and solvent relative permittivity), the researchers demonstrated remarkable control over product selectivity: C12 chain length cation produced the smaller Au15(SR)13 nanocluster, C6 cation yielded [Au25(SR)18]−, and C4 cation generated the larger [Au27(SR)13]4+. A similar reaction-directing effect can be induced by metal ions, as demonstrated by the essential role of Cd2+ in synthesizing the non-fcc-structured isomer of Au42(TBBT)26.217
Small sodium metal salts have been found to accelerate isoelectronic conversion between [Au23(p-MBA)16]− and [Au25(p-MBA)18]−.166 The degree of kinetic enhancement correlates positively with ionic strength (determined by concentration or anion valency) but remains independent of anion identity. This phenomenon is attributed to the weakening of electrostatic repulsion between negatively charged Au(I)–SR complexes and [Au23(p-MBA)16]− resulting from compression of the electric double layer. This finding illustrates how even simple ionic additives can significantly modulate reaction kinetics through electrostatic effects, providing researchers with additional tools for navigating complex nanocluster synthesis landscapes.
4.2. Condition optimization strategies
The one-factor-at-a-time (OFAT) approach represents the most commonly adopted optimization strategy in metal nanocluster synthesis. This conventional method involves identifying all relevant parameters and optimizing them sequentially, beginning with a parameter selected by researcher's decision.218 In MNC synthesis, OFAT is the strategy of choice in the optimization of various parameters discussed above, including ligand choice,102 M/L ratio,32,205 solvent,31 reducing agent,142 pH,33 and use of additives.166 However, as these parameters exhibit significant synergistic effects, achieving optimal synthesis protocols in terms of yield or purity typically requires fine-tuning of multiple parameters simultaneously. Given OFAT's inherent limitations in efficiency and accuracy when navigating multi-dimensional design spaces, researchers are turning to global optimization strategies for MNC synthesis, which will be discussed in detail in Section 5.2.5. Opportunities offered by data-driven approaches
As discussed above, the synthesis planning of MNCs inherently relies on complex structure–property (target design), precursor–product (route development) and condition–objective/product relationships (condition optimization). However, the construction of these crucial relationships is challenging. First, obtaining precise structural information can be experimentally difficult, particularly when the surface ligand is highly flexible (like water-soluble ligand-capped MNCs), ideal product purity for crystallization is difficult to achieve, and solvent-dissolved MNCs exhibit unique surface dynamics. While computational simulations provide theoretical evidence in these cases, the structural complexity of MNCs demands substantial computational resources. Second, the multidimensional parameter space created by interconnected reaction precursors and various condition parameters complicates the study of synthesis–product relationships using conventional methods. Third, most established relationships—such as core growth templates and condition parameter effects—remain largely qualitative, derived from limited datasets and semi-analytical theories with restricted applicability across the broader MNC design space.Data-driven methodologies offer promising solutions to these challenges by providing a quantitative framework where statistical models are constructed using carefully selected input and output descriptors that capture essential information about the chemical structure, properties, and reaction conditions. In this approach, complex relationships are abstracted as interactions among descriptor combinations. While property characterization data and reaction condition parameters typically serve as quantitative descriptors, chemical species and structural representations can be characterized through compositional, topological, structural, quantum-chemical, or mathematical descriptors. By integrating advanced algorithms with high-throughput data generation, this approach enables robust predictions based on high-dimensional relationship modelling with reliable accuracy, while facilitating straightforward model updates and adaptations.219,220
This section explores how data-driven methodologies can advance MNC synthesis planning through structure prediction acceleration, global condition optimization modelling, direct synthesis–property relationship modelling, and systematic model generalization and adaptation—facilitating more efficient and precise design and synthesis strategies (Table 1).
| Objectives | Methods adopted | Examples |
|---|---|---|
| Structure prediction acceleration | Random forest | Prediction of CO adsorption on Ag-doped Au nanoclusters221 |
| Convolutional neural networks | Hydride location prediction222,223 | |
| Feedforward neural networks with simulated annealing | Prediction of Au25–protein interactions224 | |
| Distance-based ML | Au38(SR)24 structure prediction at varied temperatures225 | |
| Local search algorithm | Structure prediction of metal–ligand interfaces of Au/Ag based MNCs226 | |
| Recurrent neural networks | PL property prediction of hairpin-DNA templated Ag nanoclusters227 | |
| Global condition optimization | Siamese neural networks with graph convolutional neural networks | Au25 synthesis outcome prediction46 |
| Random forest with HTE | Prediction of synthesis yield for Au–Cu bimetallic nanoclusters228 | |
| Convolutional neural networks | Composition prediction from UV-Vis absorption data229 | |
| Synthesis–property relationship modelling | Support vector machine with HTE | Precursor design for desired DNA–Ag nanocluster PL emission colour230,231 |
| Variational autoencoder with HTE | Precursor design for desired DNA–Ag nanocluster PL colour and brightness47 | |
| Extreme gradient boosting regressor | Synthesis of highly luminescent glutathione-protected Au nanoclusters232 | |
5.1. Structure prediction acceleration
When experimental determination of precise structures proves challenging, computational structure prediction provides theoretical insights that rationalize macroscopic properties of MNCs and facilitate structure–property relationship development. For example, combined molecular dynamics (MD) simulations and density functional theory (DFT) calculations have revealed dynamic interactions at the ligand–metal interface of water-dissolved m-MBA-protected Au68, explaining its simultaneous high stability and reactivity in protein conjugation reactions.233 Similarly, MD simulations identifying specific binding sites mediated by electrostatic interactions and hydrogen bonds have provided microscopic elucidation of stoichiometric binding between bovine serum albumin (BSA) and [Au25(SR)18]−, advancing the understanding of MNC–protein interactions for bio-related applications.124 However, these simulation processes typically require intensive computational resources due to the structural complexity of MNCs.ML approaches have demonstrated remarkable efficacy in accelerating structure prediction processes. For example, a ML model trained on descriptors derived from adsorbate-free and nonrelaxed structures has enabled rapid filtering of potential candidates based on predicted CO adsorption energy on Ag-doped Au nanoclusters, significantly accelerating the research process compared to exclusive reliance on time-consuming DFT calculations.221 Notably, this model performed with good accuracy for larger Au nanoclusters absent from the training data, demonstrating its generalizability. In another example, Wang et al. employed CNNs trained on existing structure libraries of hydride-doped Cu nanoclusters to model relationships between local chemical environments and hydride occupancy probability.222 Combining with symmetry constraint considerations, the authors significantly reduced the number of potential structural candidates requiring DFT optimization. However, these prediction results still require experimental verification to confirm the accuracy and validity of these ML models.
ML approaches have also advanced simulations of MNC–protein interactions. Addressing challenges including the lack of suitable force fields and the wide range of simulation timescales required for conventional modelling of dynamic MNC–protein interactions, Pihlajamäki et al. developed a ML methodology comprising a feedforward neural network (FNN) that predicts Coulomb and van der Waals contributions to interaction energies in Au nanocluster–protein complexes at a coarse-grained level.224 This prediction is followed by optimization via Monte Carlo-based structure search and refinement to atomic-scale structures. The researchers developed graph theory-based representations of Au nanoclusters and protein structures as descriptors, effectively simplifying structural complexity while preserving essential information (Fig. 14). The method was validated through subsequent MD simulations, demonstrating robust and accurate predictions of preferred binding sites between proteins and MNCs—even for larger proteins and MNCs absent from the training data. Analysis of predicted binding sites revealed the significance of electrostatic interactions between positively charged protein residues, particularly lysine (LYS) and ARG, and negatively charged MNC ligands (such as p-MBA). These results establish this method as a facile and reliable tool for studying MNC–protein interactions and dynamic properties of nano-bio interfaces at atomic scale, overcoming computational limitations of traditional approaches. However, the model could be potentially improved by incorporating hydrogen bonding when predicting interaction energies between nanoclusters and proteins, as hydrogen bonds play crucial roles in the binding of the Au25(p-MBSA)18–BSA (p-MBSA = para-mercaptobenzenesulfonic acid) complex alongside electrostatic interactions.124 This may contribute to the model's limited performance in predicting the binding behaviour of the (Au25(p-MBSA)18)–HSA (HSA = human serum albumin) complex.
