
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphobic surface-active ionic liquids as dynamic micellar phase-transfer catalysts for biphasic epoxidations†
Johannes
Luibl‡
 a,
Markus
Hegelmann‡
b,
Stephan
Schwarzinger
c,
Wolfgang
Korth
a,
Mirza
Cokoja
*b and
Andreas
Jess
*a
a,
Markus
Hegelmann‡
b,
Stephan
Schwarzinger
c,
Wolfgang
Korth
a,
Mirza
Cokoja
*b and
Andreas
Jess
*a
aFaculty of Engineering Science, Chair of Chemical Engineering, University of Bayreuth, Universitätsstraße 30, D-95447 Bayreuth, Germany. E-mail: andreas.jess@uni-bayreuth.de
bSchool of Natural Sciences, Department of Chemistry and Catalysis Research Center, Technical University of Munich, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany. E-mail: mirza.cokoja@tum.de
cNorthern Bavarian NMR Center, University of Bayreuth, Universitätsstraße 30, D-95447 Bayreuth, Germany
First published on 20th March 2025
Abstract
We report on the synthesis of amphiphobic surface-active ionic liquids (FSAILs) containing fluorinated anions. The solubility and aggregation behavior of the FSAILs was determined in both aqueous hydrogen peroxide and in organic media. The FSAILs were applied as temperature-controlled micellar and phase transfer epoxidation catalysts by mixing the FSAIL with sodium tungstate in aqueous H2O2 solution. The epoxidation of cis-cyclooctene with aqueous hydrogen peroxide was studied in terms of catalyst activity, phase behavior and recycling. To deepen insights recycability and phase transfer, especially of the catalytically active species, characteristics of the micellar catalysis were investigated in detail over different reaction time periods. High-resolution 1H-qNMR reveals a quantitative phase transfer of the FSAILs into the organic phase, while the additive remains in the aqueous phase. Additionally, the present tungsten species also remain fully in the aqueous phase as shown by ICP-MS analysis.
Introduction
Epoxides are widely used intermediates for various industrially relevant bulk chemicals, polymers and pharmaceuticals.1 Due to the high demand, numerous methods for the epoxidation of olefins have been developed, especially in homogeneous phase using molecular catalysts.2 In these cases, the workup mostly focuses on product isolation through purification methods, such as column chromatography, while catalyst recycling is not regarded as relevant. Therefore, sustainable and selective methods for the production of epoxides, particularly those that promote recycling in closed chemical cycles, are required. Phase-transfer catalysts (PTC) in biphasic organic/aqueous systems are suitable for incorporating both green oxidants and catalyst recycling. This micellar approach is also applied to various organic transformations and is used in industrial-scale C–C cross couplings and hydroformylations.3 For epoxidation systems, PTCs are used to migrate the catalyst, often a polyoxometalate (POM), to the educt/product phase, though this does not resolve the challenge of separating the catalyst from the product.4 Advancing environmentally friendly systems with aqueous hydrogen peroxide (H2O2) as the oxidant are highly promising, as it produces water as the sole byproduct.5 In this context, surface-active ionic-liquids (SAILs), most often with alkyl imidazolium cations, are versatile candidates that can function either solely as micelle-forming PTC6 or as a molecular single-component catalysts for the epoxidation of olefins in water.4 Product separation is performed via simple mechanical methods and catalyst recycling is possible by extraction or distillation.7 However, in continuous loop reactor systems, the accumulation of water dilutes the SAIL concentration below the critical micelle concentration, causing the micelles to collapse resulting in catalyst deactivation. This necessitates additional, separate distillation steps to recover the catalyst from the aqueous phase.7Research on stimuli-responsive surfactants, which adjust their aggregation behavior and phase solubility in response to factors such as temperature, pH, light, and even magnetic fields, offers a promising approach for achieving stimuli-controlled separation of the SAIL catalyst in a more elegant and cost-effective manner.8 To explore a temperature-responsive approach, cation-fluorinated imidazolium tungstate SAILs (FSAILs) were investigated.9 Fluorine functionalization at the cation notably decreased the solubility in both the aqueous and the substrate phases, while enabling temperature-dependent solubility in the aqueous phase. This principally allows for reversible temperature-controlled epoxidations; however, these catalysts are quantitatively transferred to the organic phase by the additive and the epoxidation product, resulting in quasi-homogenous conditions. While this leads to high activity and selectivity, it also prevents catalyst recycling via precipitation from the aqueous phase after reaction. Given the structural versatility of the imidazolium-SAIL motif, introducing fluorine-containing anions – such as bis((trifluoromethyl)-sulfonyl)imide – instead of functionalizing the cation results in SAILs that are not miscible with water or nonpolar organic solvents with the potential of catalyst recycling in form of a third phase.10 The goal is to design a SAIL that: (i) exhibits low solubility in aqueous media (ii) is insoluble in the organic substrate/product (iii) does not decompose H2O2 or to a minor degree, and (iv) is surface-active and solubilizes the substrate in aqueous media. To render a system with an inert SAIL catalytically active, the active species can be generated in situ by metathesis from the SAIL and the active metal salt directly within the reaction mixture, resulting in the formation of a micellar composite catalyst.11 In this work, we present a series of imidazolium FSAILs with the fluorinated bis((trifluoromethyl)sulfonyl)imide [NTf2], tetrafluoroborate [BF4] and hexafluorophosphate [PF6] anions with the aim to control the phase behavior of these amphiphobic surfactants. The solubility and micelle formation of the FSAILs were investigated in both aqueous and organic media. As reaction studies, the epoxidation of cis-cyclooctene (COE) to cyclooctene oxide (COO) was investigated with tungstate as active metal in the catalytic system (Fig. 1). FSAIL/tungstate/PPA composite systems were used to investigate the reactivity and phase behavior enabling the probing of recycling possibilities. Catalyst recycling was performed on the [DoMIM][NTf2]/Na2WO4/PPA system by thermal separation of the formed COO.
 | ||
| Fig. 1 General scheme of performed reactions (X = [NTf2], [BF4], [PF6]). | ||
Results and discussion
Synthesis and characterization of the FSAILs
All fluorinated [RMIm][X] (R = butyl [Bu], octyl [O], dodecyl [Do], hexadecyl [HexDe]; X = bis((trifluoromethyl)sulfonyl)imide [NTf2], tetrafluoroborate [BF4], hexafluorophosphate [PF6]) FSAILs were synthesized from their corresponding imidazolium bromide precursor via a anion exchange procedure (for details see the ESI†).12 Here, all imidazolium bromide precursors were eluted over a freshly activated, strongly basic ionic exchange resin. The imidazolium hydroxides were subsequently converted to the FSAILs by treatment with [Li][NTf2], [Na][BF4] or [Na][PF6], respectively. All products were obtained in high yield and purities via1H-, 11B-, 13C-, 19F- and 31P-NMR as well as elemental analysis. The absence of bromide contaminations was excluded by silver chromate tests.13 Additionally, the synthesized FSAILs were analyzed using ATR-IR spectroscopy. Increasing the chain length ([Bu] < [O] < [Do] < [HexDe]) resulted in a systematic red shift and increase of the respective intensities of the alkyl C–H stretching (ESI,† Fig. S21 and S22). This results from the inductive effect of the alkyl chain and charge redistribution among the cation–anion pairs. Notably, the hydrogen bonding between C2–H–[X] is not affected by alkyl chain variations.14 Measurements with [DoMIm][X] (X = [NTf2], [BF4], [PF6]) revealed a red shift of the C2–H vibration of [NTf2]- and [BF4]-based SAILs in comparison to the [PF6]-based one (ESI,† Fig. S23). This suggests that the cation–anion interaction of the FSAILs with fluorinated, inert anions decreases in the order [NTf2] > [BF4] > [PF6],15 which possibly impacts the formation of active composite micelles with the respective catalytically active metal salts.Aggregation studies
In biphasic liquid–liquid systems the formation of micelles is substantial to enable catalytic activity by overcoming mass transport limitations. Therefore, the critical micelle concentration (CMC) was determined at various conditions. Note that while the term CMC is standing for the formation of distinct micelles, in this work it is interchangeable with the expression critical aggregation concentration (CAC) because of the complex supramolecular nature of e.g. the [NTf2] systems. For determining the CMC/CAC value, the surface tension (ST) or electrical conductivity of the SAIL solutions was measured as function of their concentration. The concentration was varied by adding a concentrated solution of the FSAIL to the pure solvent (50% aqueous hydrogen peroxide) or by dilution of a saturated FSAIL-in-peroxide solution. The CMCs were primarily determined via tensiometry with the Wilhelmy method as well as via conductometry for underpinning the ST values. Because of the further relevance in kinetic experiments and the overall low solubility of NTf2-FSAILs, the adjacent tensiometrical CMC values are measured at 50 °C. Regarding [OMIm][NTf2], no distinct CMC value could be measured using the ST method at room temperature (RT), although reproducible values were obtained with conductometry at 1.1 mM. For [DoMIm][NTf2], the CMC is at 0.52 mM in aqueous hydrogen peroxide, whereas [OMIm][NTf2] has a notably higher CMC of about 4 mM, both values are valid for 50 °C (Fig. 2). As the alkyl chain gets longer, the CMC is decreasing, which is well-documented in the literature (ESI,† Tables S3 and S4).16–19 Compared to traditional PTCs like dodecyltrimethylammonium bromide (3.5 mM at room temperature, RT) and hexadecyltrimethylammonium bromide (0.026 mM at RT), the value is in the same range.20 This undermines the high micellar forming efficiency of [NTf2]− SAILs. Note that distinct CMC values are observed for dodecyl and octyl-substituted [NTf2]-FSAILs, while an explicit CMC could not be determined for [HexDeMIm][NTf2] and [BMIm][NTf2] (ESI,† Fig. S1 and Table S3). This results from the very low solubility of [HexDeMIm][NTf2] in water and aq. H2O2, also the short-chained alkyl chains as in [BMIm][NTf2] generally show a low tendency to form micelles (due to high CMCs) alongside a low solubility.21 The data indicate that longer alkyl side chains in [AlkylMIm][NTf2]-FSAILs promote micelle formation, thereby reducing the CMC due to the formation of favorable van der Waals (vdW) contacts.22 Although the anion often does not influence the CMC,23 the generally lower, tunable aqueous solubility of FSAILs with fluorinated anions possibly affects the micelle formation. Hence, the CMC was determined for [DoMIm][X] FSAILs with different fluorinated anions (X = [NTf2], [BF4], [PF6]) and shown in Table 1 together with their solubility in 50 wt% aq. H2O2 and cyclooctene (COE). The data highlight the solubility/CMC dependency, the FSAIL with the highest CMC is displaying the highest solubility.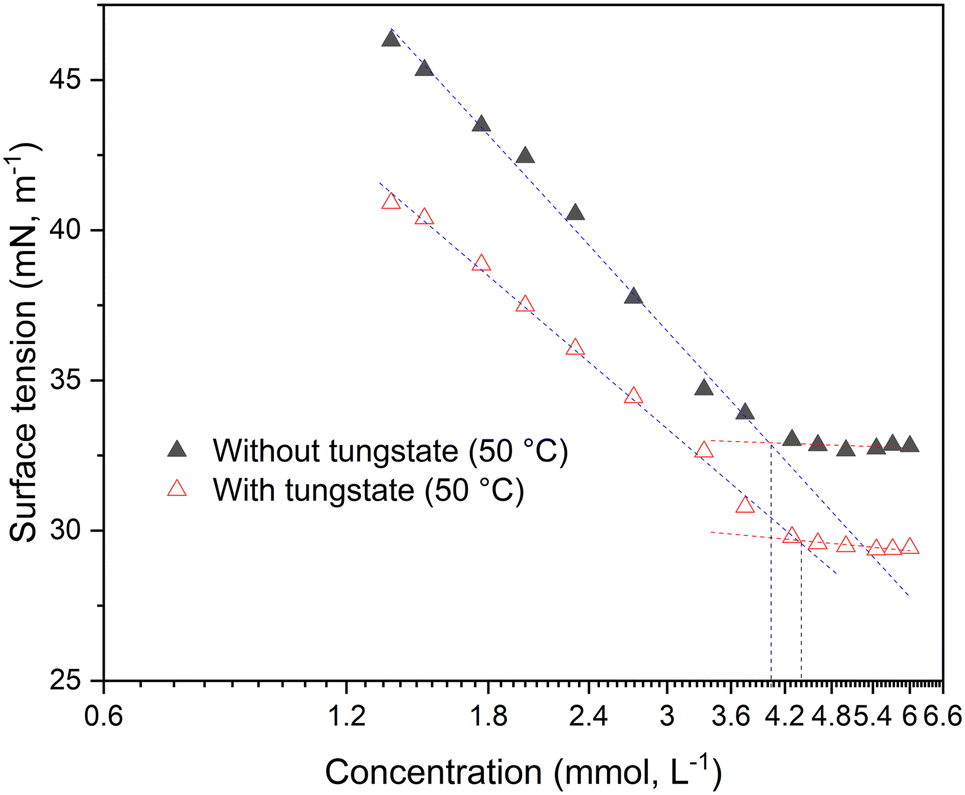 | ||
| Fig. 2 Surface tension as function of the concentration of [OMIm][NTf2] in 50% hydrogen peroxide at 50 °C with and without the addition of 18 mM of sodium tungstate CMC values are corresponding to the dashed vertical lines. | ||
| FSAIL | CMC (H2O2) (mmol L−1) | Solubility (H2O2) (mmol L−1) | Solubility (COE) (mmol L−1) |
|---|---|---|---|
| [DoMIm][NTf2] | 0.52 | 0.33 | 9.5 |
| [DoMIm][BF4] | 5.15 | >200 | 16.5 |
| [DoMIm][PF6] | 0.35 | 0.81 | 7.8 |
| [OMIm][BF4] | 65 | >1700 |
For determining the quality of the measurements, CMC values of [OMIm][BF4] and [DoMIm][BF4] measured at RT were compared with room-temperature, literature-known values in 50 wt% aq. H2O2 (ref. 21) (ESI,† Table S4), with [OMIm][BF4] exhibiting a CMC of 53 mM (48 mM)21 and [DoMIm][BF4] having a value of 4.0 mM (3.1 mM)21 respectively. Analogously, elevated temperature measurements were compared in case of [OMIm][BF4], with 65 mM measured tensiometrically at 50 °C and 75 mM via conductometry.21 Note the V-shaped plot in case of the [OMIm][BF4] sample, which is commonly seen in [HexylMIm]- and [OMIm]-based SAILs.16–18 RT conductometric values measured on site were also verified and compared with the respective tensiometric measurements (ESI,† Table S5). Concerning room temperature solubility measurements, the solubility of the FSAILs is higher in COE than in aqueous peroxide in case of [DoMIm][NTf2] and [DoMIm][PF6], whereas [DoMIm][BF4] displays a higher solubility in 50 wt% aq. H2O2 (ESI,† Tables S1 and S2). For [DoMIm][PF6], room-temperature CMC values were also determined (ESI,† Table S4). Here, as well as in the tetrafluoroborate samples, an increase of the CMC with temperature can be observed. The higher value at 50 °C is caused by the higher kinetic energy of the water molecules at higher temperatures, which reduces the interactions between water and surfactant molecules thus hindering the formation of micelles.24 Since the synthesized FSAILs are inert, a catalytically active metal salt is used for the epoxidation of olefins, and its influence on the CMC investigated. For [OMIm][NTf2], the effect of sodium tungstate on the CMC was studied at 50 °C, as shown in Fig. 2. It can be seen that the CMC remains almost unaffected upon addition of tungstate salts in the given tungstate concentration regime, which is in accordance with previous measurements involving [OMIm][BF4].21 Note that the classical L-shaped surface tension/concentration plot displaying distinct CMC values marked by the vertical dashed lines.16–18 In contrast, no clear CMC value could be determined at room temperature, owing to the respective lower solubility. The generally lower surface tension resulting from the added tungstate is likely due to impurities or altered interactions between the SAIL molecules and the solution/air interface.
The formation of micelles was verified by transition electron microscopy (TEM) and dynamic light scattering (DLS) measurements. The [NTf2]-based FSAILs form small spherical micelles (>5 nm), which tend to agglomerate to larger particles (Fig. 3 left, ESI,† Fig. S13 and S14). Note that for [NTf2]-based FSAILs DLS measurements were unsuccessful, which most likely results from the polydispersity and the presence of large non-spherical micellar agglomerates. In contrast, [BF4]-based FSAILs form solely spherical micelles (Fig. 3, ESI,† Fig. S15–S19). These primary micelles partially agglomerate towards larger spherical micellar structures, as reported in previous literature17 similar to tungstate-based SAILs.12 These results are supported by DLS, displaying a trend of decreasing size of the primary micelles (∼2 nm, ∼1.5 nm, >1 nm for [Bu], [O] and [Do], respectively) with increasing alkyl chain length (ESI,† Fig. S12). The decreasing size most likely results from increasing amounts of van der Waals (vdW) contacts, which outweigh the electronic repulsion of the head groups. In analogy to [DoMIm][BF4], similar properties were determined for the micelles of [DoMIm][PF6], which also tends to form small micelles and larger micellar agglomerates (Fig. 3, ESI,† Fig. S20).
 | ||
| Fig. 3 Top: TEM micrographs of micelles formed by [DoMIm][NTf2] (left), [DoMIm][BF4] (middle) and [DoMIm][PF6] in saturated aqueous solutions. Bottom: TEM micrographs of micellar aggregates formed by [DoMIm][NTf2] (left), [DoMIm][BF4] (middle) and [DoMIm][PF6] in saturated aqueous solutions. | ||
As micellar epoxidation catalysis is based on the solubilization of the substrate in the aqueous phase or vice versa, e.g. solubilization of the aqueous oxidant in the organic substrate/product phase to overcome mass transport limitations and enhance reaction rates, [BF4]-based SAILs were investigated under catalysis like conditions via DLS. The size distribution of the micelles of a saturated solution of [DoMIm][BF4] in 50 wt% aq. H2O2 was determined before and after the sample was treated with the model substrate COE (see ESI† for procedure details). The micelles and their aggregates exhibit a heterogeneous size distribution, with primary micelles measuring approximately 1 nm, larger agglomerates around 10 nm, and even larger ones ranging from 50 to 300 nm.
From the micrographs it is evident that these agglomerates consist of many small micelles. Upon contact with the substrate, both primary micelles and their agglomerates swell and increase in size due to substrate incorporation, as illustrated in Fig. 4. This experiment demonstrates that micellar agglomerates maintain the same desirable surface-active properties as their primary micelles, making them equally effective for catalytic applications in biphasic media. Also, the measurement indicates the disaggregation of some agglomerates to the primary micelles. An analogues measurement with [OMIm][BF4] displayed a similar trend (ESI,† Fig. S11).
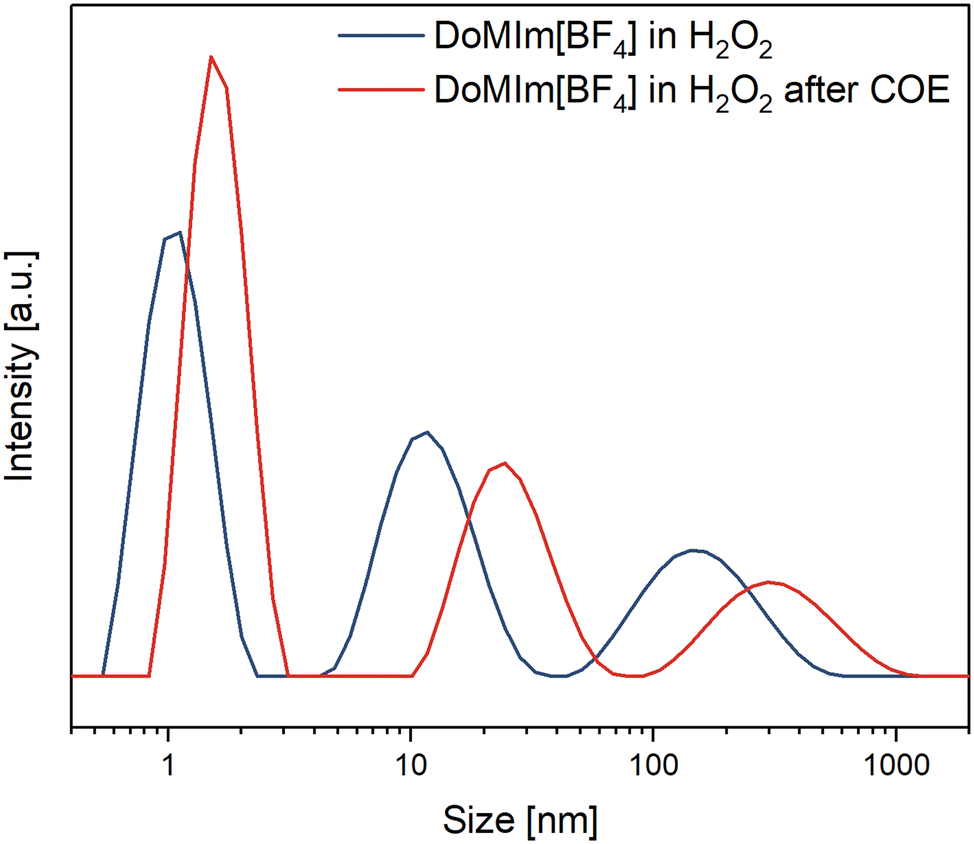 | ||
| Fig. 4 DLS measurements of [DoMIm][BF4] in H2O2 (357 mmol L−1) at room temperature prior and after treatment with COE. | ||
Catalyst screening of [RMIm][NTf2] FSAILs
The high tunability of the SAIL structure motif, including variations in alkyl chain length, enables precise control over surfactant properties, such as substrate interactions (e.g. solubilization), and offers the ability to modulate phase behaviour. Different [RMIm][X] (R = [Bu], [O], [Do], [HexDe]; X = [NTf2], [BF4], [PF6]) with fluorinated anions are paired with a suitable tungstate salt, resulting in a composite catalyst for epoxidation catalysis. These composite catalysts were investigated in detail in terms of activity as well as the phase behavior in the reaction system. In particular, [RMIm][NTf2] FSAILs were screened for the epoxidation of COE using aq. H2O2 as oxidant, Na2WO4 as catalytically active metal salt and PPA as additive, which is known to boost the activity of tungstate-based epoxidation systems.25 Note that for all catalysis experiments, all parameters, except the used FSAIL were kept constant, to determine the impact of the SAILs on the efficiency of the catalytic (H2O2:SAIL:Na2WO4:PPA) system. In analogy to [WO4]2−-based one-component catalysts, the activity increases with increasing alkyl chain length in the order of [Bu] < [O] < [Do] < [HexDe] as shown in Fig. 5.12 The data clearly shows that the activity of the [BMIm][NTf2]/tungstate composite SAILs is significantly lower (ca. 10% conversion after 90 min) compared to the other composite catalysts. The low activity results from the low solubility of [NTf2]-based ILs coupled to the low tendency of the short-chained [BMIm] cation to form micelles in aq. H2O2 and COE. The higher activity of the other three composite catalysts shows the influence of the length of the alkyl side chain on more efficient micelle formation. Here, also the increased solubility of these surfactants in organic media affects the activity (higher concentration of surfactant in solution and micelle formation in both aq. H2O2 and the substrate/product phase). However, the experimental data indicate that the effect of the side chain levels off, and a further increase from [Do] to [HexDe] only leads to a minor activity boost. This is likely due to the saturation of the reaction mixture with micelles in both the aqueous and organic phases.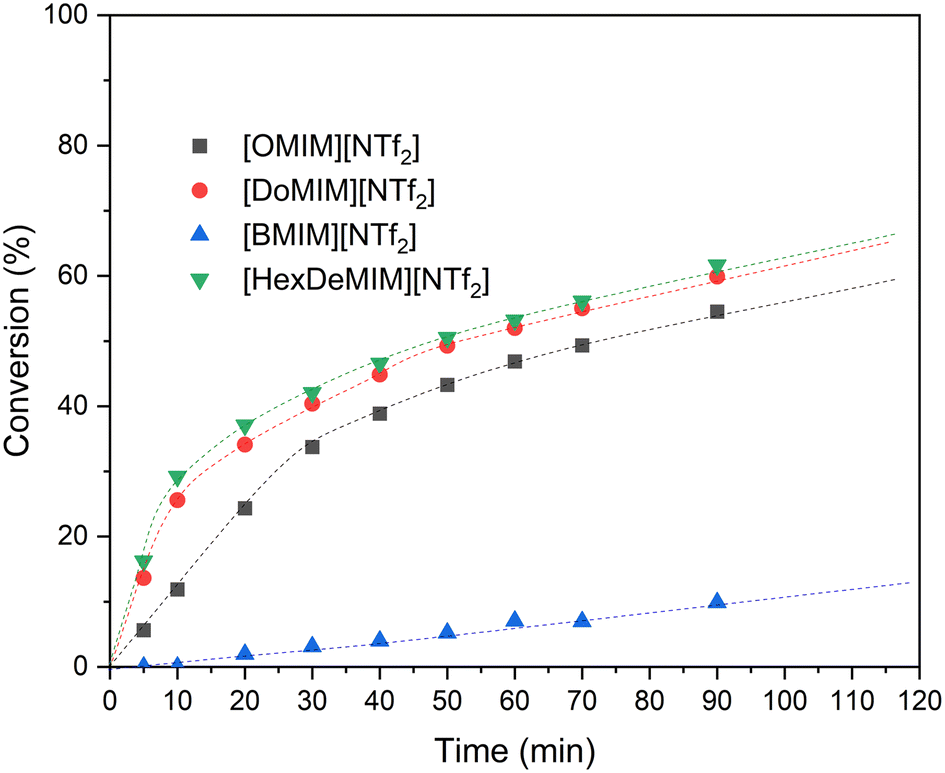 | ||
| Fig. 5 Conversion of COE to COO with different [RMIm][NTf2] SAILs (R = Bu, O, Do, HexDe) and sodium tungstate. Reaction conditions: 50 °C, 1250 rpm, 3.7 mol% SAIL (0.74 mmol, 3.7 equiv.), 165 mg sodium tungstate dihydrate (0.5 mmol, 2.5 equiv.), 158 mg PPA (1 mmol, 5.0 equiv.), 2.6 mL COE (20 mmol, 100 equiv.) and 1.4 mL 50 wt% aq. H2O2 (25 mmol, 125 equiv.). | ||
Next to the epoxidation activity, the phase behavior of the two organic catalyst components (FSAIL, PPA) in the biphasic system (COE/COO vs. aq. H2O2) was studied. Except for [BMIm][NTf2], all other ionic liquids were found almost completely (>99%) in the organic phase. This is in accordance with FSAILs with fluorinated cations, which are also transferred into the organic phase. Notably, tungstate was exclusively found in the aqueous layer. However, while in the case of FSAILs with fluorinated cations the additive PPA is quantitatively transferred into the COE/COO phase, for FSAILs where only the anion is fluorinated such a PPA transfer was not observed.8
Catalyst screening of [DoMIm][X] FSAILs
The catalytically inert anions of the FSAILs were varied using [DoMIm][X] (X = [BF4], [PF6]), as the dodecyl cation shows the most promising properties regarding its solubility and activity. The reaction parameters were kept constant as described above. Note that the [NTf2]-containing FSAIL composite catalyst yields a conversion of approximately 12% after 5 min, while the [BF4]- and the [PF6]-FSAILs show significantly higher epoxidation activities and yield full conversion after just five minutes (Fig. 6, ESI,† Fig. S26). More detailed experiments regarding the [BF4]/[PF6]-FSAILs are showing the superior activity of the [DoMIm][BF4] with a 50% conversion at approximately one minute compared to the [DoMIm][PF6] achieving 50% conversion at around two minutes as in Fig. 6 at these high catalyst concentrations. This most likely results from the lower solubility of [NTf2]-FSAILs (0.33 mmol L−1vs. >200 mmol L−1 and 0.81 mmol L−1 for [BF4] and [PF6], respectively), which subsequently results in lower micelle concentrations, thus forming less composite catalyst. Another factor could be the improved in situ ionic exchange of the active tungstate against the inert fluorinated anion in case of [BF4] and [PF6], improving the concentration of active catalyst at the reaction site. According to the IR spectroscopic characterization of [DoMIm][NTf2]-FSAILs, strong interactions between the imidazolium cation and the bistriflimide anion are hindering the ionic exchange against tungstate thus lowering the net amount of composite catalyst.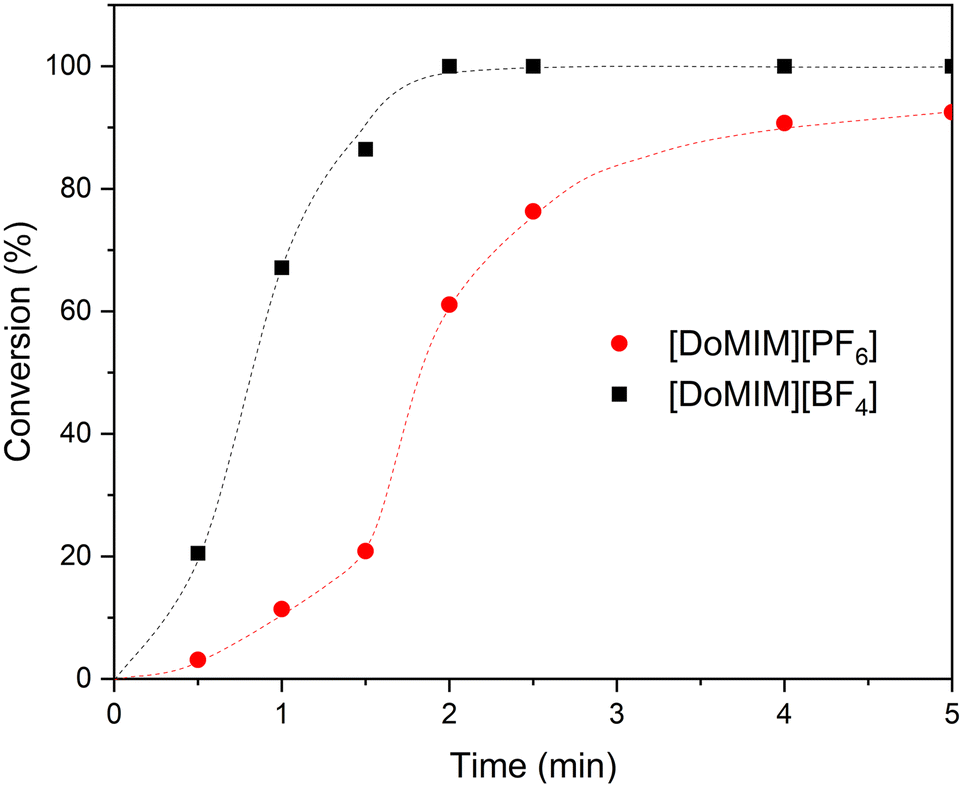 | ||
| Fig. 6 Conversion of COE to COO with various [DoMIm][X] FSAILs (X = PF6−, BF4−) and sodium tungstate. Reaction conditions: 50 °C, 1250 rpm, 3.7 mol% FSAIL (0.74 mmol, 3.7 equiv.), 165 mg sodium tungstate dihydrate (0.5 mmol, 2.5 equiv.), 158 mg PPA (1 mmol, 5.0 equiv.), 2.593 mL COE (20 mmol, 100 equiv.) and 1.423 mL 50 wt% aq. H2O2 (25 mmol, 125 equiv.). | ||
For additional kinetics regarding the [PF6] and [BF4]-FSAILs, further experiments with lowered catalyst and cocatalyst amounts were conducted (0.11 mol% of the FSAIL and PPA were used along with 0.06 mol% of sodium tungstate), while using the same amount of substrate and oxidant (ESI,† Fig. S27). Generally, both ILs show a similar activity regarding the prevailing conditions. The phase behavior was also investigated at high and low concentrations for both FSAILs (0.11 and 3.7 mol% SAIL). All [DoMIm][X]-ILs were quantitatively located in the organic phase after full conversion, indicating that the epoxidation occurs in the organic layer to some degree.
Phase behavior of the catalysts in the cyclooctene (COE)/H2O2-system
For investigating FSAIL recycling possibilities, it is crucial to analyze the phase behavior of the micelle-forming FSAIL as well as the catalytically active tungstate and the additive PPA in the biphasic system. Over 99% of the FSAILs were found in the organic phase after epoxidation, with the exception of [BMIm][NTf2] where only 20% are transferred into the COE/COO layer after conversions of 10%. This shows the low ability of [BMIm][NTf2] to form catalytically active micelles in both aqueous and organic media. Transfer of tungstate or PPA into the organic layer was not observed in any case (by 1H-qNMR). As the SAIL migrates into the organic phase, inverse micelles are formed, solubilizing the catalytically active and aq. H2O2 species therein. Thus, the system remains active. To support this, the epoxidation of COE with [OMIm][NTf2]:Na2WO4 was scaled up by a factor of ten. Here, a third phase in between the aq. oxidant phase and the organic COE/COO was visible after certain reaction times. After sufficient amounts of COE were converted, this third FSAIL phase homogenized with the organic phase, while the catalytically active species remained in the aqueous phase (ESI,† Fig. S24). This was confirmed by ICP-MS measurement of an extraction of the organic phase, which showed that no tungstate was present in the organic phase (below the detection limit of 0.16 μg L−1). Further information about the distribution of the ILs between the two phases can be gained by varying the volume/amount of the alkene reactant COE in the reaction system. Therefore, the concentration of the transferred FSAIL located in the organic phase is different due to the altered volume changing the conversion-versus-time ratio in case of FSAIL transfer. COE amounts were varied from 10 to 20 mmol, as shown in Fig. 7. When using only half of the amount of COE, the conversion almost doubles after a reaction time of 90 min. The reaction rate is higher at the experiments with reduced amount/volume of COE owing to the higher IL concentration in the organic phase. This means that the reaction is taking place in the organic layer at least partially. Note that the tungstate as well as PPA are only present in the aqueous phase after phase separation. A further indication that the micellar catalyst is subject to a phase change, is the kink in the shown plot in Fig. 7 at ca. 30 minutes in all three experiments. This kink indicates the transfer of the FSAIL into the organic layer. After the kink, the reaction rate is clearly slower indicating the lower activity of the FSAIL in the organic phase resulting from the poor solubilization of the tungstate species in the organic layer. This is underlined by plotting the reaction rate as a function of the COE conversion (ESI,† Fig. S28), which displays two distinctive regions with low conversions having the reaction in the aqueous phase and vice versa. The lower activity of the FSAIL in the organic phase is because of the catalytically active tungstate anion remaining in the aqueous phase. This is in contrast to single-compound catalysts like cationic FSAILs and [DoMIm]2[WO4] where the tungstate as well as the imidazolium cation are in one phase resulting in a higher activity. To further inquire the FSAIL phase behavior during reaction time, the influence of the cyclooctene oxide content in the organic phase was investigated. According to the screening experiments, with [BMIm][NTf2] the lowest catalytic activity is achieved. No micelles could be detected during the experiments of CMC determination. Nonetheless, the phase behavior of this FSAIL in the epoxidation system was investigated in detail for analyzing the product-initiated IL phase transfer independent of any micellar formation. Moreover, [BMIm]+-ILs are well characterized and popular in many fields of ionic liquid chemistry. The advantage of using a non-active IL is the better controllability of the organic phase COO content and therefore the SAIL biphasic behavior due to the avoidance of significant COO formation during further experiments. After a conversion of around 10%, no FSAIL was detected in the organic phase. By adding cyclooctene oxide (COO) to an aliquot of the reaction mixture and stirring the system for 15 minutes at 50 °C, a COE conversion of 50% was simulated, albeit without decomposing the hydrogen peroxide. In this mixture, 20% of the [BMIm][NTf2]-FSAIL was transferred to the organic phase (determined via1H-NMR). This results from the increased polarity of COO, due to the epoxide moiety in comparison to the corresponding oleic substrate, COE. This enhances the transfer of the relatively polar IL (ENT([BMIm][NTf2]) = 0.642 vs. ENT(ethanol) = 0.654 and ENT(water) = 1.000)26 into the now more polar, COO-enriched organic layer. | ||
| Fig. 7 Conversion of COE to COO with [OMIm][NTf2] and sodium tungstate varying the COE amount/volume. Reaction conditions: 50 °C, 1250 rpm, 355 mg FSAIL (0.74 mmol, 3.7 equiv.), 165 mg sodium tungstate dihydrate (0.5 mmol, 2.5 equiv.), 158 mg PPA (1 mmol, 5.0 equiv.), 1.309–2.593 mL COE (10.1–20 mmol, 50.5–100 equiv.) and 1.423 mL 50 wt% aq. H2O2 (25 mmol, 125 equiv.). The visible kink indicating the phase transfer is indicated with the dotted line at the experiment with 15 mmol of COE. | ||
Influence of tungstate concentration on the composite catalyst system and ion exchange in micelles
Having a composite catalyst system (inert anion-FSAIL + inorganic tungstate), the molar ratio of FSAIL cation to tungstate anion (SAIL-tungstate-ratio, STR) can be varied. In case of a tungstate-SAIL like [DoMIm]2[WO4], the STR is two. Having a composite catalyst, the minimum amount of tungstate required to have a reasonably high reaction rate of the epoxidation can be determined. Ionic exchange between an inert anion FSAIL (e.g. [DoMIm][BF4]) and an (inorganic) tungstate like Na2WO4 in a composite system can be described with the following equilibrium with the in situ formation of the active tungstate FSAIL: 2[DoMIm][BF4] + Na2WO4![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [DoMIm]2WO4 + 2NaBF4. Raising the concentration of the tungstate shifts the equilibrium to the side of the active tungstate-FSAIL. Therefore, experiments with different tungstate concentrations ranging from 0–0.025 mmol (0.006–0.06 mol%) were performed to determine the influence of tungstate concentration on the reaction rate constant (1st order of reaction).
[DoMIm]2WO4 + 2NaBF4. Raising the concentration of the tungstate shifts the equilibrium to the side of the active tungstate-FSAIL. Therefore, experiments with different tungstate concentrations ranging from 0–0.025 mmol (0.006–0.06 mol%) were performed to determine the influence of tungstate concentration on the reaction rate constant (1st order of reaction).
The activity of the composite system rises with an increasing amount of tungstate due to the equilibrium above. By plotting the reaction rates against the amount of tungstate, a linear correlation between both variables can be seen (ESI,† Fig. S29). There is no sharp point where the reaction begins at a reasonable rate as opposed to the e.g. CMC, which indicates a steep rise in reaction rate in dependence of the FSAIL concentration.
To examine the extent of micellar ion exchange, composite catalysts were compared in terms of activity with tungstate SAILs. For representative results, the used cation is [DoMIm]+ in all cases and the inert anions are [BF4]− and [PF6]− as shown in Fig. 8. No large differences between the three used catalysts are visible, so one can conclude that the equilibrium is strongly shifted to the side of the tungstate SAIL. Although the FSAIL gets transferred quantitatively into the organic phase in case of composite systems with fluorinated SAIL anions, this not the case for e.g. [DoMIm]2[WO4]. According to 1H-qNMR analysis, the tungstate-SAIL stays at the aqueous phase at the same conversion (60%), only around 10% is being transferred into the organic layer. The good performance of such composite systems is also seen on [OMIm][BF4]/sodium tungstate/PPA catalyzed epoxidation reactions,10 therefore highlighting the flexibility of such reactions regarding (co)catalyst systems.
 | ||
| Fig. 8 Conversion of COE to COO with composite [DoMIm][X]/sodium tungstate (X = BF4−, PF6−) as well as [DoMIm]2[WO4]. Reaction conditions: 50 °C, 1250 rpm, 0.11 mol% SAIL (referred to the cation, 0.05 mmol, 0.25 equiv.), 8.2 mg sodium tungstate dihydrate (0.025 mmol, 0.125 equiv.), 7.9 mg PPA (0.05 mmol, 0.25 equiv.), 2.593 mL COE (20 mmol, 100 equiv.) and 1.423 mL 50 wt% aq. H2O2 (25 mmol, 125 equiv.). | ||
FSAIL recycling in the [DoMIm][NTf2]/Na2WO4/PPA composite catalyst system
For an economic and ecologic use of catalysts the effective and energy/cost sensitive separation of the often valuable catalyst is important. Depending on the catalyst phase distribution in biphasic amphiphobic ionic liquid (SAIL) tungstate systems for the epoxidation of alkenes, multiple approaches are possible. Using previously shown phase transfer studies of the SAIL between the organic as well as the aqueous layer, a concept of SAIL-recycling was investigated in terms of the cyclooctene epoxidation system. As the SAIL gets transferred into the organic phase in nearly all cases exempt from the [DoMIm]2WO4 and the non-active [BMIm][NTf2] at reasonably high conversions, SAIL workup can be focused on the organic/product phase. To do so, a standard catalysis run was performed followed by separation of the organic layer containing the FSAIL and the product mixed with unreacted starting material. 3.7 mol% (0.74 mmol) of the FSAIL [DoMIm][NTf2] were used in conjunction with 1 mmol of PPA and 0.5 mmol of sodium tungstate at 50 °C and 1250 rpm stirrer speed in a 50 mL round bottom flask (Fig. 5). Total reaction time was 2 h followed by a phase settling period of 1–2 h to avoid excessive water and therefore sodium tungstate and PPA carryover in subsequent processing steps. The FSAIL, now dissolved in the product, can easily be separated by simple evaporation of the COO in vacuo at 50 °C and 4 × 10−2 mbar. By adding COE, 50 wt% aq. H2O2 and Na2WO4/PPA to the SAIL residue, a new catalysis run can be started again. As FSAIL, [DoMIm][NTf2] was chosen because of the reasonably high epoxidation activity and low solubility in aqueous solvents. The epoxidation experiment with the adjacent recycling procedure was carried out five times without noticeable activity decline and detection of side products (ESI,† Fig. S30). Selectivity was >99% in all recycling runs. 1H/13C-NMR analysis indicates a good stability of the [DoMIm][NTf2] after all cycles (ESI,† Fig. S31 and S32). This shows the possibility to recycle the FSAIL from the reaction mixture multiple times, allowing efficient use of such composite systems.Conclusions
We have shown that in the series of the FSAILs presented in this work, those bearing the NTf2-anion show the poorest solubility and lowest CMC, rendering them the most viable candidates for our studies. For all investigated FSAILs, in aqueous hydrogen peroxide solution spherical micelles are formed, as shown by TEM measurements. Furthermore, DLS studies reveal that all FSAIL micelles can take up cyclooctene into the aq. phase, which enables the catalytic epoxidation in water. [DoMIm][NTf2]–FSAILs are displaying lower catalytic activity compared to their [BF4]/[PF6] counterpart, resulting from their lower solubility. Similarly, to the case of the FSAILs with fluorinated cations, the product of the reaction, cyclooctene oxide and the additive phenylphosphonic acid are responsible for the transfer of the catalyst into the organic (product) phase. As the FSAIL transfer into the organic phase sharply influences catalyst activity, detailed aggregational investigations of H2O2/COO/FSAIL composite systems are to be conducted in future studies. Nevertheless, despite the catalyst and product being present in the same phase, catalyst recycling is possible by workup of the organic layer. By removal of the formed product from the organic phase via distillation, the FSAIL could be recycled for further reaction runs without any noticeable loss of activity.Experimental
A detailed description of the analytical methods, catalytic setups, synthesis procedures of all compounds and their characterization are given in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. L.: investigation (lead), data curation (lead), visualization (lead), formal analysis (lead), writing – original draft; M. H.: investigation (lead), data curation (lead), visualization (lead), formal analysis (lead), writing – original draft; S. S.: investigation, analytical expertise; W. K.: methodology, writing – review & editing; M. C.: conceptualization, resources, funding acquisition, methodology, project administration, supervision, validation, writing – review & editing; A. J.: conceptualization, resources, funding acquisition, methodology, project administration, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for financial support of this work in the frame of the DFG-project ‘Surface-active ionic liquids as catalysts of olefin epoxidation: Reaction control by switchable phase behavior’ (project no. 326649877, grant no. Je 257/24-2 and Co 1543/1-2). The authors thank Christine Benning for the ICP-MS measurement and the help by the Central Analytics Facility of the TUM Catalysis Research Center.References
- Y. Meng, F. Taddeo, A. F. Aguilera, X. Cai, V. Russo, P. Tolvanen and S. Leveneur, Catalysts, 2021, 11, 765 CrossRef CAS.
- (a) T. Katsuki and K. Barry Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS; (b) E. Santacesaria, A. Renken, V. Russo, R. Turco, R. Tesser and M. Di Serio, Ind. Eng. Chem. Res., 2012, 51, 8760–8767 CrossRef CAS.
- G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644–683 RSC.
- (a) F. Schmidt and M. Cokoja, Green Chem., 2021, 23, 708 RSC; (b) K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310 CrossRef CAS PubMed; (c) C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS; (d) Y. Ishii, K. Yamawaki, T. Yoshida, T. Ura and M. Ogawa, Chem. Eng. Technol., 1987, 52, 1868–1870 CAS.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- (a) M. Cokoja and A. Jess, et al. , ChemSusChem, 2016, 9, 1773–1776 CrossRef CAS PubMed; (b) B. Zehner, W. Korth, F. Schmidt, M. Cokoja and A. Jess, Chem. Eng. Technol., 2021, 44, 2374–2381 CrossRef CAS.
- B. Zehner, Reaktionskinetik der Epoxidierung von Cycloocten mit perrhenat- und wolframatbasierten ionischen Flüssigkeiten, PhD-Thesis, Universität Bayreuth, 2023, DOI:10.15495/EPub_UBT_00007879.
- P. Brown, C. P. Butts and J. Eastoe, Soft Matter, 2013, 9, 2365–2374 RSC.
- M. Hegelmann, J. Zuber, J. Luibl, C. Jandl, W. Korth, A. Jess and M. Cokoja, Chem. – Eur. J., 2024, e202402985 CrossRef CAS PubMed.
- H. Kan, M. Tseng and Y. Chu, Tetrahedron, 2007, 63, 1644–1653 CrossRef CAS.
- (a) J. Scheffer, M. Alber, W. Korth, M. Cokoja and A. Jess, ChemistrySelect, 2017, 2, 11891–11898 CrossRef; (b) B. Zehner, W. Korth, F. Schmidt, M. Cokoja and A. Jess, Chem. Eng. Technol., 2021, 44, 2374–2381 CrossRef CAS.
- (a) F. Schmidt, B. Zehner, W. Korth, A. Jess and M. Cokoja, Catal. Sci. Technol., 2020, 10, 4448–4457 RSC; (b) M. Hegelmann, W. F. Bohórquez, J. Luibl, A. Jess, A. Orjuela and M. Cokoja, React. Chem. Eng., 2024, 9, 2710–2717 RSC.
- S. Velasco-Lozano, F. López-Gallego, J. C. Mateos-Díaz and E. Torres, Biocatalysis, 2015, 1, 166–177 Search PubMed.
- P. Sanchora, D. K. Pandey, H. L. Kagdada, A. Materny and D. K. Singh, Phys. Chem. Chem. Phys., 2020, 22, 17687–17704 RSC.
- Y. Gao, L. Zhang, Y. Wang and H. Li, J. Phys. Chem. B, 2010, 114, 2828–2833 CrossRef CAS PubMed.
- N. M. Vaghela, N. V. Sastry and V. K. Aswal, Colloid Polym. Sci., 2011, 289, 309–322 CrossRef CAS.
- J. Bowers, C. P. Butts, P. J. Martin and M. C. Vergara-Gutierez, Langmuir, 2004, 20, 2191–2198 CrossRef CAS PubMed.
- I. Goodchild, L. Collier, S. L. Millar, I. Prokeš, J. C. D. Lord, C. P. Butts, J. Bowers, J. R. P. Webster and R. K. Heenan, J. Colloid Interface Sci., 2007, 307, 455–468 CAS.
- A. Weber, P. Isbrücker, M. Schmidt and R. Schomäcker, Chem. Ing. Tech., 2021, 93, 201–207 CrossRef CAS.
- R. M. Brito and W. L. C. Vaz, Anal. Biochem., 1986, 152, 250–255 CrossRef CAS PubMed.
- B. Zehner, F. Schmidt, W. Korth, M. Cokoja and A. Jess, Langmuir, 2019, 35, 16297–16303 CrossRef CAS PubMed.
- C. Jungnickel, J. Łuczak, J. Ranke, J. F. Fernández, A. Müller and J. Thöming, Colloids Surf., 2008, 316, 278–284 CrossRef CAS.
- N. Bin Dong, L. Li, L. Yu Zheng and T. Inoue, Langmuir, 2007, 23, 4178–4182 CrossRef PubMed.
- R. J. Hunter, Foundations of Colloid Science, Oxford University Press, Oxford, U.K., 2nd edn, 2009 Search PubMed.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto, D. Panyella and R. Noyori, Bull. Chem. Soc. Jpn., 1997, 70, 905–915 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2003 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00616j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |