
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From waste to power: utilizing barley husk as a sustainable anode active material alternative to graphite in lithium-ion batteries†
Chenghao
Yue‡
ab,
Alireza
Fereydooni‡
bc,
Puritut
Nakhanivej
 d,
Maria Balart
Murria
d,
Mingrui
Liu
a,
Yuexi
Zeng
a,
Zhijie
Wei
a,
Qiuju
Fu
e,
Xuebo
Zhao
e,
Melanie J.
Loveridge
d and
Yimin
Chao
*ab
d,
Maria Balart
Murria
d,
Mingrui
Liu
a,
Yuexi
Zeng
a,
Zhijie
Wei
a,
Qiuju
Fu
e,
Xuebo
Zhao
e,
Melanie J.
Loveridge
d and
Yimin
Chao
*ab
aNational Energy Key Laboratory for New Hydrogen-Ammonia Energy Technologies, Foshan Xianhu Laboratory, Foshan 528200, P. R. China
bSchool of Chemistry, University of East Anglia, Norwich NR4 7TJ, UK. E-mail: y.chao@uea.ac.uk
cTyndall Center for Climate Change Research, University of East Anglia, Norwich NR4 7TJ, UK
dWarwick Manufacturing Group (WMG), University of Warwick, Coventry CV4 7AL, UK
eShandong Provincial Key Laboratory of Chemistry Energy Storage and Novel Cell Technology, School of Materials Science and Engineering, Qilu University of Technology (Shandong Academy of Science), Jinan 250353, China
First published on 4th June 2025
Abstract
Barley husks (BH), an agricultural by-product rich in carbon and silica, have been utilized as a sustainable precursor for synthesizing carbon anode materials for lithium-ion batteries (LIBs). A comparative analysis with graphite, the conventional anode material, demonstrates the potential of BH-derived carbon as a viable alternative. Material characterization techniques confirmed the formation of a hard carbon structure with a porous morphology, enhanced by the silica content, which contributes to structural stability and performance. The BH-derived anode exhibited a specific capacity of 380 mA h g−1 at C/5, surpassing that of graphite with improved cycling stability and rate capability. BH represent a sustainable and promising alternative for next-generation energy storage technologies, with improved performance and eco-friendly potential.
Sustainability spotlightTo advance UN Sustainable Development Goals related to climate action, affordable, clean, and sustainable energy, this study addresses the critical need for greener alternatives to graphite anodes in lithium-ion batteries, whose production is energy-intensive and harmful to environmental. We present a green and scalable method to produce cost-effective silicon-based anode material derived entirely from barley husk, using a low-energy, chemical minimal process. The material demonstrates electrochemical performance comparable to commercial graphite, with the added benefit of renewable sourcing and reduced environmental footprint. This work advances green chemistry by promoting eco-friendly lithium-ion battery technology, renewable resource use and low-impact manufacturing. |
Introduction
The escalating concerns over global warming, the rapid depletion of fossil fuel reserves, and the pressing need to reduce carbon emissions in the electric power sector have intensified the global focus on renewable energy sources.1–3 However, the intermittent nature of these renewable sources, such as solar and wind, necessitates the development of high-performance energy storage systems. These systems are also a vital component of electric vehicles (EVs) and other electric-powered technologies that play a pivotal role in decarbonization efforts.4,5 Among these energy storage technologies, lithium-ion batteries (LIBs) have emerged as the leading solution due to their high energy density, long cycle life, and eco-friendliness.6,7 Nevertheless, to satisfy the increasing demand for higher capacity and enhanced performance in energy storage, advancements in LIBs technology are essential. Among the various components of LIBs, the anode material holds significant potential for enhancing the overall energy storage capacity.8–10 Consequently, the careful selection and optimization of anode active materials have become critical focal points in the ongoing efforts to improve the energy density and performance of LIBs.Silicon (Si) stands out as one of the most promising anode materials for high performance LIBs, primarily due to its exceptionally high theoretical capacity of around 4200 mA h g−1 for Li22Si5. Its appeal is further enhanced by its abundant availability (the second richest element in the earth's crust after oxygen), low extraction cost, low working potential, and environmentally friendly characteristics.11–14 Despite its promising potential, the use of Si as an anode material in LIBs is not without challenges. One of the primary issues is the significant volume fluctuations (∼400%) that Si undergoes during lithiation–delithiation process, leading to mechanical stress, electrode pulverization, and rapid capacity fading. Furthermore, sluggish ion diffusion kinetics (∼10−14 cm2 s−1) and poor electrical conductivity (∼10−5 S cm−1) of Si can deteriorate battery's performance.14,15 To mitigate these drawbacks, various strategies have been explored, including the incorporation of silicon into composite materials, designing nanostructured silicon, and utilizing oxidized forms of Si, such as SiO2 or silica. While SiO2 is not an active Li storage component in the same way as silicon, its presence can influence the anode material's morphology, porosity, and electrochemical properties. Zhou et al. synthesized a SiO2/Ni nanocomposite by reducing nickel silicate in an inert atmosphere, achieving a capacity of 672 mA h g−1 at 0.1 A g−1 after 50 cycles.16 Ma et al. developed a hierarchically porous multi-shell hollow SiO2 structure using a sacrificial template method with Na2SiO3 as the precursor.17 When utilized as an anode material, these hollow spheres demonstrated a capacity of 750 mA h g−1 after 500 cycles at a current density of 0.1 A g−1, which shows its ability in mitigating the volume expansion of SiO2. Zhang et al. synthesized a plum-pudding nanostructure by embedding SiO2 nanospheres within flake graphite using a hydrothermal process.18 The resulting composite demonstrated a discharge capacity of 702 mA h g−1 after 100 cycles at a current density of 0.1 A g−1, with an impressive coulombic efficiency of 99%. Based on an extensive literature review, it can be concluded that incorporating silica into a carbon matrix not only helps buffer the expansion and contraction of Si but also improves the electrical conductivity of the anode. These combined effects contribute to enhanced structural integrity and cycling stability of the anode.19–21
One of the key drivers behind the development of LIBs is the need for a cost-effective energy storage solution. Introducing high-cost materials, such as certain Si nanocomposites, could potentially undermine this objective. A promising strategy to lower the production costs of SiO2-based materials is to utilize biomass as a precursor, which also presents an opportunity to utilize agricultural waste, reduce environmental impact, and contribute to circular economy initiatives. Depending on the processing methods and the type of biomass used, biomass-derived silicon can achieve high yields, making it a sustainable and economically viable option.14 Notably, several studies have successfully employed biomass materials with silica and carbon content in the anodes of LIBs, such as rice-husk-derived composites,22–25 pinecone,26 fruit-peel,27 and other silica-rich precursors.28–32 Given the variability in agricultural environments and the differing compositions of biomass sources, it is crucial to explore new and regionally abundant biomass resources that naturally contain both silica and carbon.33 While rice husks and some other biomass sources have been widely studied and utilized for similar applications, the investigation of alternative biomass sources, such as BH, offers an opportunity to discover new sustainable materials for anodes.
Barley is the fourth largest cereal crop globally, with an annual production of approximately 150 million tons, including 7 million tons in the UK. The processing of barley, which is primarily used for animal feed (65%) and for malting, brewing, and distilling (30%), generates significant amounts of by-products, including husks. These BH, commonly utilized for animal feed, composting, or energy production through incineration, naturally contain both carbon and silica.34 While BH have been explored as a precursor for SiC-based and graphitic carbon materials in previous studies,35 their potential as a precursor for hard carbon anode materials in LIBs has received less attention.
Our study employs a simplified, energy-efficient approach to directly utilize BH for the production of hard carbon, highlighting their promise as a sustainable and practical alternative. Controlled pyrolysis produced porous hard carbon, with its structure and properties characterized using a variety of characterization techniques. Electrochemical performance, assessed via galvanostatic cycling, cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS), showed a specific capacity of 380 mA h g−1, surpassing graphite with improved cycling stability and rate capability. Unlike other biomass-derived anodes that require additional doping or complex modifications, this study utilizes naturally integrated silica into BH structure, enhancing its electrochemical properties while maintaining a cost-effective and sustainable preparation method.
Experimental
Material preparation
BH were sourced from a local brewery near Norwich City, United Kingdom. The BH were ground and washed with deionized water, then dried at 60 °C overnight. Subsequently, 35 g of the dried BH were placed in ceramic crucibles and heated in a three-zone tube furnace (Carbolite TZF 12/65/550) under a nitrogen gas flow at 1150 °C for 2 hours, with a heating rate of 5 °C min−1. After naturally cooling to room temperature, the resulting black powder (BH-SiO2/C) was mechanically ground with 1/2 inch (12.7 mm) stainless steel balls for 20 minutes using a dual-clamp high-energy ball mill (BM) (Spex Industries Digital Mixer Mill Model 8000D) at a rotating speed of 1400 rpm. The ground mixture was then immersed in 1 M HCl (purchased from Merck) at room temperature for 8 hours. This was followed by vacuum filtration and washing with deionized water until neutralized. Finally, the product was dried at 50 °C overnight, yielding a fine-tuned powder (BM-BH-SiO2/C). For comparison, the original BH-SiO2/C was also manually ground (MG) with a mortar for 5 minutes, subjected to the same acid-washing and drying steps, and the final powder was named MG-BH-SiO2/C, to provide insights into the role of ball milling in achieving a homogeneous composite structure and enhancing electrochemical properties. Additionally, commercial graphite was supplied from Acros Organics and used as active material in anodes for comparison.Materials characterization
The morphological structures of the prepared samples were examined using scanning electron microscopy (SEM, Zeiss Gemini 300 FE) and elemental mapping with energy dispersive X-ray spectroscopy (EDX, Oxford Instruments Ultim Max 170). X-ray diffraction (XRD, Rigaku Smartlab SE) with Cu Kα radiation (λ = 1.540 Å, 40 kV, 50 mA) over a 2θ range from 10° to 80°, and Raman spectroscopy (HORIBA Scientific LabRAM Odyssey Raman spectrometer) with an excitation wavelength of 532 nm were employed to investigate the phase structure of the samples. Chemical states and surface composition were analyzed via X-ray photoelectron spectroscopy (XPS, Thermo Fisher ESCALAB Xi+). Additionally, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) data were obtained using the METTLER-TOLEDO TGA/DSC1 to further investigate the sample contents. N2 adsorption–desorption isotherms were measured at −195.795 °C using a Micromeritics 3Flex 5.02 surface area analyzer to obtain the specific surface area based on the BET model and the pore size distribution using the Barrett–Joyner–Halenda (BJH) method.Electrochemical measurements
Two types of anodes were prepared using different active materials: graphite and BM-BH-SiO2/C. In each case, slurries containing the respective active material, conductive agent (carbon black, C65), and binder (22.5 g per L polyacrylic acid in water solution) in a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 were prepared using deionized water as the solvent. The working anodes were fabricated by casting the prepared slurries with an applicator gap of 200 μm onto copper-foil current collectors, followed by drying in a vacuum oven (Fistreem M Oven VAC1600) at 80 °C for 12 hours. The prepared electrodes were then punched into circular disks with a diameter of 14 mm (1.54 cm2 area and mass loading of 1.04 mg cm−2), and subsequently transferred to an argon-filled glovebox (MBRAUN UNIlab plus ECO with <0.5 ppm of O2 and H2O atmospheric conditions) to be used as anodes in 2016-coin cells. Li metal chips and microporous membranes (Celgard 2500) were used as the counter electrode and separator, respectively. Additionally, 1.2 M LiPF6 in ethylene carbonate/ethyl methyl carbonate (EC:EMC = 1:3 v/v) with 15 wt% fluoroethylene carbonate (FEC) and 3 wt% vinylene carbonate (VC) was used as the electrolyte (purchased from Shenzhen Laborxing Technology Ltd). Following the assembly of the coin cells, they were allowed to rest for 24 hours. After resting, the coin cells were removed from the glove box for testing. Electrochemical performance was evaluated using several methods. Galvanostatic charge–discharge profiles, cycle life, and rate capability tests were conducted with a battery testing system (Land CT3001A) within a voltage range of 0.01–3.0 V (vs. Li+/Li). Additionally, CV was performed at a scan rate of 0.2 mV s−1, and EIS was carried out with a voltage amplitude of 5 mV over a frequency range from 100 kHz to 0.01 Hz, both using a CHI 660E electrochemical workstation. After electrochemical cycling, selected coin cells were disassembled in the argon-filled glovebox. The anodes were carefully removed, and residual electrolyte was gently rinsed off using anhydrous dimethyl carbonate (DMC, Sigma-Aldrich). The electrodes were then dried under inert conditions within the glovebox for 12–24 hours. No thermal treatment was applied to the samples to preserve their surface morphology for subsequent postmortem SEM analysis.
:2:1 were prepared using deionized water as the solvent. The working anodes were fabricated by casting the prepared slurries with an applicator gap of 200 μm onto copper-foil current collectors, followed by drying in a vacuum oven (Fistreem M Oven VAC1600) at 80 °C for 12 hours. The prepared electrodes were then punched into circular disks with a diameter of 14 mm (1.54 cm2 area and mass loading of 1.04 mg cm−2), and subsequently transferred to an argon-filled glovebox (MBRAUN UNIlab plus ECO with <0.5 ppm of O2 and H2O atmospheric conditions) to be used as anodes in 2016-coin cells. Li metal chips and microporous membranes (Celgard 2500) were used as the counter electrode and separator, respectively. Additionally, 1.2 M LiPF6 in ethylene carbonate/ethyl methyl carbonate (EC:EMC = 1:3 v/v) with 15 wt% fluoroethylene carbonate (FEC) and 3 wt% vinylene carbonate (VC) was used as the electrolyte (purchased from Shenzhen Laborxing Technology Ltd). Following the assembly of the coin cells, they were allowed to rest for 24 hours. After resting, the coin cells were removed from the glove box for testing. Electrochemical performance was evaluated using several methods. Galvanostatic charge–discharge profiles, cycle life, and rate capability tests were conducted with a battery testing system (Land CT3001A) within a voltage range of 0.01–3.0 V (vs. Li+/Li). Additionally, CV was performed at a scan rate of 0.2 mV s−1, and EIS was carried out with a voltage amplitude of 5 mV over a frequency range from 100 kHz to 0.01 Hz, both using a CHI 660E electrochemical workstation. After electrochemical cycling, selected coin cells were disassembled in the argon-filled glovebox. The anodes were carefully removed, and residual electrolyte was gently rinsed off using anhydrous dimethyl carbonate (DMC, Sigma-Aldrich). The electrodes were then dried under inert conditions within the glovebox for 12–24 hours. No thermal treatment was applied to the samples to preserve their surface morphology for subsequent postmortem SEM analysis.
Results and discussion
SEM imaging was used to gain insight into the morphology of the obtained BH samples. Fig. S1a (ESI)† shows the SEM image of the BH-SiO2/C sample, and Fig. S1b and c (ESI)† present the corresponding EDX elemental analysis result for four selected points. EDX analysis confirms that the BH samples are primarily composed of carbon with embedded silica spots, evident as bright spots in the SEM image. Additionally, trace amounts of potassium (K), magnesium (Mg), and calcium (Ca) were detected, which is consistent with the natural origin of the sample. The natural structure of the BH is evident from the image, where silica appears randomly incorporated within the porous carbon matrix. Such distribution of silica in the BH sample indicates that further processing, such as ball milling, is required to improve the physical uniformity and the surface area exposure of the silica in the mixture of the sample. Moreover, the presence of impurities such as K, Mg, and Ca, as detected by EDX, underscores the necessity of an acid wash to remove these residual elements. Fig. 1a–e depict the SEM and EDX elemental mappings of the BH-SiO2/C sample after the acid washing stage. Based on the EDX mappings, once again it is evident that the sample primarily consists of silica incorporated into a porous carbon matrix. The EDX mappings in Fig. 1b–e clearly show the distribution of C, O, and Si across the sample, indicate that the acid wash was successful in removing impurities from the sample, as no significant traces of K, Mg, or Ca are detected post-wash, while consistent results were observed across multiple scan areas of the same sample, supporting the uniformity of the purification. Fig. S2a and b (ESI)† show the SEM images of the MG-BH-SiO2/C and BM-BH-SiO2/C samples, respectively. The SEM image in Fig. S2a (ESI)† depicts the manually grounded sample, showing a relatively coarse and uneven structure. In contrast, the SEM image in Fig. S2b (ESI)† illustrates the BM-BH-SiO2/C sample, which displays a more uniform and fine-grained structure. The ball milling process plays a crucial role in reducing the particle size and improving the physical uniformity of silica and carbon. By crushing the mixture, ball milling ensures a more uniform distribution of silica within the carbon matrix, increasing the specific surface area of the composite material, which in turn reduces the ionic diffusion distance and enhances charge transfer kinetics through charge–discharge processes. In general, ball milling facilitates better contact of the sample with the electrolyte, thereby enhancing the electrochemical reactions and overall efficiency of the anode.36–38 | ||
| Fig. 1 (a) SEM image of BH–SiO2/C powder sample after acid wash, (b) corresponding EDX elemental mapping (Si, C, O), (c) carbon mapping, (d) oxygen mapping, and (e) silicon mapping. | ||
The XPS survey spectrum and its high-resolution spectra, illustrated in Fig. 2a–d, reveal the presence of C, O, and Si. High-resolution spectra of C 1s, O 1s and Si 2p (Fig. 2b–d) were deconvoluted using Gaussian functions, showing a primary Si 2p peak at 103.2 eV, attributable to SiO2. The C 1s spectrum displays peaks at 284.8 eV, indicative of graphite-like sp2 hybridized carbon, as well as peaks at 283.65 eV and 286.4 eV, corresponding to C–Si and C–O bonds, respectively.39 These XPS results confirm that silicon is predominantly in the form of SiO2, which is embedded in a carbon matrix. This finding aligns with our SEM/EDX observations, suggesting that silicon is mainly present as oxidized silicon species within the carbon structure.
 | ||
| Fig. 2 (a) XPS survey spectra of BM-BH-SiO2/C and high-resolution spectra together with deconvoluted peaks of (b) C 1s, (c) O 1s, and (d) Si 2p. | ||
XRD measurements have been conducted to investigate crystalline properties and phase structure of BM-BH-SiO2/C sample and reported in Fig. 3a. The XRD analysis reveals a broad peak centered at approximately 23°, which can be attributed to the overlapping peaks of the (002) plane of amorphous carbon and silica. This broad peak correlates with the layer distance in the plane, which is effective for ion insertion. Additionally, a characteristic peak observed around 43° corresponds to the (100) crystal plane of carbon, indicative of the hexagonal lattice structure typically formed by the interaction of sp2 hybridized atoms in the carbon material. This peak is associated with the disordered turbostratic carbon structure.40,41 For reference, the peak positions in crystalline graphite sample (Fig. S3, ESI†) are found at approximately 26.5° and 44.5°, as indexed by JCPDS No. 75-1621. The broad and shifted nature of the peaks in the BM-BH-SiO2/C sample confirms the absence of long-range order and the presence of an amorphous carbon framework.42,43 The interlayer spacing (d002) of the BM-BH-SiO2/C sample was calculated to be 0.373 nm, based on the (002) plane characteristics using Bragg's law (Table S1, ESI†). This value is larger than that of graphite crystals (0.335 nm), suggesting a more disordered structure.44 The crystallite size along the c-axis (Lc) was determined to be approximately 2.87 nm using the Scherrer equation, which estimates crystallite size from the broadening of XRD peaks. This small Lc value highlights the presence of small, disordered crystallites, characteristic of amorphous or hard carbon structures.
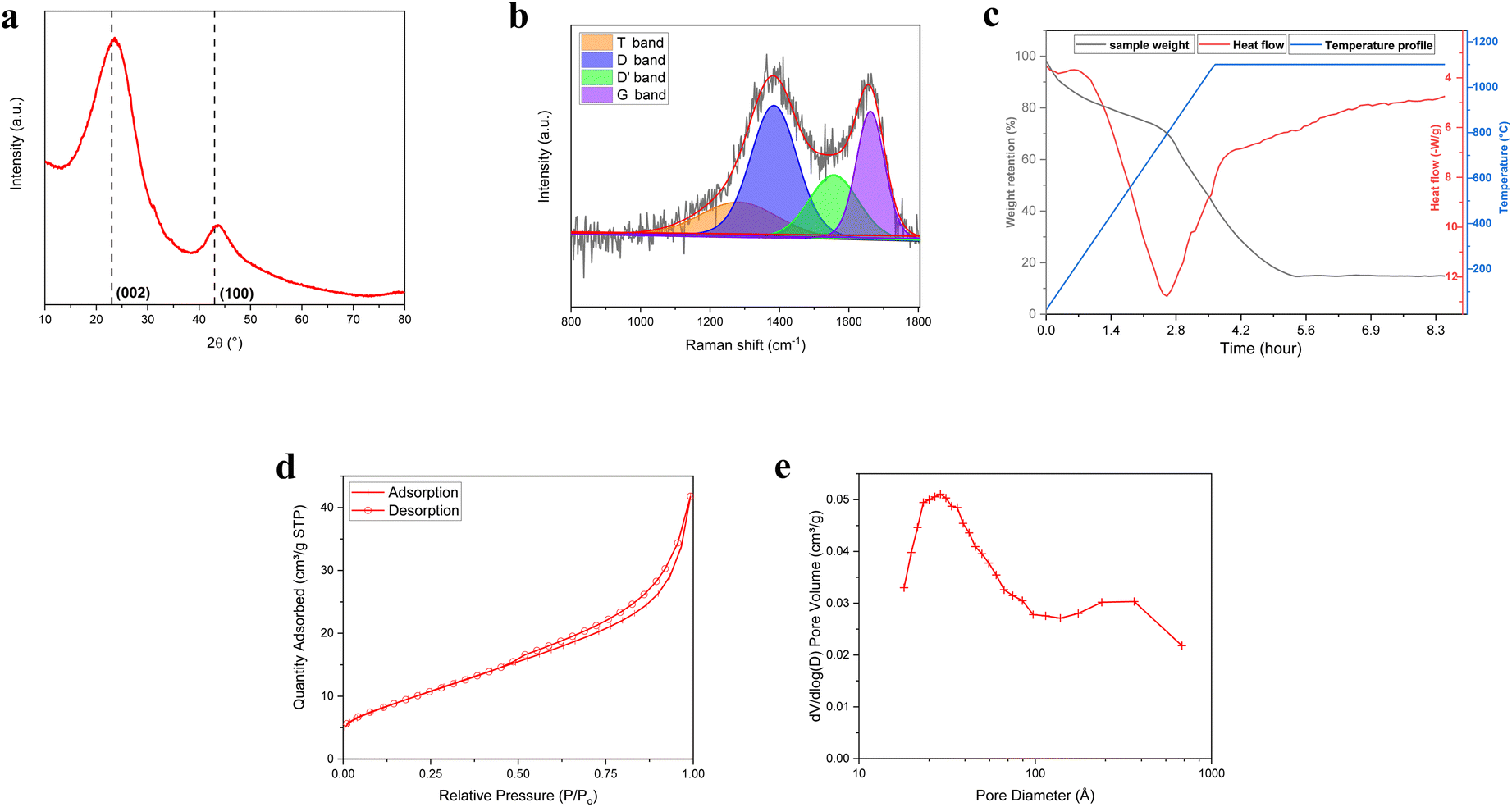 | ||
| Fig. 3 (a) XRD pattern, (b) Raman spectra, (c) TGA/DSC, (d) nitrogen adsorption–desorption isotherms, and (e) pore size distribution of BM-BH-SiO2/C sample. | ||
Using the crystallite size (Lc) with the interlayer spacing (d002), the number of carbon layers along the c-axis direction (N) was calculated to be 7.68 layers (Table S1, ESI†), indicating a limited stacking of the carbon layers. Additionally, the empirical R value, which is the ratio of peak intensity to baseline intensity (Fig. S4 and Table S1, ESI†), was found to be 2.63. This R value serves as an indicator of the relative structural organization of the carbonaceous material.44,45 Values greater than 1, such as the one observed here, suggest a moderate degree of structural organization, which may contribute to enhanced material stability during electrochemical cycling. The detailed XRD results can be found in Table S1 (ESI).†
The Raman scattering, represented in Fig. 3b, was employed to investigate the vibrational modes in BM-BH-SiO2/C sample. The spectrum exhibits two prominent peaks at approximately 1356 cm−1 and 1590 cm−1, corresponding to the disorder band (D-band) and the graphitic band (G-band) of carbon, respectively. The D-band is associated with the breathing modes of sp2 atoms in rings and indicates defects and disorder in the carbon structure, suggesting a significant amount of structural defects or amorphous carbon. The G-band however, is attributed to the in-plane stretching vibration of sp2 carbon atoms in a graphitic structure, indicating regions of ordered graphitic carbon.46–48 The intensity ratio of the D-band to the G-band (ID/IG) is approximately 1.69, indicating a substantial amount of defects and disorder in the sample. This Raman analysis supports the XRD and XPS results, confirming the amorphous and disorder nature of the sample.
The silica content of BM-BH-SiO2/C sample was determined using TGA and DSC analysis. The sample was heated to 1100 °C in synthetic air flow in a 40 μL platinum pan. Fig. 3c shows the heat flow and weight change with temperature. The TGA curve (black line) reveals a significant mass loss starting at ∼200 °C, attributed to the release of volatile components, followed by a steady degradation up to ∼700 °C, primarily due to the decomposition of carbon content into CO and CO2.49,50 A distinct weight loss event after 400 °C indicates the breakdown of more stable carbonaceous compounds. The DSC curve (red line) shows an initial endothermic peak at ∼100 °C, corresponding to the loss of adsorbed water. A sharp endothermic transition between 500 °C and 600 °C represents the energy required for the decomposition reactions. Beyond 700 °C, an accelerated weight decrease suggests further decomposition, potentially related to the oxidation of more resilient carbon structures or volatilization of additional carbonaceous material.51–53 The final residual weight (∼14% at 1100 °C) indicates the presence of inorganic residue, likely silica, as supported by XPS analysis. Additionally, the broad exothermic peak starting at ∼700 °C is attributed to the oxidation of residual carbon.
The N2 adsorption–desorption isotherms and pore size distribution of the BM-BH-SiO2/C composite are shown in Fig. 3d and e. The Type IV isotherm with a hysteresis loop from P/P0 from 0.4 to 1.0 confirms the mesoporous nature of the sample, with an adsorption quantity reaching about 35 cm3 g−1 STP. The pore size distribution reveals peaks around 2 nm, extending up to 50 nm, indicating both mesopores and some macropores. The substantial mesoporosity and high surface area enhance electrolyte penetration and ion diffusion, improving the electrochemical performance of the BM-BH-SiO2/C composite. These BET results align with SEM, EDX, XPS, XRD, and Raman findings, confirming the material's suitability for active material in anodes.
To further analyze the porosity and nanoscale structure of the BM-BH-SiO2/C sample, Small-Angle X-ray Scattering (SAXS) measurements were conducted, providing detailed insight into the microporous and mesoporous characteristics of the material. SAXS is particularly effective for identifying structural heterogeneity and pore size distributions within amorphous and carbonaceous materials, complementing findings from XRD, Raman, and BET analyses.54
The scattering intensity I(q), expressed in arbitrary units per sample mass, was plotted as a function of the scattering vector q, defined as eqn (1):
 | (1) |
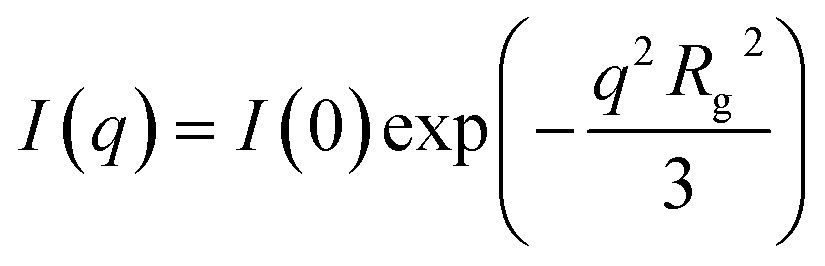 | (2) |
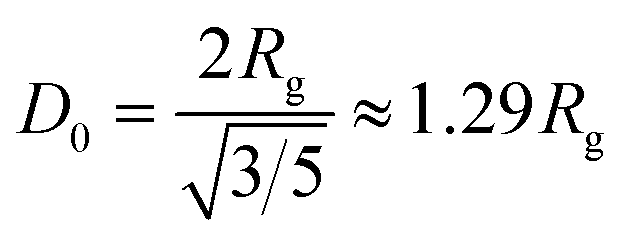 | (3) |
 | ||
| Fig. 4 (a) Log–log plot of scattering intensity I(q) vs. scattering vector q, and (b) Guinier fits showing ln(I(q)) vs. q2 for Rg determination for graphite and BM-BH-SiO2/C. | ||
The analysis revealed notable differences between the graphite and BM-BH-SiO2/C samples. For graphite, a larger Rg of 26.822 Å was obtained, corresponding to a pore diameter D0 of 69.254 Å, indicative of larger, well-ordered scattering domains. This result aligns with the XRD observations of graphite's crystalline structure, which contributes to its characteristic pore architecture. In contrast, the BM-BH-SiO2/C sample exhibited a smaller Rg of 20.079 Å and a calculated D0 of 51.844 Å, consistent with a more disordered and amorphous structure. The smaller pore size in BM-BH-SiO2/C correlates well with the broad XRD peak at 23°, the high D/G ratio in Raman analysis, and the significant mesoporosity observed in BET measurements, reinforcing the characterization of this sample as disordered carbon with fine, interconnected micropores and mesopores. The SAXS intensity profile further supports these observations, showing an overall decay in scattering intensity with increasing q, as seen in Fig. 4a. This trend reflects structural differences between the two samples, with BM-BH-SiO2/C generally exhibiting lower scattering intensity than graphite across the q-range, suggesting smaller, more disordered scattering domains. The Guinier analysis in Fig. 4b provides further quantification, with the BM-BH-SiO2/C sample showing a smaller Rg than graphite, consistent with finer, more disordered microporous structures.
The charge–discharge voltage profiles of the BM-BH-SiO2/C and graphite anodes over the first five cycles, including the initial formation cycles, are presented in Fig. 5a and b, respectively. The graphite anode displays the characteristic flat voltage plateau near 0.1 V during lithiation, typical of Li+ intercalation into a well-ordered layered structure. The profile quickly stabilizes after the first cycle, with minimal voltage hysteresis and consistent capacity, reflecting efficient solid electrolyte interphase (SEI) formation and high reversibility. In contrast, the BM-BH-SiO2/C anode exhibits a sloping voltage profile without a pronounced plateau, indicative of its disordered hard carbon-like structure. Li storage in this material occurs primarily through surface adsorption and filling of nanopores and amorphous domains.56 Over the initial five cycles, the voltage profile gradually stabilizes, with a modest increase in discharge capacity and slightly reduced hysteresis by the third cycle.
 | ||
| Fig. 5 Charge–discharge voltage profiles of (a) BM-BH-SiO2/C and (b) graphite anodes for the first five consecutive cycles at a C/5 current, (c) cycling performance and coulombic efficiency of BM-BH-SiO2/C and graphite anodes over the first 50 cycles at a current of C/5, (d) the rate performance of the BM-BH-SiO2/C and graphite anodes, (e) long-term cycling performance of BM-BH-SiO2/C and graphite anodes over 400 cycles at C/5, (f) CV curves at different cycles for BM-BH-SiO2/C anode, and (g) Nyquist plots of BM-BH-SiO2/C and graphite anodes during the first formation cycle. | ||
The BM-BH-SiO2/C anode achieves a higher specific capacity than graphite, which is attributed to its porous architecture and the intrinsic contribution of embedded silica to additional Li storage and mechanical stability. However, it also shows a greater voltage hysteresis, reflecting the more complex storage mechanisms and possible interactions at the silica–carbon interface. Based on the charge and discharge capacities of the first formation cycle, the initial coulombic efficiency (ICE) of BM-BH-SiO2/C was calculated to be around 60%, consistent with hard carbon materials containing SiO2. For comparison, the graphite anode exhibited an ICE of 89%, in line with its high reversibility and minimal irreversible capacity loss.
The cycling performance and coulombic efficiency of the BM-BH-SiO2/C and graphite anodes over first 50 cycles at a current of C/5 are shown in Fig. 5c. Both electrodes exhibit a rapid capacity drop during the first formation cycle, followed by stabilization and consistent performance. The BM-BH-SiO2/C anode delivers a higher specific capacity of approximately 380 mA h g−1 throughout most of the cycling period, compared to graphite, which stabilizes around 350 mA h g−1. This performance enhancement is attributed to the unique porous structure of the BM-BH-SiO2/C material and the contribution of silica, which enhances Li storage and structural support. The coulombic efficiency for both anodes improves rapidly after the first few cycles, reaching above 98%, indicating the formation of a stable and passivating SEI. These results highlight the BM-BH-SiO2/C anode as a promising sustainable alternative to graphite, offering both enhanced specific capacity and stable cycling performance.
The rate performance of the BM-BH-SiO2/C and graphite anodes under varying C-rates is illustrate in Fig. 5d and demonstrates distinct trends in specific capacity retention at different current densities. At lower C-rates (e.g., C/10 and C/5), both anodes exhibit relatively stable specific capacities, with the BM-BH-SiO2/C anode delivering consistently higher values (∼400 mA h g−1) compared to graphite (∼350 mA h g−1). As the C-rate increases, the specific capacities of both anodes decrease, reflecting the typical limitations in Li-ion diffusion and electronic conductivity under high current densities. Upon returning to the initial C/10 rate, both anodes recover their original capacities, indicating good structural stability and reversible Li storage behaviour. The BM-BH-SiO2/C anode consistently outperforms graphite across all C-rates, underscoring its suitability for applications requiring high power densities and fast charging capabilities while maintaining high energy storage performance.
To evaluate the long-term cycling stability, the capacity retention of BM-BH-SiO2/C and graphite anodes was assessed over 400 cycles at a current density of C/5, as shown in Fig. 5e. The BM-BH-SiO2/C anode exhibited outstanding stability, retaining 97.9%, 96.4%, 93.9%, and 87.9% of its capacity after 100, 200, 300, and 400 cycles, respectively. In comparison, the graphite anode retained 96.6%, 94.4%, 90.9%, and 87.7% at the same cycle intervals. These results highlight the excellent electrochemical durability of the BH-derived anode, which not only maintains a higher specific capacity but also exhibits slightly better retention than graphite over extended cycling. The sustained performance of BM-BH-SiO2/C is attributed to its porous morphology and the structural reinforcement provided by the embedded silica, which contributes to Li storage and mechanical resilience under repeated cycling.
The electrochemical behaviour of the as-prepared BM-BH-SiO2/C anode for LIBs was evaluated through CV to investigate the Li-storage mechanism. Although BM-BH-SiO2/C is classified as hard carbon and structurally distinct from graphite, comparing its electrochemical performance to commercial graphite remains highly relevant. Graphite serves as the industry-standard LIB anode, making it the most appropriate benchmark for evaluating alternative materials. While BM-BH-SiO2/C exhibits a disordered structure with a sloping voltage profile, its capacity, rate performance, and cycling stability must be assessed against graphite to determine its viability for practical applications. This comparison allows for a comprehensive understanding of BM-BH-SiO2/C's potential advantages and limitations. The CV test was conducted over a potential window of 0.01–3.0 V at a scan rate of 0.2 mV s−1, and the results are depicted in Fig. 5f. In the first cathodic sweep (discharge), several distinctive peaks were observed, indicating different electrochemical processes. Cathodic peaks appeared at approximately 1.85 V, 1.17 V, and 0.76 V. The peak at 0.76 V is attributed to the formation of the SEI layer, which occurs primarily during the first cycle. Additionally, the peaks around 1.85 V and 1.17 V are associated with irreversible side reactions between Li and SiO2, forming lithium silicate (Li2Si2O5) and lithium oxide (Li2O), alongside free silicon (Si), according to eqn (4), (6) and (7).57,58 These reactions involve the decomposition of SiO2 during lithiation, and their disappearance in subsequent cycles confirms their irreversible nature. These reactions are only active during the first lithiation, where they contribute to capacity loss in the initial cycle. On the anodic (charge) branches, a prominent peak is observed at around 0.9 V. This peak reflects the partial reversibility of the lithiation and delithiation of silicon, which forms lithium silicide (LixSi), as shown in eqn (5). This process accounts for the reversibility of the material and is a key contributor to the anode's long-term cycling performance. Additionally, a smaller peak at approximately 0.16 V corresponds to the reversible insertion and extraction of Li+ into the carbon matrix of the electrode.59
The findings from the CV analysis align well with the voltage profile observed for the BM-BH-SiO2/C anode. The broad voltage sloping behavior in the discharge profile corresponds to the diverse Li processes identified in the cathodic sweep, including SEI formation and the irreversible reactions of SiO2 with Li to form Li2Si2O5, Li2O, and free Si. Additionally, the anodic peak at 0.9 V, indicative of the reversible lithiation and delithiation of silicon forming LixSi, is consistent with the partial reversibility observed in the charge profile. The smaller anodic peak at 0.16 V reflects the reversible Li storage within the carbon matrix, which contributes to the sloping regions of the voltage profile.
| 5SiO2 + 4Li+ + 4e− ⇌ 2Li2Si2O5 + Si | (4) |
| Si + xLi+ + xe− ⇌ LixSi | (5) |
| SiO2 + 4Li+ + 4e− → 2LiO2 + Si | (6) |
| 2SiO2 + 4Li+ + 4e− → Li4SiO4 + Si | (7) |
The Nyquist plot in Fig. 5g shows that the BM-BH-SiO2/C anode has a smaller semicircle compared to the graphite anode, indicating lower charge transfer resistance (Rct) for BM-BH-SiO2/C. This suggests enhanced charge transfer kinetics at the electrode–electrolyte interface, likely due to the increased surface area from ball milling, which facilitates better contact with the electrolyte. Furthermore, the presence of a double semicircle suggests two distinct impedance contributions from a high-frequency semicircle likely associated with SEI resistance (RSEI) as well.60 Additionally, the reduced intercept with the Z′ axis for BM-BH-SiO2/C suggests lower overall impedance, including bulk resistance, compared to graphite. It is important to note that the ionic diffusion is influenced by multiple factors beyond surface area alone, including tortuosity, pore connectivity, electrolyte wettability, and SEI formation. The mesoporous structure observed in BM-BH-SiO2/C likely facilitates electrolyte penetration, enhancing transport kinetics. Additionally, the presence of oxygen-containing functional groups may improve wettability, further supporting ion transport. These combined properties contribute to the electrochemical performance of BM-BH-SiO2/C as the active material.
Postmortem SEM analysis was performed on both BM-BH-SiO2/C and graphite anodes before cycling and after 10 and 100 cycles at a current density of C/5, as shown in Fig. 6 and S6.† For the BM-BH-SiO2/C anode (Fig. 6a–c and S6a–c†), the fresh electrode surface shows a porous and textured morphology. After 10 cycles, the structure remains largely intact, with the formation of a thin, conformal SEI layer visible on the surface. Notably, even after 100 cycles, the electrode retains its porous network with no evidence of particle cracking, collapse, or delamination, indicating strong structural stability and mechanical integrity over extended cycling. In contrast, the graphite anode (Fig. 6d–f and S6d–f†) initially displays a smooth, flake-like morphology. After 10 cycles, surface smoothing and signs of SEI buildup are observed. By 100 cycles, the graphite surface appears more passivated, with less-defined flake edges and subtle signs of particle boundary smoothing, possibly due to prolonged SEI growth or electrolyte side reactions. However, no major degradation features such as cracking or exfoliation are present.
 | ||
| Fig. 6 Postmortem SEM images of BM-BH-SiO2/C and graphite anodes at different cycling stages under a C/5 current. (a–c) BM-BH-SiO2/C anode before cycling, after 10 cycles, and after 100 cycles, respectively. (d–f) Graphite anode before cycling, after 10 cycles, and after 100 cycles, respectively. | ||
To further investigate the charge storage mechanisms and evaluate the dynamic behavior of BM-BH-SiO2/C and graphite anodes, CV tests were conducted at varying scan rates ranging from 0.2 to 1.0 mV s−1. For BM-BH-SiO2/C (Fig. 7a), the current response with increasing scan rate was analyzed using eqn (8).61,62
| i = avb | (8) |
| log(i) = blog(v) + log(a) | (9) |
 | ||
| Fig. 7 (a) CV curves of the BM-BH-SiO2/C anode at varying scan rates (0.2, 0.5, 0.8, and 1.0 mV s−1), (b) log(i)–log(v) plot for anodic peak currents to determine the b-value, and (c) log(i)–log(v) plot for cathodic peak currents to determine the b-value. | ||
The parameter b, derived from the slope of the log(i) vs. log(v) plot (Fig. 7b and c), quantifies the charge storage mechanism. A b-value of 0.5 indicates a diffusion-controlled process, while a b-value of 1.0 signifies a surface-controlled capacitive process.63 The BM-BH-SiO2/C anode exhibits b-values of approximately 0.71 for anodic peaks (Fig. 7b) and 0.70 for cathodic peaks (Fig. 7c), highlighting a mixed but predominantly pseudocapacitive-controlled mechanism. In contrast, the graphite anode (Fig. S5, ESI,† and Fig. 7b and c) shows b-values closer to 0.5, consistent with a diffusion-controlled Li storage mechanism. These results demonstrate the superior rate capability of BM-BH-SiO2/C, attributed to its porous structure and hybrid silica–carbon composition, which facilitate rapid Li+ transport and surface-controlled charge storage. This contrasts with the slower, diffusion-limited kinetics observed in graphite, underscoring the dynamic advantages of BM-BH-SiO2/C for high-rate applications.
To better contextualize the electrochemical performance of the BM-BH-SiO2/C anode, a summary of the most recently reported Si-based anodes derived from biomass waste is provided in Table 1. The comparison includes studies using silica-rich agricultural byproducts such as rice husks and corn cobs, which have been processed through various chemical or thermal treatments to yield Si/C or SiO2/C composites. While most of these reports focus on rice-derived feedstocks, the current work utilizes barley husk as a rather unexplored biomass source. In addition to its natural silica content, the barley husk-derived composite is processed through a scalable and template-free ball milling route without the use of corrosive chemicals or exotic solvents. Compared to existing systems, the BM-BH-SiO2/C anode demonstrates competitive specific capacity, high coulombic efficiency, and superior cycling stability.
| Biomass source | Composite type and synthesis method | Electrode composition (wt%) | Specific capacity (mA h g−1) | Capacity retention | (Year) and ref. |
|---|---|---|---|---|---|
| Rice husks | Si/C, deep eutectic solvent treatment followed by carbonization | 80:10:10 |
372.5 | ∼86% after 80 cycles | (2023)64 |
| Rice husks | Si/C, acid-leaching followed by high-temperature carbonization | 80:10:10 |
345.4 | ∼89% after 80 cycles | (2023)64 |
| Corn cobs | Si/CCDHC, carbonization and ball milling | 80:5:15 |
∼690 | 87% after 100 cycles | (2022)65 |
| Rice husks | Si/C, alkali extraction, acid precipitation, followed by carbonization, ball milling, Mg reduction, and additive modification | 80:10:10 |
∼600 | 95% after 1000 cycles | (2021)31 |
| Barley husks | SiO2/C, direct pyrolysis and ball milling | 70:20:10 |
∼380 | 87.9% after 400 cycles | This work |
The straightforward process of preparing BM-BH-SiO2/C anode was chosen due to its simplicity, scalability, and alignment with industrial processing techniques for battery anode materials. While alternative synthesis methods such as chemical activation or templating could modify the material's properties, they often introduce additional synthesis complexity, costs, or chemical waste. Therefore, we prioritized a processing route that balances performance with practical feasibility. Future studies will focus on optimizing synthesis parameters, including temperature variations, activation treatments, and milling conditions, to further enhance material performance. To further improve the practical applicability of biomass-derived hard carbon, future work will also focus on prelithiation strategies, electrolyte optimization, and structural modifications to enhance initial coulombic efficiency and long-term cycling performance.
Conclusions
This study demonstrates the successful utilization of barley husks, a widely available agricultural by-product, as a sustainable precursor for the synthesis of hard carbon anode materials for LIBs. The BM-BH-SiO2/C anode exhibited a specific capacity of 380 mA h g−1, along with excellent coulombic efficiency, and long-term cycling stability, retaining 87.9% of its capacity after 400 cycles at a C/5 rate. Comprehensive material characterization revealed the porous hard carbon structure and silica content of BM-BH-SiO2/C, which contributed to its enhanced structural integrity, Li storage capacity, and dynamic electrochemical behavior.Electrochemical analyses, including galvanostatic cycling, CV, and EIS, highlighted the pseudocapacitive nature of BM-BH-SiO2/C, indicating a predominantly surface-controlled charge storage mechanism. The CV results at varying scan rates further validated the superior rate performance of BM-BH-SiO2/C compared to graphite, showcasing its ability to support rapid Li+ transport and reversible charge storage. These findings underscore the feasibility of BM-BH-SiO2/C as a cost-effective, environmentally friendly, and high-performance alternative to conventional graphite for next-generation LIBs. This work not only provides a pathway for the valorization of agricultural waste but also contributes to the advancement of sustainable and scalable energy storage solutions.
Data availability
The original contributions presented in the study are included in the article/ESI.† Further inquiries can be directed to the corresponding authors.Author contributions
Chenghao Yue and Alireza Fereydooni contributed equally to this work: conceptualization, methodology, validation, investigation, writing – original draft, writing – review & editing and formal analysis. Puritut Nakhanivej: investigation, validation, methodology, and formal analysis. Maria Balart Murria: investigation, validation, methodology, and formal analysis. Mingrui Liu: investigation, data curation, and formal analysis. Zhjie Wei: investigation, data curation, and formal analysis. Qiuju Fu: investigation, data curation, and formal analysis. Xuebo Zhao: Conceptualization, resources, and supervision. Melanie Loveridge: conceptualization, resources, and supervision. Yimin Chao: conceptualization, funding acquisition, project administration, resources, validation, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by UEA's Critical Decade for Climate Change Doctoral Training Program, funded by the Leverhulme Trust under their Doctoral Scholarship Scheme. We are also grateful to the funding from Xianhu Lab under the codes XHQD2022-001, XHR2024-001, and XHR2023-003.References
- S. Jenu, I. Deviatkin, A. Hentunen, M. Myllysilta, S. Viik and M. Pihlatie, J. Energy Storage, 2020, 27, 101023 CrossRef.
- F. Degen, M. Winter, D. Bendig and J. Tübke, Nat. Energy, 2023, 8, 1284–1295 Search PubMed.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 Search PubMed.
- N. A. Sepulveda, J. D. Jenkins, A. Edington, D. S. Mallapragada and R. K. Lester, Nat. Energy, 2021, 6, 506–516 CrossRef.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- G. Li, S. Guo, B. Xiang, S. Mei, Y. Zheng, X. Zhang, B. Gao, P. K. Chu and K. Huo, Energy Mater., 2022, 2, 200020 CrossRef CAS.
- Y. Li, A. Vasileiadis, Q. Zhou, Y. Lu, Q. Meng, Y. Li, P. Ombrini, J. Zhao, Z. Chen, Y. Niu, X. Qi, F. Xie, R. van der Jagt, S. Ganapathy, M.-M. Titirici, H. Li, L. Chen, M. Wagemaker and Y.-S. Hu, Nat. Energy, 2024, 9, 134–142 Search PubMed.
- X. Pu, H. Wang, D. Zhao, H. Yang, X. Ai, S. Cao, Z. Chen and Y. Cao, Small, 2019, 15, 32 CrossRef PubMed.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1600943 CrossRef.
- A. V. Baskar, G. Singh, A. M. Ruban, J. M. Davidraj, R. Bahadur, P. Sooriyakumar, P. Kumar, A. Karakoti, J. Yi and A. Vinu, Adv. Funct. Mater., 2023, 33, 2208349 Search PubMed.
- L. Sun, Y. Liu, R. Shao, J. Wu, R. Jiang and Z. Jin, Energy Storage Mater., 2022, 46, 482–502 CrossRef.
- P. Li, G. Zhao, X. Zheng, X. Xu, C. Yao and W. Sun, Energy Storage Mater., 2018, 15, 422–446 CrossRef.
- G. G. Eshetu, H. Zhang, X. Judez, H. Adenusi, M. Armand, S. Passerini and E. Figgemeier, Nat. Commun., 2021, 12, 5459 CrossRef CAS.
- A. Fereydooni, C. Yue and Y. Chao, Small, 2024, 20, 2307275 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- C. Tang, Y. Liu, C. Xu, J. Zhu, X. Wei, L. Zhou, L. He, W. Yang and L. Mai, Adv. Funct. Mater., 2018, 28, 3 Search PubMed.
- X. Ma, Z. Wei, H. Han, X. Wang, K. Cui and L. Yang, Chem. Eng. J., 2017, 323, 252–259 CrossRef CAS.
- H. H. Li, L. L. Zhang, C. Y. Fan, K. Wang, X. L. Wu, H. Z. Sun and J. P. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 22893–22899 RSC.
- M. Yamada, A. Ueda, K. Matsumoto and T. Ohzuku, J. Electrochem. Soc., 2011, 158, A417 CrossRef CAS.
- S. Chen, Y. Xu and H. Du, Dalton Trans., 2022, 51, 11909–11915 RSC.
- J. Ma, H. Tan, H. Liu and Y. Chao, Part. Part. Syst. Charact., 2021, 38, 1–9 CrossRef.
- L. Ma, L. Liu, X. Liu, Y. Li, Y. Feng, Y. Tian, Y. Chao, Y. Zhu and X. Wang, J. Electron. Mater., 2021, 50, 4426–4432 CrossRef CAS.
- Y. Feng, L. Liu, X. Liu, Y. Teng, Y. Li, Y. Guo, Y. Zhu, X. Wang and Y. Chao, Electrochim. Acta, 2020, 359, 136933 CrossRef CAS.
- Y. Li, X. Liu, L. Liu, W. Liu, Y. Feng, Y. Guo, Y. Zhu, J. Wang and X. Wang, J. Electrochem. Soc., 2019, 166, A2425 CrossRef CAS.
- Y. Feng, X. Liu, L. Liu, Z. Zhang, Y. Teng, D. Yu, J. Sui and X. Wang, ChemistrySelect, 2018, 3, 10338–10344 CrossRef CAS.
- T. Zhang, J. Mao, X. Liu, M. Xuan, K. Bi, X. L. Zhang, J. Hu, J. Fan, S. Chen and G. Shao, RSC Adv., 2017, 7, 41504–41511 Search PubMed.
- R. Muruganantham, F. M. Wang and W. R. Liu, Electrochim. Acta, 2022, 424, 140573 Search PubMed.
- Z. Zhu, F. Liang, Z. Zhou, X. Zeng, D. Wang, P. Dong, J. Zhao, S. Sun, Y. Zhang and X. Li, J. Mater. Chem., 2018, 6, 1513–1522 Search PubMed.
- C. d. M. Saavedra Rios, V. Simone, L. Simonin, S. Martinet and C. Dupont, Biomass Bioenergy, 2018, 117, 32–37 CrossRef CAS.
- W. Tian, L. Wang, K. Huo and X. He, J. Power Sources, 2019, 430, 60–66 Search PubMed.
- Y. Li, L. Liu, X. Liu, Y. Feng, B. Xue, L. Yu, L. Ma, Y. Zhu, Y. Chao and X. Wang, Mater. Chem. Phys., 2021, 262, 124331 Search PubMed.
- M. Saavedra, L. Simonin, C. Matei, C. Vaulot, S. Perez and C. Dupont, Fuel Process. Technol., 2022, 231, 107223 CrossRef.
- Z. Gao, Y. Zhang, N. Song and X. Li, Mater. Res. Lett., 2017, 5, 69–88 Search PubMed.
- A. Badea and C. Wijekoon, Benefits of Barley Grain in Animal and Human Diets, in Cereal grains, IntechOpen, 2021, vol. 1, DOI:10.5772/intechopen.97053.
- O. Haluska, S. M. Meščeriakovė, K. Murashko, A. Meščeriakovas, N. Kalidas, J. Rantanen, L. Liu, A. Salami, R. Lappalainen, A. Lähde, V. P. Lehto and J. Riikonen, Mater. Chem. Phys., 2023, 296, 127286 Search PubMed.
- W. Li, J. Huang, L. Feng, L. Cao, Y. Ren, R. Li, Z. Xu, J. Li and C. Yao, J. Alloys Compd., 2017, 716, 210–219 CrossRef CAS.
- D. Alvira, D. Antorán and J. J. Manyà, Chem. Eng. J., 2022, 447, 137468 Search PubMed.
- Y. Feng, L. Liu, X. Liu, Y. Teng, Y. Li, Y. Guo, Y. Zhu, X. Wang and Y. Chao, Electrochim. Acta, 2020, 359, 136933 CrossRef CAS.
- Y. Li, L. Liu, X. Liu, Y. Feng, L. Yu, Z. He, X. Cui, M. Zhang and Y. Zhu, Ionics, 2022, 151–160 CrossRef.
- W. Lee, H. Jang, M. Kim, H. Kim, J. Oh and S. Paek, J. Alloys Compd., 2019, 778, 382–390 CrossRef CAS.
- D. Bhattacharjya, H. Y. Park, M. S. Kim, H. S. Choi, S. N. Inamdar and J. S. Yu, Langmuir, 2014, 30, 318–324 Search PubMed.
- M. K. Rybarczyk, Y. Li, M. Qiao, Y. Hu, M. Titirici and M. Lieder, J. Energy Chem., 2019, 29, 17–22 Search PubMed.
- Y. Li, X. Liu, L. Liu, W. Liu, Y. Feng, Y. Guo, Y. Zhu, J. Wang and X. Wang, J. Electrochem. Soc., 2019, 166, A2425–A2430 Search PubMed.
- D. Cheng, X. Zhou, H. Hu, Z. Li, J. Chen, L. Miao, X. Ye and H. Zhang, Carbon, 2021, 182, 758–769 CrossRef CAS.
- W. Luo, Z. Jian, Z. Xing, W. Wang, C. Bommier, M. M. Lerner and X. Ji, ACS Cent. Sci., 2015, 1, 516–522 CrossRef CAS.
- Y. Feng, X. Liu, L. Liu, Z. Zhang, Y. Teng, D. Yu, J. Sui and X. Wang, ChemistrySelect, 2018, 3, 10338–10344 Search PubMed.
- H. D. Asfaw, R. Gond, A. Kotronia, C. W. Tai and R. Younesi, Sustain. Mater. Technol., 2022, 32, e00407 Search PubMed.
- L. Sbrascini, A. Staffolani, L. Bottoni, H. Darjazi, L. Minnetti, M. Minicucci and F. Nobili, ACS Appl. Mater. Interfaces, 2022, 14(29), 33257–33273 CrossRef CAS.
- R. F. Susanti, S. Alvin and J. Kim, J. Ind. Eng. Chem., 2020, 91, 317–329 Search PubMed.
- A. Jo, B. Lee, B. Guk, H. Lim, J. Tark, S. Yol, J. Kim, S. Hee, H. Jin, G. Lee, K. Baeg, B. Jeong and J. Hwan, Carbon, 2023, 201, 549–560 CrossRef CAS.
- M. lun Jiao, J. Qi, Z. qiang Shi and C. yang Wang, J. Mater. Sci., 2018, 53, 2149–2160 Search PubMed.
- T. Perveen, M. Siddiq, N. Shahzad, R. Ihsan, A. Ahmad and M. I. Shahzad, Renewable Sustainable Energy Rev., 2020, 119, 109549 CrossRef CAS.
- H. Cheng, N. Garcia-Araez and A. L. Hector, Mater. Adv., 2021, 2, 7956–7966 Search PubMed.
- V. Simone, A. Boulineau, A. de Geyer, D. Rouchon, L. Simonin and S. Martinet, J. Energy Chem., 2016, 25, 761–768 CrossRef.
- Carbon Materials for Advanced Technologies, ed. T. D. Burchell, Elsevier, 1999 Search PubMed.
- G. Wang, M. Yu and X. Feng, Chem. Soc. Rev., 2021, 50, 2388–2443 RSC.
- Z. Yang, Y. Du, G. Hou, Y. Ouyang, F. Ding and F. Yuan, Electrochim. Acta, 2020, 239, 135141 Search PubMed.
- W. Wu, M. Wang, J. Wang, C. Wang and Y. Deng, ACS Appl. Energy Mater., 2020, 3, 3884–3892 CrossRef CAS.
- J. Tu, Y. Yuan, P. Zhan, H. Jiao, X. Wang, H. Zhu and S. Jiao, J. Phys. Chem. C, 2014, 118, 7357–7362 CrossRef CAS.
- A. Fotouhi, D. J. Auger, K. Propp, S. Longo and M. Wild, Renew. Sustain. Energy Rev., 2016, 56, 1008–1021 Search PubMed.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 Search PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 Search PubMed.
- H. Yang, R. Xu, Y. Yao, S. Ye, X. Zhou and Y. Yu, Adv. Funct. Mater., 2019, 29, 1809195 CrossRef.
- C. Padwal, H. D. Pham, L. T. M. Hoang, S. Mundree and D. Dubal, Sustain. Mater. Technol., 2023, 35, e00547 CAS.
- L. Sbrascini, A. Staffolani, L. Bottoni, H. Darjazi, L. Minnetti, M. Minicucci and F. Nobili, ACS Appl. Mater. Interfaces, 2022, 14, 33257–33273 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00254k |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |