
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-step solution plasma synthesis of bifunctional CoSn(OH)6–carbon composite electrocatalysts for oxygen evolution and oxygen reduction reactions†
Sangwoo
Chae‡
a,
Akihito
Shio
b,
Taketo
Imamura
b,
Kouki
Yamamoto
b,
Yuna
Fujiwara
b,
Gasidit
Panomsuwan
 c and
Takahiro
Ishizaki
*d
c and
Takahiro
Ishizaki
*d
aSIT Research Laboratories, Shibaura Institute of Technology, Tokyo 135-8548, Japan
bMaterials Science and Engineering, Graduate School of Engineering and Science, Shibaura Institute of Technology, Toyosu, Koto-Ku, Tokyo, 135-8548, Japan
cDepartment of Materials Engineering, Faculty of Engineering, Kasetsart University, Bangkok 10900, Thailand
dCollege of Engineering, Shibaura Institute of Technology, Tokyo 135-8548, Japan. E-mail: ishizaki@shibaura-it.ac.jp
First published on 29th May 2025
Abstract
Development of efficient bifunctional catalysts for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) is highly required for the application in rechargeable metal–air batteries. Many research groups continue to develop active materials that enhance ORR and/or OER, aiming to improve the electrocatalytic properties and durability of electrodes in metal–air batteries. Currently, the most commonly used materials for ORR/OER catalysts are precious metals, that need to be replaced by low-cost catalysts with comparable performance. We have successfully synthesized non-precious metal-based catalytic composite materials composed of perovskite hydroxide, CoSn(OH)6 (CSO), and carbon materials via the solution plasma process (SPP). SPP realized the single step synthesis of carbon composite materials with the formation of CSO nanoparticles and provides excellent control over the nanostructure of the catalysts. The process can induce unique surface properties due to the plasma environment, potentially enhancing catalytic activity. The synthesized CSO and carbon composite catalysts exhibited promising catalytic properties for both ORR and OER. For ORR, the CSO and Ketjen Black (KB) composites, synthesized at pH 12, achieved the highest potential value at a current density of −3 mA cm−2. In OER, the same CSO and KB composite material synthesized at pH 12 reached the lowest potential value at a current density of 10 mA cm−2, surpassing the performance of RuO2. This study demonstrated the potential to customize and manufacture high-performance and low-cost bifunctional electrocatalysts for energy conversion systems by single-step synthesis, offering a sustainable materials alternative to commercialized precious metal-based electrocatalysts.
1. Introduction
As air pollution exacerbated by the overuse of fossil fuels intensifies, the imperative for new and renewable energy sources grows increasingly evident across numerous countries. A metal–air battery is a device for electrochemically converting chemical energy into electrical energy. It has a high theoretical energy conversion efficiency and does not emit any pollutants while converting energy, earning it recognition as a next-generation environmentally friendly energy source.1,2 The main characteristic of this metal–air battery is that, unlike the existing internal combustion engine, which uses oxygen for fuel combustion, it oxidizes and reduces oxygen electrochemically.3The theoretical energy density of a metal–air battery, predicated on the maximal utilization of the lithium anode, can reach up to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 W h kg−1.4 On the other hand, the energy density of a conventional Li-ion battery, including an insertion compound anode, hardly exceeds 1000 W h kg−1.5 However, at the present technology level, the inadequate durability and performance of metal–air batteries limit the actual use of these metal–air batteries due to the performance limitations of conventional catalysts typically used in ORR and OER.6,7
800 W h kg−1.4 On the other hand, the energy density of a conventional Li-ion battery, including an insertion compound anode, hardly exceeds 1000 W h kg−1.5 However, at the present technology level, the inadequate durability and performance of metal–air batteries limit the actual use of these metal–air batteries due to the performance limitations of conventional catalysts typically used in ORR and OER.6,7
At the present stage, there is no commercially available bifunctional catalyst with practical battery performance. Under state-of-the-art technology, precious metals and alloys such as Pt, Pt–Au, and Pt–Pd have been studied and developed as the highest-performance catalysts for metal–air batteries.8–11 However, their limited availability and high cost restrict practical application in metal–air batteries. Consequently, the development of low-cost, corrosion-resistant, and highly active bifunctional catalysts for both ORR and OER in metal–air cells is of paramount importance.12
Recent reports indicate that perovskite-based materials with an ABO3 structure (A = alkaline earth metal, B = transition metal, O = oxygen) can be formed using relatively inexpensive elements and exhibit high electrocatalytic activity for OER.13 Perovskite oxides such as Pr0.5Ba0.5CoO3−δ (PBCO)14 and Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF)15 have been proposed to produce high OER activity comparable to those of RuO2. A transition metal oxide, such as Co3O4, is also known as a catalyst for OER because the charge transfer of the energy levels of 3d orbitals of transition metal and 2p orbitals of oxide ions can easily occur.14 In addition, Co-based compounds such as spinel-type CoFe2O4, perovskite-type SrCoO3, and CoSn(OH)6 (CSO) are also known to have high catalytic performance for OER and be stable in an alkaline solution.16–18 Therefore, a study on these compounds has been carried out to understand the catalytic mechanism for OER and improve their catalytic performance. Among them, CSO has been reported to have excellent OER performance.19 However, the ORR activity of the CSO is relatively lower compared to other catalysts. Thus, technical development is essential to improve the electrocatalytic activity of CSO for ORR and OER. Song et al. reported that an electrochemical preconditioning greatly improved the electrocatalytic activity of the CSO for OER through 80 cyclic voltammetry scans (CVs) and galvanostatic activation.19 However, the ORR performance of the CSO could not be improved. The composite of the CSO with the catalytic material having ORR activity, such as carbon materials, will be necessary to impart the ORR activity to the CSO. The composite catalytic materials can exhibit synergistic effects and are expected to perform much better performance.20,21 Thus, the process of developing and synthesizing a catalytic composite material is vitally necessary.
A newly developed discharge process in the liquid phase, solution plasma process (SPP), could simply synthesize metal nanoparticles and heteroatom-doped carbon materials under atmospheric conditions at room temperature.22–28 The nitrogen-doped carbon materials synthesized by SPP showed good electrocatalytic activity for ORR.28 Panomsuwan et al. reported that catalytic carbon composite materials composed of carbon nanofiber (CNF) and nitrogen-doped carbon materials was synthesized by SPP, and the catalytic composite carbon materials showed excellent electrocatalytic activity for ORR comparable to commercial Pt/C.27 Our group has also reported that SPP has enabled the synthesis of the CSO, and the electrocatalytic properties of the CSO synthesized under specific conditions for OER were excellent.29 SPP can also produce composite materials such as metal nanoparticles-incorporated carbon in a single-step process.30 The carbon material could act as a support and conductor, improving the electrocatalytic properties of the composite materials. In addition, the plasma process could promote the formation of CSO nanoparticles. Thus, the SPP holds significant potential for synthesizing catalytic carbon composite materials. One of the most superior characteristics for SPP is to enable to synthesis the composite materials at single-step process.
In recent years, various wet-chemical methods, such as hydrothermal or co-precipitation routes, have been explored for synthesizing metal hydroxide-based electrocatalysts. However, these methods often require multiple steps, surfactants, and post-processing. Compared to hydrothermal and chemical co-precipitation methods, the solution plasma process (SPP) enables a rapid, surfactant-free, and single-step synthesis with in situ hybridization of nanocrystalline hydroxides and carbon, enhancing interfacial integration and electrocatalytic performance.31,32
In this study, we report on the SPP synthesis and characteristics of composite catalytic materials composed of CSO and carbon materials. The ORR and OER activities of the synthesized composite catalytic materials were also investigated. In addition, ORR and OER activities of the composite catalytic materials synthesized by SPP and coprecipitation methods were compared.
2. Experimental methods
2.1 Preparation of the solution
Cobalt chloride (CoCl2, purity > 97.0%) and tin(IV) chloride pentahydrate (SnCl4·5H2O, purity > 98.0%), ethanol (C2H5OH, purity > 95.0%), and 1 M potassium hydroxide (KOH) aqueous solution were purchased from Kanto Chemical Co., Inc. Nafion® solution (5 wt% in a mixture of lower aliphatic alcohols and water) was purchased from Sigma-Aldrich. Ultrapure water (18.2 MΩ cm) was obtained from an RFD250NB Aquarius water purification system. Carbon nanotubes (CNT), graphene (G), and Ketjen black (KB: EC600JD) were purchased from GSI Co., Inc., Sigma Aldrich, and Lion Specialty Chemicals Co., Ltd, respectively. All reagents were analytical grade and used without further purification.2.2 Synthesis of CSO and carbon composite materials by SPP and coprecipitation methods
0.01 M CoCl2 and 0.01 M SnCl4·5H2O mixed aqueous solutions were prepared by dissolving them in 100 mL of ultrapure water to synthesize CoSn(OH)6 (CSO). The pH of the mixed aqueous solution was 1.3. The solution pH was adjusted to 10 or 12 using a 0.1 M sodium hydroxide aqueous solution. Then, 25 mg of carbon nanotubes (CNT), graphene (G), or Ketjen black (KB) were added to the mixed solution. For SPP synthesis, the mixed solution at pH = 10 or 12 was poured into the SPP reactor. In the SPP reactor, two tungsten electrodes (W, purity > 99.95%, Nilaco, 1.0 mm in diameter) were fixed to face each other, and a side surface of each electrode was covered with an insulator by a ceramic tube. The gap distance between the electrodes is fixed at 0.5 mm. Fig. 1 shows the schematic illustration of the set-up for SPP. Synthesis was performed by generating plasma in a precursor solution using a bipolar pulse power supply (MPP-HV04 Bipolar-DC pulsed power supply, Kurita Seisakusho Co., Ltd, Japan). The pulse voltage between the electrodes, the repetition frequency, and the pulse width were set at 1.2 kV, 50 kHz, and 1.0 μs, respectively. The discharge was maintained for 20 minutes under these conditions. Table S1† shows the SPP conditions for synthesizing CSO and carbon composite materials. In case of coprecipitation methods, the mixed solution pH was adjusted to 12 using a 0.1 M sodium hydroxide aqueous solution and the mixed solution was then stirred for 2 h.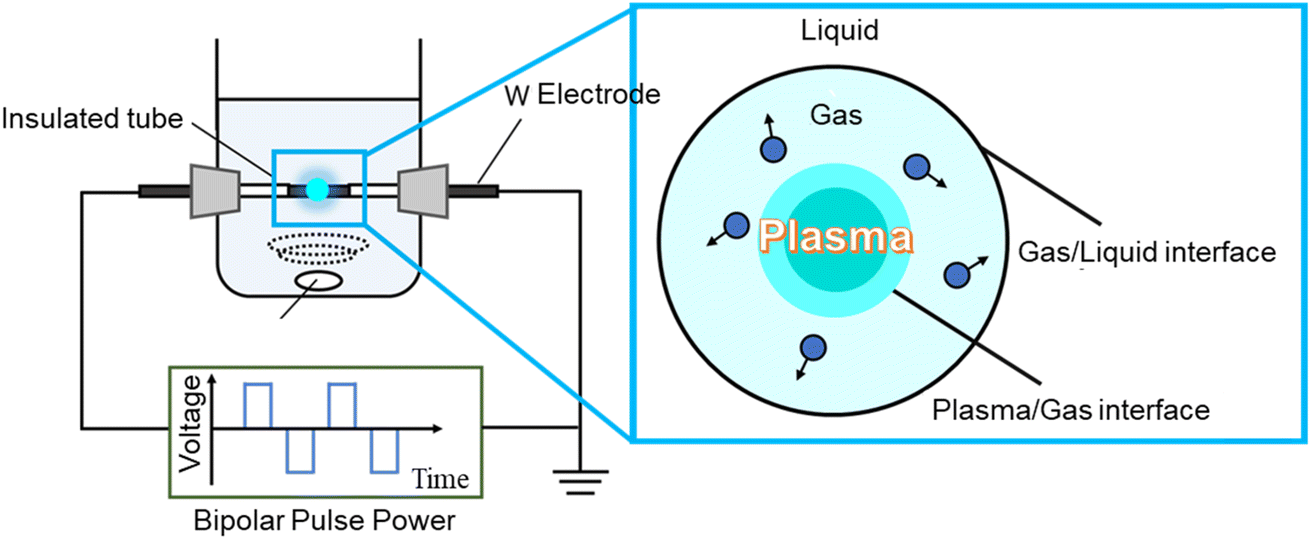 | ||
| Fig. 1 Schematic diagram of SPP process set-up. | ||
After synthesizing composite materials by SPP and coprecipitation methods, the samples were collected by suction filtration using a hydrophilic PTFE membrane filter with a pore size of 0.1 μm (JVWP04700, Merck Millipore) and then washed with ultrapure water and ethanol, subsequently. After washing, the sample was dried in an electric furnace at 60 °C for 12 hours.
Hereafter, the sample names of composite materials composed of CoSn(OH)6 and CNT, G, or KB synthesized at pH = 10 and 12 by SPP are referred to as CSO_CNT_pH10, CSO_G_pH10, CSO_KB_pH10, CSO_CNT_pH12, CSO_G_pH12, and CSO_KB_pH12, respectively. The sample names of composite materials composed of CoSn(OH)6 and CNT, G, or KB synthesized at pH = 12 by coprecipitation method are referred to as CSO_CNT_pH12(co), CSO_G_pH12(co), and CSO_KB_pH12(co), respectively.
2.3 Characterization
The shape of the synthesized sample was observed with a field emission scanning electron microscope (FE-SEM, JEOL Ltd, JSM-7100F, Tokyo, Japan) at an accelerating voltage of 15 kV. The atomic composition of the synthesized samples was analyzed using energy-dispersive X-ray spectroscopy (EDS, JEOL Ltd, JED-2300F, Tokyo, Japan) at an accelerating voltage of 20 kV. The microstructures of the synthesized sample were observed with a transmission electron microscope (TEM, JEOL Ltd, JEM-2100, Tokyo, Japan) at an accelerating voltage of 200 kV. The crystal structures of the synthesized samples were identified by X-ray diffraction (XRD, Rigaku Co., Smart Lab, Tokyo, Japan) measurement with Cu Kα radiation (λ = 0.154 nm) operating at 40 kV and 40 mA (1.6 kW). X-ray photoelectron spectroscopy (XPS, JEOL Ltd, JPS-9010 MC, Tokyo, Japan) with monochromatic Mg Kα radiation (1253.6 eV) as an excitation source under ultra-high vacuum conditions was used to investigate the chemical bonding states. The operating emission current and anode voltages were set to be 25 mA and 10 kV, respectively. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method in the relative pressure (P/P0) range of 0.05–1.00 using a specific surface area/pore distribution measuring equipment (Micromeritics Instruments Co., TriStar II 3020 Tokyo, Japan). As a pretreatment for the measurement, 50 mg of the sample was degassed at 100 °C for 12 hours. After degassing, the sample cell was cooled with liquid nitrogen and then measured.2.4 Electrochemical measurements
The electrocatalytic performance of the synthesized samples for ORR and OER was evaluated by linear sweep voltammetry (LSV). The LSV was conducted on a computer-controlled ALS-CH instrument electrochemical analyzer model 704ES (BAS, Co.) equipped with a rotating ring disk electrode rotator apparatus (RRDE-3A, ALS Co.). A Pt coil (ALS, Co., Ltd) and an Ag/AgCl electrode (ALS, Co., Ltd) filled with a saturated aqueous KCl solution were used as counter and reference electrodes, respectively. A suspension of 5.0 mg of the synthesized samples in 480 μL ultrapure water, 480 μL ethanol, and 40 μL Nafion® aqueous solution was sonicated until a homogeneous dispersion was obtained (5 mg mL−1). Next, 7.5 μL of homogeneous suspension was applied to a glassy carbon disk (GC) of a rotating-ring disk electrode (RRDE) to prepare the working electrode and allowed to dry in the air. A 4 mm diameter glassy carbon electrode (area: 0.126 cm2) was used as the working electrode for all measurements. All electrochemical measurements were performed at room temperature in 1 M KOH aqueous solution. N2 gas has flowed for more than 30 min to remove the dissolved O2 gas before the measurements. Then, O2 gas has flowed for 20 min to estimate the electrocatalytic activity of the OER. The O2 gas was passed on the solution level in the electrochemical cell during the measurements. ORR measurements were conducted in O2-saturated 1 M KOH using a rotating disk electrode at 1600 rpm with a scan rate of 10 mV s−1. OER measurements were performed in the same electrolyte under ambient conditions at 1600 rpm with a scan rate of 10 mV s−1. The onset potentials for ORR and OER were determined as the potential at which the current density reaches 1.0 mA cm−2. Electrochemically active surface area (ECSA) was determined by measuring the capacitive current associated with double-layer charging in the scan rate dependence of CVs. To this end, the potential window for CV was 1.22–1.32 V for RHE. The scan rates were 20, 50, 100, 200, 400, 600, 800, and 1000 mV s−1. The double-layer capacitance (Cdl) was estimated by plotting RHE for ΔJ = (Ja − Jc) versus scan rates at 1.27 V (Ja: anodic current density, Jc: cathodic current density). The linear slope is twice the double-layer capacitance Cdl. The chronoamperometry at an overpotential of 320 mV was performed to evaluate the long-term stability of the synthesized composite carbon materials.3. Results and discussion
Fig. 2a and b show the XRD patterns of the composite materials synthesized at pH = 10 and 12 by SPP, respectively. In Fig. 2a, some peaks corresponding to the 111, 200, 220, 310, 311, 222, 400, 331, 420, 422, 440, 531, 442, and 620 reflections of CSO (CoSn(OH)6, JCPDS card no. 13-0356) were observed at 2θ = 19.98°, 23.08°, 32.77°, 36.94°, 38.32°, 40.47°, 47.12°, 51.59°, 52.91°, 58.27°, 68.36°, 71.91°, 73.19°, and 77.77°.34 These peaks are indicative of the perovskite hydroxide structure. In addition to these peaks, a few weak peaks attributable to CoOOH (at 2θ = 20.2° (003), JCPDS card no. 07-0169), Co3O4 (at 2θ = 44.4° (400), JCPDS card no. 78-1969), and SnO2 (at 2θ = 33.7° (101) and 61.9° (221)) were also identified in the samples synthesized at pH 10 (Fig. 2a).35 These results indicate that the samples synthesized at pH = 10 include compound different from CSO. In contrast, Fig. 2b demonstrates that all samples synthesized at pH 12 had sharp and high-intensity peaks originating from CSO, indicating high crystallinity of CSO. No peak attributed to SnO2 was observed in the samples synthesized at pH 12. It was found that the composite of various carbon materials by SPP did not induce any observable changes in the crystal phase of CSO. Although the precursor ratios were fixed at 1:1 (metal salt:carbon), the final compositions may vary slightly due to differences in plasma-induced crystallization and the formation of amorphous secondary phases. This could partly account for the observed variations in catalytic activity among the samples.
 | ||
| Fig. 2 The XRD patterns of the samples synthesized at (a) pH = 10 and (b) pH = 12. | ||
Fig. S1 (in ESI†) shows XRD patterns of the composite materials synthesized at pH = 12 by coprecipitation methods. Some peaks corresponding to the 111, 220, 310, 311, 222, 400, 331, 420, 422, 440, 531, 442, and 620 reflections of CSO (CoSn(OH)6, JCPDS card no. 13-0356) were clearly observed at 2θ = 19.98°, 32.77°, 36.94°, 38.32°, 40.47°, 47.12°, 51.59°, 52.91°, 58.27°, 68.36°, 71.91°, 73.19°, 77.77°, and 82.42°, indicating high crystallinity of the perovskite hydroxide structured CSO. No peak attributed to other compounds such as SnO2 and Co3O4 was observed in the samples synthesized by coprecipitation method at pH 12. These results show that the composite of various carbon materials by coprecipitation method also did not induce any observable changes in the crystal phase of CSO.
Fig. 3a–f shows FE-SEM images of the composite materials composed of CSO and carbon materials ((a) CSO_CNT_pH10, (b) CSO_G_pH10, (c) CSO_KB_pH10, (d) CSO_CNT_pH12, (e) CSO_G_pH12, and (f) CSO_KB_pH12). In Fig. 3a and d, some CNTs and granular particles with sizes of 10 to 100 nm were observed. In addition, the aggregation of the particles and the particles attached to CNTs were also seen. The attachment of the particles to the CNT may induce the improvement of the electrocatalytic activity for ORR or OER because the electronic states of the synthesized materials can be changed by the composites. In Fig. 3b and e, the angulated substances with flat surfaces and granular particles with sizes of 10 to 100 nm were observed. The angulated substances could be graphene, which was added as the raw material. In addition, the attachment of the particles to the angulated substances can be observed slightly. In Fig. 3c and f, amorphous substances with sub-micron sizes and granular particles with 10 to 100 nm sizes were also observed. Moreover, in all Fig. 3a–f, the presence of the substances with cubic-like structures was confirmed. It has been reported that the shape of crystalline CSO showed a cubic-like structure.33 The crystalline CSO samples synthesized at pH 10 and 12 by SPP exhibited similar structures (Fig. S2 in ESI†).29 These results suggest that our SPP method can synthesize carbon-based composite materials without altering the inherent cubic shape of CSO.
 | ||
| Fig. 3 FE-SEM images of (a) CSO_CNT_pH 10, (b) CSO_G_pH 10, (c) CSO_KB_pH 10, (d) CSO_CNT_pH 12, (e) CSO_G_pH 12, and (f) CSO_KB_pH 12. | ||
Fig. S3† show FE-SEM images of the composite materials synthesized at pH = 12 by coprecipitation methods. In Fig. S3,† some CNTs and granular particles with sizes of 10 to 100 nm were observed. In addition, the aggregation of the particles and the particles attached to CNTs were also observed as well as the composite materials synthesized by SPP. The shapes of the composite materials observed in Fig. S3a–c† were also similar to ones synthesized by SPP. From these results, it was found that no change in the crystallinity and shape was observed in spite of the different synthesis method.
Fig. 4a–f show TEM images of (a) CSO_CNT_pH10, (b) CSO_G_pH10, (c) CSO_KB_pH10, (d) CSO_CNT_pH12, (e) CSO_G_pH12, and (f) CSO_KB_pH12. STEM, and elemental mapping images of (a) CSO_CNT_pH10, (b) CSO_G_pH10, (c) CSO_KB_pH10, (d) CSO_CNT_pH12, (e) CSO_G_pH12, and (f) CSO_KB_pH12 are also shown in Fig. S5–S10.† Table S2† summarizes the atomic concentrations for all samples, as determined by TEM-EDS analysis. The TEM observations revealed that all samples contained a mixture of amorphous and crystalline phases, consistent with findings from FE-SEM observation and XRD measurements. The crystalline phase was identified as CSO, as characterized by a cubic morphology. These agree well with many studies, indicating that most Sn-based or Co–Sn-based oxides or hydroxides exhibit cubic structures.19,29,33–36 This cubic structures were observed for both CSO_pH10 and CSO_pH12, confirming that the CSO structure remains unaffected by the composite of carbon materials (as shown in Fig. S4†). Elemental mapping images (Fig. S5–S10†) of CSO_pH10 and CSO_pH12 demonstrated the uniform distribution of Co, Sn, O, and C elements, succeeding in the formation of CSO and carbon composite materials. Table S2† revealed that the atomic concentration ratios of Co and Sn in all samples were nearly identical to those in the synthesized CSO_pH10 and CSO_pH12 before compositing. EDS quantitative analysis further confirmed that the structural integrity of CSO was maintained after the composite of carbon materials. These findings on the morphological and structural properties suggest that the synthesized samples comprise amorphous materials (Co compounds and SnO2) and crystalline CSO. The average crystallite sizes of CSO samples for pH 10 and pH 12 were calculated using the Scherrer equation and were found to be 23.4 nm and 17.1 nm, respectively. This result aligns with the TEM-based morphological analysis and suggests that the smaller crystallites at pH 12 lead to a larger active surface area, contributing to the superior electrocatalytic performance.
 | ||
| Fig. 4 TEM images of (a) CSO_CNT_pH10, (b) CSO_G_pH10, (c) CSO_KB_pH10, (d) CSO_CNT_pH12, (e) CSO_G_pH12, and (f) CSO_KB_pH12. | ||
Fig. S11–S14† depicts the wide, Co 2p, Sn 3d, and O 1s XPS spectra of the carbon composite samples synthesized via the SP method, respectively. Quantitative analysis results for the surface composition of each sample are summarized in Table 1. From Fig. S11–S14,† peaks corresponding to Co, Sn, and O were identified in all samples, indicating their presence. In the high-resolution Co 2p XPS spectrum, two prominent peaks at approximately 782.8 eV and 799.1 eV correspond to Co 2p3/2 and Co 2p1/2, respectively. The Co 2p3/2 spin–orbit doublet can be further deconvolved into two separate peaks at 782.7 eV and 784.7 eV, attributed to Co3+ 2p3/2 and Co2+ 2p3/2, respectively. Similarly, the Co 2p1/2 spin–orbit doublet can be deconvolved into two peaks at 798.6 eV and 799.8 eV, associated with Co3+ 2p1/2 and Co2+ 2p1/2, respectively.36 Additionally, two satellite peaks at 788.7 eV and 804.4 eV, alongside the Co 2p3/2 and Co 2p1/2 spin–orbit doublets, confirm the presence of cobalt oxides.36 In previous our report for CSO synthesized by SPP,29 the Co 2p spectra were deconvoluted into two distinct peaks and two weak satellites. The two distinct peaks corresponding to Co 2p3/2 and Co 2p1/2, respectively, were assigned to cobalt oxides located at 780.7 and 796.7 eV and the spin–orbit doublet of Co 2p3/2 can be deconvolved into two peaks at 780.5 and 782.1 eV assinged to the Co3+ 2p3/2 and Co2+ 2p3/2 configurations, respectively. The Co 2p1/2 spin-orbital doublet can also be deconvoluted into two separate peaks located at binding energies of 796.4 and 797.5 eV. Compared to previously reported CSO samples synthesized at pH 10 and 12 (CSO_pH10 and CSO_pH12),29 the peak positions of Co 2p were shifted to higher binding energy. This means that the CSOs were slightly oxidized by the composite of carbon materials. This change in oxidization states could change the charge density of Co atoms. The change in the charge density can change the electrocatalytic activity for ORR or OER. Thus, it is expected that the composite of CSO and carbon materials can improve the electrocatalytic activity for ORR or OER, compared to CSO. For the Sn 3d high-resolution XPS spectrum, the peaks at approximately 486.1 eV and 494.5 eV are designated to Sn 3d5/2 and Sn 3d3/2, respectively, with a peak separation of 8.4 eV, indicating the presence of Sn in its Sn4+ state.37,38 In the high-resolution XPS Sn 3d spectra of the CSO samples, two peaks at 486.1–486.5 and 494.5–494.9 eV attributed to Sn 3d5/2 and 3d3/2, respectively were observed.29 The energy separation of the observed spin was found to be almost 8.4 eV. These values on the peak potions and energy separation agrees with the reported value of the CSO samples synthesized by SPP.29 This indicates that the remarkable change in the chemical bonding states for Sn occur hardly by the composite of CSO and carbon materials. The O 1s high-resolution XPS spectrum reveals the peaks primarily associated with metal–oxygen (metal–O at 530.6 eV) and metal–hydroxide (metal–OH at 531.7 eV) bonds, alongside evidence of adsorbed oxygen and water on the sample surface.39,40 The analysis of elemental composition and chemical bonding states confirms Co, Sn, and O as the main components in all samples. Quantitative surface analysis results, as shown in Table 1, also revealed that no change in the Co–Sn presence ratio were observed when compared to CSO_pH10 and CSO_pH12.29 Consequently, it is found that the composite of carbon materials changed the chemical bonding states of Co 2p in CSO. The Sn-rich surface detected by XPS is likely due to segregation of amorphous SnO2 species during SPP treatment. This enrichment plays a role in modulating the local electronic structure and could facilitate the formation of Co3+–O species favorable for OER. Similar surface enrichment and modulation of Co3+ active sites by metal dopants (e.g., Sn, Pd, Zn) have been shown to improve OER efficiency through changes in surface electronic configuration and oxygen intermediate binding energy.41–43
| O | Co | Sn | W | |
|---|---|---|---|---|
| CSO_CNT_pH10 | 19.82 | 14.23 | 60.85 | 5.09 |
| CSO_G_pH10 | 19.83 | 12.69 | 62.98 | 4.50 |
| CSO_KB_pH10 | 17.36 | 17.72 | 60.52 | 4.40 |
| CSO_CNT_pH12 | 21.76 | 13.59 | 59.71 | 4.94 |
| CSO_G_pH12 | 21.41 | 13.93 | 59.77 | 4.89 |
| CSO_KB_pH12 | 19.47 | 13.94 | 61.86 | 4.73 |
The electrocatalytic activity of the synthesized samples for ORR was initially assessed via cyclic voltammetry (CV) measurements in a 1 M KOH solution saturated with either N2 or O2 gas at room temperature (Fig. S15†). The CV profiles of all catalysts exhibited featureless voltammetric currents in the N2-saturated solution (represented by dashed lines), which indicates a stable electrochemical environment in the absence of ORR. In contrast, a pronounced ORR peak emerged at around 0.6–0.7 V vs. NHE in the O2-saturated solution (illustrated by solid lines), confirming the ORR activity of all composite samples.
Fig. 5a and b illustrate the results of linear sweep voltammetry (LSV) measurements for the samples synthesized at pH 10 and 12, respectively, providing insights into the kinetics of the electrochemical reactions. The results of linear sweep voltammetry (LSV) measurements for the composite samples synthesized by coprecipitation method at pH 12 are also shown in Fig. S16.† Table S3† compiles the ORR onset potentials, current densities at 0.464 V vs. RHE, electron transfer numbers, and hydrogen peroxide production rates for each sample, offering a comprehensive electrocatalytic properties of the materials synthesized in this study. For benchmarking purposes, the ORR onset potentials, current densities at 0.464 V vs. RHE, electron transfer numbers, and hydrogen peroxide production rates for commercial Pt/C was also described in Tables S3.† Among the samples synthesized at pH 10, CSO_KB_pH10 exhibited the most positive ORR onset potential at 0.843 V vs. RHE and demonstrated the highest current density at 0.464 V vs. RHE, indicating superior catalytic activity. Similarly, CSO_KB_pH12 presented the most positive ORR onset potential at 0.915 V vs. RHE among the samples synthesized at pH 12, along with the highest current density at the specified potential, suggesting that the pH conditions during synthesis have a pronounced effect on the electrocatalytic properties of the materials. Compared to the composite materials synthesized by coprecipitation method, the onset potentials and limiting current density values at 0.464 V of the composit materials synthesized by SPP are relatively superior. In particular, the limiting current values of the samples synthesized by SPP are more than two times higher than those synthesized by coprecipitation method. This means that the samples synthesized by SPP have much more active sites than those synthesized by coprecipitation method.
 | ||
| Fig. 5 LSV curves using an RRDE electrode in an O2-saturated 1 M KOH solution at a scan rate of 10 mV s−1. The rotation speed was 1600 rpm; (a) sample synthesized at pH = 10 and 20 wt% Pt/C, (b) sample synthesized at pH = 12 and 20 wt% Pt/C, (c) electron transfer number (n), and (d) HO2− yield derived from the disk and ring currents at the potential range from 0.30 to 0.60 V. | ||
Fig. 5c and d depict the variation in electron transfer numbers and hydrogen peroxide production rates for samples synthesized at pH 10 and 12, respectively, as a function of potential. The electron transfer numbers for CSO_G_pH10 and CSO_G_pH12 were approximately 3.4 at all potentials, suggesting 4- and 2-electron ORR processes occurred simultaneously. This functional activity could be leveraged in energy conversion and storage systems that benefit from both reaction pathways. The electron transfer numbers for the other composite samples hovered around 3.7, indicating a predominance of the 4-electron ORR pathway, which is typically associated with higher energy efficiency and less corrosive product formation. Notably, the electron transfer number for CSO_KB_pH12 was found to be 3.9, underscoring a significant dominance of the 4-electron reaction, which is the desired pathway for fuel cell applications due to its direct production of water without intermediate peroxide species. Compared to the composit materials synthesized by coprecipitation method, the electron transfer numbers is much higher, indicating that the electrocatalytic activity of the composite materials synthesized by SPP is greatly superior to those by coprecipitation method.
Compared to the commercial 20 wt% Pt/C catalyst, the CSO_KB_pH12 exhibited a slightly lower onset potential but a higher limiting current density, indicating comparable electrocatalytic performance for the ORR. In addition, CSO_KB_pH12 demonstrated an electron transfer number (n = 3.93) closer to 4 and a lower H2O2 yield than Pt/C (n = 3.7), suggesting that the catalyst predominantly follows the more favorable 4-electron ORR pathway rather than the less efficient 2-electron route associated with peroxide formation. This higher electron transfer number is mainly attributed to the structural properties of Ketjen Black, including its high surface area, abundant oxygen-containing functional groups, and microporous architecture, which facilitate O2 adsorption and OOH conversion, consistent with previous studies on carbon-based ORR catalysts.44,45
From these results, it was concluded that the composite of the carbon materials to the CSO significantly improved ORR onset potential and limiting current density across all composite samples, which could be attributed to the synergistic effect of combined conductive networks, suitable electronic states and increase in the active sites for ORR.
The OER catalytic properties of the synthesized samples and a commercial RuO2 catalyst were also assessed using linear sweep voltammetry (LSV). The electrocatalytic activity of all samples was examined in a 1 M KOH solution. Catalytic inks, comprising either the composite material or RuO2 as the active component, were drop-casted onto vitreous carbon electrodes.39 Cyclic voltammetry (CV) was conducted at a scan rate of 10 mV s−1 as an electrochemical preconditioning step to stabilize the electrode surface prior to the LSV measurements. Fig. 6a and c display the LSV curves at 1500 rpm for samples synthesized by SPP at pH 10 and pH 12, respectively. The LSV curves of the samples synthesized by coprecipitation method at pH 12 are also shown in Fig. S17.†Fig. 6b and d present the Tafel plots following the onset of OER for samples synthesized at pH 10 and 12. As depicted in Fig. 6, the overpotentials of all samples were lower than those of RuO2 when achieving a current density of 10 mA cm−2. The OER onset potentials, overpotentials at 10 mA cm−2, and Tafel slope values for each sample are listed in Table S4.† Notably, the OER onset potential for CSO_KB_pH12 (1.322 V vs. RHE) was the lowest values among all synthesized samples, exhibiting a 148 mV increase compared to RuO2 (1.470 V vs. RHE). Furthermore, CSO_KB_pH12 demonstrated that the potential at a current density of 10 mA cm−2 (1.548 V vs. RHE) was the lowest among all samples, surpassing RuO2 by 0.111 V (1.659 V vs. RHE). The samples synthesized at pH 12 exhibited lower onset potential and overpotential values at 10 mA cm−2 than those synthesized at pH 10, suggesting an improvement in OER catalytic properties with increasing pH of the synthesis condition. The composite samples synthesized at pH 12 by SP showed lower onset potentials and overpotentials at 10 mA cm−2 than those synthesized by coprecipitation method, suggesting an improvement in OER catalytic properties with different synthesis process. The Tafel slope for CSO_CNT_pH10 was estimated to be 77 mV per decade, and was the smallest among all synthesized samples by SPP. In addition, it was significantly less than that of commercial RuO2 (162 mV per decade). On the other hand, the Tafel slope values for CSO_KB_pH10 and CSO_KB_pH12 were estimated to be 126 and 144 mV per decade, respectively. Their Tafel slope values are relatively large among all samples synthesized by SPP although their onset potentials and overpotentials at 10 mA cm−2 are relatively positive. This indicates that the OER can occur easily on the CSO_KB_pH10 and CSO_KB_pH12, but the reaction rate is not so fast. These findings imply that the OER catalytic performance of CSO_KB_pH10 and CSO_KB_pH12 is more superior than commercial RuO2 and the CSO_KB_pH12 were the most advantageous electrocatalytic properties for OER among all samples synthesized in this study.
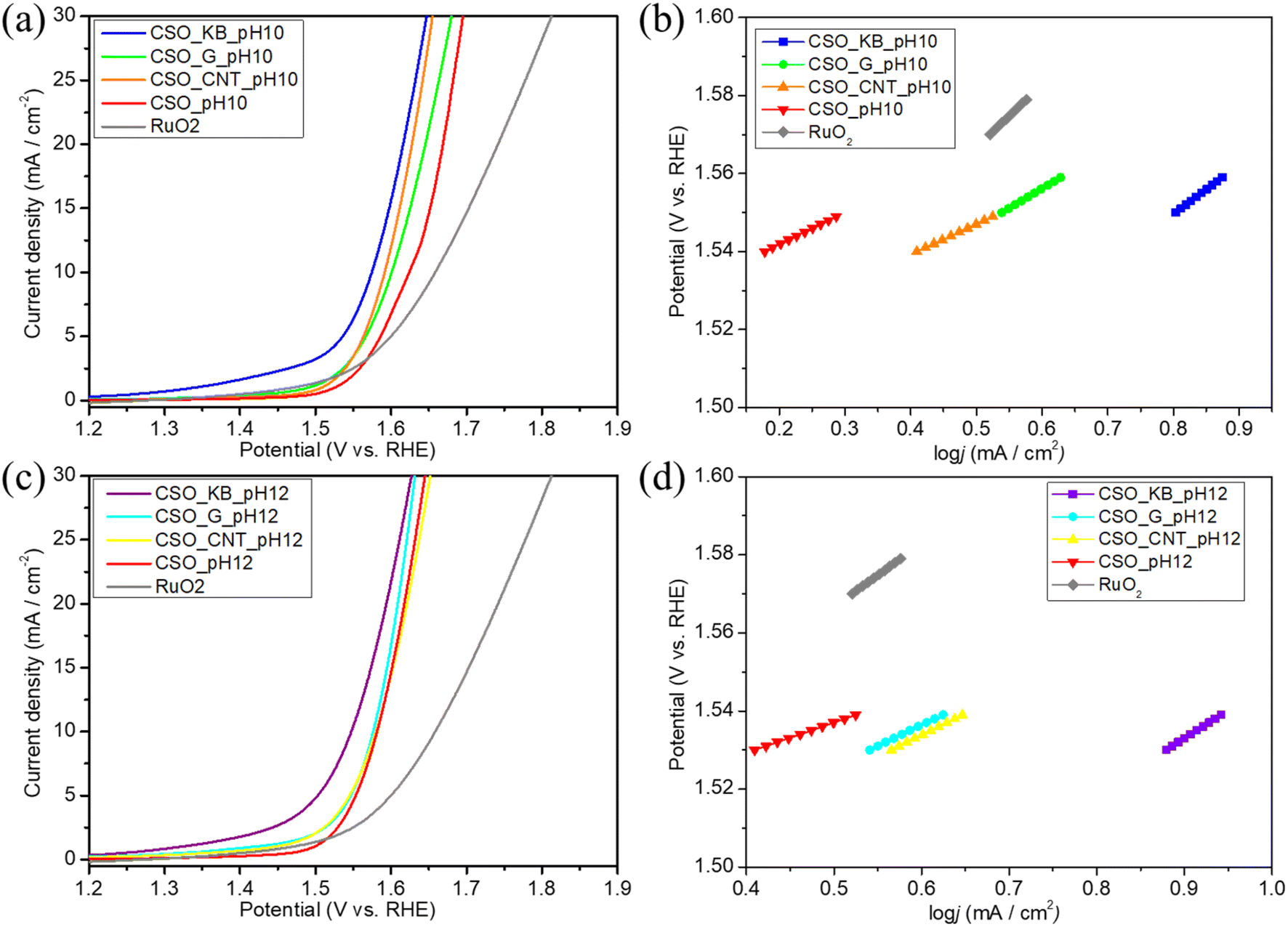 | ||
| Fig. 6 (a) LSV curves and (b) Tafel plots of the composite samples synthesized at pH = 10 and commercial RuO2. (c) LSV curves and (d) Tafel plots of the composite samples synthesized at pH = 12 and commercial RuO2. All measurements were performed in 1 M KOH electrolyte at a scan rate of 10 mV s−1. | ||
The electrochemical double-layer capacitance (Cdl) was measured to estimate the electrochemical surface area (ECSA). ECSA was evaluated only for the CSO_KB_pH10 and CSO_KB_pH12 samples, as these exhibited the best electrocatalytic performance in both ORR and OER. ECSA was estimated by measuring the double-layer capacitance (Cdl) from CV curves at varying sweep rates (20–1000 mV s−1). Although some protocols recommended slower scan rates, recent studies using CoSn(OH)6-based catalysts have applied sweep rates up to 1000 mV s−1 to reliably extract Cdl, assuming linear capacitive behavior and stable current response.19,29 The circulating potential–current diagrams at the various scan speeds of CSO_KB_pH10 and CSO_KB_pH12 were shown in Fig. 7a and b, respectively, and the Cdl values were calculated. As shown in Fig. 7c, the Cdl of CSO_KB_pH10 and CSO_KB_pH12 were found to be 4.15 and 4.50 mF cm−2, respectively, and the ECSA values of CSO_KB_pH10 and CSO_KB_pH12 were estimated to be 104.03 and 120.12, respectively. This indicates that the active surface areas for oxygen evolution of CSO_KB_pH12 are larger than those of CSO_KB_pH10. This result agrees well with the results of OER properties obtained by electrochemical measurements. Catalytic activity and long stability for OER are two important parameters for OER. The chronoamperometric (CA) curve of CSO_KB_pH12 was measured at an overpotential of 320 mV for 45000 s.
 | ||
| Fig. 7 (a) CSO_KB_pH10 and (b) CSO_KB_pH12 measured in 1 M KOH at scan rates of 0.02 to 0.1 V s−1 (c) Cdl at 1.27 V vs. RHE of CSO_KB_pH10 and CSO_KB_pH12. (d) The chronoamperometric (CA) curve of CSO_KB_pH12 at an overpotential of 320 mV. The duration time for CA is 45000 s. | ||
The CA curve is shown in Fig. 7d. No change in the CA curve can be observed with time evolution, indicating the long stability of the CSO_KB_pH12 for OER is excellent. XPS and XRD measurements were performed to investigate the change in the surface state and crystallinity of CSO_pH12sp before and after CA measurement for 45000 s. High-resolution XPS Co 2p, Sn 5d, and O 1s spectra before and after CA measurement are shown in Fig. S18.† No significant peak change between before and after the CA measurement in this high-resolution XPS was observed. However, the relative ratio of Co3+ content increased slightly after the CA measurement. An increase in the Co3+ ratio can be caused by electrochemical oxidation. Co3+ is known to play an essential role in OER activities and has been reported to contribute to improvement of the durability. In addition, the XRD patterns of CSO_KB_pH12 before and after CA measurement are also shown in Fig. S19.† No change in the crystal phase was observed from the XRD patterns. These results confirm that CSO_KB_pH12 has excellent long-term durability in a 1 M KOH solution. Therefore, CSO_KB_pH12 had a low onset potential, low overpotential at 10 mA cm−2, and a high Cdl, so it is likely to be developed as a better catalyst for OER than the other samples shown in Table 2.
| Sample | Overpotentials at reaching 10 mA cm−2 [V vs. RHE] | Tafel slope [mV dec.−1] | References |
|---|---|---|---|
| CSO_KB pH12 | 0.318 | 144 | This work |
| CSO_KB_pH10 | 0.345 | 126 | This work |
| CoSn(OH)6/CoOx | 0.274 | — | 19 |
| Co(OH)2 | 0.310 | 69 | 52 |
| Co3O4/NiCo2O4 | 0.340 | 88 | 53 |
| Co3O4/nanoporous carbon | 0.310 | 70 | 54 |
| χ-CoOOH nanosheet | 0.300 | 38 | 55 |
| Hollow Co3O4 microtubes | 0.290 | 84 | 56 |
| Co3O4 nanosheet | 0.300 | 68 | 57 |
| Co2P nanoneedle | 0.310 | 50 | 46 |
| NiCo LDH | 0.367 | 40 | 58 |
| CoMn LDH | 0.324 | 43 | 59 |
| Fe-doped Mo/Te | 0.300 | 47 | 60 |
| NiTe2/TM | 0.315 | 82 | 61 |
| Co–P film | 0.345 | 47 | 62 |
| N-Doped graphene–CoO | 0.340 | 71 | 63 |
| Ni0.95Mn0.05O/CNT | 0.293 | 56 | 64 |
The synergistic interaction between CSO and carbon significantly enhanced the electrocatalytic properties for both ORR and OER. Carbon's high electrical conductivity and large surface area can facilitate electron transfer. This improves the accessibility to active sites on CSO, leading to more efficient catalytic reactions and thereby boosting ORR and OER performance.47,48 In addition, as shown in Co 2p XPS spectra, the binding energy was shifted to higher energy by the composite of CSO and carbon materials. The change in the oxidization states of CSO could affect the electrocatalytic properties for both ORR and OER due to the change in charge density of CSO. Moreover, the mechanism of Co-based OER catalysts involves the formation of a Co-oxide intermediate from CoOOH to CoO2. The critical oxidation of this intermediate, culminating in O2 generation, plays a pivotal role in enhancing OER performance.49 It has been observed that an increase in the ratio of Co-containing compounds, facilitated by adjusting the synthesis pH from 10 to 12, leads to a higher number of active sites for OER. This adjustment is crucial for optimizing the electrocatalytic efficiency of the system.
Unlike other carbon materials, Ketjen Black's (KB) porous structure facilitates a high dispersion of CoSn(OH)6 particles and promotes effective mass transfer of reactants and products, offering substantial benefits for catalytic applications. Synthesis at pH 12 is particularly advantageous for generating well-dispersed CSO nanoparticles on the Ketjen Black surface,50 as alkaline conditions favor the hydrolysis and subsequent precipitation of cobalt and tin hydroxides into cobalt stannate oxide upon calcination.51 These conditions ensure uniform nucleation and growth of CSO particles, resulting in a more evenly distributed layer of catalytically active sites on the KB substrate.
In conclusion, the synergistic interaction between CSO nanoparticles and carbon materials like KB not only enhances the electrocatalytic properties for ORR and OER but also underlines the importance of material composition and synthesis conditions in catalytic performance optimization. This comprehensive approach to catalyst design opens new avenues for research and development in efficient and sustainable energy conversion technologies.
The specific surface area and pore structure characteristics of the synthesized samples were analyzed using the Brunauer–Emmett–Teller (BET) theory. Fig. S20† illustrates the N2 adsorption and desorption isotherm curves for CSO_CNT_pH10, CSO_G_pH10, CSO_KB_pH10, CSO_CNT_pH12, CSO_G_pH12, and CSO_KB_pH12. These curves displayed a Type IV profile with a hysteresis loop, as categorized by the International Union of Pure and Applied Chemistry (IUPAC), indicative of mesoporous structures. The estimated specific surface areas from the isotherm curves are listed in Table S5.† The specific surface area values for CSO_pH10, CSO_CNT_pH10, CSO_G_pH10, CSO_KB_pH10, CSO_pH12, CSO_CNT_pH12, CSO_G_pH12, and CSO_KB_pH12 were estimated to be 248.17, 229.56, 212.43, 298.77, 146.20, 203.20, 208.62, and 169.88 m2 g−1, respectively. It is evident from Fig. S20† that all samples exhibit BET-type adsorption isotherms.65 From the results of Table S5,† it is revealed that the samples synthesized at pH 10 possess larger surface areas compared to those synthesized at pH 12, with CSO_KB_pH10 exhibiting the highest specific surface area among all samples. Conventionally, an increase in surface area is associated with an enhancement in catalytic active sites, potentially improving catalytic performance.66 Contrary to this expectation, the samples synthesized at pH 12 with lower surface areas demonstrated superior OER catalytic performance in this study, indicating no direct correlation between surface area and catalytic efficiency. This anomaly is attributed to the composition of CSO_pH10, which contains a significant proportion of SnO2. Although SnO2 contributes to the overall surface area, it is comparatively less active in OER processes than the components present in CSO_pH12. Additionally, it was reported that catalysts of CoOOH/CoSn(OH)6, Co3O4, CoSnO2 and Co3O4/SnO2 have lower OER catalytic activity compared to CSO.19 Consequently, the presence of Co compounds and SnO2 within the composite materials is hypothesized to diminish the electrocatalytic properties of OER.
The ORR–OER bifunctional catalytic performance of all samples was evaluated using LSV. The LSV measurement results (ORR–OER) are shown in Fig. 8. Table 3 also shows the potential values (Ej = −3) at reaching −3 mA cm−2, the potential values (Ej = 10) at reaching 10 mA cm−2, and the potential gap (ΔE = Ej = 10 − Ej = −3) values of the composite materials synthesized in this study and reported values. The ΔE value of CSO_KB_pH12 was the smallest value among all samples synthesized and was found to be 0.835. From these results, it can be concluded that all synthesized composite materials have bifunctional catalytic performance for ORR and OER and the CSO_KB_pH12 had the most superior bifunctional catalytic activity among all synthesized samples in this study. The Ej = 10 and Ej = −3 values of CSO_KB_pH12 showed superior values compared to the reported values.67,68 However, compared to these values of P, N Co-doped graphene, our values were inferior. This is due to that the performance of our carbon materials for ORR was lower than that of P, N Co-doped graphene. This indicates that our composite materials synthesized by SPP have a potential to improve the electrocatalytic activity of the composite materials for ORR by doping heteroatoms to the synthesized composite material composed of CSO and carbon materials. Based on this concept, further study will required to improve the electrocatalytic activity.
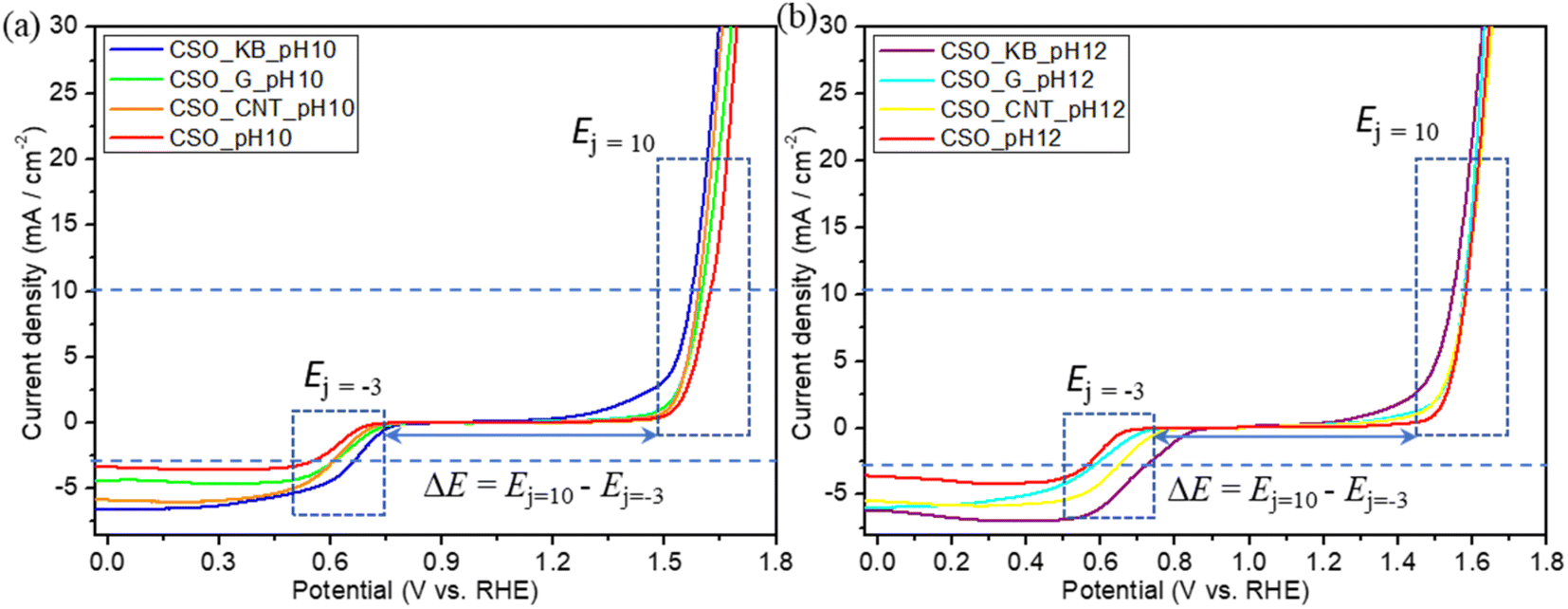 | ||
| Fig. 8 LSV measurement results (ORR–OER) of the samples synthesized at pH = (a) 10 and (b) 12. | ||
| Sample | E j =−3 [V vs. RHE] | E j = 10 [V vs. RHE] | ΔE [V vs. RHE] | Ref. |
|---|---|---|---|---|
| CSO_pH10 | 0.545 | 1.625 | 1.080 | This work |
| CSO_CNT_pH10 | 0.605 | 1.592 | 0.987 | This work |
| CSO_G_pH10 | 0.603 | 1.601 | 0.998 | This work |
| CSO_KB_pH10 | 0.660 | 1.575 | 0.915 | This work |
| CSO_pH12 | 0.554 | 1.582 | 1.028 | This work |
| CSO_CNT_pH12 | 0.641 | 1.580 | 0.939 | This work |
| CSO_G_pH12 | 0.572 | 1.577 | 1.005 | This work |
| CSO_KB_pH12 | 0.713 | 1.548 | 0.835 | This work |
| 20 wt% Ir/C | 0.690 | 1.610 | 0.920 | 67 |
| ZnCo2O4 | 0.700 | 1.640 | 0.940 | 68 |
| P, N Co-doped graphene | 0.845 | 1.550 | 0.705 | 69 |
The enhanced catalytic activity of CSO–carbon composites, especially in the case of CSO_KB_pH12, is attributed to a synergistic interaction between the CSO nanoparticles and the carbon support. This interaction is supported by several observations: first, TEM analysis showed that the CSO nanoparticles were uniformly distributed across the carbon surface without agglomeration, ensuring maximum exposure of active sites and improving electron mobility.70 Second, XPS analysis revealed a shift in the Co 2p and Sn 3d binding energies in the composite compared to pristine CSO, indicating strong interfacial electronic coupling. This charge transfer between the metal hydroxide and the conductive carbon facilitates the adsorption and activation of O2 and OH− intermediates during ORR and OER.71,72 Finally, the superior catalytic performance of CSO_KB_pH12 over other composites and even RuO2 in OER confirmed the presence of a cooperative mechanism between the components.73 Similar synergistic effects in metal hydroxide–carbon systems have been reported in the literature, where interfacial charge redistribution and nanoconfinement significantly improved bifunctional catalytic activity.74
Therefore, the observed performance enhancement arises from not only from the individual properties of CSO or carbon, but from their integrated architecture created by the SPP method.
4. Conclusions
CSO and carbon composite materials for ORR/OER catalysis were successfully synthesized via the single-step SPP method. The characteristics of CSO_carbon were thoroughly analyzed using XRD, FE-SEM, XPS, BET, and TEM. FE-SEM and TEM analyses confirmed the crystalline nature of CSO, which possesses a cubic shape with an approximate diameter of 150 nm. The synthesized CSO_carbon composites exhibited bifunctional catalytic activity for both ORR and OER. Notably, the CSO_KB_pH12 sample demonstrated superior performance, achieving the highest potential at −3 mA cm−2 for ORR, and the lowest overpotential at reaching 10 mA cm−2 for OER. The CSO_KB_pH10 sample also showed commendable performance, with a low overpotential. These findings indicate that the presence of cobalt and perovskite compounds significantly enhances OER performance. The comparison of the composite materials synthesized at pH 12 by SPP and coprecipitation method revealed that the ORR and OER electrocatalytic performance of the composite materials synthesized by SPP was much superior to those synthesized by coprecipitation method. Compared to pure CSO, the CSO_carbon composite enhanced catalytic efficiency by facilitating electron transfer, which is attributed to the support provided by carbon materials to CSO particles.The CSO_KB_pH12 sample exhibited a potential gap (ΔE = Ej = 10 − Ej =−3) of 0.835 V, marking it as the most promising ORR/OER bifunctional catalyst among all samples synthesized in this study. Specifically, CSO_KB_pH12 and CSO_CNT_pH10 showcased outstanding OER catalytic properties. This research paves the way for the development of a new class of non-precious metal electrocatalysts exhibiting exceptional performance for applications in fuel cells, electrochemical water-splitting systems, and metal–air batteries. The application of SPP technology holds the potential to revolutionize the development of advanced electrocatalytic materials.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts fo declare.Acknowledgements
This work was supported by the Strategic International Collaborative Research Program (SICORP) grant number JPMJSC18H1, from the Japan Science and Technology Agency (JST), and Grant-in-Aid for Scientific Research (B) (No. 24K01182) and Grant-in-Aid for Early-Career Scientists (No. 23K13565) from the Japan Society for the Promotion of Science (JSPS).References
- J.-S. Lee, H.-S. Kim and W.-H. Ryu, Appl. Surf. Sci., 2019, 466, 562–567 CrossRef.
- W. Yang, J. Salim, C. Ma, Z. Ma, C. Sun, J. Li, L. Chen and Y. Kim, Electrochem. Commun., 2013, 28, 13–16 CrossRef.
- W. Yang, J. Salim, S. Li, C. Sun, L. Chen, J. B. Goodenough and Y. Kim, J. Mater. Chem., 2012, 22, 18902–18907 RSC.
- M. Rahman, X. Wang and C. Wen, J. Appl. Electrochem., 2014, 44, 5–22 CrossRef.
- M. S. Whittingham, Chem. Rev., 2020, 120, 6328–6330 CrossRef.
- Y. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, ACS Nano, 2017, 11, 11594–11601 CrossRef PubMed.
- J.-S. Lee, G. S. Park, H. I. Lee, S. T. Kim, R. Cao, M. Liu and J. Cho, Nano Lett., 2011, 11, 5362–5366 CrossRef.
- B. Lim, X. Lu, M. Jiang, P. H. Camargo, E. C. Cho, E. P. Lee and Y. Xia, Nano Lett., 2008, 8, 4043–4047 CrossRef PubMed.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef PubMed.
- B. Lim, T. Yu and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 9819–9820 CrossRef PubMed.
- Z. Chen, M. Waje, W. Li and Y. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef PubMed.
- Z. Chen, A. Yu, D. Higgins, H. Li, H. Wang and Z. Chen, Nano Lett., 2012, 12, 1946–1952 CrossRef.
- J. G. Lee, J. Hwang, H. J. Hwang, O. S. Jeon, J. Jang, O. Kwon, Y. Lee, B. Han and Y.-G. Shul, J. Am. Chem. Soc., 2016, 138, 3541–3547 CrossRef PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef.
- T. Jia, Z. Zeng, H. Lin, Y. Duan and P. Ohodnicki, RSC Adv., 2017, 7, 38798–38804 RSC.
- V. Sagar, V. S. R. Atluri, A. Tomitaka, P. Shah, A. Nagasetti, S. Pilakka-Kanthikeel, N. El-Hage, A. McGoron, Y. Takemura and M. Nair, Sci. Rep., 2016, 6, 1–11 CrossRef.
- X. Lin, Y. Gao, M. Jiang, Y. Zhang, Y. Hou, W. Dai, S. Wang and Z. Ding, Appl. Catal., B, 2018, 224, 1009–1016 CrossRef CAS.
- F. Song, K. Schenk and X. L. Hu, Energy Environ. Sci., 2016, 9, 473–477 RSC.
- Y. Bu, G. Nam, S. Kim, K. Choi, Q. Zhong, J. Lee, Y. Qin, J. Cho and G. Kim, Small, 2018, 14, 1802767 CrossRef PubMed.
- C. Sun, F. Li, C. Ma, Y. Wang, Y. Ren, W. Yang, Z. Ma, J. Li, Y. Chen and Y. Kim, J. Mater. Chem. A, 2014, 2, 7188–7196 RSC.
- T. Morishita, T. Ueno, G. Panomsuwan, J. Hieda, A. Yoshida, M. A. Bratescu and N. Saito, Sci. Rep., 2016, 6, 1–13 CrossRef.
- X. Hu, X. Shen, O. Takai and N. Saito, J. Alloys Compd., 2013, 552, 351–355 CrossRef CAS.
- S. Chae, G. Panomsuwan, M. A. Bratescu, K. Teshima and N. Saito, ACS Appl. Nano Mater., 2019, 2, 1350–1355 CrossRef CAS.
- S. Chae, P. Q. Phan, G. Panomsuwan, M. A. Bratescu, T. Hashimoto, K. Teshima and N. Saito, ACS Appl. Nano Mater., 2020, 3, 10183–10189 CrossRef CAS.
- G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972–9981 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, ACS Appl. Mater. Interfaces, 2016, 8, 6962–6971 CrossRef CAS PubMed.
- G. Panomsuwan, N. Saito and T. Ishizaki, Carbon, 2016, 98, 411–420 CrossRef CAS.
- M. Narahara, S. Y. Lee, K. Sasaki, K. Fukushima, K. Tanaka, S. Chae, X. Hu, G. Panomsuwan and T. Ishizaki, Sustainable Energy Fuels, 2023, 7, 2582–2593 RSC.
- P. Q. Phan, S. Chae, P. Pornaroontham, Y. Muta, K. Kim, X. Wang and N. Saito, RSC Adv., 2020, 10, 36627–36635 RSC.
- D. Pradhan, B. K. Satpathy and C. R. Raj, SSRN, 2022, 4147461 Search PubMed.
- C. Dong, X. Zhang, S. Zhang, S. Zhao, X. Lin, X. Wang and F. Huang, Green Energy Environ., 2023, 8, 1417–1428 CrossRef CAS.
- Y. Shu, B. Li, Q. Xu, P. Gu, X. Xiao, F. Liu, L. Yu, H. Pang and X. Hu, Sens. Actuators, B, 2017, 241, 528–533 CrossRef CAS.
- S. Subbarayan, M. Natesan and S.-M. Chen, New J. Chem., 2020, 44, 11271–11281 RSC.
- B. G. S Raj, J. J. Wu, A. M. Asiri and S. Anandan, RSC Adv., 2016, 6, 33361–33368 RSC.
- X. Deng, S. Zhu, F. He, E. Liu, C. He, C. Shi, Q. Li, J. Li, L. Ma and N. Zhao, Electrochim. Acta, 2018, 283, 1269–1276 CrossRef.
- B. Li, P. Gu, G. Zhang, Y. Lu, K. Huang, H. Xue and H. Pang, Small, 2018, 14, 1702184 CrossRef.
- N. Kumar, D. Mishra, S. Y. Kim and S. H. Jin, Mater. Lett., 2020, 262, 127173 CrossRef.
- J. Zhou, Y. Zhao, L. Qin, C. Zeng and W. Xiao, Funct. Mater. Lett., 2016, 9, 1642009 Search PubMed.
- R. Sahoo, A. K. Sasmal, C. Ray, S. Dutta, A. Pal and T. Pal, ACS Appl. Mater. Interfaces, 2016, 8, 17987–17998 CrossRef.
- G. S. Rocha, A. L. Silva, L. P. Silva, F. B. Passos and N. M. Carvalho, Energy Fuels, 2022, 36, 12719–12728 CrossRef.
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, ACS Appl. Energy Mater., 2020, 3, 5439–5447 CrossRef.
- R. M. Ramsundar, V. K. Pillai and P. A. Joy, Phys. Chem. Chem. Phys., 2018, 20, 29452–29461 RSC.
- P. Cui, L. Zhao, Y. Long, L. Dai and C. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218269 CrossRef PubMed.
- J. Shui, M. Wang, F. Du and L. Dai, Sci. Adv., 2015, 1, e1400129 CrossRef.
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, ACS Energy Lett., 2016, 1, 169–174 CrossRef.
- J. Wu, Y. Xue, X. Yan, W. S. Yan, Q. M. Cheng and Y. Xie, Nano Res., 2012, 5, 521–530 CrossRef.
- A. Karczmarska, M. Adamek, S. E. Houbbadi, P. Kowalczyk and M. Laskowska, Crystals, 2022, 12, 584 CrossRef.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- P. Manikanta, R. R. Nikam, S. Sandeep and B. M. Nagaraja, Mater. Chem. Phys., 2023, 306, 128131 CrossRef CAS.
- V. Premkumar, G. Sivakumar, S. Dinesh and S. Barathan, J. Mater. Sci.: Mater. Electron., 2017, 28, 4780–4787 CrossRef CAS.
- H. F. Liu, D. Y. Guo, W. Zhang and R. Cao, J. Mater. Res., 2018, 33, 568–580 CrossRef CAS.
- H. Hu, B. Y. Guan, B. Y. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 6321 CrossRef PubMed.
- Y. P. Zhu, T. Y. Ma, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 1324–1328 CrossRef CAS.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., 2016, 128, 5363–5367 CrossRef.
- H. Liang, F. Meng, M. Caban-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS.
- R. He, M. Li, W. Qiao and L. Feng, Chem. Eng. J., 2021, 423, 130168 CrossRef CAS.
- M. Sadaqat, L. Nisar, F. Hussain, M. N. Ashiq, A. Shah, M. F. Ehsan, M. Najam-Ul-Haq and K. S. Joya, J. Mater. Chem. A, 2019, 7, 26410–26420 RSC.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., 2015, 127, 6349–6352 CrossRef.
- S. Mao, Z. Wen, T. Huang, Y. Hou and J. Chen, Energy Environ. Sci., 2014, 7, 609–617 RSC.
- L. Luo, H. Huang, Y. Yang, S. Gong, Y. Li, Y. Wang, W. Luo and Z. Li, Appl. Surf. Sci., 2022, 575, 151699 CrossRef CAS.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- Z. Wang, Z. Wang, H. Wu and X. W. D. Lou, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- J. O. Abe, A. Popoola, E. Ajenifuja and O. M. Popoola, Int. J. Hydrogen Energy, 2019, 44, 15072–15086 CrossRef.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 RSC.
- B. Wang, C. Jin, S. Shao, Y. Yue, Y. Zhang, S. Wang and X. Li, Green Energy Environ., 2023, 8, 1128–1140 CrossRef.
- Y. Liu, R. Jiang, H. Xiang, Z. Huang and Y. Yu, Electrochim. Acta, 2022, 425, 140746 CrossRef.
- K. Wang, Y. Tan, P. Li and J. Sun, Electrochim. Acta, 2020, 353, 136538 CrossRef.
- O. Boeva, E. Kudinova, I. Vorakso, K. Zhavoronkova and A. Antonov, Int. J. Hydrogen Energy, 2022, 47, 4759–4765 Search PubMed.
- S. Lu, Q. Liu, R. Han, J. Shi, M. Guo, C. Song and D. Ma, Chem. Eng. J., 2021, 409, 128194 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00370a |
| ‡ Present address: Institutes of Innovation for Future Society Designated, Nagoya University, Japan. |
| This journal is © The Royal Society of Chemistry 2025 |