 | ||
| Fig. 14 The schematic illustration of the ML integrated framework for nanocluster–protein interaction simulations. Reprinted with permission from ref. 224. Copyright 2024, Wiley-VCH. | ||
When high-quality datasets are already available, a statistical modelling-based approach enables rapid structure predictions with accuracy close to the quantum mechanical level. For instance, a model trained to correlate the local atomic environment of hydrogen in Cu- and Pd-doped [Au25(SR)18]− with the hydrogen–nanocluster interaction energy for catalyst design in electrocatalytic hydrogen evolution reactions achieved predicted energies within 0.1 eV difference compared to DFT calculation results.234
5.2. Modelling for global condition optimization
The multidimensional nature of synthesis parameters poses significant challenges for conventional optimization approaches in MNC synthesis. Traditional OFAT methodologies, which optimize condition parameters sequentially (Fig. 15a), demonstrate inherent limitations in identifying global optima and elucidating synergistic relationships among interdependent parameters.218 Furthermore, the expansive design space renders comprehensive experimentation across all possible parameter combinations prohibitively resource-intensive. Statistical modelling approaches, therefore, offer a methodologically robust alternative that effectively balances the critical trade-off between experimental efficiency and optimization accuracy. | ||
| Fig. 15 (a) Schematic illustration of the OFAT optimization strategy for parameter space exploration, where each dot represents an experiment. Reprinted with permission from ref. 218. Copyright 2023, American Chemical Society. (b) Decision tree generated by statistical modelling for the prediction of [Au25(SR)18]− synthesis outcome in the aqueous phase, where ovals represent decision nodes and rectangles represent reaction-outcome bins (where the reaction outcome is represented as 1 for success and 0 for failure and number of reaction examples correctly and incorrectly classified are denoted in parentheses before and after the slash respectively). Triangles depict excised subtrees due to both extra small examples in that branch and chemical intuition. Reprinted with permission from ref. 46. Copyright 2019, Wiley-VCH. | ||
Li et al. pioneered the application of statistical modelling to predict MNC synthesis outcomes (success or failure) based on specific parameter sets.46 Their innovative approach employed a Siamese neural network (SNN) stacked with a graph convolutional neural network (GCNN) classification model, trained to predict whether particular parameter combinations would yield monodisperse [Au25(SR)18]− protected by thiols with diverse ligand bodies. After training, the model was deployed to predict synthesis outcomes for all parameter combinations within the design space, enabling the construction of a decision tree to provide simple guidance on subsequent nanocluster synthesis, albeit with acknowledged precision limitations (Fig. 15b). Extending this data-driven methodology to bimetallic systems, Tang et al. developed a condition–yield relationship model utilizing the random forest algorithm.228 The team constructed a HTE platform capable of conducting up to 264 simultaneous reactions to facilitate data collection. The model exhibits good prediction accuracy for samples with <50% yield but low accuracy in the >50% region. To further improve on such models, additional data collection in less represented regions is required through an iterative process of data collection, model updating, and experiment suggestion—a methodology known as active learning.
Active learning methodologies have demonstrated remarkable success in organic synthesis optimization, notably through Bayesian optimization algorithms.40 In a typical optimization process, a statistical surrogate model (e.g., Gaussian process) is initially constructed using a set of preliminary experimental data, mapping the design space with expected mean values and variances. An acquisition function (e.g., expected improvement, which typically prioritizes conditions with the highest expected improvement compared to the current best) then guides the selection of subsequent experiments for model refinement. To avoid convergence on local optima, the algorithm strategically balances between exploiting areas of high predicted performance and exploring regions with high uncertainty. This iterative process continues until identifying the globally optimal parameter set. By modifying either the model output or the acquisition function, optimization can address multiple objectives simultaneously.219,235 HTE platforms can significantly accelerate this data collection process, enhancing efficiency. Upon successful optimization, the resulting surrogate model—trained with comprehensive experimental data—provides an accurate representation of condition–objective relationships, enabling clear visualization of synergistic effects among reaction parameters through response surface modelling and facilitating reaction kinetics and mechanistic analysis (Fig. 16a).
 | ||
| Fig. 16 (a) Graphs depicting the objective values versus experiment number (top left), three-dimensional plots of objective values (top right) and condition parameter values experimented (bottom left), and the response surface modelling the relationship between yield and condition parameters generated by the Gaussian process models trained with experimental data (bottom right) after a multi-objective organic synthesis optimization campaign. Reprinted with permission from ref. 219. Copyright 2022, American Chemical Society. (b) Predicted relative abundance (green bars) based on UV-Vis absorption spectra (shown in insets) and actual relative abundance (orange bars) of the compositions in polydisperse Au nanocluster samples. Reprinted with permission from ref. 229. Copyright 2023, Springer Nature Limited. | ||
Exploration through the design space beyond single-product regions enables the construction of comprehensive condition–product relationships, providing deeper insights into the roles of various synthesis parameters. Du et al. demonstrated this approach in polyoxometalate synthesis, where phase diagrams derived from design space exploration and condition optimization revealed the complex interplay among reaction parameters in crystal formation and intermediate linker length determination, ultimately dictating product identity.236 This methodology is particularly relevant for MNC synthesis, where many nanoclusters are produced through kinetically controlled processes in which minor parameter adjustments can significantly alter product outcomes. Well-constructed phase diagrams reveal valuable kinetic and mechanistic information while providing clear guidance for protocol optimization and rational design strategies.
The overlapping regions in the phase diagram, where multiple kinetically controlled species coexist, present opportunities for advanced kinetic modelling approaches. Utilizing ML algorithms, Li et al. demonstrated the possibility of predicting the composition of a MNC mixture solution and the relative abundance of the respective compositional species in the ESI-MS spectrum from UV-Vis absorption results (Fig. 16b).229 As such, once a model is well-trained with in situ UV-Vis absorption and real-time ESI-MS measurements, it is technically possible to reduce the reliance on the labour-intensive time-course ESI-MS measurements while still enabling compositional evolution monitoring for kinetic model development. Such models would allow prediction the relative abundance of byproducts under similar reaction conditions, enabling synthetic route ranking based on byproduct profiles in optimized synthesis products. This information becomes invaluable during the route development stage of the synthesis planning process, facilitating more informed decision-making based on quantitative predictions rather than qualitative assessments.
5.3. Synthesis–property relationship modelling
When the product structural information is difficult to determine due to either polydispersity or difficulty in obtaining single crystals, rational MNC target design can instead be anchored to reaction precursors or synthetic condition parameters to construct direct synthesis–property (in terms of precursor- or condition-property) relationship models. The construction of these high-dimensional relationships has become increasingly feasible through statistical modelling approaches.The precursor–property relationship modelling approach has demonstrated notable success in designing DNA-stabilized Ag nanoclusters with targeted fluorescence properties, even without precise structural characterization. Copp et al. demonstrated this approach by training a classification model using Ag nanocluster products synthesized with 1432 distinct DNA oligomers to predict product colour, categorized by fluorescence spectra peak wavelength (Fig. 17a).230 In this system, the DNA nucleobase sequence determines the cluster size and, consequently, its photoluminescence characteristics. The researchers parameterized DNA sequences as arrays of approximately 120 binary descriptors, each representing the presence or absence of specific base patterns in the DNA oligomer. This model successfully generated DNA sequences for synthesizing products in the desired “green” and “very red” colour classes with selectivity enhancements of 81% and 330%, respectively, compared to the training data. The authors noted that many products targeting green emission exhibited brightness below the detection threshold, contributing to relatively low selectivity in this category.
 | ||
| Fig. 17 (a) Schematic illustration of the methodology for designing DNA-stabilized Ag nanoclusters with specific colour classes via DNA sequence design using ML algorithms. Reprinted with permission from ref. 230. Copyright 2018, American Chemical Society. (b) Schematic illustration of the synthesis of GSH-protected photoluminescent Au nanoclusters (top) and the workflow for the construction of the synthetic phase diagram by ML algorithms. Reproduced with permission from ref. 232. Copyright 2023, Royal Society of Chemistry. | ||
To enhance the precision and accuracy of this precursor–property relationship model, the same research group incorporated chemical insights obtained from the recently resolved crystal structure of DNA-stabilized Ag16.147 This advancement enabled refinement of the feature engineering protocol to include both adjacent and nonadjacent base patterns, accounting for the three-dimensional relationship between the Ag core and the DNA ligand.231 Based on this improved methodology, the authors increased the selectivity by 12.3 times targeting bright near-infrared nanoclusters with promising deep tissue bioimaging capabilities. Furthermore, to address the challenge of insufficient brightness in some products, the team upgraded their model to enable simultaneous colour and brightness selection, which successfully reduced the occurrence of products with inadequate brightness while increasing the selectivity of those with both desired colour and brightness characteristics.47 Through this stepwise approach, the precursor–property relationship model was progressively refined to guide target design with increasingly specific performance requirements. While the current model successfully generates DNAs for nanoclusters above a brightness threshold, future enhancements could focus on ranking DNA candidates based on predicted brightness values. This advancement would not only facilitate the identification of nanoclusters with the highest emission intensity—desirable for bioimaging and biosensing applications—but also reduce the candidate pool size, thereby enhancing the efficiency of identifying suitable DNA sequences for potential highly performing metal nanocluster candidates.
Similarly, condition–property relationship modelling has proven valuable for designing photoluminescent glutathione-stabilized Au nanoclusters. Due to the significant roles played by the various condition parameters in the synthesis of photoluminescent glutathione-stabilized Au nanoclusters and the unavailability of precise structural information, Jin et al. developed a condition–property relationship model for target design to optimize QY (Fig. 17b).232 Their investigation focused on a three-dimensional design space comprising M/L ratio, reaction temperature and synthesis duration. The model's accuracy was validated by low error between the experimental and predicted QY values at 10 randomly selected points in the design space. Analysis of the trained model revealed complex parameter interactions, with high QY products obtained under specific conditions: low M/L ratio (0.5–0.63), short reaction time (3.3–10.2 h) and high temperatures (85–95 °C). These insights enabled the researchers to successfully reverse-engineer highly photoluminescent glutathione-stabilized Au nanoclusters with optimized properties. However, the model can be further enhanced by incorporating additional condition parameters such as the solvent used and the pH of the solution to cover a higher proportion of the entire synthesis design space.
These direct modelling approaches significantly facilitate the design of target MNCs whose structural information is difficult to obtain or whose monodispersity is challenging to achieve, thereby circumventing the labour-intensive process of structural determination for numerous less qualified MNC candidates. By establishing quantitative relationships between readily controllable synthesis parameters and desired functional properties, these methodologies provide an efficient pathway for rational MNC design in cases where traditional structure-guided approaches are not readily available.
5.4. Model generalization and adaptation
Current principles and models in metal nanocluster (MNC) synthesis planning are typically restricted to specific MNC subgroups due to limited dataset sizes, constraining predictability for new systems.57,232 This reflects a fundamental trade-off between model complexity and generalizability: while complex models can capture intricate synthesis relationships essential for accurate MNC prediction, they require extensive data to generalize effectively and risk overfitting in new chemical spaces with limited training examples. Conversely, simpler models may generalize more readily but may miss critical nonlinear relationships governing synthesis outcomes.Data-driven approaches strategically navigate this trade-off through generalization techniques that enable the deployment of appropriate complex models while maintaining broad applicability.46,225,226,231 The effectiveness of model adaptation depends primarily on the chemical similarity between source and target domains. When similarity is high, transfer learning provides a powerful framework for leveraging knowledge from data-rich domains to guide synthesis in related but data-limited domains.237,238 This transfer can be implemented through several complementary approaches: instance-based methods reweight samples (such as species or ligand molecules) to account for domain differences; feature-based approaches utilize descriptors exhibiting minimal numeric variations between domains; parameter-based approaches directly transfer model parameters (such as regression model coefficients) or chemical insights; and relational-based methods map connections between different chemical spaces to apply prior knowledge.
For example, models initially developed for condition–purity relationships of a specific MNC species can be systematically expanded along precursor dimensions through feature-based transfer learning. This expansion enables researchers to probe the complex roles of different precursor types (such as ligand) in synergism with the condition parameters during synthesis while preserving established relationships. Furthermore, by establishing correlations between synthesis condition parameters, reaction precursors, and product identity and purity, researchers can elucidate the complex synthesis design space of MNCs (e.g., MNC size and structural dependencies on ligand and metal precursors) and develop comprehensive synthesis protocols with systematic understanding of parameter interactions.
For systems characterized by low domain similarity, combining transfer learning with active learning offers iterative performance refinement through strategic data incorporation.220,239 These updates can integrate both literature-derived information and in-house experimental results, though data quality remains critical—requiring representative sampling and consistent descriptor usage across datasets. While literature data integration faces challenges including publication bias toward successful reactions and inconsistent descriptors, HTE platforms provide an effective solution by efficiently generating systematic, unbiased datasets in-house, specifically designed to improve model adaptability across diverse MNC systems. These datasets can be deliberately structured to address the gaps in existing knowledge bases, enhancing the robustness of predictive models across chemical space.
The continued development of these modelling efforts, coupled with coordinated data collection strategies, will progressively build a comprehensive MNC data library shared throughout the research community. This collaborative approach facilitates the development of increasingly general and reliable predictive models for precise MNC design, ultimately bridging the gaps between targeted properties, MNC structures and optimal synthesis conditions. As these models evolve to incorporate a broader range of MNC systems, they will enable more rapid adaptation to emerging research directions and accelerate the discovery of novel MNCs with tailored functionalities.
6. Conclusion and outlook
In summary, this perspective has examined the fundamental considerations in the rational design and synthesis of MNCs within a comprehensive synthesis planning framework. We have systematically explored the design principles governing target core and ligand shell composition and architectures, critical factors in synthetic route selection, and the profound effects of various condition parameters on successful MNC synthesis outcomes. Data-driven approaches offer transformative opportunities to enhance both efficiency and precision throughout the synthesis planning process and have demonstrated capabilities in accelerating structure prediction to deepen our understanding of complex MNC structures and establishing quantitative relationship models that provide predictive capabilities beyond traditional qualitative approaches. Moreover, model generalization and adaptation through transfer learning and active learning methodologies enables knowledge transfer from priorly studied domains to new target problems. However, several challenges must be addressed for effective data-driven approach adoption in MNC synthesis planning.Firstly, the existence of uncertainties arising from both noisy training data and model ignorance can affect model performance. Noisy data pose difficulties in model training that result in low accuracy and unreliable predictions. Effective data preprocessing—including smoothing for spectral data,240 and feature engineering techniques such as normalization and dimensionality reduction—can mitigate aleatoric uncertainty effects. Algorithm selection critically impacts uncertainty handling. While Gaussian-based models intrinsically account for uncertainties, approaches like autoencoders provide dimensionality reduction benefits. Besides, uncertainty quantification techniques such as ensemble-based methods which involve training multiple model replicates and calculating predictions as arithmetic means,241 and mean-variance estimation (MVE) incorporating additional variance prediction neurons can be incorporated for more robust and reliable predictions.242 However, further research is needed to identify optimal approaches for specific problems. These techniques enhance prediction confidence and enable informed decision-making while providing foundations for active learning-based model refinement.
Secondly, beyond relationship modelling and prediction, model explainability should be addressed to reveal underlying prediction mechanisms. Post-hoc explanation methods such as local interpretable model-agnostic explanations (LIME) and shapley additive explanations (SHAP) provide model-agnostic tools.243,244 For example, SHAP analysis identified gas hourly space velocity and temperature as critical features for higher alcohol synthesis catalyst development.245 However, SHAP treats features independently, ignoring causal relationships. Partial dependence plots can probe synergistic feature interactions when informative descriptors are employed.246 For deep learning models, attention mechanisms highlight crucial input data regions affecting predictions.247 The incorporation of these techniques can not only enhance prediction reliability, but also reveal synthetic and mechanistic insights that guide future research directions.
The foundation of successful modelling approaches relies on high-quality data collection. It is crucial to develop unbiased datasets with descriptors carefully designed not only for the specific relationships under investigation but also selected with foresight toward their utility in future model generalization and cross-domain adaptability. HTE significantly enhances efficiency by enabling systematic exploration of parameter spaces that would be prohibitively time-consuming through traditional methods—a capability particularly valuable when confronting the vast synthetic design space inherent in MNC systems.248 Standardized reporting protocols for experimental design, characterization, data collection, featurization, and model training should be promoted to facilitate data sharing, enhance reproducibility, and establish collective databases.249–251 Such practices alleviate data preprocessing workloads, enhance efficiency through facilitating automated literature data mining using ML tools including large language models (LLM),252–254 and provide high-quality data for robust model training.
Looking forward, the continued expansion of the MNC library, accompanied by robust property characterization data, will progressively enrich comprehensive MNC databases, enabling adoption of powerful yet data-demanding tools such as generative models for inverse design of novel materials with desired properties.255,256 These approaches have demonstrated promising capabilities across diverse fields, from crystalline inorganic materials generation under chemistry or property constraints,257 to applications in organic molecules,258 drug discovery,259 metal–organic frameworks (MOFs),36 and plasmonic nanoparticles.260 However, ensuring synthesizability of computationally predicted materials remains a critical challenge requiring substantial research effort. This growing knowledge repository allows for increasingly sophisticated structure–property relationship modelling, advancing the field toward the ambitious goal of “dial-a-MNC” capabilities where on-demand MNCs can be synthesized with precisely tailored properties. Through strategic integration of computational approaches with experimental advances, the rational design and predictive synthesis of metal nanoclusters with tailored properties for specific applications becomes increasingly achievable, opening new frontiers in nanomaterial science and technology.
Author contributions
J. Xie supervised this work. J. Xie and J. Lyu conceived the idea and wrote the manuscript. J. Lyu conducted the literature survey. J. Qian and J. Lyu participated in visualization. J. Qian and Z. Yang commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
We acknowledge the Ministry of Education, Singapore, for their financial support, through the Academic Research Fund (AcRF) Grant No. A-8002406-00-00. We would also like to acknowledge the support from the National Natural Science Foundation of China (22071174).Notes and references
- T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, J. Med. Chem., 2020, 63, 8667–8682 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- H. Lin, X. Song, O. J. H. Chai, Q. Yao, H. Yang and J. Xie, Adv. Mater., 2024, 36, 2401002 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, O. J. H. Chai, N. Ding, W. Xu, S. Zang and J. Xie, Angew. Chem., Int. Ed., 2020, 59, 9934–9939 CrossRef CAS PubMed.
- Z. Liu, L. Luo and R. Jin, Adv. Mater., 2024, 36, 2309073 CrossRef CAS PubMed.
- C. Liu, Y. Zhao, T.-S. Zhang, C.-B. Tao, W. Fei, S. Zhang and M.-B. Li, Nat. Commun., 2023, 14, 3730 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- Y. Li, H. K. Kim, R. D. McGillicuddy, S.-L. Zheng, K. J. Anderton, G. J. Stec, J. Lee, D. Cui and J. A. Mason, J. Am. Chem. Soc., 2023, 145, 9304–9312 CrossRef CAS PubMed.
- C. Zhu, Z.-L. Chen, H. Li, L. Lu, X. Kang, J. Xuan and M. Zhu, J. Am. Chem. Soc., 2024, 146, 23212–23220 CrossRef CAS PubMed.
- S. Masuda, K. Sakamoto and T. Tsukuda, Nanoscale, 2024, 16, 4514–4528 RSC.
- F. Xiao, Y. Chen, J. Qi, Q. Yao, J. Xie and X. Jiang, Adv. Mater., 2023, 35, 2210412 CrossRef CAS PubMed.
- K. Zheng and J. Xie, Acc. Mater. Res., 2021, 2, 1104–1116 CrossRef CAS.
- G. Yang, Z. Wang, F. Du, F. Jiang, X. Yuan and J. Y. Ying, J. Am. Chem. Soc., 2023, 145, 11879–11898 CrossRef CAS PubMed.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed.
- Y. Li, M. Zhou and R. Jin, Adv. Mater., 2021, 33, 2006591 CrossRef CAS PubMed.
- Y. Niihori, S. Miyajima, A. Ikeda, T. Kosaka and Y. Negishi, Small Sci., 2023, 3, 2300024 CrossRef CAS PubMed.
- Q. Yao, M. Zhu, Z. Yang, X. Song, X. Yuan, Z. Zhang, W. Hu and J. Xie, Nat. Rev. Mater., 2025, 10, 89–108 CrossRef CAS.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Small, 2021, 17, 2004381 CrossRef CAS PubMed.
- F. Bertorelle, K. D. Wegner, M. Perić Bakulić, H. Fakhouri, C. Comby-Zerbino, A. Sagar, P. Bernadó, U. Resch-Genger, V. Bonačić-Koutecký, X. Le Guével and R. Antoine, Chem. – Eur. J., 2022, 28, e202200570 CrossRef CAS PubMed.
- J.-H. Huang, H. Zhang, Z.-Y. Wang, J.-H. Hu, J. Li, J. Cai and S.-Q. Zang, J. Am. Chem. Soc., 2025, 147, 16593–16601 CrossRef CAS PubMed.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- H. Shan, J. Shi, T. Chen, Y. Cao, Q. Yao, H. An, Z. Yang, Z. Wu, Z. Jiang and J. Xie, ACS Nano, 2023, 17, 2368–2377 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- T. Chen, H. Lin, Y. Cao, Q. Yao and J. Xie, Adv. Mater., 2021, 34, 2103918 CrossRef PubMed.
- J. Qian, Z. Yang, J. Lyu, Q. Yao and J. Xie, Precis. Chem., 2024, 2, 495–517 CrossRef CAS PubMed.
- Y. Cao, T. Chen, Q. Yao and J. Xie, Acc. Chem. Res., 2021, 54, 4142–4153 CrossRef CAS PubMed.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- Y. Yu, Q. Yao, K. Cheng, X. Yuan, Z. Luo and J. Xie, Part. Part. Syst. Charact., 2014, 31, 652–656 CrossRef CAS.
- H. Qian and R. Jin, Chem. Mater., 2011, 23, 2209–2217 CrossRef CAS.
- Y. Yu, X. Chen, Q. Yao, Y. Yu, N. Yan and J. Xie, Chem. Mater., 2013, 25, 946–952 CrossRef CAS.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- M. Zhu, Q. Yao, Z. Liu, B. Zhang, Y. Lin, J. Liu, M. Long and J. Xie, Angew. Chem., Int. Ed., 2022, 61, e202115647 CrossRef CAS PubMed.
- Z. Yao, B. Sánchez-Lengeling, N. S. Bobbitt, B. J. Bucior, S. G. H. Kumar, S. P. Collins, T. Burns, T. K. Woo, O. K. Farha, R. Q. Snurr and A. Aspuru-Guzik, Nat. Mach. Intell., 2021, 3, 76–86 CrossRef.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- H. Zhao, W. Chen, H. Huang, Z. Sun, Z. Chen, L. Wu, B. Zhang, F. Lai, Z. Wang, M. L. Adam, C. H. Pang, P. K. Chu, Y. Lu, T. Wu, J. Jiang, Z. Yin and X.-F. Yu, Nat. Synth., 2023, 2, 505–514 CrossRef CAS.
- J.-F. Masson, J. S. Biggins and E. Ringe, Nat. Nanotechnol., 2023, 18, 111–123 CrossRef CAS PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed.
- A. A. Volk, R. W. Epps, D. T. Yonemoto, B. S. Masters, F. N. Castellano, K. G. Reyes and M. Abolhasani, Nat. Commun., 2023, 14, 1403 CrossRef CAS PubMed.
- B. C. Haas, D. Kalyani and M. S. Sigman, Sci. Adv., 2025, 11, eadt3013 CrossRef PubMed.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk, A. Merchant, H. Kim, A. Jain, C. J. Bartel, K. Persson, Y. Zeng and G. Ceder, Nature, 2023, 624, 86–91 CrossRef CAS PubMed.
- Z. Yang, A. Shi, R. Zhang, Z. Ji, J. Li, J. Lyu, J. Qian, T. Chen, X. Wang, F. You and J. Xie, ACS Nano, 2024, 18, 27138–27166 CrossRef CAS PubMed.
- J. Li, T. Chen, K. Lim, L. Chen, S. A. Khan, J. Xie and X. Wang, Adv. Intell. Syst., 2019, 1, 1900029 CrossRef.
- E. Sadeghi, P. Mastracco, A. Gonzàlez-Rosell, S. M. Copp and P. Bogdanov, ACS Nano, 2024, 18, 26997–27008 CrossRef CAS PubMed.
- S. Li, N.-N. Li, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Rev., 2024, 124, 7262–7378 CrossRef CAS PubMed.
- H. Tao, T. Wu, M. Aldeghi, T. C. Wu, A. Aspuru-Guzik and E. Kumacheva, Nat. Rev. Mater., 2021, 6, 701–716 CrossRef.
- L. Rao, Y. Yuan, X. Shen, G. Yu and X. Chen, Nat. Nanotechnol., 2024, 19, 1769–1781 CrossRef CAS PubMed.
- K. Kwak, Q. Tang, M. Kim, D.-E. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- M. Zhou, T. Higaki, Y. Li, C. Zeng, Q. Li, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2019, 141, 19754–19764 CrossRef CAS PubMed.
- Y. Li, Y. Song, X. Zhang, T. Liu, T. Xu, H. Wang, D.-E. Jiang and R. Jin, J. Am. Chem. Soc., 2022, 144, 12381–12389 CrossRef CAS PubMed.
- Y. Zhang, P. Song, T. Chen, X. Liu, T. Chen, Z. Wu, Y. Wang, J. Xie and W. Xu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10588–10593 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2022, 61, e202211771 CrossRef CAS PubMed.
- K. Kwak, V. D. Thanthirige, K. Pyo, D. Lee and G. Ramakrishna, J. Phys. Chem. Lett., 2017, 8, 4898–4905 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS PubMed.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- W. W. Xu, B. Zhu, X. C. Zeng and Y. Gao, Nat. Commun., 2016, 7, 13574 CrossRef CAS PubMed.
- L. Xiong, S. Yang, X. Sun, J. Chai, B. Rao, L. Yi, M. Zhu and Y. Pei, J. Phys. Chem. C, 2018, 122, 14898–14907 CrossRef CAS.
- C. Zeng, Y. Chen, C. Liu, K. Nobusada, N. L. Rosi and R. Jin, Sci. Adv., 2015, 1, e1500425 CrossRef PubMed.
- W.-Q. Shi, L. Zeng, R.-L. He, X.-S. Han, Z.-J. Guan, M. Zhou and Q.-M. Wang, Science, 2024, 383, 326–330 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2014, 136, 11922–11925 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS PubMed.
- S. Yang, J. Chai, Y. Song, J. Fan, T. Chen, S. Wang, H. Yu, X. Li and M. Zhu, J. Am. Chem. Soc., 2017, 139, 5668–5671 CrossRef CAS PubMed.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-E. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS PubMed.
- A. Dass, T. Jones, M. Rambukwella, D. Crasto, K. J. Gagnon, L. Sementa, M. De Vetta, O. Baseggio, E. Aprà, M. Stener and A. Fortunelli, J. Phys. Chem. C, 2016, 120, 6256–6261 CrossRef CAS.
- S. M. Reilly, T. Krick and A. Dass, J. Phys. Chem. C, 2010, 114, 741–745 CrossRef CAS.
- L. Xiong, B. Peng, Z. Ma, P. Wang and Y. Pei, Nanoscale, 2017, 9, 2895–2902 RSC.
- Y. Wang, C. G. Gianopoulos, Z. Liu, K. Kirschbaum, D. Alfonso, D. R. Kauffman and R. Jin, JACS Au, 2024, 4, 1928–1934 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- Z. Ma, P. Wang and Y. Pei, Nanoscale, 2016, 8, 17044–17054 RSC.
- W. W. Xu, Y. Li, Y. Gao and X. C. Zeng, Nanoscale, 2016, 8, 7396–7401 RSC.
- S. Takano, S. Yamazoe, K. Koyasu and T. Tsukuda, J. Am. Chem. Soc., 2015, 137, 7027–7030 CrossRef CAS PubMed.
- Z. Ma, P. Wang, G. Zhou, J. Tang, H. Li and Y. Pei, J. Phys. Chem. C, 2016, 120, 13739–13748 CrossRef CAS.
- X. Liu, W. W. Xu, X. Huang, E. Wang, X. Cai, Y. Zhao, J. Li, M. Xiao, C. Zhang, Y. Gao, W. Ding and Y. Zhu, Nat. Commun., 2020, 11, 3349 CrossRef CAS PubMed.
- H. Dong, L. Liao, S. Zhuang, C. Yao, J. Chen, S. Tian, M. Zhu, X. Liu, L. Li and Z. Wu, Nanoscale, 2017, 9, 3742–3746 RSC.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430–8434 CrossRef CAS PubMed.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- X.-K. Wan, Z.-W. Lin and Q.-M. Wang, J. Am. Chem. Soc., 2012, 134, 14750–14752 CrossRef CAS PubMed.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560–10564 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-E. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS PubMed.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS PubMed.
- Z. Liu, M. Zhou, L. Luo, Y. Wang, E. Kahng and R. Jin, J. Am. Chem. Soc., 2023, 145, 19969–19981 CrossRef CAS PubMed.
- Y. Liu, X. Chai, X. Cai, M. Chen, R. Jin, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2018, 57, 9775–9779 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- C. Liu, T. Li, G. Li, K. Nobusada, C. Zeng, G. Pang, N. L. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 9826–9829 CrossRef CAS PubMed.
- E. L. Albright, T. I. Levchenko, V. K. Kulkarni, A. I. Sullivan, J. F. DeJesus, S. Malola, S. Takano, M. Nambo, K. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2024, 146, 5759–5780 CrossRef CAS PubMed.
- S.-F. Yuan, C.-Q. Xu, W.-D. Liu, J.-X. Zhang, J. Li and Q.-M. Wang, J. Am. Chem. Soc., 2021, 143, 12261–12267 CrossRef CAS PubMed.
- J.-H. Huang, Y. Cui, Z.-Y. Wang and S.-Q. Zang, Acc. Chem. Res., 2025, 58, 1249–1261 CrossRef CAS PubMed.
- R. Xiao, J. Jia, R. Wang, Y. Feng and H. Chen, Acc. Chem. Res., 2023, 56, 1539–1552 CrossRef CAS PubMed.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS PubMed.
- D. M. Chevrier, L. Raich, C. Rovira, A. Das, Z. Luo, Q. Yao, A. Chatt, J. Xie, R. Jin, J. Akola and P. Zhang, J. Am. Chem. Soc., 2018, 140, 15430–15436 CrossRef CAS PubMed.
- X. Yuan, N. Goswami, I. Mathews, Y. Yu and J. Xie, Nano Res., 2015, 8, 3488–3495 CrossRef CAS.
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603–3609 CrossRef CAS PubMed.
- X.-K. Wan, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2017, 56, 11494–11497 CrossRef CAS PubMed.
- X.-K. Wan, Q. Tang, S.-F. Yuan, D.-E. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS PubMed.
- A. Das, T. Li, G. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, Nanoscale, 2014, 6, 6458–6462 RSC.
- Y. Song, S. Wang, J. Zhang, X. Kang, S. Chen, P. Li, H. Sheng and M. Zhu, J. Am. Chem. Soc., 2014, 136, 2963–2965 CrossRef CAS PubMed.
- S. Chen, L. Xiong, S. Wang, Z. Ma, S. Jin, H. Sheng, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2016, 138, 10754–10757 CrossRef CAS PubMed.
- J.-J. Li, Z.-J. Guan, Z. Lei, F. Hu and Q.-M. Wang, Angew. Chem., Int. Ed., 2019, 58, 1083–1087 CrossRef CAS PubMed.
- Z. Liu, Y. Li, E. Kahng, S. Xue, X. Du, S. Li and R. Jin, ACS Nano, 2022, 16, 18448–18458 CrossRef CAS PubMed.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS PubMed.
- Q. Yao, X. Yuan, Y. Yu, Y. Yu, J. Xie and J. Y. Lee, J. Am. Chem. Soc., 2015, 137, 2128–2136 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, S. Y. Yoon, G. Ramakrishna and D. Lee, Nanoscale, 2016, 8, 20008–20016 RSC.
- Y. Cao, S. Malola, M. F. Matus, T. Chen, Q. Yao, R. Shi, H. Häkkinen and J. Xie, Chem, 2021, 7, 2227–2244 CAS.
- Y. Zhong, J. Zhang, T. Li, W. Xu, Q. Yao, M. Lu, X. Bai, Z. Wu, J. Xie and Y. Zhang, Nat. Commun., 2023, 14, 658 CrossRef CAS PubMed.
- H. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2024, 57, 3194–3205 CrossRef CAS PubMed.
- Q. Yao, L. Liu, S. Malola, M. Ge, H. Xu, Z. Wu, T. Chen, Y. Cao, M. F. Matus, A. Pihlajamäki, Y. Han, H. Häkkinen and J. Xie, Nat. Chem., 2023, 15, 230–239 CrossRef CAS PubMed.
- T. Chen, S. Yang, J. Chai, Y. Song, J. Fan, B. Rao, H. Sheng, H. Yu and M. Zhu, Sci. Adv., 2017, 3, e1700956 CrossRef PubMed.
- X. Wang, B. Yin, L. Jiang, C. Yang, Y. Liu, G. Zou, S. Chen and M. Zhu, Science, 2023, 381, 784–790 CrossRef CAS PubMed.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan, L. Li, X. Mu, J. Wang, W. Mi, J. Luo, J. Xie and X.-D. Zhang, Adv. Mater., 2019, 31, 1901015 CrossRef CAS PubMed.
- X. Wang, M. B. Liisberg, G. L. Nolt, X. Fu, C. Cerretani, L. Li, L. A. Johnson, T. Vosch and C. I. Richards, ACS Nano, 2023, 17, 12862–12874 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- X. Jiang, B. Du, Y. Huang, M. Yu and J. Zheng, Bioconjugate Chem., 2020, 31, 1522–1528 CrossRef CAS PubMed.
- A. I. Sullivan, E. A. Steele, S. Takano, E. Zeinizade, J. Chen, S. Malola, K. Siddhant, H. Häkkinen, K. G. Stamplecoskie, T. Tsukuda, G. Zheng and C. M. Crudden, J. Am. Chem. Soc., 2025, 147, 4230–4238 CrossRef CAS PubMed.
- B. Zhang, M. F. Matus, Q. Yao, X. Song, Z. Wu, W. Hu, H. Häkkinen and J. Xie, J. Am. Chem. Soc., 2025, 147, 6404–6414 CrossRef CAS PubMed.
- A. Mathew, G. Natarajan, L. Lehtovaara, H. Häkkinen, R. M. Kumar, V. Subramanian, A. Jaleel and T. Pradeep, ACS Nano, 2014, 8, 139–152 CrossRef CAS PubMed.
- H. Deng, K. Huang, L. Xiu, W. Sun, Q. Yao, X. Fang, X. Huang, H. A. A. Noreldeen, H. Peng, J. Xie and W. Chen, Nat. Commun., 2022, 13, 3381 CrossRef CAS PubMed.
- Y. Zhang, S.-R. He, Y. Yang, T.-S. Zhang, Z.-M. Zhu, W. Fei and M.-B. Li, J. Am. Chem. Soc., 2023, 145, 12164–12172 CrossRef CAS PubMed.
- P. N. Gunawardene, J. F. Corrigan and M. S. Workentin, J. Am. Chem. Soc., 2019, 141, 11781–11785 CrossRef CAS PubMed.
- P. N. Gunawardene, J. Martin, J. M. Wong, Z. Ding, J. F. Corrigan and M. S. Workentin, Angew. Chem., Int. Ed., 2022, 61, e202205194 CrossRef CAS PubMed.
- V. Rück, N. K. Mishra, K. K. Sørensen, M. B. Liisberg, A. B. Sloth, C. Cerretani, C. B. Mollerup, A. Kjaer, C. Lou, K. J. Jensen and T. Vosch, J. Am. Chem. Soc., 2023, 145, 16771–16777 CrossRef PubMed.
- Z. Liu, H. Tan, B. Li, Z. Hu, D.-E. Jiang, Q. Yao, L. Wang and J. Xie, Nat. Commun., 2023, 14, 3374 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, S. Szymkuć, K. Molga, A. Aspuru-Guzik, F. Glorius and B. A. Grzybowski, J. Am. Chem. Soc., 2024, 146, 11005–11017 CAS.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- Q. Yao, X. Yuan, V. Fung, Y. Yu, D. T. Leong, D.-E. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef PubMed.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS PubMed.
- S. Wang, H. Abroshan, C. Liu, T.-Y. Luo, M. Zhu, H. J. Kim, N. L. Rosi and R. Jin, Nat. Commun., 2017, 8, 848 CrossRef PubMed.
- Y. Lin, Y. Cao, Q. Yao and J. Xie, Chem. Sci., 2022, 13, 4598–4607 RSC.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS PubMed.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar, R. L. Whetten, W. G. Cullen, P. N. First, C. Gutiérrez-Wing, J. Ascensio and M. J. Jose-Yacamán, J. Phys. Chem. B, 1997, 101, 7885–7891 CrossRef CAS.
- V. L. Jimenez, D. G. Georganopoulou, R. J. White, A. S. Harper, A. J. Mills, D. Lee and R. W. Murray, Langmuir, 2004, 20, 6864–6870 CrossRef CAS PubMed.
- A. Ghosh, J. Hassinen, P. Pulkkinen, H. Tenhu, R. H. A. Ras and T. Pradeep, Anal. Chem., 2014, 86, 12185–12190 CrossRef CAS PubMed.
- Y. Negishi, S. Hashimoto, A. Ebina, K. Hamada, S. Hossain and T. Kawawaki, Nanoscale, 2020, 12, 8017–8039 RSC.
- C. Cerretani, H. Kanazawa, T. Vosch and J. Kondo, Angew. Chem., Int. Ed., 2019, 58, 17153–17157 CrossRef CAS PubMed.
- V. Rück, V. A. Neacsu, M. B. Liisberg, C. B. Mollerup, P. H. Ju, T. Vosch, J. Kondo and C. Cerretani, Adv. Opt. Mater., 2024, 12, 2301928 CrossRef.
- C.-Y. Liu, S.-F. Yuan, S. Wang, Z.-J. Guan, D.-E. Jiang and Q.-M. Wang, Nat. Commun., 2022, 13, 2082 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- H. Qian, M. Zhu, U. N. Andersen and R. Jin, J. Phys. Chem. A, 2009, 113, 4281–4284 CrossRef CAS PubMed.
- W. Du, S. Jin, L. Xiong, M. Chen, J. Zhang, X. Zou, Y. Pei, S. Wang and M. Zhu, J. Am. Chem. Soc., 2017, 139, 1618–1624 CrossRef CAS PubMed.
- T. A. Dreier and C. J. Ackerson, Angew. Chem., Int. Ed., 2015, 54, 9249–9252 CrossRef CAS PubMed.
- Y. Cao, T. Liu, T. Chen, B. Zhang, D.-E. Jiang and J. Xie, Nat. Commun., 2021, 12, 3212 CrossRef CAS PubMed.
- Y. Cao, V. Fung, Q. Yao, T. Chen, S. Zang, D.-E. Jiang and J. Xie, Nat. Commun., 2020, 11, 5498 CrossRef CAS PubMed.
- C. Yan, Y. Li, E. Wang and W. W. Xu, J. Phys. Chem. Lett., 2023, 14, 7632–7637 CrossRef CAS PubMed.
- M. A. Tofanelli, K. Salorinne, T. W. Ni, S. Malola, B. Newell, B. Phillips, H. Häkkinen and C. J. Ackerson, Chem. Sci., 2016, 7, 1882–1890 RSC.
- T. Dainese, S. Antonello, S. Bogialli, W. Fei, A. Venzo and F. Maran, ACS Nano, 2018, 12, 7057–7066 CrossRef CAS PubMed.
- S. Yang, S. Chen, L. Xiong, C. Liu, H. Yu, S. Wang, N. L. Rosi, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2018, 140, 10988–10994 CrossRef CAS PubMed.
- B. Zhang, C. Chen, W. Chuang, S. Chen and P. Yang, J. Am. Chem. Soc., 2020, 142, 11514–11520 CrossRef CAS PubMed.
- X. S. Han, X. Luan, H. F. Su, J. J. Li, S. F. Yuan, Z. Lei, Y. Pei and Q. M. Wang, Angew. Chem., Int. Ed., 2020, 59, 2309–2312 CrossRef CAS PubMed.
- A. Das, C. Liu, H. Y. Byun, K. Nobusada, S. Zhao, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140–3144 CrossRef CAS PubMed.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed.
- A. Dass, T. C. Jones, S. Theivendran, L. Sementa and A. Fortunelli, J. Phys. Chem. C, 2017, 121, 14914–14919 CrossRef CAS.
- M. P. Maman, A. S. Nair, H. Cheraparambil, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2020, 11, 1781–1788 CrossRef CAS PubMed.
- Q. Yao, V. Fung, C. Sun, S. Huang, T. Chen, D.-E. Jiang, J. Y. Lee and J. Xie, Nat. Commun., 2018, 9, 1979 CrossRef PubMed.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS PubMed.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- Y. Negishi, K. Munakata, W. Ohgake and K. Nobusada, J. Phys. Chem. Lett., 2012, 3, 2209–2214 CrossRef CAS PubMed.
- L. V. Nair, S. Hossain, S. Takagi, Y. Imai, G. Hu, S. Wakayama, B. Kumar, W. Kurashige, D.-E. Jiang and Y. Negishi, Nanoscale, 2018, 10, 18969–18979 RSC.
- Q. Li, K. J. Lambright, M. G. Taylor, K. Kirschbaum, T.-Y. Luo, J. Zhao, G. Mpourmpakis, S. Mokashi-Punekar, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2017, 139, 17779–17782 CrossRef CAS PubMed.
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 9511–9514 CrossRef CAS PubMed.
- Z.-H. Gao, K. Wei, T. Wu, J. Dong, D.-E. Jiang, S. Sun and L.-S. Wang, J. Am. Chem. Soc., 2022, 144, 5258–5262 CrossRef CAS PubMed.
- T.-H. Chiu, J.-H. Liao, F. Gam, Y.-Y. Wu, X. Wang, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2022, 144, 10599–10607 CrossRef CAS PubMed.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS PubMed.
- S. Yamazoe, W. Kurashige, K. Nobusada, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2014, 118, 25284–25290 CrossRef CAS.
- H. Qian, D.-e Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS PubMed.
- M. Sera, S. Hossain, S. Yoshikawa, K. Takemae, A. Ikeda, T. Tanaka, T. Kosaka, Y. Niihori, T. Kawawaki and Y. Negishi, J. Am. Chem. Soc., 2024, 146, 29684–29693 CrossRef CAS PubMed.
- C. Kumara and A. Dass, Nanoscale, 2012, 4, 4084–4086 RSC.
- Y. Niihori, Y. Koyama, S. Watanabe, S. Hashimoto, S. Hossain, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 4930–4934 CrossRef CAS PubMed.
- Y. Niihori, S. Hashimoto, Y. Koyama, S. Hossain, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2019, 123, 13324–13329 CrossRef CAS.
- C. Yao, Y.-j Lin, J. Yuan, L. Liao, M. Zhu, L.-h Weng, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 15350–15353 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- K. Zheng, V. Fung, X. Yuan, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 18977–18983 CrossRef CAS PubMed.
- Q. Yao, Y. Feng, V. Fung, Y. Yu, D.-E. Jiang, J. Yang and J. Xie, Nat. Commun., 2017, 8, 1555 CrossRef PubMed.
- Q. Li, T.-Y. Luo, M. G. Taylor, S. Wang, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- Q. Li, S. Wang, K. Kirschbaum, K. J. Lambright, A. Das and R. Jin, Chem. Commun., 2016, 52, 5194–5197 RSC.
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS PubMed.
- M. Zhu, P. Wang, N. Yan, X. Chai, L. He, Y. Zhao, N. Xia, C. Yao, J. Li, H. Deng, Y. Zhu, Y. Pei and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 4500–4504 CrossRef CAS PubMed.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS PubMed.
- M. Neumaier, A. Baksi, P. Weis, E. K. Schneider, P. Chakraborty, H. Hahn, T. Pradeep and M. M. Kappes, J. Am. Chem. Soc., 2021, 143, 6969–6980 CrossRef CAS PubMed.
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., Int. Ed., 2014, 53, 4623–4627 CrossRef CAS PubMed.
- J.-H. Huang, Z.-Y. Wang, S.-Q. Zang and T. C. W. Mak, ACS Cent. Sci., 2020, 6, 1971–1976 CrossRef CAS PubMed.
- H. Shen, Z. Xu, M. S. A. Hazer, Q. Wu, J. Peng, R. Qin, S. Malola, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2021, 60, 3752–3758 CrossRef CAS PubMed.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS PubMed.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS PubMed.
- Z. Lei, J.-J. Li, Z.-A. Nan, Z.-G. Jiang and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 14415–14419 CrossRef CAS PubMed.
- B. M. Barngrover and C. M. Aikens, J. Am. Chem. Soc., 2012, 134, 12590–12595 CrossRef CAS PubMed.
- J. S. Kim, H. Chang, S. Kang, S. Cha, H. Cho, S. J. Kwak, N. Park, Y. Kim, D. Kang, C. K. Song, J. Kwag, J.-S. Hahn, W. B. Lee, T. Hyeon and J. Park, Nat. Commun., 2023, 14, 3201 CrossRef CAS PubMed.
- Y. G. Srinivasulu, N. Goswami, Q. Yao and J. Xie, J. Phys. Chem. C, 2021, 125, 4066–4076 CrossRef CAS.
- T. Chen, Q. Yao, X. Yuan, R. R. Nasaruddin and J. Xie, J. Phys. Chem. C, 2017, 121, 10743–10751 CrossRef CAS.
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138–1139 CrossRef CAS PubMed.
- X. Meng, Z. Liu, M. Zhu and R. Jin, Nanoscale Res. Lett., 2012, 7, 277 CrossRef PubMed.
- O. Cybulski, C. Quintana, M. Siek and B. A. Grzybowski, Small, 2024, 20, 2400306 CrossRef CAS PubMed.
- T. Chen, Z. Luo, Q. Yao, A. X. H. Yeo and J. Xie, Chem. Commun., 2016, 52, 9522–9525 RSC.
- L. Liao, S. Zhuang, C. Yao, N. Yan, J. Chen, C. Wang, N. Xia, X. Liu, M.-B. Li, L. Li, X. Bao and Z. Wu, J. Am. Chem. Soc., 2016, 138, 10425–10428 CrossRef CAS PubMed.
- L. Liao, S. Zhuang, P. Wang, Y. Xu, N. Yan, H. Dong, C. Wang, Y. Zhao, N. Xia, J. Li, H. Deng, Y. Pei, S.-K. Tian and Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 12644–12648 CrossRef CAS PubMed.
- X. Lin, J. Tang, C. Zhu, L. Wang, Y. Yang, R. A. Wu, H. Fan, C. Liu and J. Huang, Chem. Sci., 2023, 14, 994–1002 RSC.
- Q. Yao, Y. Yu, X. Yuan, Y. Yu, J. Xie and J. Y. Lee, Small, 2013, 9, 2696–2701 CrossRef CAS PubMed.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- T. Chen, Q. Yao, Y. Cao and J. Xie, Cell Rep. Phys. Sci., 2020, 1, 100206 CrossRef.
- Z. Wu, M. A. MacDonald, J. Chen, P. Zhang and R. Jin, J. Am. Chem. Soc., 2011, 133, 9670–9673 CrossRef CAS PubMed.
- F. Bertorelle, I. Russier-Antoine, C. Comby-Zerbino, F. Chirot, P. Dugourd, P.-F. Brevet and R. Antoine, ACS Omega, 2018, 3, 15635–15642 CrossRef CAS PubMed.
- A. Ghosh, T. Udayabhaskararao and T. Pradeep, J. Phys. Chem. Lett., 2012, 3, 1997–2002 CrossRef CAS.
- S. Zhuang, L. Liao, M.-B. Li, C. Yao, Y. Zhao, H. Dong, J. Li, H. Deng, L. Li and Z. Wu, Nanoscale, 2017, 9, 14809–14813 RSC.
- S. Zhuang, L. Liao, Y. Zhao, J. Yuan, C. Yao, X. Liu, J. Li, H. Deng, J. Yang and Z. Wu, Chem. Sci., 2018, 9, 2437–2442 RSC.
- S. Zhuang, L. Liao, J. Yuan, N. Xia, Y. Zhao, C. Wang, Z. Gan, N. Yan, L. He, J. Li, H. Deng, Z. Guan, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 4510–4514 CrossRef CAS PubMed.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- A. M. K. Nambiar, C. P. Breen, T. Hart, T. Kulesza, T. F. Jamison and K. F. Jensen, ACS Cent. Sci., 2022, 8, 825–836 CrossRef CAS PubMed.
- N. I. Rinehart, R. K. Saunthwal, J. Wellauer, A. F. Zahrt, L. Schlemper, A. S. Shved, R. Bigler, S. Fantasia and S. E. Denmark, Science, 2023, 381, 965–972 CrossRef CAS PubMed.
- G. Panapitiya, G. Avendaño-Franco, P. Ren, X. Wen, Y. Li and J. P. Lewis, J. Am. Chem. Soc., 2018, 140, 17508–17514 CrossRef CAS PubMed.
- S. Wang, Z. Wu, S. Dai and D.-E. Jiang, Angew. Chem., Int. Ed., 2021, 60, 12289–12292 CrossRef CAS PubMed.
- S. Wang, T. Liu and D.-E. Jiang, ACS Appl. Mater. Interfaces, 2021, 13, 53468–53474 CrossRef CAS PubMed.
- A. Pihlajamäki, M. F. Matus, S. Malola and H. Häkkinen, Adv. Mater., 2024, 36, 2407046 CrossRef PubMed.
- A. Pihlajamäki, J. Hämäläinen, J. Linja, P. Nieminen, S. Malola, T. Kärkkäinen and H. Häkkinen, J. Phys. Chem. A, 2020, 124, 4827–4836 CrossRef PubMed.
- S. Malola, P. Nieminen, A. Pihlajamäki, J. Hämäläinen, T. Kärkkäinen and H. Häkkinen, Nat. Commun., 2019, 10, 3973 CrossRef PubMed.
- F. Zhai, Y. Guan, Y. Li, S. Chen and R. He, ACS Appl. Nano Mater., 2022, 5, 9615–9624 CrossRef CAS.
- L. Tang, L. Wang, B. Wang, Y. Pei and S. Wang, Chem. – Eur. J., 2024, 30, e202302602 CrossRef CAS PubMed.
- T. Chen, J. Li, P. Cai, Q. Yao, Z. Ren, Y. Zhu, S. Khan, J. Xie and X. Wang, Nano Res., 2023, 16, 4188–4196 CrossRef CAS.
- S. M. Copp, A. Gorovits, S. M. Swasey, S. Gudibandi, P. Bogdanov and E. G. Gwinn, ACS Nano, 2018, 12, 8240–8247 CrossRef CAS PubMed.
- P. Mastracco, A. Gonzàlez-Rosell, J. Evans, P. Bogdanov and S. M. Copp, ACS Nano, 2022, 16, 16322–16331 CrossRef CAS PubMed.
- K. Jin, W. Wang, G. Qi, X. Peng, H. Gao, H. Zhu, X. He, H. Zou, L. Yang, J. Yuan, L. Zhang, H. Chen and X. Qu, Nanoscale, 2023, 15, 15358–15367 RSC.
- T.-R. Tero, S. Malola, B. Koncz, E. Pohjolainen, S. Lautala, S. Mustalahti, P. Permi, G. Groenhof, M. Pettersson and H. Häkkinen, ACS Nano, 2017, 11, 11872–11879 CrossRef CAS PubMed.
- A. Pihlajamäki, S. Malola, T. Kärkkäinen and H. Häkkinen, J. Phys. Chem. C, 2023, 127, 14211–14221 CrossRef.
- J. A. G. Torres, S. H. Lau, P. Anchuri, J. M. Stevens, J. E. Tabora, J. Li, A. Borovika, R. P. Adams and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 19999–20007 CrossRef CAS PubMed.
- M.-H. Du, Y. Dai, L.-P. Jiang, Y.-M. Su, M.-Q. Qi, C. Wang, L.-S. Long, L.-S. Zheng and X.-J. Kong, J. Am. Chem. Soc., 2023, 145, 23188–23195 CrossRef CAS PubMed.
- C. Cai, S. Wang, Y. Xu, W. Zhang, K. Tang, Q. Ouyang, L. Lai and J. Pei, J. Med. Chem., 2020, 63, 8683–8694 CrossRef CAS PubMed.
- C. Jiang, H. He, H. Guo, X. Zhang, Q. Han, Y. Weng, X. Fu, Y. Zhu, N. Yan, X. Tu and Y. Sun, Nat. Commun., 2024, 15, 6301 CrossRef CAS PubMed.
- E. Shim, J. A. Kammeraad, Z. Xu, A. Tewari, T. Cernak and P. M. Zimmerman, Chem. Sci., 2022, 13, 6655–6668 RSC.
- X. Xue, H. Sun, M. Yang, X. Liu, H.-Y. Hu, Y. Deng and X. Wang, Anal. Chem., 2023, 95, 13733–13745 CrossRef CAS PubMed.
- Z. Yang, Y.-M. Zhao, X. Wang, X. Liu, X. Zhang, Y. Li, Q. Lv, C. Y.-C. Chen and L. Shen, Nat. Commun., 2024, 15, 8148 CrossRef CAS PubMed.
- L.-Y. Chen and Y.-P. Li, Nat. Commun., 2025, 16, 3262 CrossRef CAS PubMed.
- X. Zhong, B. Gallagher, S. Liu, B. Kailkhura, A. Hiszpanski and T. Y.-J. Han, npj Comput. Mater., 2022, 8, 204 CrossRef.
- B. B. Mendes, Z. Zhang, J. Conniot, D. P. Sousa, J. M. J. M. Ravasco, L. A. Onweller, A. Lorenc, T. Rodrigues, D. Reker and J. Conde, Nat. Nanotechnol., 2024, 19, 867–878 CrossRef CAS PubMed.
- M. Suvarna, T. Zou, S. H. Chong, Y. Ge, A. J. Martín and J. Pérez-Ramírez, Nat. Commun., 2024, 15, 5844 CrossRef CAS PubMed.
- A. Rao and M. Grzelczak, Chem. Mater., 2024, 36, 2577–2587 CrossRef CAS PubMed.
- T.-S. Vu, M.-Q. Ha, D.-N. Nguyen, V.-C. Nguyen, Y. Abe, T. Tran, H. Tran, H. Kino, T. Miyake, K. Tsuda and H.-C. Dam, npj Comput. Mater., 2023, 9, 215 CrossRef.
- G. Wu, H. Zhou, J. Zhang, Z.-Y. Tian, X. Liu, S. Wang, C. W. Coley and H. Lu, Nat. Synth., 2023, 2, 515–526 CrossRef CAS.
- M. D. Wilkinson, M. Dumontier, I. J. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J.-W. Boiten, L. B. da Silva Santos, P. E. Bourne, J. Bouwman, A. J. Brookes, T. Clark, M. Crosas, I. Dillo, O. Dumon, S. Edmunds, C. T. Evelo, R. Finkers, A. Gonzalez-Beltran, A. J. G. Gray, P. Groth, C. Goble, J. S. Grethe, J. Heringa, P. A. C. Hoen, R. Hooft, T. Kuhn, R. Kok, J. Kok, S. J. Lusher, M. E. Martone, A. Mons, A. L. Packer, B. Persson, P. Rocca-Serra, M. Roos, R. van Schaik, S.-A. Sansone, E. Schultes, T. Sengstag, T. Slater, G. Strawn, M. A. Swertz, M. Thompson, J. van der Lei, E. van Mulligen, J. Velterop, A. Waagmeester, P. Wittenburg, K. Wolstencroft, J. Zhao and B. Mons, Sci. Data, 2016, 3, 160018 CrossRef PubMed.
- H. Wang, T. Fu, Y. Du, W. Gao, K. Huang, Z. Liu, P. Chandak, S. Liu, P. Van Katwyk, A. Deac, A. Anandkumar, K. Bergen, C. P. Gomes, S. Ho, P. Kohli, J. Lasenby, J. Leskovec, T.-Y. Liu, A. Manrai, D. Marks, B. Ramsundar, L. Song, J. Sun, J. Tang, P. Veličković, M. Welling, L. Zhang, C. W. Coley, Y. Bengio and M. Zitnik, Nature, 2023, 620, 47–60 CrossRef CAS PubMed.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 23414–23436 CrossRef CAS PubMed.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, J. Am. Chem. Soc., 2023, 145, 18048–18062 CrossRef CAS PubMed.
- J. Dagdelen, A. Dunn, S. Lee, N. Walker, A. S. Rosen, G. Ceder, K. A. Persson and A. Jain, Nat. Commun., 2024, 15, 1418 CrossRef CAS PubMed.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- A. Ilnicka and G. Schneider, Nat. Comput. Sci., 2023, 3, 922–933 CrossRef PubMed.
- C. Zeni, R. Pinsler, D. Zügner, A. Fowler, M. Horton, X. Fu, Z. Wang, A. Shysheya, J. Crabbé, S. Ueda, R. Sordillo, L. Sun, J. Smith, B. Nguyen, H. Schulz, S. Lewis, C.-W. Huang, Z. Lu, Y. Zhou, H. Yang, H. Hao, J. Li, C. Yang, W. Li, R. Tomioka and T. Xie, Nature, 2025, 639, 624–632 CrossRef CAS PubMed.
- F. Therrien, E. H. Sargent and O. Voznyy, Nat. Commun., 2025, 16, 4301 CrossRef CAS PubMed.
- Y. Zheng, J. C. Schupp, T. Adams, G. Clair, A. Justet, F. Ahangari, X. Yan, P. Hansen, M. Carlon, E. Cortesi, M. Vermant, R. Vos, L. J. De Sadeleer, I. O. Rosas, R. Pineda, J. Sembrat, M. Königshoff, J. E. McDonough, B. M. Vanaudenaerde, W. A. Wuyts, N. Kaminski and J. Ding, Nat. Biomed. Eng., 2025 DOI:10.1038/s41551-025-01423-7.
- T. Rahman, A. Tahmid, S. E. Arman, T. Ahmed, Z. T. Rakhy, H. Das, M. Rahman, A. K. Azad, M. Wahadoszamen and A. Habib, Nanoscale Adv., 2025, 7, 634–642 RSC.
| This journal is © The Royal Society of Chemistry 2025 |