
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive polymers for cell support: poloxamers as a case study of promise and challenge
Shane Clerkin a,
Krutika Singhb,
Danielle Winningb,
Ivan Krupaa,
John Crean†
a,
Dermot F. Brougham*b and
Jacek K. Wychowaniec*bc
a,
Krutika Singhb,
Danielle Winningb,
Ivan Krupaa,
John Crean†
a,
Dermot F. Brougham*b and
Jacek K. Wychowaniec*bc
aConway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland
bSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: dermot.brougham@ucd.ie
cAO Research Institute Davos, Clavadelerstrasse 8, 7270, Davos, Switzerland. E-mail: jacek.wychowaniec@aofoundation.org
First published on 8th July 2025
Abstract
Thermoresponsive biomaterials have the potential to improve the complexity of in vitro models, to generate dynamically controlled extracellular microenvironments and act as in situ forming drug delivery systems. Due to its known biocompatibility and ease of use, poloxamer 407 (P407), also known as pluronic F127, has attracted significant attention as a component for next-generation cell culture and biomedical applications. P407 display rapid gelation into hydrogels with facile ease-of-handling, and which possess good shear-thinning properties that enable 3D printability with high fidelity. Although P407 has been extensively used as a support matrix for cell proliferation, differentiation and the on-demand release of biomolecules and drugs, significant issues relating to mechanical stability under physiological conditions limit its application. Multiple protocols report the use of P407 ‘hydrogel’ for a variety of applications but often do not emphasise its inherent limitations at the concentrations described. Here we emphasise the disparity between written protocols and what specifically constitutes a hydrogel, showing selected examples from the literature and suggesting clarifications in the language used in describing P407 supports. We describe progress in the field, which is accelerating in part due to development of multi-network hydrogels that include P407 as a stabiliser, for shear-thinning and as a sacrificial component aiding 3D printing. We also contrast P407 to a panel of other promising thermoresponsive systems that have emerged as alternative biomaterials. Finally, we briefly discuss challenges and new opportunities in the field. This includes evaluation of the relative merits of current thermoresponsive polymer systems as they are formulated for use, also by advanced manufacturing, in next-generation 4D-responsive functional hydrogel networks for cell culture automation and as components in responsive-release devices.
1. Introduction
Hydrogels are self-supporting networks of physically entangled or chemically crosslinked polymers that are composed of at least 90% (by weight) of water and are self-supporting solid-like materials with viscous-like characteristics. Thermoreversible polymers are commonly used components of physical hydrogels, currently being explored for a variety of purposes, including in bioprinting1,2 in advanced cell culture3 and as injectable materials for sustained drug delivery in vivo.4 Aqueous temperature-responsive polymer solutions undergo reversible thermogelation at a lower critical solution temperature (LCST), resulting in a phase transition from miscible fluid to viscous hydrogel. This responsive phenomenon arises from a thermally induced entropically-driven conformational shift associated with interacting polymer subunits, resulting in collapse and structural rearrangement of the hydrophobic–hydrophilic groups. A variety of materials undergo such sol–gel transitions close to physiological temperatures (usually ∼25–37 °C), depending on the polymer concentration. Responsive gelation behaviours may enable next-generation cell culture technologies, with minor alterations in temperature close to the LCST providing externally switchable control over the polymer network, including temporal alteration to stiffness profiles and generation of growth factor release gradients between the support material and encapsulated cells.5,6 The physicochemical properties of thermoreversible materials may also be favourable for bioprinting, with thermally induced gelation of cell-laden material occurring in situ either immediately following, or during, extrusion to generate complex three-dimensional (3D) constructs.7–9 Similarly, for in vivo cell or drug delivery purposes, polymer solutions loaded with entrapped therapeutic materials may be utilised to gel in situ upon contact with the body and provide a reservoir for prolonged release of agents.10,11Poloxamers, sold under various registered trademark names including pluronic®, synperonic® or lutrol®, are a family of synthetic thermoreversible copolymers composed of hydrophilic poly(ethylene) oxide (PEO) and hydrophobic poly(propylene) oxide (PPO) blocks of the general ABA chemical formula (PEO)X–(PPO)Y–(PEO)X, Fig. 1A. Depending on block chain length, poloxamers of variable molecular weights with distinct thermogelation profiles may be obtained.12 Among these, poloxamer 407 (P407), also commonly known as pluronic F127, is one of the most extensively studied thermoreversible polymers, Fig. 1B. P407 is unique among the broader family of poloxamers due to its favourable thermogelling behaviour that occurs at physiological temperature in concentrated aqueous solutions. This spontaneous self-assembly under mild temperature conditions has made P407 the current gold standard biomaterial of its kind,13 with a broad range of biomedical applications.14 The amphiphilic properties of P407 render it a model scaffold for encapsulation of a wide variety of hydrophobic and hydrophilic substances including drugs,15,16 cells17 and nanomaterials.7 Additionally, P407 is approved by the food and drug administration (FDA) as a drug delivery excipient and is highly versatile with a wide range of pharmacological routes of administration and formulation types. P407 is currently listed as an inactive ingredient in 16 different FDA-approved ophthalmic, oral, and topical drugs, among others, Fig. 1C and D. A notable pharmaceutical use example among these is Durasite® (InSite Vision, Alameda, California, U. S. A) a patented ophthalmic drug delivery platform that incorporates P407 in its formulation.18,19 In this case, P407, in combination with other polymers, enabled prolonged sustained release of drugs from eye drops, i.e. besivance® (0.6% besifloxacin) and AzaSite® (1% azithromycin).20 Aside from use as excipients in drug delivery formulations, thermosensitive poloxamer hydrogels have also been used for controlled release of gene vectors, potentially enabling spatiotemporal specification of gene therapy delivery. A recent preclinical gene therapy example for cartilage repair included P407 in a formulation applied in an in vivo animal model.21,22 Recent in vitro studies also demonstrated the potential of P407 (and similar temperature sensitive micelle or hydrogel forming polymers) as temperature-triggered drug delivery systems.23–25
 | ||
| Fig. 1 P407: an extensively studied and biocompatible thermoreversible hydrogel. (A) The general chemical structure of poloxamers. The hydrophobic core polymer region (Y) is flanked by two hydrophilic polymer chains of equal length (X). (B) Number of publications that reference poloxamer 407 or pluronic F127 in the title or abstract from 1990–2024 using the PubMed search engine. (C) Distribution of routes by which current FDA-approved drugs containing P407 as an excipient are administered and (D) the formulation type of these drugs. | ||
However, despite their favourable thermogelling properties, P407 hydrogels suffer from significant limitations. In this review we highlight the structural and physicochemical behaviour of P407 and describe how stability under physiological conditions affects both cell culture scaffold and drug delivery applications. We identify literature focused on improving the physicochemical properties of the bulk material using double and interpenetrating networks. We also emphasise the need for improved clarity when describing P407 as a hydrogel and provide a performance comparison with other thermogelling polymers commonly used in biomedical research.
There are two commonly utilised criteria for defining hydrogels. First, from the rheological perspective, hydrogels have storage modulus (G′) higher than their loss modulus (G′′) in the linear viscoelastic region, in which case the materials exhibit solid-like behaviour.26,27 Whilst there is no defined threshold for how much higher G′ should be, it is commonly accepted that the difference should be at least an order of magnitude.28 However some materials will technically not meet this criterion, often depending on the testing conditions, but are commonly regarded as hydrogels. Hence a second, more practical method, known as the the inverted vial test, is also often utilised for P407-based materials.9,29 Hydrogelation is considered to have occurred when a sample does not exhibit flow under gravity, on inverting the tube containing it, over a period as short as 5 min,29 and up to 72 hours.9 We note that adherence to these criteria is dependent on both the concentration of the polymer used, as well as the environmental conditions (including pH, temperature, salt concentrations) all of which should be, but are not always, specified. In more detailed studies phase diagrams are constructed to better understand the gelation behaviour. We recommend that researchers properly evaluate their materials and report their findings, to distinguish true hydrogels from viscous solutions. We return to this point below.
Current challenges for thermoresponsive hydrogels include achieving a balance between printability and mechanical stability, as many systems exhibit excellent shear-thinning behaviour but insufficient structural integrity post-printing under physiological conditions. Additionally, ensuring long-term biocompatibility and minimizing potential cytotoxic effects of degradation products remain critical hurdles for their successful translation into biomedical applications. We anticipate that this review will serve as an up-to-date practical guide, in a rapidly evolving field, to the technical capabilities of thermoresponsive polymers, while also providing clarity on the terminology needed to promote more predictable and reproducible outcomes in their development as biomaterials.
2. Self-assembly and gelation of P407
P407 is an ABA tri-block copolymer with a purified molecular weight of ∼12.6 kDa and an average (PEO)100–(PPO)65–(PEO)100 chemical structure, which is thermoreversible in aqueous solution, Fig. 2A.30 At low temperatures dispersed unimers are stabilised by hydrogen bonding between the hydrophilic PEO blocks and surrounding water molecules. On increasing the temperature above the LCST these weak interactions are destabilised prompting a structural rearrangement of the tri-block, driven by its amphiphilic properties. Micelles form spontaneously, the PPO copolymer blocks form an inner hydrophobic core enveloped by an outer hydrophilic shell of PEO units and the aqueous solution, Fig. 2A. A minimum concentration of amphiphilic molecules, the critical micelle concentration (CMC), is required at the transition temperature, hence the LCST and CMC values are inter-dependent. Further increasing the temperature enhances the stability of the hydrophobic core favouring inter-micelle packing, self-assembly and hydrogel formation, above critical gelation concentration (CGC). | ||
| Fig. 2 P407 micellisation, gelation, and physical properties. (A) Chemical structure of P407 (PEO–PPO–PEO) blocks and micellisation after critical phase transition temperature. Critical amount of polymer monomers above critical micellisation temperature leads to formation of well-defined hydrogels. (B) Thermal analysis results for P407 concentrations ranging from 1 to 35%, performed at a heating rate of 10 °C min−1. (a) Thermograms (offset by fixed factor of 2 for clarity) as a function of temperature. (b) Peak temperatures and their respective enthalpies. The curves show results for triplicate runs. (C) Static SNS measurements at (a) 5 °C and (b) 37 °C for concentrations of 10 to 30% P407. (B) and (C) are reproduced under a Creative Commons Attribution 4.0 International (CC BY 4.0) license from ref. 31. | ||
The favourable thermoreversible behaviour of P407 at physiological temperatures can principally be attributed to the appropriate chain length ratio of PEO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPO subunits. Although the most commonly used poloxamers are all freely soluble in aqueous solution, those with greater fraction of hydrophilic PEO blocks possess more pronounced intermolecular interaction with the surrounding aqueous solution and require higher temperatures to destabilise the copolymer configuration and facilitate micellisation.32 There is a direct relationship between CMC and the number of PEO units within the copolymer and a consequential inverse relationship with the number of hydrophobic PPO units. For example, poloxamer 188 (P188), also known as pluronic F68 (PF68), is composed of 80% PEO hydrophilic residues12,33 and possesses significantly higher sol–gel transition temperatures compared to P407 at comparable w/v concentrations.34,35 Indeed, blends containing various mixtures of P407 and P188 have been shown to modify critical gelation temperatures (CGT) and improve hydrogel characteristics for specific applications.36
:PPO subunits. Although the most commonly used poloxamers are all freely soluble in aqueous solution, those with greater fraction of hydrophilic PEO blocks possess more pronounced intermolecular interaction with the surrounding aqueous solution and require higher temperatures to destabilise the copolymer configuration and facilitate micellisation.32 There is a direct relationship between CMC and the number of PEO units within the copolymer and a consequential inverse relationship with the number of hydrophobic PPO units. For example, poloxamer 188 (P188), also known as pluronic F68 (PF68), is composed of 80% PEO hydrophilic residues12,33 and possesses significantly higher sol–gel transition temperatures compared to P407 at comparable w/v concentrations.34,35 Indeed, blends containing various mixtures of P407 and P188 have been shown to modify critical gelation temperatures (CGT) and improve hydrogel characteristics for specific applications.36
The transition temperature of P407 is strongly dependent on polymer concentration, as noted, but also on the presence of salts, proteins, drugs, or other additives.9,37–40 Consequently micellization, as well as gelation, is affected. These effects arise from changes in hydrophobic interactions and polymer–solvent affinity, which can either promote or hinder micelle formation and network stability, depending on the additive's nature and concentration. In general, hydrophobic molecules tend to partition into the micelle core, potentially disrupting its structure and reducing micelle stability, which can delay or inhibit gelation.9 In contrast, hydrophilic additives may interfere with inter-micellar interactions or hydration shells, weakening network formation, increasing the CGT or reducing gel strength.9 The P407 composition dependence of the transition temperature has been observed calorimetrically, with only concentrations quite close to 35% w/v displaying the endothermic transition at close to the often reported value of 4 °C. At lower concentrations values approaching ∼20 °C are possible, Fig. 2B. The temperature dependence has been studied for several poloxamers and in all cases the particular structure and concentration (often above 10% w/v) affect the transition temperature.31,41 For P407 small angle neutron scattering (SANS) revealed the presence of well-defined micelles at low temperatures of 5 °C, transforming to a para-crystal structure approaching 37 °C, Fig. 2C. The scattering signal was found to be proportional to polymer concentration, and the inter-micelle lattice spacing was found to decrease, indicating higher packing density which was also shown by shifts of the q*, or ‘Bragg’ peaks to higher q-values, Fig. 2C. At higher concentrations P407 forms a face centred cubic packed structure, rendering it a functional hydrogel,31,42,43 and the typically reported CGC values lie in the 15–25% w/v range.38,44,45
Evaluating CGC values for pluronics based hydrogels is inherently challenging, due to the significant variability in reported values for ostensibly similar formulations. Fig. 3A–C show differing phase boundaries and gelation regimes for similar concentration and temperature ranges, reflecting sensitivity to many factors such as polymer source, preparation methods, and measurement conditions.46 Fig. 3D further emphasizes this inconsistency, with CGT values and viscosity minima varying widely even for comparable P407 concentrations (15–25 wt%) as measured in this case by NMR relaxometry. Fig. 3E shows a comparison study from Jalaal and co-workers48 that examined previously published work.40,49–54 The phase diagram reported demonstrates the difficulties in identifying the gelation transition, with shifts in CGT values falling within the 15–25% w/v in the 18–30 °C ranges. In a more recent study, Paxton et al. visually assessed printable formulations of P407 and assigned gelation transitions based on examination of extruded strands.55 Between 20 and 25% w/v increasing printability and shape retention was observed (Fig. 3F), which is a pragmatic view but again the extrusion parameters used are also most likely to affect the outcome.9 Overall, the noted disparities in reporting complicate the extraction of quantitative physical thresholds like the exact CMC or CGC, or for that matter LCST, or any viscoelastic transition points, which are highly context dependent. Nonetheless, the ease of manipulation of P407 hydrogels has rendered it one of the most useful systems studied in cell culture systems, as we will discuss in Section 3.
 | ||
| Fig. 3 Phase diagrams and rheological transitions of common pluronic systems. Phase diagrams for (A) P85, (B) P123 and (C) their 50:50% mixture. Reproduced with permission from ref. 46. Copyright © 2009 Elsevier Ltd. (D) A diagram demonstrating concentration and temperature ranges of “solid-like” Gaussian 1H T2 relaxation of –CH3 protons in PPO blocks (black central circles) and CH2 protons of PEO blocks (black outer coils) of the pluronic. Reproduced Under a Creative Commons Attribution 4.0 International (CC BY 4.0) license from ref. 47. (E) P407 system CGT values obtained in ref. 48, as compared to other studies using a variety of experimental techniques.40,49–53 The shaded region indicates temperatures within which the rheology of P407 is unsteady, and below which they behaved as simple yield-stress fluids. The vertical solid (red) and dashed (yellow) lines indicate the temperature ranges where microstructural analyses suggest that well-ordered micelle lattice forms a stiff hydrogel (from ref. 51 and 54, respectively.). Reproduced from ref. 48. Copyright © 2017 AIP Publishing. (F) (a) Fibre formation and (b) layer stacking results for Nivea Crème and P407 samples, demonstrating that concentrations of P407 below 25 wt% are unsuitable for printing due to their inability to form fibres or maintain shape fidelity on extrusion. Scale bar = 1 mm. Reproduced under Creative Commons Attribution 4.0 International (CC BY 4.0) license from ref. 55. | ||
Due to the micellisation process and formation of a hydrophobic core, P407 micelles can be used as carriers of hydrophobic and biologically instructive molecules.56–58 Specifically at much lower concentrations (0.5–10% w/v), the micelles can be used directly as injectable suspensions with the cargo.57 Morita et al. explained that the high drug loading efficiency commonly observed stems from the long-range attractive forces observed for instance for cyclosporin A encapsulation, as revealed by small angle X-ray scattering (SAXS). Many similar examples over the past two decades have established P407 as a successful carrier for multiple applications, and we will highlight some key aspects below. The micellar transition of P407 directly influences gel homogeneity and injectability by determining how uniformly micelles pack and indeed interact. If micellization occurs too rapidly it can lead to phase separation and inhomogeneous gel networks, compromising mechanical performance and application. Conversely, a controlled and gradual micellar transition, traversed with good homogeneous temperature control, promotes uniform micelle packing, resulting in smoother gelation and better injectability.59 Additionally, delayed or disrupted micellization (e.g. due to additives) may increase injection force requirements or cause gelation to occur prematurely within delivery devices, limiting practical usability.60
3. Applications of thermoreversible P407 hydrogels
3.1. Cell culture applications
Responsive hydrogels represent the next generation of advanced functional biomaterials and a significant progression towards more sophisticated and physiologically relevant cell culture platforms that can more accurately mimic the dynamic and heterogeneous nature of native cellular microenvironments. Cells within human tissues are exposed to distinct extracellular biomechanical cues which manifest as dynamic alterations to the morphological, viscoelastic and tensile properties of the cells themselves.61 Materials that can undergo tuneable modulation of their physicochemical properties may facilitate local control over the biomechanical and biochemical environment to which cells are exposed. Such manipulation permits greater recapitulation of in vivo interfaces and microarchitectures implicated both for normal organ functionality and in disease states. In the following subsections we will discuss approaches to achieving this complexity and the utility of P407 in these.Traditional in vitro cell culture methods have provided some understanding of molecular and cell biology phenomena, including differentiation and morphogenesis. However, given the profound complexity and context-dependence of the in vivo ECM, the limitations of conventional two-dimensional culture methods have become increasingly apparent.72,73 Adherent monocultures grown on rigid plastic or glass surfaces, although convenient, result in unphysiological morphologies and loss of 3D cell–cell and cell-ECM contextual signalling and so usually provide unrepresentative cell behaviour in vitro. Considering current drug development attrition rates, there is a growing need for more authentic and physiologically relevant in vitro models to improve the reproducibility and translatability of basic research.74
Hydrogel-based scaffolds have emerged as effective 3D, biomimetic materials to bridge the gap between traditional cultures and native microenvironments. Hydrogels capture intrinsic features of native ECM and allow cells to adopt realistic morphologies, rather than simply polarised lateral cell–cell junctional associations observed in monolayer cultures. Importantly, the highly porous hydrogel architecture facilitates nutrient, oxygen, soluble factor and waste material permeability to support appropriate cell maintenance and growth.75,76 Additionally, the molecular porosity of the hydrogels may be altered, through polymer and crosslinker concentrations, to influence the viscoelastic properties/mechanical growth environment and so mimic the stiffness profiles of different tissues. Such approaches have provided new insights into cellular mechano-transduction cascades and the influence of the biophysical environment on cellular behaviours, including stem cell differentiation.77,78 Additionally, integrating mechanical tunability into established 3D cellular formats, such as spheroids and organoid technologies, could provide new tools to probe and determine cellular self-organisation and fate.79 Key hydrogel design features to be considered for cell culture and tissue engineering applications are shown in Table 1.
| Design feature | Significance for cell culture and biomedical applications |
|---|---|
| Biocompatibility | • Non-toxic to cells |
| • Can be sterilized | |
| • Non-immunogenic | |
| • Contain cell binding motifs | |
| Porosity | • Sufficient to facilitate appropriate mass transport (ions, nutrients, waste products) |
| • Sufficient to facilitate appropriate cell–cell interactions, proliferation, and migration | |
| Stiffness/viscoelasticity | • Mimic dynamic nature of the ECM |
| • Sufficient mechanical strength under physiological conditions over defined periods of time | |
| • Predictable swelling behaviours | |
| Crosslinking | • Avoid use of cytotoxic crosslinking agents |
| • Gelation should occur under mild conditions of temperature and pH to limit any effect on cell viability | |
| Degradability | • Matching dynamic matrix remodelling over time |
| • Resorbed and degraded into non-toxic monomeric units that can be metabolised by local environment or safely excreted |
Hydrogels for in vivo or clinical use typically must possess all these features to fulfil their application. However, specific property or function targets for different tissue types or target applications, such as stiffness/viscoelasticity, which vary significantly from tissue to tissue,76,80,81 must be specified. With resorbable implants as an example, tissue remodelling is highly dependent on the specific values of each feature/parameter and how they evolve over time. Together these help direct the fate of residential and peripheral cells, and influence systemic immune responses,82 the overall migration and adhesion of cells, and ultimately the remodelling of local ECM microenvironment.76 More importantly, in soft biomaterials the last three features, porosity, stiffness/viscoelasticity and crosslinking, are intrinsically inter-linked.81,83,84
Considering the dynamic nature of biological systems, the amendable physical properties of thermogelling polymers may be exploited as a means to temporally alter the extracellular microenvironment to facilitate controlled release of signalling molecules, which may be particularly useful in modifying the differentiation potential of stem cells over time.90 This type of control over the extracellular environment may also enable improved cell culture complexity for dynamically specifying cellular behaviours, Fig. 4A. Thermoreversible hydrogels have shown significant utility for bioprinting technologies, as support scaffolds for cells both during extrusion, and subsequently in the spatially-organised cell culture.91 P407 has attracted attention as it is commercially available as a consistent synthetic product, with good cell viability,17,44 that can undergo thermogelation at close to physiological temperature and pH, and which has good shear thinning and recoverability properties providing extrudability and good shape fidelity7,92 poloxamers have indeed been utilised in a variety of biofabrication strategies. Examples include bulk encapsulation,38,93,94 bioprinting of cell-laden bioinks,17 and utilisation of P407 as a sacrificial bioink for printing of complex 3D structures88,89,95 (Fig. 4B). However, it remains a significant challenge to combine these properties for bioprinting.96
 | ||
| Fig. 4 Towards next-generational in vitro microenvironments. (A). Opportunities to increase physiological relevance and complexity of in vitro models with poloxamer hydrogels. (i) Cells traditionally cultured on plastic or glass experience junctional cell–cell interactions and adopt unphysiological morphologies. (ii) Cells cultured within 3D hydrogels experience biochemical and biomechanical stimuli either from the matrix itself or from neighbouring cells. (iii) Multicellular organoids cultured within compliant 3D hydrogel scaffolds generate structural organisation and may reveal microenvironment influence on morphogenesis and cell fate determination. (iv) Responsive biomaterials, such as P407, may be used to temporally alter the microenvironment (mimicking the dynamic nature of the in vivo ECM) or to facilitate controlled release. (B). Examples of 3D bio-fabrication modalities possible with poloxamer hydrogels. (i) Traditional bulk encapsulation of cells or organoids within 3D hydrogels can be performed manually (encapsulation lacks defined or physiologically relevant geometries). (ii) Bioprinting of cell + hydrogel ‘inks’ allows definition of precise cell-laden patterns in 3D. (iii) Thermoreversible hydrogels such as P407 may also be utilised as sacrificial inks that may be removed after hydrogel deposition leaving well-defined physiologically relevant 3D patterns.88,89 Created with https://BioRender.com. | ||
3.2. In situ drug and cell delivery applications
P407 has been widely investigated as an injectable drug delivery system due to its favourable gelation properties at body temperature.97 The liquid nature of P407 just below room temperature enables straightforward loading and minimally invasive delivery by injection. P407 has been described in controlled release of cancer therapeutics for which its potential for slow, sustained and targeted release is particularly useful.98 It has also been investigated as a means of improving the release profile of CTLA-4 blockade therapies.99 P407 hydrogels loaded with human IgG antibodies were shown to mediate tumour growth inhibition to an extent comparable with the equivalent oil-based treatment using incomplete freunds adjuvant (IFA), but without cytotoxicity associated with the latter. While these are interesting examples we point the reader to a recent review describing advances in P407 for drug delivery and return to our central theme.1004. Thermoresponsive P407 hydrogels – stability under physiological conditions
A range of thermosensitive hydrogels possessing distinct critical gelation properties may be generated from poloxamers. As noted above, depending on concentration, P407 is capable of sol-to-gel transition at a wide variety of temperatures and with variable gelation times to produce a broad range of gel viscosities.13,31,41 P407 has been investigated both as a single component network former and as a component in double-network scaffolds for maintaining and inducing cell proliferation and differentiation. Although P407 possesses advantageous thermogelling properties and its applications are extensive, significant discrepancies are present in the literature. Some groups report the successful maintenance of cells over periods of days to weeks encapsulated within P407. Conversely, other groups point to the rapid dissolution of P407 under physiological conditions,101 with dissolution of 25% w/v P407 hydrogels reported within even 4 hours.102,103 Others conclude that the high permeability of these materials render the unmodified P407 structurally unstable, as compared to P407-dimethacrylate.104In one of the first reports describing cellular encapsulation within P407, HepG2 (human liver carcinoma), HMEC-1 (endothelial) and L6 (muscle) cells were grown in P407 of concentrations ranging from 15 to 20% (w/w) (17.6–25% w/v).38 Viability studies over 5 days of culture revealed complete cell death by the last day 5 for all studied lines. Higher cellular viability was only retained when membrane-stabilizing agents (hydrocortisone, glucose, and glycerol) were added. Similarly, Fedorovich et al. reported low cell survival of multipotent stromal cells seeded both directly on top of and encapsulated within P407.93 After 3 days of encapsulation cell survival was significantly reduced in this case, even in the presence of hydrocortisone and glycerol. However, it is unclear how 3D growth was retained over the quoted experimental timeline as the authors describe P407 dissolution within minutes in the presence of culture medium.
On the other hand Vashi et al. reported viability and successful differentiation of bone marrow derived mesenchymal stem cells (BM-MSCs) into adipocytes when encapsulated within 20% w/v P407.94 The authors suggest stability of the scaffold was maintained in culture over a 14-day period, facilitating 3D growth and adipogenic differentiation of the BM-MSCs. Similarly, Diniz et al. reported the culture of dental pulp derived stem cells, and control BM-MSCs in 20% w/v P407, demonstrating high stem cell viability (>90% viability) and proliferation following 7 days in culture using two separate viability assays.105 While the authors acknowledge an 85% degradation of the hydrogel in culture over the 14-day period, complete dissolution of P407 at similar concentrations has been reported in the literature. Gioffredi et al., for example, prepared P407 hydrogels ranging from 16–40% w/v, from initial formulations in different solvents, and characterised their stability using rheological measurements including flow curves, frequency sweeps and viscosity recovery.44 Interestingly, although the original solvent used had minimal influence on P407 LCST, none of the final hydrogels were sufficiently stable to facilitate subsequent cell viability studies. In the same work, although mouse embryonic fibroblast cells encapsulated within 25% w/v P407 (dissolved in medium) were reported to be viable immediately following extrusion through a 200 μm print head nozzle, long-term cell culture studies were not possible as hydrogel dissolution was reported within minutes in the presence of culture medium.44 It is also worth mentioning that other effects of cellular exposure on the matrix include rapid internalisation of P407 micelles into HeLa and U87 cancer cells.106
Issues related to long-term stability of P407 have also been reported when used as a platform for drug delivery. A comparison between injectable formulations of pluronic P407 and a poly(ethylene glycol)-β-polycaprolactone diblock copolymer (HC) in the delivery of 5-fluorouracil (Fu) and doxorubicin (DOX) demonstrated the excellent release properties of P407 but also highlighted issues with its long-term stability.107 Both formulations were free-flowing at room temperature and both were described as almost immediately gelling at 37 °C, despite the viscosity of the P407 formulation at this temperature being ∼60 times greater. Whilst the overall release of Fu and DOX was greater for pluronics based injectables, the long-term stability at 37 °C, monitored using viscosity measurements rapidly decreased after ∼15 hours, apparently due to weak association of the hydrophobic PPO segments. The PEG-based HC formulation, with which the comparison was made, preserved its structure over the 50-hour time-period investigated, enabling more sustained release of Fu and DOX. More sustained release, compared to pure P407, has also been noted for a composite hydrogel block copolymer formed from poly(ethylene glycol)–poly(εcaprolactone)–poly(ethylene glycol) (PEG-PCL-PEG, or PECE) and P407.108 The composition of PECE:P407 (100:0, 80:20, 60:40, 40:60, 20:80 and 0:100% w/w were used) was found to adjust in vitro release at 37 °C of vitamin B12, honokiol and bovine serum albumin, with higher P407 content composites demonstrating rapid release with little to no control. Scanning electron microscopy images obtained before and after drug release showed cracks in the fast-degrading P407 hydrogel structure, suggesting degradation at the molecular level caused the uncontrolled release. Additionally, cytotoxicity studies for PECE showed similar human HEK293 cell viability to P407 at all compositions, making it a promising alternative.
Pluronic-based nanocarriers also have the ability to coat amphiphilic molecules through hydrophobic interactions, enabling delivery of siRNA molecules and chemotherapeutic drugs.109 However, due to the physical nature of the association, such carriers provide little control over loading and release. More recently, siRNA molecules have been covalently bound to the surface of the pre-formed pluronic 108 micelles using redox-sensitive linkers, enabling extremely efficient loading and improved control over release for the purpose of transfecting cells.110 The micelles were found to mediate tissue factor silencing and so increase the survival of MSCs for prospective regenerative applications. Poor structural integrity of P407 under physiological conditions, an issue for bulk hydrogels, can also be problematic for injectable micellar-drug complexes.
In multiple reports, including several that feature in this article, the term ‘hydrogel’ is used, when the P407 concentration applied was below the nominal CGC of ∼20% w/v. While in most cases the cells are at least seeded into gels, that undoubtedly changes over time. So many descriptions of successful encapsulation of cells in 3D culture over the course of days to weeks more likely reflect cell viability in developing bio-broths. Researchers should accurately document their observations, to avoid misunderstanding in cell culture handling protocols (which may include, for instance, multiple media changes) to enable attempts to reproduce outcomes. For extended time-course studies, it is not sufficient to simply define the original unconditioned material as a ‘hydrogel’. The biomaterial state over time and conditioning should be evaluated (rheological characterisation, or inverted vial tests as a minimum) for full characterisation and to enable reproducibility. For example, recently rheological amplitude testing was performed over extended culture time/media changes for kidney organoid specification. When the hydrogel support matrix was formed using peptide hydrogels, it was found that depending on the β-sheet peptide sequence the retention of G′ varied significantly over 15 days.111 The time-dependent changes in the physical properties of the different matrices had significant impact in these cases on cell lineage and maturation. On the other hand, chemically crosslinked matrices, such as gelatin methacrylate (GelMA), offer unchanging stiffness of the overall construct over organoid culturing times, providing the opportunity to study maturation as a function of concentration and hence storage modulus.112 In the next section we illustrate the necessity for improvements in experimental design and documentation for the potential of P407 as a platform for controlled delivery and cell and tissue engineering to be realised.
5. Approaches to improving the stability of P407
In this section we briefly describe the many strategies have been proposed to overcome instability issues associated with P407. We also include other, previously not categorised, approaches such as chemical stabilisation of the PEO blocks within micelles. A selection of approaches for generation of double network and interpenetrating hydrogels networks, incorporating P407, for cell culture applications is also provided, and the advantages of additive manufacturing enabled by P407 are described.5.1. Modification of components and double-network hydrogels
The poor mechanical stability of P407 in aqueous environments renders it unsuitable for long-term in vitro and in vivo studies, limiting translational applications of the advantageous temperature-responsiveness. This is principally because micelles are inherently susceptible to dissociation when encountering physiological environments with complex and variable pH and salt concentrations, proteins, lipids and other biomolecules and cells. A significant decrease in concentration to below the CMC, e.g. in culture, results in rapid micelle dissolution and uncontrolled release of encapsulated drugs. Modifications have been introduced both to improve the physicochemical properties and to generate more favourable biological characteristics. These include chemical alterations to the P407 backbone113–117 and integration of other hydrophilic polymers.118–121Composite hydrogels often possess superior biological and/or physical properties. By compositing P407 with other, typically hydrophilic, biocompatible polymers by either physical mixing118,120 or covalent attachment,121,122 multiple thermoreversible composites have been prepared, often with superior characteristics.123 Physical mixing is denoted here with a ‘/’ between the mixed components, while covalent linkages are denoted with a ‘-’. A wide variety of hydrophilic polymers have been used to modify the thermoresponse, to enhance the mechanical properties, to improve cell viability and to provide improved drug release profiles.124 Any covalent modification made to P407 renders the polymer a novel excipient, in which case application125 would require extensive toxicity and safety screens which are usually not described. Hence physical mixtures, each of which is known to be biocompatible/well-tolerated in humans, may be advantageous. In fact, this is the most common approach, perhaps also because it is less synthetically challenging. However, one can anticipate a limit to the stability possible for ‘physical’ gels, so ‘chemical’ gels probably retain some advantages.
A great many strategies have been employed to improve the inherent properties of P407. To save space and facilitate comparisons a selection of the most promising examples of P407 composites are listed in Table 2, some of these are discussed in the subsequent sections. In most cases P407 is the major component, in the 15–25% w/v range, and gelation is driven by physical interactions between micelles. Improvements to the physicochemical properties of the hydrogel are highlighted in addition to any demonstrated improvements for biological or drug delivery outcomes.
| Species composited with P407 [ref.]a | Reported matrix characteristics |
|---|---|
| a Physical mixing is denoted with a ‘/’ between components, covalent linkages are denoted with a ‘-‘. | |
| P407/collagen type 194 | Adipogenic permissiveness of BM-MSC in this 3D environment. |
| Hyaluronic acid/P407-Heparin126 | Improved mechanical strength with good resistance to rapid dissolution in media. |
| Release of TGF-β1 induced chondrogenic differentiation of human adipose-derived stem cells (ASCs). | |
| Implanted scaffold in rabbit showed ASC differentiation into chondrogenic cells and healing of cartilage injury. | |
| Hyaluronic acid/P407 (HA/P407)118 | Critical gelation concentration at 37 °C was 13 wt% for HA/P407 composite vs. 17 wt% for P407 control. 1.4-fold higher maximum viscous modulus in vs. P407 control. |
| No in vitro cellular cytotoxicity studies were performed. | |
| Articular cavity injection of piroxicam (PX) hydrogels into male beagle dogs. PX-loaded P407 had a cumulative drug release in vitro of ∼80% by day 10 vs. ∼50% for HA/P407 hydrogel. | |
| T1/2 of PX-loaded HP in vivo was 86 hours vs. 24 hours for commercial PX formulation. | |
| HA-P407*122 | No in vitro cellular cytotoxicity studies were performed. |
| *By catechol–thiol reaction of thiol-terminated P407 with HA-dopamine. | CGC of HA-P407 was substantially lower at ∼7 wt% than P407 control. |
| 16 wt% HA-P407; remained stable in culture for >30 days, with slight swelling and retained at site of injection for 21 days (78.4 wt% dry). For P407 control; complete dissolution by day 8 and no hydrogel remained at injection site after 1 day in vivo. | |
| HA-P407 | No in vitro cellular cytotoxicity, or in vivo studies performed. |
| Gelatin-P407127 | HA-P407 hydrogels at a 1:80 (v/v) had greater stability and elastic modulus (1–5 fold vs. P407), very high CGT at high HA concentration (phase separation). |
| Elastic modulus minimally affected by addition of glucose and NaCl (isotonic conditions), but CGT decreased at higher concentrations (salting-out). | |
| P407/N,N,N-trimethyl chitosan (TMC)/Polyethylene glycolated hyaluronic acid (PEG-HA*, formed by carbodiimide chemistry)119 | No in vitro cellular cytotoxicity, or in vivo studies performed. |
| P407, TMC and PEG-HA loaded with gallic acid at a drug:polymer ratio of 1:9 (w/w) improved composite mechanical stability (rheology). |
|
| In vitro cumulative drug release was higher after 5 days for the P407/N,N,N-trimethyl chitosan (TMC)/polyethylene glycolated hyaluronic acid (PEG-HA) (88%±1) vs. P407 (75%±1). |
|
| Chitosan (CS)/P407 | Addition of 1% w/v chitosan doubled gelation time at 20% P407. |
| Chitosan (CS)-P407*120 | Viability of seeded chondrocytes was unchanged at >100% by day 7. |
| *Crosslinked with sodium tripolyphosphate (TPP) | CS-P407 showed 50% reduced release of dexamethasone as compared to P407. |
| Near IR signal, observed at the injection site in mice for up to 35 days for CS-P407, was lost by day 14 for P407, indicating extended implant survival. | |
| (Chitosan) CS-P407* | The gelation time of a 7.5% w/v CS-P407 at 37 °C was noted within 2.5 minutes. |
| CS-P407/PLGA1128 | |
| *Composite formed by carbodiimide/N-hydroxysuccinimide chemistry. | Complete dissolution of CS-P407/PLGA and CS-P407 was noted after 18 and 14 days in culture as compared to 4 days for CS/P407. |
| 1PLGA nanoparticles were loaded with the model vaccine antigen ovalbumin (OVA) and adjuvants monophosphoryl lipid A (MPL) and quil A (QA). | CS-P407/PLGA soluble vaccine or PLGA nanoparticle vaccine induced cellular and humoral responses lasting up to 49 days post injection. |
| P407/Alginate* (P407/Alg)121 | Tissue from infected wounds in animals treated with GSNO-P407/Alg, histologically resembled normal dermal tissue on day 11, unlike untreated, GSNO alone and PL/AL. |
| *S-nitrosoglutathione (GSNO) was synthesised and incorporated by mixing into the composite as a source of NO. | Wound area on day 11 was measured as 6 ± 2% for GSNO-P407/Alg treated animals vs. 33 ± 4% and 34 ± 8% for GSNO and P407/Alg. |
| P407/Alg*129 | 0.7% w/w alginate and 14% w/w P407 were required for gelation above RT. |
| *Pilocarpine was added to the polymer solutions by mixing | 0.1% w/w alginate and 14% w/w P407 administered ocularly to rabbits extended pharmacological response to pilocarpine vs. P407 control. |
| P407/Carboxymethyl hexanoyl chitosan (P407/CA) | CGT and elastic modulus decreased with addition of CA. |
| P407/CA-glutaraldehyde (GA) * (P407/CA-GA)130 | P407/CA/GA-encapsulated L929 cells remained alive during the 5-day incubation. |
| *Glutaraldehyde was also added to the mixture | |
| P407/d-alpha tocopheryl polyethyene glycol 1000 succinate/Docetaxel nanocrystals loaded (P407/TPGS/DOC-NCs)131 | Increasing elastic modulus and CGT observed with increasing TPGS concentration. |
| P407/TPGS/DOC-NCs showed increased tumour growth rate inhibition, compared with intravenous (5-fold) and intratumorally injected DOC (1.8-fold) solution. | |
| P407-fibrinogen | CGT reduced with addition of fibrinogen. PEGDA nanocomposites degraded faster than P407-fibrinogen |
| PEG-diacrylate (PEGDA)-fibrinogen was used as a control132 | The susceptibility of this P407-fibrinogen to protease degradation utilizing dermal fibroblasts and consequent cell-mediated remodelling was controlled by the pluronic®F127 constituent. |
| P407-fibrinogen supported cell spreading, chemical conjugation did not interfere with fibrinogen promoted cell adhesion or cell-mediated proteolysis. | |
| P407/Polycaprolactone (PCL)/porcine acellular bladder submucosa matrix (BSM) (P407/PCL/BSM)133 | P407/PCL/BSM was generated for cell-mediated analysis of upper-urinary-tract-derived urine stem cells (uUSCs). Tumorigenicity of the P407/PCL/BSM was evaluated by implantation into kidney subcapsular space in ICR mice for 8 weeks. |
| Elastic modulus and tensile strength of P407/PCL/BSM decreased with increasing BSM. Enhanced cell adhesion was noted in P407/PCL/BSM as compared to P407/PCL control. | |
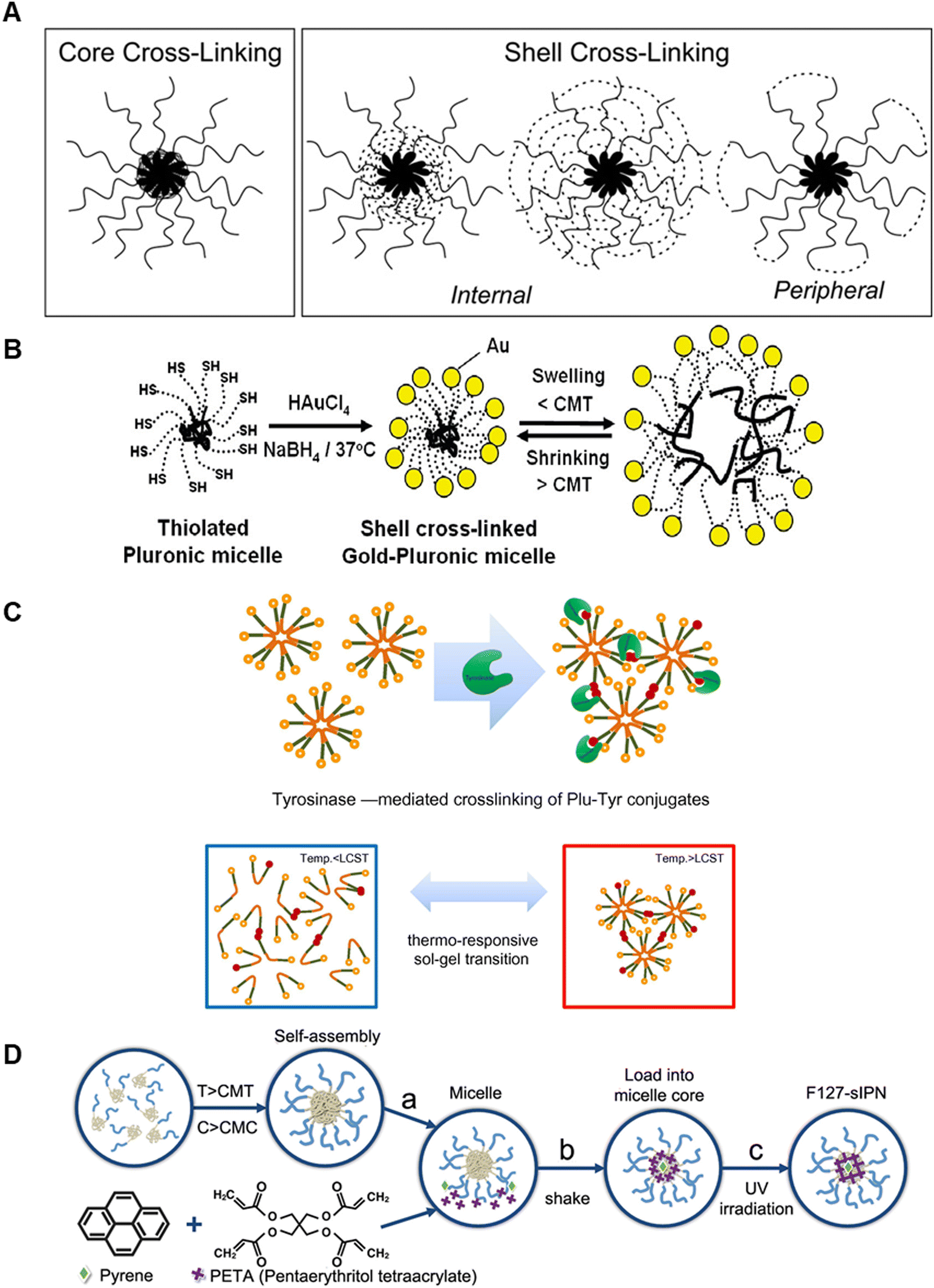 | ||
| Fig. 5 Schematics of P407 stabilisation by inter-micelle covalent modifications. (A) Core and shell (internal and peripheral) crosslinking for enhancing micelle stabilisation. Reproduced with permission from ref. 116 Copyright © 2014, Springer Science Business Media New York (B) Au nanoparticle crosslinking onto thiolated micelle shells for covalent stabilisation. Reproduced with permission from ref. 113. Copyright © 2006, American Chemical Society (C) enzyme-mediated covalent crosslinking of tyramine to the tri-block termini with augmented micelle bridging above LCST. Reproduced with permission from ref. 117. Copyright © 2010 Acta Materialia Inc. Published by Elsevier Ltd. (D) Covalent crosslinking of the hydrophobic core: (a) micelle formation; (b) loading of PETA and micelle into the core; and (c) final UV photo-crosslinking of the core. Reproduced with permission from ref. 134. Copyright © 2018, AIP Publishing. | ||
Lippens et al., also modified the PEO hydroxyl end groups of the copolymer to reduce its degradation and generate an appropriate 3D scaffold for supporting MC3T3-E1 osteoblastic cells.114 Building on work first reported by Swennen et al.,115 the hydroxyl end groups were converted to α-bromo esters and the modified tri-block was coupled to N-methacryloyl-alanine to incorporate a UV cross-linkable depsipeptide unit composed of L-lactic acid and alanine into the end-product (Plu ALA-L). UV exposure (λ = 365 nm) in the presence of Irgacure-2959 induced radical polymerisation of the network. The degradation rate in PBS could be altered through the Plu ALA-A content. In all conditions, following 15 days of culture, only ∼10% of the 20, 25 and 30% w/v hydrogels had degraded. It should be noted that the incubation temperature, ideally 37 °C, was not specified. Subsequent viability studies demonstrated a dramatic effect of chemical stabilisation on MC3T3-E1 viability, which was >80% at day 5 of culture for cells encapsulated in both 10 and 15% w/v Plu ALA-L crosslinked hydrogels, as compared to ∼20% for P407 control at that time.
More recently Wakaskar et al. used ethylenediamine to peripherally crosslink P407 micelles to investigate the effect on the release profile and anti-tumorigenic potential of an encapsulated hydrophobic drug, combretastatin A4 (CA4).116 Hydrogels containing micelles with 76% PEO peripheral crosslinking increased both drug blood residence time and drug efficacy against primary murine breast tumours, following IV administration. Peripheral crosslinking was shown to increase tumour growth inhibition; 5-days post injection tumour volume decreased to 471 ± 54 mm3 for peripherally crosslinked P407, compared to 710 ± 71 mm3 for non-crosslinked P407. Peripheral shell crosslinking may be a straightforward approach to improve P407 stability and release profiles over extended periods.
Lee et al. used a similar strategy to produce a P407-based hydrogel with enhanced mechanical stability by covalently modifying the PEO block with tyramine to, again, strengthen inter-micelle interactions, Fig. 5C.117 Conjugation of tyramine to the terminal ends of the tri-block (P407-Tyr) in the presence of tyrosinase was shown to induce enzymatic oxidation that crosslinked P407-Tyr residues, generating covalent inter-micelle contacts. 10% w/w P407-Tyr hydrogels treated with tyrosinase demonstrated enhanced rheological properties compared to P407-Tyr alone. When measured at 37 °C with frequency sweep at 1 Hz, the elastic modulus (G′) of the enzyme-crosslinked hydrogels was 14.5 times greater. Importantly, the rapid and reversible sol–gel and gel–sol properties of P407 were retained following crosslinking and the hydrogel could undergo rapid (<5 s) transitions on temperature cycling between 4 and 37 °C. An in vitro dissolution test further highlighted improved structural stability of the enzymatically crosslinked hydrogel. 30% enzymatically crosslinked P407-Tyr hydrogel remained intact after almost 2 weeks at 37 °C in PBS solution, while control 20% w/w P407 gels demonstrated complete dissolution within 3 days. Due to their slower dissolution, these gels also demonstrated superior sustained release of FITC-labelled dextran over the period studied. We also note that P407-Tyr could in principle be crosslinked by other approaches, for instance by horseradish peroxidase in the presence of H2O2, an established approach for stabilising biopolymers135,136 and enabling cell tethering to intrinsically non-cell binding materials.137
Crosslinking of the inner micelle core has also been investigated, Fig. 5D.134,138 One approach was to use pyrene and pentaerythritol tetraacrylate (PETA), in the presence of UV light, above the CGC, to generate the core crosslinks, which was found to stabilise the gels to lower temperature. The viscoelastic response was also altered, with slow and fast viscoelastic relaxation becoming apparent. These were shown to affect cellular behaviour in different ways.139 Core crosslinking is an under-developed approach; it may reduce changes in hydrophilicity apparent on PEO modification (which may have the advantages140 of retaining the native LCST while improving stability). Further studies, particularly into the effects of core modification on viscoelasticity, long-term stability in culture and into any effects on cellular behaviour are required.
5.1.2.1. ‘Physical’ composite P407 hydrogels. Simple physical mixing of P407 (at ∼20% w/v, so micellisation drives gelation) with high-molecular weight hyaluronic acid (HA, Mw ∼1000 kDa, at ∼1% w/v) was described by Jung et al.118 HA, a linear glycosaminoglycan that is a structural and active signalling component in native ECM, has been shown to possess advantages for tissue engineering and drug delivery.141–144 The composited ‘physical’ hydrogel (HA/P407) was shown to have a sol–gel phase transition of 30–31 °C, very similar to P407, but with viscous modulus 1.4-fold higher. Dynamic light scattering, at concentration below the CMC, demonstrated significant differences in hydrodynamic size, dhyd, below the LCST; P407 micelles had dhyd ∼15 nm as compared to ∼160 nm for HA/P407, and for the latter dhyd increased to ∼400 nm above the transition. The authors suggested that the presence of HA increases both the packing density and cohesive association with P407 within micelles, increasing size and so improving mechanical stability. The release profile of piroxicam (PX), a non-steroidal anti-inflammatory drug (NSAID) was measured in vitro over a 10-day period. HA/P407 had a more sustained release profile than P407, with 50 and 20% wt PX, respectively, released within 50 hours, which is consistent with prolonged micelle cohesion.
Alginate is another linear polysaccharide that has been investigated as a composite for improving P407 stability.121,129,145–147 Commercial alginate is derived from both brown algae and bacteria, with molecular weight and physicochemical properties dependent on its source.148 Alginate is known to be well tolerated in humans and so has been investigated for a wide range of biomedical applications.148 Cao et al. investigated the potential of P407 and alginate as a ‘physical’ nitric oxide (NO)-releasing composite with applications for wound healing.121 The composition range was similar to the previous (HA) example, with 20% w/v P407 and 1% w/v alginate included along with S-nitrosoglutathione (GSNO, which an NO source, mediated both by its S–NO bond cleavage and its external conditions-dependent decomposition over time). The CGT of P407/Alg was shown to be slightly lower, at 24.2 ± 0.3 °C, than for P407, at 26.4 ± 0.2 °C, the value was lower again for the loaded ‘physical’ nanocomposite GSNO/P407/Alg at 23.4 ± 0.2 °C. The effectiveness of the material for inducing healing following topical administration on bacterial-infected murine burn wounds was investigated. Wound areas treated with GSNO/P407/Alg were shown to histologically resemble normal dermal tissue on day 11, while untreated, GSNO alone and P407/Alg maintained typical features of infected wound tissue. The authors suggest the physical composites as an effective means of administering NO-based therapeutics for improved wound healing.
Chitosan is a linear polysaccharide whose polymer self-assembly and mechanical properties are heavily pH-dependent.149 García-Couce et al. prepared ‘physical’ composite P407/chitosan hydrogels for controlled release of dexamethasone (DMT).120 The composite showed initial ‘burst’ release, comparable to 25% w/v P407 control, but subsequently an extended release profile with ∼50% released by 120 hours as compared to ∼100% for control. Mathematical modelling led the authors to the view that the altered response was due to reduced chain relaxation and diffusion rates, which are rapid (due to micelle disentanglement) in non-composited P407. In vivo, the residence time of the sodium tripolyphosphate (TPP)-crosslinked 1% w/v chitosan/25% w/v P407 composite was shown, using NIR florescence imaging, to be significantly enhanced at the articular knee joint injection site in an osteoarthritic mouse model. 14 days-post injection no fluorescence was observed in PBS control, while significant signal was detectable after 35 days at the composite injection site. The authors did not however include a 25% w/v P407 control injection, which would have been a useful non-composited comparison.
To obtain shorter gelation times, enhanced stability in culture medium and reduced cytotoxicity P407 (at ∼20% w/v) was composited with carboxymethyl hexanoyl chitosan (CA, in the range 0.5–1.5% w/v) and glutaraldehyde (GA 0.1% v/v, which crosslinks the CA) for encapsulating fibroblasts (L-929). The P407 component largely determines the gelation, and the crosslinked CA again contributes stability. As the interactions between the two polymers are physical, we label this material P407/CA-GA. Yap et al., reported that gelation time increases significantly with increasing CA content due to decreased hydrophobic interactions in P407. Subsequently cell viability was shown for 5-days by encapsulating L929 cells in the different formulations, which remained in the gelled state in DMEM. Cells in P407/CA-GA proliferated during encapsulation. The results show that the material has good cytocompatibility, with at least 90% cell viability after a 5-day incubation, with the lowest viability for 1.5% CA as compared to 0.5 and 1.0%. Hence P407/CA-GA may have potential as an injectable scaffold.130
In a study from Gao et al., docetaxel nanocrystals (DOCNC at 2 mg mL−1, comminuted into the nano-range) were incorporated at low temperature into liquid 15 or 20% w/v P407, which had been chemically modified with d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), forming a room temperature thermosensitive physical hydrogel. We again classify DOCNC/P407-TPGS as a physical gel, as gelation is driven by non-covalent micelle contacts. The goal of the study was to reverse drug resistance due to in P-glycoprotein (P-gp)-overexpression in human liver cancer SMMC-7721 cells. The 0.1% TPGS modified hydrogels showed significantly higher in vitro inhibition of SMMC-7721/RT compared with DOC in solution and P407 containing DOCNCs. Furthermore DOCNC/P407-TPGS inhibited P-gp function, which was attributed to action of TPGS, providing good anti-tumour efficacy in SMMC-7721/RT tumour bearing mice.131 DOCNC/P407-TPGS retained the thermosensitive properties of unmodified P407 and preserved physical stability of DOCNCs without changing particle size during gelation. The composite is a locally injectable carrier with potential to overcome P-gp overexpression.
A similar study was reported by Jung et al., in which temperature-induced gelation of P407 was again exploited.126 Glycolide-based functionalisation of P407 (to form P407F) provided functional groups for sequential covalent attachment of heparin (P407FH) and subsequently transforming growth factor-β1, or TGF-β1, (P407FH-TGF). Physical gels of P407FH-TGF (at ∼20% w/v) with crosslinked hyaluronic acid (X-HA, Mw 1600 Da at ∼10% w/v, to provide stability and biodegradability) and with dexamethasone (Dex, another chondrogenic factor which was also physically included) were formed. These are labelled here as P407FH-TGF/X-HA/Dex. Again the gels retained the thermoresponsiveness of the pluronics component with added stability from HA. Three composites were evaluated for encapsulating human adipose stem cells (ASCs); (i) P407F/X-HA as a control; (ii) P407F/TGF/X-HA/Dex with free TGF-β1 and free Dex, and; (iii) P407FH-TGF/X-HA/Dex with linked heparin-bound TGF-β1 and free Dex. It was found that at day 3 ASC viability decreased to 50–60% in vitro. X-HA/PF407 hydrogels were then injected into the defects of a rabbit knee articular cartilage model. After 4 weeks it was found that; PF407/X-HA eroded significantly and was poorly securing the defect site, whereas; P407F/TGF/X-HA/Dex maintained its original volume with evidence of cartilaginous matrix deposition, and; P407FH-TGF/X-HA/Dex also maintained its original mass and there were indicators of differentiation into the chondrocyte lineage within the hydrogel.126
5.1.2.1. ‘Chemical’ composite P407 hydrogels. Lee et al. composited a HA-dopamine (∼5% w/v) conjugate with thiol end-capped P407 (P407SH), using a Michael-type catechol–thiol reaction, to generate chemically crosslinked composites (HA-P407SH),122 with covalent links between the two components. The CGC was shown to be significantly lower, at 7 wt%, than that of P407, at 16 wt%. The authors hypothesised that this difference may be due to enhanced stability of the minimally crosslinked composites, which enabled facile micelle self-assembly. Indeed, stabilisation of micelles by covalent crosslinking has previously been described to manifest as a decrease in CMC.140 Lee et al. additionally demonstrated that resistance of the composite to erosion at 37 °C in PBS solution (pH 7.4) over the course of several weeks was highly dependent on the thiolated poloxamer content, pinpointing the role of the covalent linkages in stabilising the hydrogel. HA-P407SH containing 16 wt% P407SH remained stable for over a month in culture, while those formed with 12 wt% P407SH eroded by 8 days, and those with pure 16 or 12 wt% P407 by 2–3 days.122 In vivo, after subcutaneous injection of the composite into mice, 78 wt% retention of the original dry weight 21 days post injection was noted and the material showed advantageously strong adhesion to nearby tissue surfaces. Furthermore, maintenance of adhesion to a mouse skin biopsy was shown to be 414% higher in HA-P407SH, as compared to the equivalent HA/P407 physical mixture. The authors suggested that the improved adhesion arose from unreacted catechol moieties in the hydrogel which adhere to tissue surfaces.122
Shachaf et al. described a different approach of crosslinking a chemically-modified, P407 network.132 Physical hydrogel formation was observed for unimers of fibrinogen conjugated pluronic-diacrylate (labelled here P407DA-Fib) and the system retained thermoreversibility prior to UV crosslinking. Interestingly, reducing the temperature during cross-linking progressively increased the G′ values, showing how chain dynamics determines the final network properties and enabling control over the physical properties for the same nominal composition. P407DA-Fib hydrogels provide biocompatibility, associated with the fibrinogen component, and enhanced control over the stiffness and degradation rate, as compared to P407. Cell viability studies using human dermal fibroblasts showed some potentially useful modulus-dependent cell-spreading.
5.2. Use of P407 as a sacrificial ink in biofabrication strategies
As shown in the previous section P407 composites can be used as cell culture supports, in which it plays a key role in controlling matrix properties. Here we describe how the same favourable characteristics can also be harnessed to enable fabrication of complex constructs. These can more closely recapitulate the environment of native tissues, vasculature, and different components of ECM. In the first instance, due to its shear thinning properties55,150 and ease of handling and preparation, P407 was used in developing formulations for 3D printing both as a component of the final matrix,7,92,151 and as a fugitive, or sacrificial, ink.88,152As previously noted by Kolesky et al., heterogeneous structures can be printed from multiple bioinks (usually using multiple print heads/nozzles) while including fugitive inks. These can subsequently be removed to generate channels which can, for instance, mimic vascularisation, Fig. 6A.89 Due to its thermogelation behaviour and excellent shear-thinning properties P407 is a common choice for this approach.153 At room temperature P407 composite inks are typically stiff and solid-like with storage modulus, G′ > loss modulus, G′′, while at low temperature G′′ > G′, and they flow readily, Fig. 6B. As noted in previous sections, the exact transition temperature depends on the P407 concentration and solvent constituents (e.g. salts), but in general P407 > 25% w/v is used for 3D printing.7,55,92 The shear-thinning behaviour provides printability and the thermogelation allows removal of the P407 component ink as a perfusable liquid, by cycling the temperature from high to low and back. While many 3D-printable inks, including those with fugitive P407, have reasonable recovery after shear thinning, good shape fidelity in the final constructs typically requires surrounding supports, which are often composed of PCL,154 or other crosslinked hydrogels.89 P407 has become a benchmark for printable extrusion-based polymers with guidelines on printability assessment described by Paxton et al.55
 | ||
| Fig. 6 Schematic views of. (A) A 3D bioprinting approach (left), in which vasculature, cells, and ECM are co-printed to yield engineered tissue constructs composed of heterogeneous subunits (right). (B) Thermally reversible gelation, with the corresponding temperature-dependent shear elastic (G′) and loss moduli (G′′) for P407 fugitive ink. (C)–(F) Representations and optical and fluorescent images of embedded vascular networks that are printed, evacuated, and perfused with a water-soluble fluorescent dye. (F) Optical image of a representative microchannel within a HUVEC suspension perfused 2D vascular network. Reproduced with permission from ref. 89. © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In Table 3 we present prominent examples of the use of P407 as a sacrificial ink and note how its use enabled better recapitulation of tissue. Increasingly complex 3D architectures become possible, as compared to conventional homogeneous cell culture. For instance, the favourable properties of P407 have enabled printing of complex structures with channels in 1, 2 or 3 dimensions using multi-nozzle extrusion printing. As an example, the structures shown in Fig. 6C–E were formed using sacrificial P407 printed in one printhead and GelMA in another.89 The resulting microchannel networks could be perfused with cells, in this case HUVEC cells were seeded into a millimetre scale vascular chip, Fig. 6F.89 Modifications of inks for generating similar ‘vascular’ structures have also been noted. For example P407 was combined into a composite ink with GelMA, allowing single nozzle printing of millimetre scale structures that, due to the ability to wash away P407, could subsequently be perfused.155 Other studies have shown that perfusable chips of this type can be used successfully for stem cell support and growth factor infiltration.88
| Molecule/polymer composited with P407 | Physicochemical formation response/benefits of P407 | Printing conditions | Biological culture done? | Ref. |
|---|---|---|---|---|
| (i) P407 diacrylate (AC) with (ii) hyaluronan methacrylate (HAMA) | To overcome the instability of P407 in cell culture and increase biocompatibility, diacrylated P407 was mixed with unmodified P407 at printable concentration for subsequent cell viability assay. | Needle ID 300 μm; print head temperature;37 °C; print bed temperature 35 and 38 °C; pressure 2 bar; feed rate 100 mm min−1 | Cell viability was tested for bovine chondrocytes in AC (20% w/v) and P407 (17% w/v)/AC (3% w/v); 88% viability on day 14, only 62% for AC (20%). | 151 |
| Single layers 160 μm thick | HAMA (0.1% and 0.5% w/v) was added to P407 (17% w/v)/AC (3% w/v) giving a minor decrease in cell viability to 80%, due to decrease in pore size/reduced nutrient diffusion | |||
| N-isopropylacrylamide (NIPAM) with poly(ethylene glycol) diacrylate (PEGDA), with P407 as a fugitive ink | Multilayer structures were printed consisting of (i) and (ii) combined with P407 as sacrificial ink. After removing P407 at 4 °C, NIPAM/PEGDA showed characteristic volume change behaviour while maintaining fidelity. | ID diameter 200 μm; pressure; 1.2–2.8 bar; feed rate 60–240 mm min−1 | None reported | 152 |
| NIPAM/PEGDA | Thickness not noted | |||
| Poly(ε-caprolactone) (PCL) (internal scaffold) sacrificial P407 (external scaffold); gelatin, fibrinogen, HA and glycerol for cell support, used locally designed 4-headed extrusion “integrated tissue–organ printer (ITOP)” system. | To improve tissue sized prints structural strength, PCL internal and F127 external (sacrificial) scaffolds were used. | Cell-laden hydrogel: ID 300 μm; pressure 0.5–0.8 bar. | Mandible and calvarial bone (Human AFSCs) ear cartilage (rabbit primary auricular chondrocytes) and skeletal muscle (C2C12 myoblasts) | 154 |
| P407: ID 250 μm; pressure 2–3 bar. | ||||
| PCL: ID 250 μm; pressure 8 bar; head temperature 92.5 °C | ||||
| Cell-laden GelMA and P407 | P407 as a support material for printing, which after subsequent crosslinking is removed by cooling to 4 °C. | ID 30, 100, and 200 μm; pressure 1.3–4.1 bar; feed rate 60–600 mm min−1 | Green fluorescent protein-expressing human neonatal | 89 |
| Dermal fibroblast cells (GFP-HNDFs) and 10T1/2 fibroblast cells | ||||
| P407 and gelatin–fibrin ink printed with a custom-designed multi-material 3D bioprinter. | P407 as a sacrificial print support. Following P407 liquefication/removal, open conduits were present in the gelatin-fibrin inks. | ID 100 to 410 μm; pressure 0.7–9.6 bar; printing speed 6–300 cm min−1 | Human mesenchymal stem cells (hMSCs) and human neonatal dermal fibroblasts (hNDFs) and human umbilical vein endothelial cells (HUVECs) | 88 |
| Pluronic monocarboxylated (P407-MP) and GelMA reacted in different mass ratios (P407-MP:GelMA 1:2, 1:1.5, 1:1, 1.5:1 and 1:2) |
To fabricate complex structures, dual-nozzle printing was used in which one dual nozzle (P407-GelMA and print pluronic as support material) printing. Cured and cold water washed removed the sacrificial material. | ID 335 μm; pressure 3–5 bar; feed rate 500 mm min−1; head temperature 30 ± 3 °C | L2959 mouse fibroblast cells were used for biocompatibility and toxicity tests. All formulations supported cell proliferation | 155 |
| Carboxylated agarose (CA), P407 | After bioprinting, incubated at 4 °C solubilised P407 and consolidated the CA hydrogel phase, providing stable cylinders within a hollow vessel. | ID 230, 150 and 100 μm; pressure 2 bar | None reported | 156 |
Kang et al. demonstrated printing of mandible and calvarial bone, ear cartilage and skeletal muscle using an integrated tissue-organ printer.154 A range of materials were evaluated to provide the characteristics needed to support the different cell types. These included; gelatine (to provide thermo-sensitivity); fibrinogen (to improve stability, cell adhesion and induction of cell proliferation), and; HA and glycerol (to enhance printing uniformity and reduce nozzle clogging).154 Furthermore, to create micro-channels in the printed constructs with structural strength, PCL was used internally as a support and P407 as an external sacrificial scaffold. Fig. 7 depicts the process of printing PCL-supported P407-based constructs, for generating functional muscle tissue mimics. Arslan et al., described fabrication of modular 3D structures which can show complex motions through assembly of stimulus responsive components.152 This approach relies on printing of low viscosity precursor formulations with shear-thinning properties comprised of; N-isopropylacrylamide (NIPAM, providing volume changes at its LCST that could have value for robotic actuation); poly(ethylene glycol) diacrylate (PEGDA, as the matrix former), and; P407 (as the fugitive ink). 3D structures were created with anisotropic actuation using two types of hydrogels with different swelling/shrinking properties, which provide twisting and bending motions upon thermal stimulus (25 °C ↔ 40 °C). In principle this modular approach could enable programming of many types of soft devices with bio-inspired motions.152
 | ||
| Fig. 7 In vitro bioprinted muscle. (A) Schematic fiber bundle structure for muscle organisation. PCL pillars (green) were used to maintain the structure and to induce the compaction phenomenon for cell alignment. (B) Visualised motion program for 3D printing muscle construct. (C) 3D patterned muscle construct (left) before and after removing the sacrificial P407. The construct was cross-linked with thrombin to gel fibrinogen, and the uncross-linked sacrificial material was dissolved with cold medium. (D) and (E) The PCL pillar structure is essential to stabilise the print and to induce compaction of the patterned cell-laden hydrogel generating longitudinal cell alignment; without PCL pillar (D) and with PCL pillar (E). The latter showed consistent unidirectionally cellular alignment, in contrast with random orientations without PCL pillar. (F) Live/dead (green/red) staining of encapsulated cells indicates high cell viability after printing. (G) Immunofluorescent staining for myosin heavy chains after 7-days differentiation; encapsulated myoblasts aligned along the longitudinal direction of the fibers. Reproduced with permission from ref. 154. Copyrights © 2016 Nature America, Inc. | ||
In summary, while P407 does not possess sufficient long-term stability when used as the sole matrix former for cell culture applications, it can provide improved printability and good structural support to 3D printed structures and can enable fabrication of hierarchical structures when used as a sacrificial template. In the next section some emerging and some well-known alternative thermoresponsive/thermogelling polymers are described, these could fill some of the roles currently played by P407 while providing additional favourable properties.
6. Alternative thermoresponsive polymers for cell culture
In this section we briefly describe alternative thermoresponsive (LCST) polymers, other than micelles, that have demonstrated applications in cell culture and biomedical drug delivery. We finish the section with a comparison of the degradation profiles of these materials with those of P407.6.1. Cell recovery applications
First, cell recovery from culture platforms usually relies on mechanical, chemical, or enzymatic treatments, each of which has disadvantages including difficulties in scaling and avoiding cell damage. A significant opportunity lies in the use of thermoresponsive polymers for detachment/recovery which may, in principle, address both of these issues. As discussed previously, at temperatures above the LCST, water is expelled, and the network is in a collapsed hydrophobic state, supporting attachment, see Fig. 8A. On decreasing temperature to below the transition, water enters, the polymer chains adopt an expanded coil conformation with increased hydrophilicity, enabling detachment. Hence the ideal LCST is a little below 37 °C. This approach works when cell and protein-adhesion is primarily to hydrophobic, as opposed to the hydrophilic, regions of the polymers.157,158 Examples of polymers that have these characteristics are given in Table 4 and discussed below. | ||
| Fig. 8 (A) Schematic of reversible cell attachment/detachment using thermoresponsive polymers. At 37 °C polymer chains are hydrophobic and cells adhere to the surface. At ambient temperature, the polymer chains are hydrated and adopt a hydrophilic expanded coil conformation, the cells detach. (B) Schematic of cell storage and recovery using a thermogel. Mixing is achieved in the sol-state, on increasing temperature above the LCST transition, cells are encapsulated in the gel. Cells can be recovered by centrifugation on decreasing temperature. Reproduced with permission from ref. 159. Copyright © 2021, American Chemical Society (C) Stability of a nanodiamond (ND) chitosan-gelatin nanocomposite loaded over a 21 day period, at varying ND amounts (0–0.02% w/v). Reproduced with permission from ref. 160. © 2017 Acta Materialia Inc. Published by Elsevier Ltd. (D) Macroscopic images of a cartilage defect site taken at 1 and 12 weeks with no treatment (a and c) and HA-pNIPAM treated (b and d). Reproduced with permission from ref. 161. Copyright © 2016 Wiley Periodicals, Inc. | ||
| Application | Molecule/polymer | Physicochemical formation response | Biological culture done? | Degradation mechanism & timeframea | Ref. |
|---|---|---|---|---|---|
| a Timeframes are estimations with the following meaning: short: days–weeks, medium: weeks–months, long: months–years, or stable/non-degradable. | |||||
| As cellular matrices | P(DEGMA-b-PEGMA-b-DEGMA) | LCST 30–40 °C (dependent on chain length of PDEGMA and PPEGMA units) | Cell viability of P(DEGMA70-b-PEGMA260-b-DEGMA70) with HeLa cells and PC3 confirmed | Non-enzymatic (oxidative) | 167 |
| Long | |||||
| PCL–PEG–PPG | LCST 5–50 °C dependent upon amount of PPG in copolymer and copolymer concentration | CD4+ T-cell lymphocyte | Enzymatic and hydrolytic (mainly PCL) | 168 | |
| Medium | |||||
| Poly(N-vinylcaprolactam) | Sol gel temperature between 25 and 37 °C. Gelation time dependent upon MW. | Chondrocytes (CHs) and mesenchymal stem cells, ∼90% viability | Non-enzymatic; minimal degradation | 179 | |
| Long | |||||
| As delivery systems for tissue engineering | Chitosan–gelatin nanocomposite | 35–39 °C | Vascular endothelial growth factor (VEGF) | Enzymatic (lysozyme, collagenase) | 160 |
| Short to medium | |||||
| Hyaluronan acid – pNIPAm copolymer | ∼30 °C | Stromal cell derived factor (SDF-1), bovine IVD | Partially enzymatic (HA by hyaluronidase), pNIPAm non-enzymatic | 161 and 169–171 | |
| Short to medium (HA: days–weeks, pNIPAm: months–years) | |||||
| For cell storage and recovery | Polypetide poly(ethylene glycol)−poly(L-alanine) (PEG−PA) | Sol–gel transition 10–25 °C | Tonsil-derived mesenchymal stem cells | Enzymatic (protease cleavage of polypeptide block) | 159 |
| The poly(L-alanine) block is enzymatically degradable (by proteases), while PEG is stable | |||||
| Medium (weeks to months) | |||||
| Poly(N-isopropylacrylamide) – modified silica beads | LCST 34 °C | Chinese hamster ovary cells, cells adhered to the surface of the beads at 37 °C, whilst detached at 25 °C | Non-enzymatic | 166 | |
| Long | |||||
| Poly(N-isopropylacrylamide) surfaces | LCST 31 °C | Bovine aortic endothelial cells (BAEC), cells adhered to the surface at 37 °C. At RT, cell sheet began to detach after 10 min (comparison to no detachment under same conditions from control TCPS surface) | Non-enzymatic | 162 | |
| Long | |||||
| Poly(PEGMA188)-PET fibrous scaffolds | LCST 22.5–25.2 °C | Human corneal stromal stem cells (hCSSC), comparable cell attachment and proliferation at 37 °C to TCPS control. Cell detachment at ambient temperature (17 °C) from thermoresponsive scaffold. | Non-enzymatic (oxidative possible) | 163 | |
| Long | |||||
Amongst the thermoresponsive materials reported for use in 2D cell culture surfaces are poly(N-isopropylacrylamide) (pNIPAm), poly(PEGMA) and a copolymer of 2-(acryloyloxyethyl) trimethylammonium chloride (AEtMA-Cl) and 2-(diethylamino)ethyl acrylate (DEAEA).162–165 For each of these materials, when used as cell contact surfaces, comparable cell attachment was demonstrated at 37 °C as for the control surface (TCPS or Matrigel), while cell detachment at ambient temperature (<LCST) was induced only for those with a thermoresponsive layer.
For example, 100 μm silica beads surface grafted with thermoresponsive pNIPAm, (prepared with a grafting-from RAFT polymerisation to be of varying chain length, Mn 6200–23700), were investigated as 3D cell culture scaffolds.166 Cells successfully adhered to the beads at 37 °C for all thermoresponsive layer thicknesses studied. However, cell detachment at 37 °C was only observed for beads with pNIPAm Mn > 10000. The suggestion was that lower Mn grafts were less hydrated in the swollen (low temperature) state impacting temperature-induced changes in the hydrophilic/phobic properties of the layers.
Long term storage and transportation of cells typically requires cryopreservation techniques which can result in damage to cells. Storage of cells at ambient temperatures can be achieved through the incorporation of cells within a thermoresponsive polymer gel, Fig. 8B. The temperature induced sol–gel transition allows suspension of cells within the hydrogel after pre-mixing of cells with the thermoresponsive gel in the sol state. A polypeptide based poly(ethylene glycol)–poly(L-alanine) (PEG-PA) ‘chemical’ composite was used to demonstrate significantly greater cell recovery of 64%, as compared to 26% obtained using a conventional cryopreservation technique,159 and <1% for cells stored in PBS with P407 at the same concentration. The sol–gel transition of this particular hydrogel could be controlled from 10–25 °C through the polymer concentration.
6.2. Delivery applications
Thermoresponsive polymers are commonly used as components in delivery systems for tissue engineering. A triblock copolymer formed from poly(ethylene glycol) methyl ether methacrylate (PEGMA) and diethylene glycol methyl ether methacrylate (DEGMA) was evaluated as a cell culture scaffold.167 The LCST was tuned through variation in chain length of the hydrophilic and hydrophobic segments and polymer concentration. Gelation was physical in nature and so was temperature reversible. Dynamic glide force measurements showed better ambient temperature injectability, at 7.98 N, than for P407, at 36.44 N. In addition, low cytotoxicity and good cell viability after 24-hour incubation, was shown for hydrogels formed at 20% and 25% wt. However, hydrogel stability over longer times was not described.Similarly a poly(caprolactone-)–poly(ethylene/propylene glycol) (PCC–PEG–PPG) copolymer was synthesised for use as an in situ gelling system with enzyme-triggered cell delivery.168 CD4+ T-cell lymphocytes were encapsulated within the copolymer gels. As a key goal was to improve biodegradability, degradation was monitored by 1H NMR in the presence of lipase as a model. The ester linkages of the copolymer were shown to be enzymatically degradable, with the extent of degradation dependent on enzyme activity. Preliminary results suggest no cytotoxicity within 5 days, however more detailed studies would be needed to establish cell viability.
Naturally occurring thermoresponsive polymers can provide improved compatibility with biological environments, for instance when implanted, however limitations can arise dur to their fast degradability. An injectable nanodiamond (ND) chitosan–gelatin nanocomposite demonstrated controlled release of growth factors at 37 °C.160 This material was found to show no cytotoxicity in vitro over 24 hours across the ND concentration range investigated (0.005–0.02% w/v). At an ND concentration of 0.01% w/v the stiffness of the gel improved (Youngs modulus 7.3 kPa, compared to 6.3 kPa for ND free) which was ascribed to increased polar interactions with the polymer matrix. This effect on the stiffness was limited to a surprisingly narrow ND concentration range c.0.01% w/v, outside of which there was no improvement. Compositing with NDs also enhanced long-term hydrogel stability in vitro over a 14-day period, however no additional stability was observed at longer times compared to ND-free gels, Fig. 8C. Additionally, hydrogels containing ND's showed good biocompatibility in vivo after 21 days following subcutaneous implantation of the nanocomposite hydrogels into the dorsal skin of Wister rats.
Conjugation of HA with pNIPAm has been reported by several groups as an approach that provides injectable cellular delivery system with temperature-induced gelation, typically c.30 °C, for formulations in the 2–10% w/v range.161,169–171 The mechanical properties of the hydrogel can be improved by extending the pNIPAm chain length, an effect usually ascribed to increased pNIPAm inter-chain association, enabling stronger/more persistent entanglement. High grafting densities of pNIPAm were found to compromise mechanical strength.172 Thus a balance between chain length and grafting density must be considered in material design for cell culture. Sustained in vitro release of stromal cell derived factor (SDF-1) over a 7-day period was achieved for copolymerised HA-pNIPAm at 10% w/v, with no evidence of hydrogel degradation.171 Similarly, an injectable copolymer HA-pNIPAm brush copolymer (10% w/v, with pNIPAM chains covalently linked to HA backbone) demonstrated superior properties in cartilage repair. The hydrogel showed fluid behaviour at room temperature with a rapid, <1 min, transition to a soft-gel type behaviour c.30 °C.161 No degradation was observed after 12 weeks of injection and cartilage healing was promoted in the presence of the hydrogel, as evaluated using the ICRS Cartilage Repair Assessment System, Fig. 8D. However, the non-covalent nature of the copolymer chain associations forming the gel, and thus the mechanical strength, was identified as a potential limitation. Furthermore, a composite network of crosslinked HA and copolymer HA-pNIPAm demonstrated exceptional rheological properties compared to alternative single network hydrogels (elastic modulus greater than in agarose, HA or HA-pNIPAm formulations alone).169 These HA/HA-pNIPMam mixtures had good cell retention after 7 days however some degradation was described.
Thermoresponsive polymers, such as PEGMA-DEGMA, PCC-PEG-PPG, and HA-pNIPAm, show significant potential for inclusion in formulations for injectable cell-delivery systems. However, despite their demonstrated favourable cytotoxicity profiles and tuneable mechanical/gelation properties, challenges remain in ensuring long-term stability, biodegradability, and retention of optimal mechanical strength, especially in the case of natural polymers or their composites.
6.3. Degradation of thermoresponsive polymers
Pluronics, including poloxamer 407, are chemically stable and are not readily enzymatically degraded in the human body or under physiological conditions.173,174 The stability contributes to their utility but the lack of biodegradability can be limiting for application. In the human body, it is expected that pluronics will be cleared via renal excretion, rather than being broken down by enzymes.175,176 In contrast to P407, many of the alternative thermoresponsive systems demonstrate a range of degradation mechanisms and timescales, which are increasingly being leveraged to improve material clearance, remodelling, and in vivo functionality (Table 4).On the other hand, the physical nature of the interactions that drive hydrogelation renders these types of hydrogels prone to disruption. In bio-mimetic environments, such as cell culture media or simulated body fluids, P407 undergoes rapid dissolution and erosion, particularly under dilute conditions or in the presence of serum proteins, which can disrupt micelle packing and destabilise the gel matrix.177 Several examples of this are described in Section 4. Many P407 hydrogels gradually disintegrate, over hours to days, in aqueous or physiological conditions, even when originally prepared at high concentrations (>30% w/v). This limits the long-term integrity of the objects, often making them unsuitable for sustained structural applications without reinforcement.
In contrast, polyesters like PCL–PEG–PPG form more stable networks with far slower (many weeks) degradation kinetics, arising from covalent or secondary structure-based interactions. pNIPAm- or poly(N-vinylcaprolactam)-based materials are generally more resistant to dissolution due to stronger hydrophobic associations and chain entanglements. However, they too can swell and eventually disperse depending on molecular weight, architecture, and medium composition.178 Notably, some hydrogels, such as HA-based copolymers, may initially maintain structural integrity, but rapidly lose mass upon enzymatic cleavage.161,169–171 Others polymers, such as P(DEGMA-b-PEGMA-b-DEGMA), remain intact for extended periods but may undergo slow erosion due to oxidative side-chain degradation.167 Thus, the dissolution profile of thermoresponsive polymers in biological settings plays a critical role in determining their implantation potential, residence time, and functional lifespan in vivo. This highlights the need for careful tuning of polymer structure to balance thermoresponsiveness with the desired mechanical and degradation behaviour.
In the context of in vivo degradability, natural polymer-based hydrogels such as chitosan–gelatin composites,160 and HA–pNIPAm copolymers,161,169–171 undergo relatively rapid enzymatic degradation via lysozyme or hyaluronidase, albeit the pNIPAm component itself follows non-enzymatic degradation routes. Similarly, co-polymer systems like PEG–poly(L-alanine) polypeptide degrade over weeks to months through proteolytic cleavage, and stable excretion of scissored PEG chains.159 Other synthetic polymers, such as PCL–PEG–PPG show slower enzymatic and hydrolytic degradation driven by cleavage of ester bonds in the PCL block.168 Whereas thermoresponsive systems like poly(N-vinylcaprolactam), pNIPAm, and P(DEGMA-b-PEGMA-b-DEGMA) are largely non-biodegradable,167,179 with only limited susceptibility to oxidative degradation of PEG-based side chains over months to years. The latter materials, much like P407, are thus more persistent in vivo from the polymer chain perspective and may require careful consideration regarding long-term clearance or potential accumulation, particularly in implantable or injectable contexts. Compared to these, P407 is uniquely challenged by its lack of mechanical robustness at physiological temperature, leading to rapid physical dissolution even in the absence of chemical degradation—a feature which, while limiting its long-term structural use, supports its role as a sacrificial matrix or delivery vehicle in short-term biomedical applications.
7. Conclusions and future perspectives
While P407 is a versatile and promising biomaterial, difficulties arise from its intrinsic ‘softness’, including; dissolution in biological settings (which is often under-represented and under-reported in the literature), and; limiting viscoelastic properties of the gels (related to degradability). Currently, these challenges present barriers to the translational utility of P407 as a clinically relevant biomaterial. Degradation of P407 has been reported in vivo.174 While the safety of Durasite® drug formulations containing P407, namely AzaSite® and Besivance® has also been called into question.180 The effects of sterilisation, typically by autoclaving, on poloxamer hydrogel rheological properties and injectability may also be a practical hurdle. These aspects need detailed/full evaluation, while existing literature for example is limited and conflicting.25,181,182 The costs of effective and suitable sterilisation methods, such as electron beam irradiation, might be a challenge for future clinical applications.182Approaches used to address these limitations and design a close to ideal thermoresponsive system (Fig. 9), while retaining the favourable physicochemical properties and balancing the need for controllable degradability in longer-term cell culture and delivery applications, include: (i) chemical (covalent) stabilisation of core, shell, or of inter-micelle contacts; (ii) copolymerisation of natural polymers with more stable synthetic polymers and incorporation of mediating nanomaterials, and; (iii) inclusion of other (often natural) polymers to form inter-penetrating or double networks with emergent bulk properties that combine the advantages of both components. In parallel there has been significant activity on the use of P407; (iv) as a modifier during, usually extrusion-based, advanced manufacturing of bulk structures/devices (with subsequent removal of the pluronics component), and; (v) to confer thermoresponsive and thermogelling properties for multiple biomedical applications. Thermogelation is generally retained in printable ‘physical inks’ based on both P407 and pNIPAm, enabling 3D printing (and other applications requiring rapid gelation) at elevated temperatures. The effectiveness of thermogelation is heavily dependent on the concentration of the polymer and the presence of other additives (e.g. solvents, crosslinkers, or stabilisers). Thermogelation can also be retained in printable ‘chemical inks’. In the case of copolymers, this is usually the case once chemical links are not formed between chain types, leading to stiffening of the network. More generally it is found once the modifications do not disrupt the physical mixture. Subsequent photo-crosslinking of the printed inks fixes the structures. The chemical approach can also be favourable as chemical crosslinking can be less sensitive to temperature variations, than is the case for purely physical gelation. The minimum concentrations required to achieve thermogelation vary, but they are in a similar range. For P407 an LCST is typically observed at c.18–20% (w/v) in aqueous solutions, with use of co-polymers and additives pushing that down. For pNIPAm systems the LCST can also vary with the degree of polymerisation, but c.10–20% (w/v) is required for stable thermogelation. As expected, the gelation concentration is generally lower for ‘chemical ink’ formulations. These can provide more flexibility in terms of concentration, allowing for lower polymer loads without compromising gel formation, however, they may require complicated chemistry. P407 is generally superior to pNIPAm in applications requiring rapid, reversible gelation without the need for chemical modification, or when it is desirable to subsequently remove the ‘sacrificial’ component. It is particularly useful in physical inks for 3D printing, where the focus is on temperature-triggered gelation. On the other hand, pNIPAm excels in situations where a more robust gelation mechanism is required, especially in chemical inks or when crosslinking is part of the formulation. pNIPAm-based systems offer better control over the gel's mechanical properties, which may be advantageous in bioengineering or drug delivery applications.
 | ||
| Fig. 9 Ideal physical properties enabling therapeutic functions for thermoresponsive poloxomer 407 (or similar) hydrogels. The factors listed in this diagram can be used singly or in combination in design of functional thermoresponsive hydrogel systems. Created with https://Biorender.com. | ||
In summary, the current state-of-the-art in P407 compositing research is quite advanced. However, exploitation of the kaleidoscope of responsive matrices, described in the literature and outlined above, is limited by the absence of reliable (commercial) sources of all but the simplest matrices. Challenges yet to be addressed include combinatorial approaches for developing next-generation 4D-responsive functional hydrogel networks for cell culture automation and as components in responsive-release devices.183,184 In this context it is critical when reporting novel thermoresponsive matrices, that the dissolution and degradation behaviour of the materials studied are clearly described. This is not only a concern for P407 but also for pNIPAm and other thermoresponsive systems. All these materials undergo physical or chemical breakdown depending on the environmental conditions which affect stability and function. It is essential to distinguish whether the observed dissolution or degradation is due to changes in the gels physical state (e.g. thermoreversible sol-to-gel transition) or chemical breakdown (e.g. polymer cleavage, crosslinker degradation), but this is rarely reported. Clear specification of these details in publications would facilitate reproducibility and enable interpretation the time-dependent material properties described. Combination of standard used dry-weight degradation methods, as well as chromatographic (e.g. LC-MS) evaluation as routine would be hugely advantageous.
Author contributions
S. C.: conceptualization, data curation, formal analysis, investigation, validation, visualization, writing – original draft. K. S.: conceptualization, data curation, formal analysis, investigation, validation, visualization, writing – original draft. D. W.: conceptualization, data curation, formal analysis, investigation, validation, visualization, writing – original draft. I. K.: data curation, formal analysis, validation, funding acquisition, visualization, writing – review & editing. J. C.: conceptualization, project administration, supervision, funding acquisition, validation, writing – review & editing. D. F. B.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, writing – original draft, writing – review & editing. J. K. W.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest. All research data supporting this publication are directly available within this publication. ChatGPT tools were used to improve the readability and language of the manuscript.Data availability
All data supporting the findings of this review are derived from previously published studies, which are fully cited within the manuscript. The data are publicly available through the respective sources referenced throughout the text.Acknowledgements
The authors acknowledge support from Science Foundation Ireland (16/IA/4584). This work was supported by the European Union's Horizon 2020 (H2020-MSCA-IF-2019) research and innovation programme under the Marie Skłodowska-Curie grant agreement 893099—ImmunoBioInks and by AO Foundation to J. K. W. I. K. would like to acknowledge the Irish Research Council grant GOIPG/2023/3604. Graphical abstract has been created with https://BioRender.com.References
- R. Suntornnond, J. An and C. K. Chua, Macromol. Mater. Eng., 2017, 302, 1600266 CrossRef.
- S. Santoni, M. Sponchioni, S. G. Gugliandolo, B. M. Colosimo and D. Moscatelli, Proc. CIRP, 2022, 110, 348–353 CrossRef.
- K. Uto, C. K. Arakawa and C. A. DeForest, Cold Spring Harbor Perspect. Biol., 2020, 12(9), a035691 CrossRef PubMed.
- M. Sponchioni, U. Capasso Palmiero and D. Moscatelli, Mater. Sci. Eng., C, 2019, 102, 589–605 CrossRef CAS PubMed.
- M. Kuzucu, G. Vera, M. Beaumont, S. Fischer, P. Wei, V. P. Shastri and A. Forget, ACS Biomater. Sci. Eng., 2021, 7, 2192–2197 CrossRef CAS PubMed.
- J. Liu, W. Xu, Z. Kuang, P. Dong, Y. Yao, H. Wu, A. Liu and F. Ye, J. Mater. Chem. C, 2020, 8, 12092–12099 RSC.
- P. Monks, J. K. Wychowaniec, E. McKiernan, S. Clerkin, J. Crean, B. J. Rodriguez, E. G. Reynaud, A. Heise and D. F. Brougham, Small, 2021, 17, e2004452 CrossRef PubMed.
- A. Schwab, R. Levato, M. D'Este, S. Piluso, D. Eglin and J. Malda, Chem. Rev., 2020, 120, 11028–11055 CrossRef CAS PubMed.
- K. Singh, J. K. Wychowaniec, C. J. C. Edwards-Gayle, E. G. Reynaud, B. J. Rodriguez and D. F. Brougham, J. Colloid Interface Sci., 2024, 660, 302–313 CrossRef CAS PubMed.
- K. Zhang, K. Xue and X. J. Loh, Gels, 2021, 7, 77 CrossRef CAS PubMed.
- T. Sarwan, P. Kumar, Y. E. Choonara and V. Pillay, Front. Mater., 2020, 7, 73 CrossRef.
- P. Zarrintaj, J. D. Ramsey, A. Samadi, Z. Atoufi, M. K. Yazdi, M. R. Ganjali, L. M. Amirabad, E. Zangene, M. Farokhi, K. Formela, M. R. Saeb, M. Mozafari and S. Thomas, Acta Biomater., 2020, 110, 37–67 CrossRef CAS PubMed.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2020, 31, 2008123 CrossRef.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- A. M. Bodratti and P. Alexandridis, J. Funct. Biomater., 2018, 9, 11 CrossRef PubMed.
- H. Abdeltawab, D. Svirskis and M. Sharma, Expert. Opin Drug Delivery, 2020, 17, 495–509 CrossRef CAS PubMed.
- S. D. Dutta, J. Bin, K. Ganguly, D. K. Patel and K. T. Lim, RSC Adv., 2021, 11, 20342–20354 RSC.
- K. A. Soliman, K. Ullah, A. Shah, D. S. Jones and T. R. R. Singh, Drug Discovery Today, 2019, 24, 1575–1586 CrossRef CAS PubMed.
- L. Bowman and S. Pham, US Pat., 8501800, U.S. Patent and Trademark Office, Washington, DC, 2013.
- Z. K. Al-Qaysi, I. G. Beadham, S. L. Schwikkard, J. C. Bear, A. A. Al-Kinani and R. G. Alany, Expert Opin. Drug Delivery, 2023, 20, 905–919 CrossRef CAS PubMed.
- H. Madry, L. Gao, A. Rey-Rico, J. K. Venkatesan, K. Müller-Brandt, X. Cai, L. Goebel, G. Schmitt, S. Speicher-Mentges, D. Zurakowski, M. D. Menger, M. W. Laschke and M. Cucchiarini, Adv. Mater., 2020, 32, 1906508 CrossRef CAS PubMed.
- M. Cucchiarini and H. Madry, Nat. Rev. Rheumatol., 2019, 15, 18–29 CrossRef PubMed.
- X. Guo, D. Li, G. Yang, C. Shi, Z. Tang, J. Wang and S. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 8549–8559 CrossRef CAS PubMed.
- Y. Yu, Y. Cheng, J. Tong, L. Zhang, Y. Wei and M. Tian, J. Mater. Chem. B, 2021, 9, 2979–2992 RSC.
- F. Sun, Y. Wang, Y. Wei, G. Cheng and G. Ma, J. Bioact. Compat. Polym., 2014, 29, 301–317 CrossRef.
- D. K. Baby, in Rheology of Polymer Blends and Nanocomposites, ed. S. Thomas, C. Sarathchandran and N. Chandran, Elsevier, 2020, pp. 193–204 DOI:10.1016/B978-0-12-816957-5.00009-4.
- G. Stojkov, Z. Niyazov, F. Picchioni and R. K. Bose, Gels, 2021, 7(4), 255 CrossRef CAS PubMed.
- N. R. Richbourg, M. K. Rausch and N. A. Peppas, Polymer, 2022, 258, 125316 CrossRef CAS.
- M.-Y. Yeh, J.-Y. Zhao, Y.-R. Hsieh, J.-H. Lin, F.-Y. Chen, R. D. Chakravarthy, P.-C. Chung, H.-C. Lin and S.-C. Hung, RSC Adv., 2017, 7, 21252–21257 RSC.
- A. Fakhari, M. Corcoran and A. Schwarz, Heliyon, 2017, 3, e00390 CrossRef PubMed.
- B. Shriky, A. Kelly, M. Isreb, M. Babenko, N. Mahmoudi, S. Rogers, O. Shebanova, T. Snow and T. Gough, J. Colloid Interface Sci., 2020, 565, 119–130 CrossRef CAS PubMed.
- H. Abdeltawab, D. Svirskis, A. G. Hill and M. Sharma, Gels, 2022, 8, 484 CrossRef CAS PubMed.
- N. Cui, C.-Y. Dai, X. Mao, X. Lv, Y. Gu, E.-S. Lee, H.-B. Jiang and Y. Sun, Gels, 2022, 8(6), 360 CrossRef CAS PubMed.
- N. Hirun, P. Kraisit and V. Tantishaiyakul, Polymers, 2022, 14(9), 1836 CrossRef CAS PubMed.
- C. S. Yong, J. S. Choi, Q.-Z. Quan, J.-D. Rhee, C.-K. Kim, S.-J. Lim, K.-M. Kim, P.-S. Oh and H.-G. Choi, Int. J. Pharm., 2001, 226, 195–205 CrossRef CAS PubMed.
- Z. M. A. Fathalla, A. Vangala, M. Longman, K. A. Khaled, A. K. Hussein, O. H. El-Garhy and R. G. Alany, Eur. J. Pharm. Biopharm., 2017, 114, 119–134 CrossRef CAS PubMed.
- M. Bohorquez, C. Koch, T. Trygstad and N. Pandit, J. Colloid Interface Sci., 1999, 216, 34–40 CrossRef CAS PubMed.
- S. F. Khattak, S. R. Bhatia and S. C. Roberts, Tissue Eng., 2005, 11, 974–983 CrossRef CAS PubMed.
- N. Pandit, T. Trygstad, S. Croy, M. Bohorquez and C. Koch, J. Colloid Interface Sci., 2000, 222, 213–220 CrossRef CAS PubMed.
- N. K. Pandit and J. Kisaka, Int. J. Pharm., 1996, 145, 129–136 CrossRef CAS.
- M. Zhang, M. Djabourov, C. Bourgaux and K. Bouchemal, Int. J. Pharm., 2013, 454, 599–610 CrossRef CAS PubMed.
- D. C. Pozzo and L. M. Walker, Macromolecules, 2007, 40, 5801–5811 CrossRef CAS.
- J. Jiang, C. Li, J. Lombardi, R. H. Colby, B. Rigas, M. H. Rafailovich and J. C. Sokolov, Polymer, 2008, 49, 3561–3567 CrossRef CAS.
- E. Gioffredi, M. Boffito, S. Calzone, S. M. Giannitelli, A. Rainer, M. Trombetta, P. Mozetic and V. Chiono, Proc. CIRP, 2016, 49, 125–132 CrossRef.
- M. Müller, J. Becher, M. Schnabelrauch and M. Zenobi-Wong, J. Vis. Exp., 2013, e50632, DOI:10.3791/50632.
- G. E. Newby, I. W. Hamley, S. M. King, C. M. Martin and N. J. Terrill, J. Colloid Interface Sci., 2009, 329, 54–61 CrossRef CAS PubMed.
- M. Shaikhullina, A. Khaliullina, R. Gimatdinov, A. Butakov, V. Chernov and A. Filippov, J. Mol. Liq., 2020, 306, 112898 CrossRef CAS.
- M. Jalaal, G. Cottrell, N. Balmforth and B. Stoeber, J. Rheol., 2017, 61, 139–146 CrossRef CAS.
- G. Wanka, H. Hoffmann and W. Ulbricht, Macromolecules, 1994, 27, 4145–4159 CrossRef CAS.
- B. Stoeber, Y. Zhihao, D. Liepmann and S. J. Muller, J. Microelectromech. Syst., 2005, 14, 207–213 Search PubMed.
- R. K. Prud'homme, G. Wu and D. K. Schneider, Langmuir, 1996, 12, 4651–4659 CrossRef.
- V. Lenaerts, C. Triqueneaux, M. Quartern, F. Rieg-Falson and P. Couvreur, Int. J. Pharm., 1987, 39, 121–127 CrossRef CAS.
- M. Malmsten and B. Lindman, Macromolecules, 1992, 25, 5440–5445 CrossRef CAS.
- K. Mortensen and Y. Talmon, Macromolecules, 1995, 28, 8829–8834 CrossRef CAS.
- N. Paxton, W. Smolan, T. Bock, F. Melchels, J. Groll and T. Jungst, Biofabrication, 2017, 9, 044107 CrossRef PubMed.
- L. Djekic, B. Čalija and Đ. Medarević, J. Mol. Liq., 2020, 303, 112639 CrossRef CAS.
- T. Morita, S. Mukaide, Z. Chen, K. Higashi, H. Imamura, K. Moribe and T. Sumi, Nano Lett., 2021, 21, 1303–1310 CrossRef CAS PubMed.
- K. H. Ullah, F. Raza, S. M. Munawar, M. Sohail, H. Zafar, M. I. Zafar and T. Ur-Rehman, Polymers, 2021, 13(19), 3376 CrossRef CAS PubMed.
- M. Bercea, M. Constantin, I.-A. Plugariu, M. Oana Daraba and D. Luminita Ichim, J. Mol. Liq., 2022, 362, 119717 CrossRef CAS.
- A. C. Marques, P. C. Costa, S. Velho and M. H. Amaral, Gels, 2024, 10(5), 307 CrossRef CAS PubMed.
- M. Hu, Z. Ling and X. Ren, J. Biol. Eng., 2022, 16, 13 CrossRef CAS PubMed.
- R. Loganathan, B. J. Rongish, C. M. Smith, M. B. Filla, A. Czirok, B. Bénazéraf and C. D. Little, Development, 2016, 143, 2056–2065 CrossRef CAS PubMed.
- J. K. Kular, S. Basu and R. I. Sharma, J. Tissue Eng., 2014, 5, 2041731414557112 CrossRef PubMed.
- P. Lu, K. Takai, V. M. Weaver and Z. Werb, Cold Spring Harbor Perspect. Biol., 2011, 3(12), a005058 Search PubMed.
- A. S. Williams, L. Kang and D. H. Wasserman, Trends Endocrinol. Metab., 2015, 26, 357–366 CrossRef CAS PubMed.
- J. Winkler, A. Abisoye-Ogunniyan, K. J. Metcalf and Z. Werb, Nat. Commun., 2020, 11, 5120 CrossRef CAS PubMed.
- T. R. Cox and J. T. Erler, Dis. Models Mech., 2011, 4, 165–178 CrossRef CAS PubMed.
- M. Urbanczyk, S. L. Layland and K. Schenke-Layland, Matrix Biol., 2020, 85–86, 1–14 CrossRef CAS PubMed.
- P. Romani, I. Brian, G. Santinon, A. Pocaterra, M. Audano, S. Pedretti, S. Mathieu, M. Forcato, S. Bicciato, J. B. Manneville, N. Mitro and S. Dupont, Nat. Cell Biol., 2019, 21, 338–347 CrossRef CAS PubMed.
- C. C. Kumar, Oncogene, 1998, 17, 1365–1373 CrossRef CAS PubMed.
- R. J. Khadilkar, K. Y. L. Ho, B. Venkatesh and G. Tanentzapf, Curr. Biol., 2020, 30, 3316–3329 CrossRef CAS PubMed.
- K. Stock, M. F. Estrada, S. Vidic, K. Gjerde, A. Rudisch, V. E. Santo, M. Barbier, S. Blom, S. C. Arundkar, I. Selvam, A. Osswald, Y. Stein, S. Gruenewald, C. Brito, W. van Weerden, V. Rotter, E. Boghaert, M. Oren, W. Sommergruber, Y. Chong, R. de Hoogt and R. Graeser, Sci. Rep., 2016, 6, 28951 CrossRef CAS PubMed.
- A. Riedl, M. Schlederer, K. Pudelko, M. Stadler, S. Walter, D. Unterleuthner, C. Unger, N. Kramer, M. Hengstschlager, L. Kenner, D. Pfeiffer, G. Krupitza and H. Dolznig, J. Cell Sci., 2017, 130, 203–218 CAS.
- A. A. Seyhan, Transl. Med. Commun., 2019, 4, 18 CrossRef.
- N. Gjorevski, A. Ranga and M. P. Lutolf, Development, 2014, 141, 1794–1804 CrossRef CAS PubMed.
- C. M. Walsh, J. K. Wychowaniec, D. F. Brougham and D. Dooley, Pharmacol. Ther., 2021, 108043, DOI:10.1016/j.pharmthera.2021.108043.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- M. Hofer and M. P. Lutolf, Nat. Rev. Mater., 2021, 6, 402–420 CrossRef CAS PubMed.
- N. Gjorevski, N. Sachs, A. Manfrin, S. Giger, M. E. Bragina, P. Ordóñez-Morán, H. Clevers and M. P. Lutolf, Nature, 2016, 539, 560–564 CrossRef CAS PubMed.
- A. Šećerović, M. Pusic, P. Kostešić, M. Vučković, R. Vukojević, S. Škokić, B. Sasi, A. Vukasović, D. Hudetz, D. Vnuk, D. Maticic, I. Urlić, M. Mumme, I. Martin and A. Ivkovic, Am. J. Sports Med., 2021, 49, 3635465211014190 Search PubMed.
- A. Elosegui-Artola, A. Gupta, A. J. Najibi, B. R. Seo, R. Garry, C. M. Tringides, I. de Lazaro, M. Darnell, W. Gu, Q. Zhou, D. A. Weitz, L. Mahadevan and D. J. Mooney, Nat. Mater., 2023, 22, 117–127 CrossRef CAS PubMed.
- J. K. Wychowaniec, E. I. Bektas, A. J. Vernengo, M. Mürner, M. Airoldi, P. S. Tipay, J. Sapudom, J. Teo, D. Eglin and M. D'Este, Biomater. Adv., 2024, 214166, DOI:10.1016/j.bioadv.2024.214166.
- K. Salma-Ancane, A. Sceglovs, E. Tracuma, J. K. Wychowaniec, K. Aunina, A. Ramata-Stunda, V. Nikolajeva and D. Loca, Int. J. Biol. Macromol., 2022, 208, 995–1008 CrossRef CAS PubMed.
- F. Zheng, B. Derby and J. Wong, Biofabrication, 2021, 13, 035006 CrossRef CAS PubMed.
- M. Mahinroosta, Z. Jomeh Farsangi, A. Allahverdi and Z. Shakoori, Mater. Today Chem., 2018, 8, 42–55 CrossRef CAS.
- W. He, M. Reaume, M. Hennenfent, B. P. Lee and R. Rajachar, Biomater. Sci., 2020, 8, 3248–3269 RSC.
- K. Varaprasad, G. M. Raghavendra, T. Jayaramudu, M. M. Yallapu and R. Sadiku, Mater. Sci. Eng., C, 2017, 79, 958–971 CrossRef CAS PubMed.
- D. B. Kolesky, K. A. Homan, M. A. Skylar-Scott and J. A. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3179 CrossRef CAS PubMed.
- D. B. Kolesky, R. L. Truby, A. S. Gladman, T. A. Busbee, K. A. Homan and J. A. Lewis, Adv. Mater., 2014, 26, 3124–3130 CrossRef CAS PubMed.
- M. Patel, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomaterials, 2018, 159, 91–107 CrossRef CAS PubMed.
- F.-Y. Hsieh, H.-H. Lin and S.-H. Hsu, Biomaterials, 2015, 71, 48–57 CrossRef CAS PubMed.
- E. Rani Aluri, E. Gannon, K. Singh, S. Kolagatla, K. Kowiorski, S. Shingte, E. McKiernan, C. Moloney, K. McGarry, L. Jowett, B. J. Rodriguez, D. F. Brougham and J. K. Wychowaniec, J. Colloid Interface Sci., 2022, 611, 533–544 CrossRef CAS PubMed.
- N. E. Fedorovich, I. Swennen, J. Girones, L. Moroni, C. A. van Blitterswijk, E. Schacht, J. Alblas and W. J. A. Dhert, Biomacromolecules, 2009, 10, 1689–1696 CrossRef CAS PubMed.
- A. V. Vashi, E. Keramidaris, K. M. Abberton, W. A. Morrison, J. L. Wilson, A. J. O’Connor, J. J. Cooper-White and E. W. Thompson, Biomaterials, 2008, 29, 573–579 CrossRef CAS PubMed.
- R. N. Shamma, R. H. Sayed, H. Madry, N. S. El Sayed and M. Cucchiarini, Tissue Eng., Part B, 2021, 28, 451–463 CrossRef PubMed.
- Z. Fu, L. Ouyang, R. Xu, Y. Yang and W. Sun, Mater. Today, 2022, 52, 112–132 CrossRef CAS.
- M. S. H. Akash and K. Rehman, J. Controlled Release, 2015, 209, 120–138 CrossRef CAS PubMed.
- J.-J. Xuan, Y.-D. Yan, D. H. Oh, Y. K. Choi, C. S. Yong and H.-G. Choi, Drug Delivery, 2011, 18, 305–311 CrossRef CAS PubMed.
- C. K. Chung, M. F. Fransen, K. van der Maaden, Y. Campos, J. Garcia-Couce, D. Kralisch, A. Chan, F. Ossendorp and L. J. Cruz, J. Controlled Release, 2020, 323, 1–11 CrossRef CAS PubMed.
- R. Jaquilin, O. S. Oluwafemi, S. Thomas and A. O. Oyedeji, J. Drug Delivery Sci. Technol., 2022, 72, 103390 CrossRef.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS PubMed.
- H.-R. Lin and K. C. Sung, J. Controlled Release, 2000, 69, 379–388 CrossRef CAS PubMed.
- A. Sosnik, D. Cohn, J. S. Román and G. A. Abraham, J. Biomater. Sci., Polym. Ed., 2003, 14, 227–239 CrossRef CAS PubMed.
- I. M. A. Diniz, C. Chen, X. Xu, S. Ansari, H. H. Zadeh, M. M. Marques, S. Shi and A. Moshaverinia, J. Mater. Sci.: Mater. Med., 2015, 26, 153 CrossRef PubMed.
- A. Arranja, A. P. Schroder, M. Schmutz, G. Waton, F. Schosseler and E. Mendes, J. Controlled Release, 2014, 196, 87–95 CrossRef CAS PubMed.
- D. Y. Kim, D. Y. Kwon, J. S. Kwon, J. H. Park, S. H. Park, H. J. Oh, J. H. Kim, B. H. Min, K. Park and M. S. Kim, Biomaterials, 2016, 85, 232–245 CrossRef CAS PubMed.
- C. Y. Gong, S. Shi, P. W. Dong, X. L. Zheng, S. Z. Fu, G. Guo, J. L. Yang, Y. Q. Wei and Z. Y. Qian, BMC Biotechnol., 2009, 9, 8 CrossRef PubMed.
- S. H. Lee, S. H. Choi, S. H. Kim and T. G. Park, J. Controlled Release, 2008, 125, 25–32 CrossRef CAS PubMed.
- V. K. Rangasami, G. Nawale, K. Asawa, S. Kadekar, S. Samanta, B. Nilsson, K. N. Ekdahl, S. Miettinen, J. Hilborn, Y. Teramura, O. P. Varghese and O. P. Oommen, Biomacromolecules, 2021, 22, 1980–1989 CrossRef CAS PubMed.
- N. J. Treacy, S. Clerkin, J. L. Davis, C. Kennedy, A. F. Miller, A. Saiani, J. K. Wychowaniec, D. F. Brougham and J. Crean, Bioact. Mater., 2023, 21, 142–156 CAS.
- S. Clerkin, K. Singh, J. L. Davis, N. J. Treacy, I. Krupa, E. G. Reynaud, R. M. Lees, S. R. Needham, D. MacWhite-Begg, J. K. Wychowaniec, D. F. Brougham and J. Crean, Biomaterials, 2025, 123349, DOI:10.1016/j.biomaterials.2025.123349.
- K. H. Bae, S. H. Choi, S. Y. Park, Y. Lee and T. G. Park, Langmuir, 2006, 22, 6380–6384 CrossRef CAS PubMed.
- E. Lippens, I. Swennen, J. Gironès, H. Declercq, G. Vertenten, L. Vlaminck, F. Gasthuys, E. Schacht and R. Cornelissen, J. Biomater. Appl., 2013, 27, 828–839 CrossRef PubMed.
- I. Swennen, V. Vermeersch, M. Hornof, E. Adriaens, J. P. Remon, A. Urtti and E. H. Schacht, J. Controlled Release, 2006, 116, e21–e24 CrossRef CAS PubMed.
- R. R. Wakaskar, S. P. R. Bathena, S. B. Tallapaka, V. V. Ambardekar, N. Gautam, R. Thakare, S. M. Simet, S. M. Curran, R. K. Singh, Y. Dong and J. A. Vetro, Pharm. Res., 2015, 32, 1028–1044 CrossRef CAS PubMed.
- S. H. Lee, Y. Lee, S.-W. Lee, H.-Y. Ji, J.-H. Lee, D. S. Lee and T. G. Park, Acta Biomater., 2011, 7, 1468–1476 CrossRef CAS PubMed.
- Y.-s Jung, W. Park, H. Park, D.-K. Lee and K. Na, Carbohydr. Polym., 2017, 156, 403–408 CrossRef CAS PubMed.
- S. Chatterjee, P. C. Hui, C. W. Kan and W. Wang, Sci. Rep., 2019, 9, 11658 CrossRef PubMed.
- J. García-Couce, M. Tomás, G. Fuentes, I. Que, A. Almirall and L. J. Cruz, Gels, 2022, 8, 44 CrossRef PubMed.
- J. Cao, M. Su, N. Hasan, J. Lee, D. Kwak, D. Y. Kim, K. Kim, E. H. Lee, J. H. Jung and J. W. Yoo, Pharmaceutics, 2020, 12, 926 CrossRef CAS PubMed.
- Y. Lee, H. J. Chung, S. Yeo, C.-H. Ahn, H. Lee, P. B. Messersmith and T. G. Park, Soft Matter, 2010, 6, 977–983 RSC.
- S. Utech and A. R. Boccaccini, J. Mater. Sci., 2016, 51, 271–310 CrossRef CAS.
- C. Sheffield, K. Meyers, E. Johnson and R. M. Rajachar, Gels, 2018, 4, 51 CrossRef PubMed.
- M. A. Abou-Shamat, J. Calvo-Castro, J. L. Stair and M. T. Cook, Macromol. Chem. Phys., 2019, 220, 1900173 CrossRef.
- H. H. Jung, K. Park and D. K. Han, J. Controlled Release, 2010, 147, 84–91 CrossRef CAS PubMed.
- D. Bhatnagar, N. Mehandru, Y. Sun, J. Nanda and M. Rafailovich, J. Chem. Biol. Interfaces, 2013, 1, 1–7 CrossRef.
- S. Bobbala, B. Gibson, A. B. Gamble, A. McDowell and S. Hook, Immunol. Cell Biol., 2018, 96, 656–665 CrossRef CAS PubMed.
- H.-R. Lin, K. C. Sung and W.-J. Vong, Biomacromolecules, 2004, 5, 2358–2365 CrossRef CAS PubMed.
- L.-S. Yap and M.-C. Yang, Colloids Surf., B, 2016, 146, 204–211 CrossRef CAS PubMed.
- L. Gao, X. Wang, J. Ma, D. Hao, P. Wei, L. Zhou and G. Liu, Colloids Surf., B, 2016, 140, 307–316 CrossRef CAS PubMed.
- Y. Shachaf, M. Gonen-Wadmany and D. Seliktar, Biomaterials, 2010, 31, 2836–2847 CrossRef CAS PubMed.
- Y.-J. Jang, S. Y. Chun, G. N. Kim, J. R. Kim, S. H. Oh, J. H. Lee, B. S. Kim, P. H. Song, E. S. Yoo and T. G. Kwon, Acta Biomater., 2014, 10, 3117–3125 CrossRef CAS.
- X. Li, E.-k Park, K. Hyun, L. Oktavia and M. Kwak, J. Rheol., 2018, 62, 107–120 CrossRef CAS.
- C. Loebel, S. E. Szczesny, B. D. Cosgrove, M. Alini, M. Zenobi-Wong, R. L. Mauck and D. Eglin, Biomacromolecules, 2017, 18, 855–864 CrossRef CAS PubMed.
- M. Isik, B. O. Okesola, C. C. Eylem, E. Kocak, E. Nemutlu, E. Emregul, M. D'Este and B. Derkus, Biomacromolecules, 2022, 23, 4254–4267 CrossRef CAS PubMed.
- T. Kamperman, S. Henke, J. F. Crispim, N. G. A. Willemen, P. J. Dijkstra, W. Lee, H. L. Offerhaus, M. Neubauer, A. M. Smink, P. de Vos, B. J. de Haan, M. Karperien, S. R. Shin and J. Leijten, Adv. Mater., 2021, 33, e2102660 CrossRef PubMed.
- L. Oktavia and M. Kwak, Master's thesis, Pukyong National University, 2016 DOI:10.13140/RG.2.2.19347.32809.
- M. Walker, E. W. Pringle, G. Ciccone, L. Oliver-Cervelló, M. Tassieri, D. Gourdon and M. Cantini, Adv. Healthcare Mater., 2024, 13, 2302571 CrossRef CAS PubMed.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef PubMed.
- N. Maghsoudnia, R. B. Eftekhari, M. Roshandel, M. Ahmadi, M. Edalat, M. Hajimolaali, S. Milani, M. K. Shal and F. A. Dorkoosh, in Polysaccharide-based Biomaterials, The Royal Society of Chemistry, 2022, pp. 126–171 10.1039/9781839166235-00126.
- Q. Zhang, X. Wei, Y. Ji, L. Yin, Z. Dong, F. Chen, M. Zhong, J. Shen, Z. Liu and L. Chang, J. Mater. Chem. B, 2020, 8, 5441–5450 RSC.
- Y. W. Ding, Z. Y. Wang, Z. W. Ren, X. W. Zhang and D. X. Wei, Biomater. Sci., 2022, 10, 3393–3409 RSC.
- R. Silva-Carvalho, T. Leao, A. I. Bourbon, C. Goncalves, L. M. Pastrana, P. Parpot, I. Amorim, A. M. Tomas and F. M. Gama, Biomater. Sci., 2022, 10, 1952–1967 RSC.
- N. Xu, J. Xu, X. Zheng and J. Hui, ChemistryOpen, 2020, 9, 451–458 CrossRef CAS PubMed.
- P. Díaz-Rodríguez, A. Rey-Rico, H. Madry, M. Landin and M. Cucchiarini, Int. J. Pharm., 2015, 496, 614–626 CrossRef PubMed.
- W. L. Stoppel, J. C. White, S. D. Horava, S. R. Bhatia and S. C. Roberts, Acta Biomater., 2011, 7, 3988–3998 CrossRef CAS PubMed.
- R. Abka-khajouei, L. Tounsi, N. Shahabi, A. K. Patel, S. Abdelkafi and P. Michaud, 2022, 20.
- H. Xu and S. Matysiak, Chem. Commun., 2017, 53, 7373–7376 RSC.
- P. T. Smith, A. Basu, A. Saha and A. Nelson, Polymer, 2018, 152, 42–50 CrossRef CAS.
- M. Müller, J. Becher, M. Schnabelrauch and M. Zenobi-Wong, Biofabrication, 2015, 7, 035006 CrossRef PubMed.
- H. Arslan, A. Nojoomi, J. Jeon and K. Yum, Adv. Sci., 2019, 6, 1800703 CrossRef PubMed.
- R. N. Shamma, R. H. Sayed, H. Madry, N. S. El Sayed and M. Cucchiarini, Tissue Eng., Part B, 2022, 28(2), 451–463 CrossRef CAS PubMed.
- H.-W. Kang, S. J. Lee, I. K. Ko, C. Kengla, J. J. Yoo and A. Atala, Nat. Biotechnol., 2016, 34, 312–319 CrossRef CAS PubMed.
- R. Suntornnond, E. Y. S. Tan, J. An and C. K. Chua, Sci. Rep., 2017, 7, 16902 CrossRef PubMed.
- A. Forget, T. Derme, D. Mitterberger, M. Heiny, C. Sweeney, L. Mudili, T. R. Dargaville and V. P. Shastri, Emergent Mater., 2019, 2, 233–243 CrossRef CAS.
- C. Xue, B.-C. Choi, S. Choi, P. V. Braun and D. E. Leckband, Adv. Funct. Mater., 2012, 22, 2394–2401 CrossRef CAS.
- M. E. Nash, W. M. Carroll, N. Nikoloskya, R. Yang, C. O. Connell, A. V. Gorelov, P. Dockery, C. Liptrot, F. M. Lyng, A. Garcia and Y. A. Rochev, ACS Appl. Mater. Interfaces, 2011, 3, 1980–1990 CrossRef CAS PubMed.
- Z. Piao, J. K. Park, S. J. Park and B. Jeong, Biomacromolecules, 2021, 22, 5390–5399 CrossRef CAS PubMed.
- S. Pacelli, F. Acosta, A. R. Chakravarti, S. G. Samanta, J. Whitlow, S. Modaresi, R. P. H. Ahmed, J. Rajasingh and A. Paul, Acta Biomater., 2017, 58, 479–491 CrossRef CAS PubMed.
- M. D'Este, C. M. Sprecher, S. Milz, D. Nehrbass, I. Dresing, S. Zeiter, M. Alini and D. Eglin, J. Biomed. Mater. Res., Part A, 2016, 104, 1469–1478 CrossRef PubMed.
- H. E. Canavan, X. Cheng, D. J. Graham, B. D. Ratner and D. G. Castner, Langmuir, 2005, 21, 1949–1955 CrossRef CAS PubMed.
- A. M. Aladdad, M. H. Amer, L. Sidney, A. Hopkinson, L. J. White, C. Alexander and F. R. A. J. Rose, Acta Biomater., 2019, 95, 427–438 CrossRef CAS PubMed.
- M. A. Cooperstein and H. E. Canavan, Langmuir, 2010, 26, 7695–7707 CrossRef CAS PubMed.
- J. Litowczenko, J. Gapinski, R. Markiewicz, A. Wozniak, J. K. Wychowaniec, B. Peplinska, S. Jurga and A. Patkowski, Mater. Sci. Eng., C, 2021, 118, 111507 CrossRef CAS PubMed.
- B. R. Park, Y. Nabae, M. Surapati, T. Hayakawa and M.-A. Kakimoto, Polym. J., 2013, 45, 210–215 CrossRef CAS.
- T. Li, F. Huang, D. Diaz-Dussan, J. Zhao, S. Srinivas, R. Narain, W. Tian and X. Hao, Biomacromolecules, 2020, 21, 1254–1263 CrossRef CAS PubMed.
- K. Brewer, B. Gundsambuu, P. Facal Marina, S. C. Barry and A. Blencowe, Polymers, 2020, 12(2), 367 CrossRef CAS PubMed.
- W. Guo, L. Douma, M. H. Hu, D. Eglin, M. Alini, A. Secerovic, S. Grad, X. Peng, X. Zou, M. D'Este and M. Peroglio, Carbohydr. Polym., 2022, 277, 118828 CrossRef CAS PubMed.
- M. D’Este, M. Alini and D. Eglin, Carbohydr. Polym., 2012, 90, 1378–1385 CrossRef PubMed.
- C. L. Pereira, R. M. Gonçalves, M. Peroglio, G. Pattappa, M. D'Este, D. Eglin, M. A. Barbosa, M. Alini and S. Grad, Biomaterials, 2014, 35, 8144–8153 CrossRef CAS PubMed.
- D. Mortisen, M. Peroglio, M. Alini and D. Eglin, Biomacromolecules, 2010, 11, 1261–1272 CrossRef CAS PubMed.
- B. Erlandsson, Polym. Degrad. Stab., 2002, 78, 571–575 CrossRef CAS.
- H. Feng, J. Sun and P. Jiang, Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi, 2008, 22, 28–31 Search PubMed.
- X. Y. Xiong, K. C. Tam and L. H. Gan, Macromolecules, 2004, 37, 3425–3430 CrossRef CAS.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- N. Hirun, P. Kraisit and S. Soontaranon, Polymers, 2023, 15(11), 2465 CrossRef CAS PubMed.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- R. L. Sala, M. Y. Kwon, M. Kim, S. E. Gullbrand, E. A. Henning, R. L. Mauck, E. R. Camargo and J. A. Burdick, Tissue Eng., Part A, 2017, 23, 935–945 CrossRef CAS PubMed.
- P. J. Ness, N. Mamalis, L. Werner, S. Maddula, D. K. Davis, E. D. Donnenfeld and R. J. Olson, Am. J. Ophthalmol., 2010, 150, 498–504.e491 CrossRef CAS PubMed.
- M. C. Beard, L. H. Cobb, C. S. Grant, A. Varadarajan, T. Henry, E. A. Swanson, S. Kundu and L. B. Priddy, J. Biomed. Mater. Res., Part B, 2021, 109, 338–347 CrossRef CAS PubMed.
- A. De Lauretis, A. Eriksson Agger, A. Pal, J. Skov Pedersen, S. M. Szostak, R. Lund, S. P. Lyngstadaas, J. E. Ellingsen, D. Linke and H. J. Haugen, Regener. Biomater., 2025, 12, rbaf005 CrossRef PubMed.
- C. de Kergariou, G. J. Day, A. W. Perriman, J. P. K. Armstrong and F. Scarpa, Soft Matter, 2024, 20, 4021–4034 RSC.
- M. Shahbazi, H. Jäger, R. Ettelaie, A. Mohammadi and P. Asghartabar Kashi, Addit. Manuf., 2023, 71, 103598 CAS.
Footnote |
| † Prof. John Crean has passed away on the 10th of May 2025, before the resubmission of the revised version of this manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |