
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manganese catalysed dehydrogenative synthesis of polyureas from diformamide and diamines†
Angus
McLuskie‡
a,
Claire N.
Brodie‡
 a,
Michele
Tricarico
b,
Chang
Gao
a,
Gavin
Peters
a,
Aaron B.
Naden
a,
C. Logan
Mackay
c,
Jin-Chong
Tan
*b and
Amit
Kumar
*a
a,
Michele
Tricarico
b,
Chang
Gao
a,
Gavin
Peters
a,
Aaron B.
Naden
a,
C. Logan
Mackay
c,
Jin-Chong
Tan
*b and
Amit
Kumar
*a
aSchool of Chemistry, University of St. Andrews, North Haugh, St. Andrews, KY169ST, UK. E-mail: ak336@st-andrews.ac.uk
bDepartment of Engineering Science, University of Oxford, Parks Road, Oxford, OX13PJ, UK. E-mail: jin-chong.tan@eng.ox.ac.uk
cSchool of Chemistry, University of Edinburgh, EH93FJ, UK
First published on 15th May 2023
Abstract
We report here the synthesis of polyureas from the dehydrogenative coupling of diamines and diformamides. The reaction is catalysed by a manganese pincer complex and releases H2 gas as the only by-product making the process atom-economic and sustainable. The reported method is greener in comparison to the current state-of-the-art production routes that involve diisocyanate and phosgene feedstock. We also report here the physical, morphological, and mechanical properties of synthesized polyureas. Based on our mechanistic studies, we suggest that the reaction proceeds via isocyanate intermediates formed by the manganese catalysed dehydrogenation of formamides.
Introduction
Polyureas are useful plastics with applications in the construction, coating, and biomedical industries.1–5 Their global market is £780 million per year and is expected to grow to £1.4 billion by 2030.6 Polyureas are industrially made from the reaction of diamines with diisocyanates; diisocyanates are made from phosgene gas which is extremely toxic and hazardous to human health and the environment. Thus, there is an urgent need to develop greener methods that avoid toxic feedstock for the synthesis of polyureas.Indeed, a few methods that avoid diisocyanates feedstock have been reported for the synthesis of polyureas. For example, polyureas can be made from the condensation of diamines with CO2, however, either a dehydrating agent or a harsh reaction condition (e.g. 100 bar and 180 °C) is needed for this process.7–9 The use of other carbonylating agents such as urea,10 carbamate11 or biscarbamate12 instead of diisocyanates has also been demonstrated for the synthesis of polyureas.13 However, they suffer from the issues of limited substrate scope, low selectivity, or the use of harsh reaction conditions or specialty solvents.
Catalytic dehydrogenation is a green and atom-economic approach to the synthesis of organic compounds.14,15 The synthesis of urea derivatives has been reported from the dehydrogenative coupling of amines and methanol using ruthenium and iron pincer catalysts by Hong16 and Bernskoetter,17 respectively. Milstein has reported the synthesis of urea derivatives by the dehydrogenative coupling of formamide with amines in the presence of a ruthenium pincer catalyst.18 Gunanathan has also reported the synthesis of urea derivatives from the coupling of N,N′-dimethylformamide and amine where dimethylamine was observed as a by-product in the presence of a ruthenium-pincer catalyst.19
We have recently expanded the concept of the dehydrogenative synthesis of urea derivatives and reported a new method for the synthesis of polyureas from the dehydrogenative coupling of diamines and methanol using ruthenium20 and manganese21 pincer complexes. Liu has also reported the synthesis of polyureas from the dehydrogenative coupling of diamines and methanol using analogous manganese pincer complexes.22 The discovered method substitutes diisocyanates with methanol which is relatively much less toxic. Additionally, methanol is cheaper and can be made from renewable feedstock which makes the process potentially more sustainable.23 However, this method has two limitations: (a) molecular weight issue: the need for high temperature (150 °C) makes it difficult to use methanol under open conditions due to its low boiling point (65 °C) because of which reactions were conducted in a sealed system. This limits the polymer chain length or their molecular weight due to the accumulation of H2 gas in the system which can disfavour the dehydrogenation reaction. (b) Functionality issue: only one type of functionality (e.g. either aliphatic or aromatic) can be incorporated in the polyurea using this method whereas the conventional methodology can make polyurea containing two different functionalities – one coming from diisocyanate and the other from diamine. This is particularly useful for various applications where a rigid/hard segment (from aromatic groups) and a soft/flexible segment (from aliphatic chains) are needed in the polymer. The presence of both hard and soft segments in polyureas leads to a unique microphase separation in the polymer microstructure that leads to desired macroscopic properties, such as stability, high strength, and aging resistance.4,24
An alternative approach to avoid diisocyanates as well as overcome these limitations is to make polyureas from the dehydrogenative coupling of diformamides and diamines. This approach has been recently demonstrated by Robertson using a ruthenium pincer catalyst (Fig. 1).25 However, the use of precious metals such as ruthenium raises concerns of sustainability due to their high cost and low abundance on the earth's crust. The use of a catalyst based on an earth-abundant metal such as manganese, which is the third most abundant transition metal on earth's crust, can make the process more cost-effective and sustainable.26 Indeed, several (de)hydrogenative transformations catalysed by manganese complexes have been reported in the recent past.27–30 We report here that a manganese pincer complex Mn(PNHP-iPr)(CO)2Br, 1, (PNHP-iPr = N,N′-bis(diisopropylphosphinoethyl)amine) is an effective precatalyst for the dehydrogenative coupling of diformamides and diamines to form polyureas (Fig. 1). We also present their chemical (spectroscopy, mass spectrometry), physical (melting temperatures, decomposition temperatures), morphological (SEM) and mechanical (indentation modulus, hardness) properties of the synthesized polyureas.
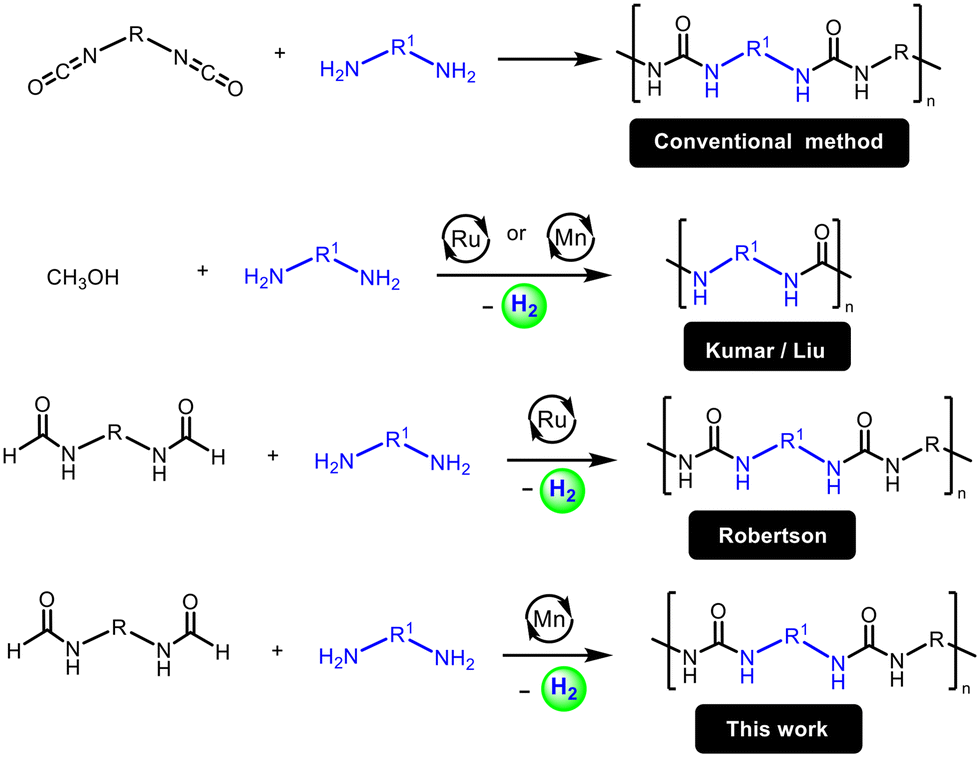 | ||
| Fig. 1 Synthesis of polyureas using conventional and dehydrogenative routes. | ||
Results and discussion
We started our investigation by studying the reaction of N,N′-(1,4-phenylene)diformamide (DF1, 0.5 mmol) with diaminooctane (DA1, 0.5 mmol) in the presence of a manganese pincer complex 1 (2 mol%) and KOtBu (8 mol%) in THF solvent in a sealed system. Performing the reaction at 150 °C, in THF under sealed conditions for 24 h led to the isolation of an off-white solid in 82% yield (Table 1, entry 1). The product did not dissolve in common solvents such as water, THF, toluene, DCM, and CHCl3. It exhibited partial solubility in TFA (trifluoroacetic acid), therefore d-TFA was used for the NMR analysis. 1H and 13C{1H} NMR spectra showed signals corresponding to both aliphatic and aromatic protons that would come from (aliphatic) diamines and (aromatic) diformamides. The end group analysis of the partially soluble sample showed the number average molecular weight (Mn) to be 1657 Da at the end of a 24 h reaction period. In the case of Robertson's report on the ruthenium catalysed synthesis of polyureas from diformamides and diamines, the molecular weight (Mn) of a polyurea was found to significantly increase after 1 day of reaction time (e.g. 3700 Da after day 1; 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 Da after day 2; and 32800 Da after day 3) in a manner expected for a step growth polymerisation mechanism.25 However, in our case, the formation of insoluble/poorly soluble material at the end of polymerisation reaction has made the estimation of molecular weight difficult. The IR spectrum of the polyurea (entry 1) showed a signal at 1680 cm−1 corresponding to the carbonyl stretching frequency and 1576 cm−1 corresponding to N–H bending both characteristics to a urea functional group. Interesting insights were obtained from MALDI FT-ICR mass spectrometry studies that showed the presence of repeating units corresponding to polyurea formed from aromatic diformamide (DF1) and aliphatic diamine (DA1). Mass spectrometry data also showed that the formed polymers contained all combinations of end groups: amine, amine; formyl, formyl; and formyl, amine. More interestingly, the mass spectrometry data revealed that the formed material is a mixture of copolyurea (formed from DF1 + DA1) as well as homopolyurea (resulting only from DA1). We speculate that the formation of the latter occurs via decarbonylation (vide infra) or/and the transformylation of DA1 with DF1 with the elimination of p-phenylenediamine (Fig. 2). Such transformylation reaction has been reported by us and others in the past.21,31,32 Considering the insoluble nature of the majority of the isolated material it was not possible to determine the exact ratio of homopolyurea and copolyurea. However, for an approximate comparison in different reaction conditions, we have provided the ratio of the integration of aliphatic vs. aromatic signals from the 1H NMR spectra (Table 1, see ESI† Fig. S1). These need to be used with caution as the isolated materials are only partially soluble in d-TFA. TGA (thermogravimetric analysis) and DSC (differential scanning calorimetry) studies were conducted to estimate the decomposition temperature (Td) and melting temperature (Tm) of the polyurea which were found to be 246 °C and 221 °C respectively in this case.
300 Da after day 2; and 32800 Da after day 3) in a manner expected for a step growth polymerisation mechanism.25 However, in our case, the formation of insoluble/poorly soluble material at the end of polymerisation reaction has made the estimation of molecular weight difficult. The IR spectrum of the polyurea (entry 1) showed a signal at 1680 cm−1 corresponding to the carbonyl stretching frequency and 1576 cm−1 corresponding to N–H bending both characteristics to a urea functional group. Interesting insights were obtained from MALDI FT-ICR mass spectrometry studies that showed the presence of repeating units corresponding to polyurea formed from aromatic diformamide (DF1) and aliphatic diamine (DA1). Mass spectrometry data also showed that the formed polymers contained all combinations of end groups: amine, amine; formyl, formyl; and formyl, amine. More interestingly, the mass spectrometry data revealed that the formed material is a mixture of copolyurea (formed from DF1 + DA1) as well as homopolyurea (resulting only from DA1). We speculate that the formation of the latter occurs via decarbonylation (vide infra) or/and the transformylation of DA1 with DF1 with the elimination of p-phenylenediamine (Fig. 2). Such transformylation reaction has been reported by us and others in the past.21,31,32 Considering the insoluble nature of the majority of the isolated material it was not possible to determine the exact ratio of homopolyurea and copolyurea. However, for an approximate comparison in different reaction conditions, we have provided the ratio of the integration of aliphatic vs. aromatic signals from the 1H NMR spectra (Table 1, see ESI† Fig. S1). These need to be used with caution as the isolated materials are only partially soluble in d-TFA. TGA (thermogravimetric analysis) and DSC (differential scanning calorimetry) studies were conducted to estimate the decomposition temperature (Td) and melting temperature (Tm) of the polyurea which were found to be 246 °C and 221 °C respectively in this case.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Diformamide | Diamine | Base | Solvent | N2 | Yield/% | M n (MALDI) | Al:Ar |
T d/°C | T m/°C |
|
a Catalytic conditions: diamine (0.5 mmol), diformamide (0.5 mmol), solvent (2 mL), complex 1 (2 mol%), and KOtBu (8 mol%). Al:Ar = aliphatic:aromatic NMR integral relationship. Mn (MALDI, g mol−1): value is estimated as the maximum signal observed in MALDI FT-ICR mass spectrometry. Td (decomposition temperature) was recorded at 5% mass loss. Tm stands for melting temperature.
b
M
n of 44000 g mol−1 was estimated using GPC.
|
||||||||||
| 1 | DF1 | DA1 | KOtBu | THF | Sealed | 82 | 1877 | 1.67 | 246 | 221 |
| 2 | DF1 | DA1 | KOtBu | Anisole | Sealed | 77 | 2111 | 1.03 | 246 | 220 |
| 3 | DF1 | DA1 | KOtBu | Anisole | Open | 32 | 2111 | 1.67 | 237 | 201 |
| 4 | DF1 | DA1 | KOtBu | Diglyme | Open | 79 | 1904 | 2.01 | 238 | 212 |
| 5 | DF1 | DA1 | KOtBu | DMSO | Open | 44 | — | 4.88 | 266 | 211 |
| 6 | DF1 | DA1 | K2CO3 | Anisole | Open | 30 | 1770 | 1.73 | 242 | 204 |
| 7 | DF2 | DA1 | KOtBu | Anisole | Open | 85 | 2111 | N/A | 253 | 216 |
| 8 | DF2 | DA1 | KOtBu | Diglyme | Open | 25 | 2110 | N/A | 213 | 210 |
| 9 | DF2 | DA1 | KOtBu | THF | Open | 78 | 2594 | N/A | 247 | 215 |
| 10 | DF1 | DA2 | KOtBu | Anisole | Open | 94 | 3140b | 1.16 | 223 | — |
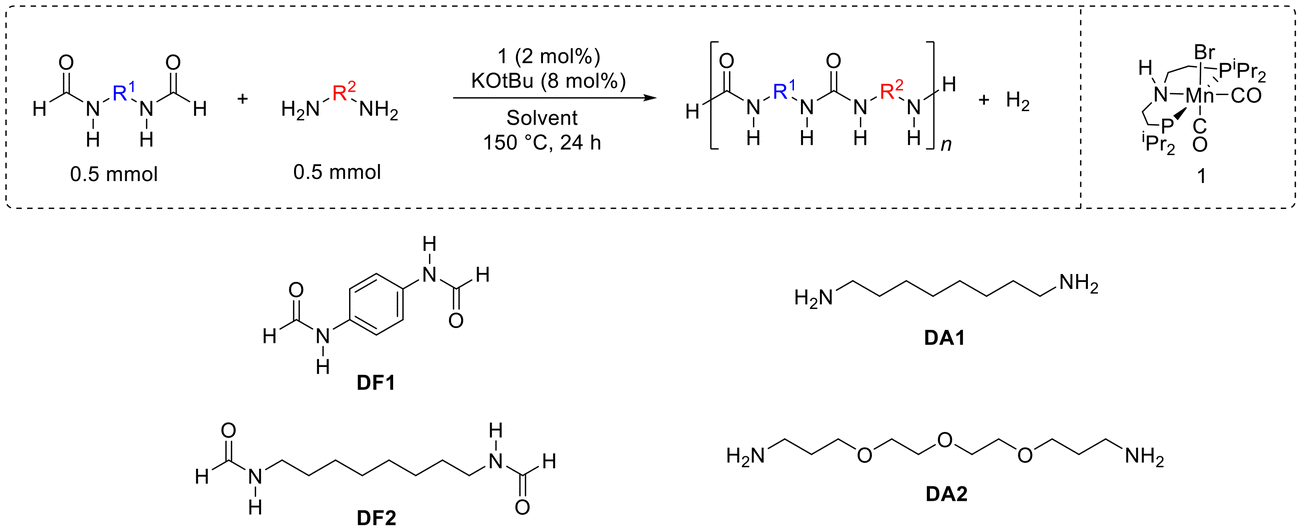
 | ||
| Fig. 2 Proposed pathway for the formation of alternate copolyurea (A), homopolyurea (B) and random copolyurea (C) from the manganese catalysed dehydrogenative coupling of diformamides and diamines. | ||
We also studied the effect of solvents on the polymerisation reaction as a high boiling point solvent can allow the reaction to occur under open conditions (flow of N2 or Ar) and at high temperature, and the polarity of solvent can influence the solubility of oligomers. Both these factors can affect the polymer chain length. Changing the solvent to anisole keeping the remaining conditions the same led to the observation of similar composition of polymers, albeit at a slightly reduced yield (77%, Table 1, entry 2) with respect to that in the case of THF (entry 1). Performing the reaction in an open system using anisole as a solvent resulted in a drop in yield (32%; entry 3). However, this issue is overcome when the solvent is changed to diglyme and the catalysis is carried out in open conditions under the flow of N2 (entry 4), the product can be isolated in 79% yield. MALDI FT-ICR mass spectrometry showed a higher abundance of signals corresponding to copolyurea than those to homopolyurea from the transformylation reaction (see ESI,† Table S2). Performing the reaction in DMSO (entry 5), results in a significant reduction in yield (44%). An experiment carried out in anisole with a change in base to K2CO3 led to a low yield of 30% (entry 6). Additionally, conducting control experiments in the absence of either a base or complex 1 (remaining conditions as per described in Table 1, entry 1) did not lead to the isolation of any product and starting materials were recovered at the end of the reaction. Attempting to synthesise polyurea from DF1 with a secondary amine, piperazine, was also unsuccessful (see ESI,† Table S1, entry 15).
We next studied the coupling of diamine DA1 with another diformamide DF2 (N,N′-(octane-1,8-diyl)diformamide). The “R” groups of DF2 and DA1 are identical, so, regardless of transformylation, a reaction of these together would generate polymers of the same chemical structure, and identical to the polyurea made from the dehydrogenative coupling of 1,8-diaminooctane and methanol as previously reported by us using a manganese pincer complex 1.20 The catalytic studies were conducted in anisole, diglyme, and THF under the open flow of nitrogen. Anisole and THF led to higher yields (85% and 78%) whereas lower yields were obtained in the case of diglyme as described in Table 1 (entries 7–9). Td (253 °C) and Tm (216 °C) in the case of polyurea made from DF2 and DA1 (entry 7) were found to be higher than that of polyurea made from DA1 and methanol (Td, 190 °C and Tm, 89 °C) under the reaction condition: complex 1 (2 mol%), KOtBu (8 mol%), 150 °C, 24 h THF.21
To resolve the difficulty in determining polyurea molecular weight because of poor solubility (vide supra), we have also performed the polymerisation of DF1 with a diamine which has a glycol-rich backbone, DA2. To our satisfaction, this reaction yielded polyurea in high yield (94%) that is water-soluble. While MALDI-MS returned the highest molecular weight fragment at m/z 3140 g mol−1, end-group analysis of the 1H NMR spectrum of this soluble sample in D2O estimates Mn to be ∼19000 g mol−1. Furthermore, GPC analysis of this sample (H2O eluant, relative to monodisperse PEG/PEO standards) estimates a higher molecular weight still, Mn 44000 g mol−1, with polydispersity, Đ = 1.4. Despite narrow dispersity measured, the GPC curve obtained for this sample does exhibit bimodality (see Fig. S86†), possibly due to the presence of both alternate copolyureas and random copolyureas, as suggested by MALDI-MS. Taking the molecular weight information obtained from end-group analysis and GPC for this soluble polymer, it is likely, therefore, that the molecular weights obtained from MALDI-MS for other polyurea samples here are underestimated. Indeed, observation of polymers with molecular weight >5000 g mol−1 is known to be difficult by MALDI-MS.33,34 We also studied the morphology of polyureas made from DF1 and DA1 (containing both aromatic and aliphatic groups, Table 1, entry 4) as well as from DF2 and DA1 (containing only aliphatic groups, Table 1, entry 7). The data obtained from the scanning electron microscope showed that the aliphatic polyurea (from DF2 and DA1, Table 1, entry 7) is composed of small sheet-like features typically ∼1–3 μm long and ∼50–70 nm thick whereas the aromatic polyurea (from DF1 and DA1; Table 1, entry 7) exhibits a more porous and less densely packed microstructure within the larger agglomerations (see ESI,† Fig. S83). Energy dispersive X-ray spectroscopy (EDX) provides a convenient means of rapidly assessing the elemental composition of the samples. The EDX spectra for both samples confirmed the presence of C, N, and O.
To get an understanding of the mechanical properties of polyureas made from this method, we performed nanoindentation analysis of both aromatic (from DF1 and DA1; Table 1, entry 4) as well as aliphatic polyurea (from DF2 and DA1; Table 1, entry 7). The tests were carried out using a KLA iMicro nanoindenter, equipped with a 50 mN force actuator and a Berkovich tip. Continuous stiffness measurements (CSM) were performed, allowing to measure the indentation modulus E* (E* = E/1 − ν2) which can be used in place of Young's modulus E when the Poisson's ratio ν is unknown. Our studies showed a high indentation modulus of 4.25 ± 0.72 GPa and a hardness of 252 ± 64 MPa for aromatic polyurea (from DF1 and DA1, entry 4) as well as for the aliphatic polyurea (indentation modulus: 4.93 ± 0.63 GPa; hardness: 291 ± 59 MPa made from DF2 and DA1, entry 7, see ESI,† Section S12). The elastic modulus of a commercial polyurea XS-350 has been reported to be less than what we observe here (100 MPa).35
In our recent work on the manganese catalysed synthesis of urea derivatives from formamides and amines, we reported a mechanism based on the DFT computation where the manganese complex 2 dehydrogenates a formamide to form an isocyanate followed by its subsequent reaction with an amine to form a urea derivative.21 We speculate that the synthesis of polyureas from diformamides and diamines as reported here proceeds via a similar pathway. To probe further, we monitored the reaction under stoichiometric and catalytic conditions using NMR spectroscopy and mass spectrometry. The reaction of complex 1 with KOtBu (1.2 equivalents) in an NMR tube in toluene-d8 resulted in the formation of the amido-complex 2 as also previously reported (Fig. 3).36 Addition of 2 equivalents of formamide (HCONH2) or formanilide (PhCONH2) to the in situ formed complex 2 resulted in the formation of a new complex as evidenced by the complete consumption of the signal corresponding to 2 (δP 113.0 ppm) in 10 minutes (room temperature) and the concomitant appearance of a new signal in the 31P{1H} NMR spectra. Analysis of the 1H and 13C{1H} NMR spectra as well as HRMS (EI) data suggests the formation of complexes 3 and 5 through N–H activation of formamide and formanilide via metal–ligand cooperation.37 The N–H activation of formanilide by complex 2 to form complex 5via metal–ligand cooperation has been recently reported by Boncella and Tondreau.38 Interestingly, heating the NMR tube containing the in situ formed complex 3 at 110 °C for 4 days led to the quantitative formation of a new complex (31P{1H} NMR: δP 88.6 ppm) which was characterised as the manganese-isocyanate complex 4 using NMR spectroscopy and mass spectrometry. The 1H NMR spectrum also shows a signal at δH 4.51 corresponding to dissolved H2 that would be produced during the formation of isocyanate upon dehydrogenation of formamide. These results agree with a recent report by Milstein where a ruthenium-PNP pincer complex was found to activate and dehydrogenate formamide to form a ruthenium-coordinated isocyanate complex, analogous to 4.18 Heating complex 5 at 110 °C slowly formed a new complex as observed by a clean signal in 31P{1H} NMR spectrum at δP 89.2 ppm after 9 days. NMR spectroscopy and HRMS data suggested the formation of complex 6 featuring the Mn–N–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N cycle that would be formed from the dehydrogenation of complex 5. Indeed, dissolved H2 gas was observed in the 1H NMR spectrum (δH 4.51) of the reaction mixture. The 13C{1H} NMR chemical shift of the carbonyl carbon in complex 6 shifts further upfield (δC 156.8) in comparison to that of complex 5 (δC 171.2) as would be expected upon decreasing electron density of the carbonyl carbon on becoming a urea moiety. Additionally, a 1H,13C-HMBC (toluene-d8) NMR spectrum of complex 6 shows the correlation between the carbonyl carbon (δC 156.8 ppm) and the C–H protons of the ligand backbone (δH 2.22–2.12 ppm) supporting the proposed structure of complex 6 (Fig. S100, see ESI†). This is similar to a recent report by Milstein where an analogous ruthenium-pincer complex activates the formanilide and dehydrogenates to form a complex featuring Ru–N–(CO)–C cycle.18 Analogous manganese complex featuring Mn–N–(CO)–O cycle has also been recently reported by Milstein from the activation of CO2 across a manganese–amido bond.39 GC-MS analysis of the reaction mixture upon heating complex 5 (110 °C, 9 days) showed the formation of aniline and diphenylurea. We speculate that under this condition formanilide can undergo decarbonylation to form aniline which could then react with isocyanate to form diphenylurea.19,40 This could be an alternative pathway to the formation of homopolyurea in addition to the transformylation route proposed in Fig. 2. Conducting a catalytic reaction of diformamide (DF1, 0.5 mmol) in the absence of diamine (remaining conditions described as per Table 1 entry 7), resulted in the formation of homopolyurea in ∼5% yield. It is possible that both pathways (transformylation and decarbonylation) occur under the catalytic conditions. Furthermore, the formation of complexes from the N–H activation of diformamide and the corresponding dehydrogenation product analogous to complexes 5 and 6 were also obtained in catalyst speciation studies when we measured the NMR spectra for a sample taken after 1 h from the catalytic experiment as described in Table 1, entry 3 (see ESI,† Fig. S103). These observations support our hypothesis that the manganese complex 2 is capable of dehydrogenating formamide and forming an isocyanate intermediate that can further react with an amine to form a urea functional group. Our efforts to grow crystals suitable for single-crystal X-ray diffraction for complexes 4 and 6 were not successful. Interestingly, the reaction of complex 6 with aniline (toluene, 110 deg, 24 h) did not lead to the formation of any urea derivative suggestive of complex 6 to be an off-cycle intermediate. This is consistent with a recent report by Sanford and co-workers (that was published during the communication of this manuscript)41 where the reaction of an analogue of complex 6 with cyclohexanol doesn't yield any carbamate derivative. They have suggested that the O-coordinated isocyanate rather than the N-coordinated one (like in complex 6) is the active species which might also be possible in our case.
O)–N cycle that would be formed from the dehydrogenation of complex 5. Indeed, dissolved H2 gas was observed in the 1H NMR spectrum (δH 4.51) of the reaction mixture. The 13C{1H} NMR chemical shift of the carbonyl carbon in complex 6 shifts further upfield (δC 156.8) in comparison to that of complex 5 (δC 171.2) as would be expected upon decreasing electron density of the carbonyl carbon on becoming a urea moiety. Additionally, a 1H,13C-HMBC (toluene-d8) NMR spectrum of complex 6 shows the correlation between the carbonyl carbon (δC 156.8 ppm) and the C–H protons of the ligand backbone (δH 2.22–2.12 ppm) supporting the proposed structure of complex 6 (Fig. S100, see ESI†). This is similar to a recent report by Milstein where an analogous ruthenium-pincer complex activates the formanilide and dehydrogenates to form a complex featuring Ru–N–(CO)–C cycle.18 Analogous manganese complex featuring Mn–N–(CO)–O cycle has also been recently reported by Milstein from the activation of CO2 across a manganese–amido bond.39 GC-MS analysis of the reaction mixture upon heating complex 5 (110 °C, 9 days) showed the formation of aniline and diphenylurea. We speculate that under this condition formanilide can undergo decarbonylation to form aniline which could then react with isocyanate to form diphenylurea.19,40 This could be an alternative pathway to the formation of homopolyurea in addition to the transformylation route proposed in Fig. 2. Conducting a catalytic reaction of diformamide (DF1, 0.5 mmol) in the absence of diamine (remaining conditions described as per Table 1 entry 7), resulted in the formation of homopolyurea in ∼5% yield. It is possible that both pathways (transformylation and decarbonylation) occur under the catalytic conditions. Furthermore, the formation of complexes from the N–H activation of diformamide and the corresponding dehydrogenation product analogous to complexes 5 and 6 were also obtained in catalyst speciation studies when we measured the NMR spectra for a sample taken after 1 h from the catalytic experiment as described in Table 1, entry 3 (see ESI,† Fig. S103). These observations support our hypothesis that the manganese complex 2 is capable of dehydrogenating formamide and forming an isocyanate intermediate that can further react with an amine to form a urea functional group. Our efforts to grow crystals suitable for single-crystal X-ray diffraction for complexes 4 and 6 were not successful. Interestingly, the reaction of complex 6 with aniline (toluene, 110 deg, 24 h) did not lead to the formation of any urea derivative suggestive of complex 6 to be an off-cycle intermediate. This is consistent with a recent report by Sanford and co-workers (that was published during the communication of this manuscript)41 where the reaction of an analogue of complex 6 with cyclohexanol doesn't yield any carbamate derivative. They have suggested that the O-coordinated isocyanate rather than the N-coordinated one (like in complex 6) is the active species which might also be possible in our case.
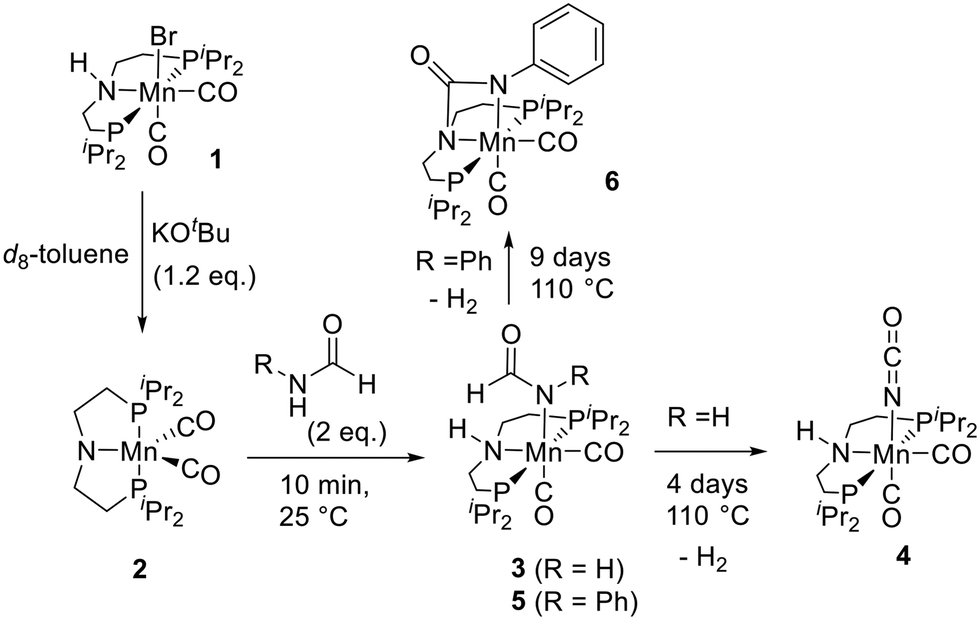 | ||
| Fig. 3 Reaction of formamide with in situ generated complex 2. | ||
Conclusions
In conclusion, we report here the synthesis of polyureas from the manganese catalysed dehydrogenative coupling of diformamides with diamines. Polyureas have been chemically characterized by NMR and IR spectroscopies as well as MALDI FT-ICR mass spectrometry. Further insights into the physical properties (decomposition temperature and melting temperature), morphology (SEM, EDX), and mechanical properties (indentation modulus, and hardness using nanoindentation) have also been provided. Based on our experimental studies reported here (Fig. 3) and previous report by us using DFT computation,21 we propose that the polymerization reaction proceeds by the dehydrogenation of diformamides to form diisocyanates followed by their reaction with diamines to form polyureas. Additionally, our studies also suggest the possibility of competitive pathways based on transformylation (Fig. 2) and decarbonylation (vide supra) that would lead to the formation of homopolyurea or random copolyurea as observed by the MALDI FT-ICR mass spectrometry in this case. The presented methodology replaces the use of diisocyanates (which can be toxic) with a safer feedstock – diformamides. The use of an earth-abundant metal-based catalyst is an added advantage that makes the process more sustainable.Data availability
The raw research data supporting this publication can be accessed at the University of St Andrews Research Portal: https://doi.org/10.17630/36c115ac-4dbb-4a87-a3e9-c042f8f1a91e.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from UKRI Future Leaders Fellowship (MR/W007460/1). A. M. thanks the University of St. Andrews (Impact & Innovation Fund) for funding. M. T. and J. C. T. thank the ERC Consolidator Grant (PROMOFS grant agreement 771575) for funding the research. ABN gratefully acknowledges funding from the EPSRC through grant numbers EP/L017008/1, EP/R023751/1 and EP/T019298/1. We thank Dr. Pavel Kulyabin for his assistance with the MALDI-MS technique.References
- M. Tripathi, S. Parthasarathy and P. K. Roy, J. Appl. Polym. Sci., 2020, 137, 48573 CrossRef CAS.
- B. Shojaei, M. Najafi, A. Yazdanbakhsh, M. Abtahi and C. Zhang, Polym. Adv. Technol., 2021, 32, 2797–2812 CrossRef CAS.
- P. Rocas, C. Cusco, J. Rocas and F. Albericio, Curr. Drug Delivery, 2018, 15, 37–43 CAS.
- R. Zhang, W. Huang, P. Lyu, S. Yan, X. Wang and J. Ju, Polymer, 2022, 14, 2670 CAS.
- C. I. Chiriac and F. Tanasaã, in Ullmann's Encyclopedia of Industrial Chemistry, 2016, pp. 1–5 Search PubMed.
- Polyurea Coatings Market Global Forecast to 2025|MarketsandMarkets, https://www.marketsandmarkets.com/Market-Reports/polyurea-coatings-market-152676861.html?gclid=Cj0KCQjw8rT8BRCbARIsALWiOvRwzrAtBwMcCRV3eXvOYKIdHfFk5OpjWw5rp5oVMIpe50yRVEX5LHcaAkN0EALw_wcB, (accessed 19 October 2020).
- P. X. Wu, H. Y. Cheng, R. H. Shi, S. Jiang, Q. F. Wu, C. Zhang, M. Arai and F. Y. Zhao, Adv. Synth. Catal., 2019, 361, 317–325 CrossRef CAS.
- S. Jiang, R. Shi, H. Cheng, C. Zhang and F. Zhao, Green Energy Environ., 2017, 2, 370–376 CrossRef.
- S. Jiang, H. Y. Cheng, R. H. Shi, P. X. Wu, W. W. Lin, C. Zhang, M. Arai and F. Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47413–47421 CrossRef CAS PubMed.
- J. M. Sirrine, S. A. Schexnayder, J. M. Dennis and T. E. Long, Polymer, 2018, 154, 225–232 CrossRef CAS.
- N. Kébir, M. Benoit, C. Legrand and F. Burel, Eur. Polym. J., 2017, 96, 87–96 CrossRef.
- H. Y. Chen, W. C. Pan, C. H. Lin, C. Y. Huang and S. A. Dai, J. Polym. Res., 2012, 19, 9754 CrossRef.
- M. Soccio, R. Mazzoni, C. Lucarelli, S. Quattrosoldi, A. Cingolani, M. Fiorini, N. Lotti and T. Tabanelli, ACS Sustainable Chem. Eng., 2020, 8, 15640–15650 CrossRef CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- A. Kumar and C. Gao, ChemCatChem, 2021, 13, 1105–1134 CrossRef CAS.
- S. H. Kim and S. H. Hong, Org. Lett., 2016, 18, 212–215 CrossRef CAS PubMed.
- E. M. Lane, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2018, 9, 4003–4008 RSC.
- J. Bruffaerts, N. Von Wolff, Y. Diskin-Posner, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 16486–16493 CrossRef CAS PubMed.
- V. Krishnakumar, B. Chatterjee and C. Gunanathan, Inorg. Chem., 2017, 56, 7278–7284 CrossRef CAS PubMed.
- A. Kumar, D. Armstrong, G. Peters, M. Nagala and S. Shirran, Chem. Commun., 2021, 57, 6153–6156 RSC.
- A. E. Owen, A. Preiss, A. McLuskie, C. Gao, G. Peters, M. Bühl and A. Kumar, ACS Catal., 2022, 12, 6923–6933 CrossRef CAS.
- J. Guo, J. Tang, H. Xi, S.-Y. Zhao and W. Liu, Chin. Chem. Lett., 2023, 34, 107731 CrossRef CAS.
- Q. I. Roode-Gutzmer, D. Kaiser and M. Bertau, ChemBioEng Rev., 2019, 6, 209–236 CrossRef CAS.
- T. Choi, D. Fragiadakis, C. M. Roland and J. Runt, Macromolecules, 2012, 45, 3581–3589 CrossRef CAS.
- C. R. Langsted, S. W. Paulson, B. H. Bomann, S. Suhail, J. A. Aguirre, E. J. Saumer, A. R. Baclasky, K. H. Salmon, A. C. Law, R. J. Farmer, C. J. Furchtenicht, D. S. Stankowski, M. L. Johnson, L. G. Corcoran, C. C. Dolan, M. J. Carney and N. J. Robertson, J. Appl. Polym. Sci., 2022, 139, 52088 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, 2014, DOI:10.1201/b17118.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180–1223 CAS.
- J. Yin, J. Zhang, C. Cai, G.-J. Deng and H. Gong, Org. Lett., 2019, 21, 387–392 CrossRef CAS PubMed.
- T. Lebleu, H. Kotsuki, J. Maddaluno and J. Legros, Tetrahedron Lett., 2014, 55, 362–364 CrossRef CAS.
- H. C. M. Byrd and C. N. McEwen, Anal. Chem., 2000, 72, 4568–4576 CrossRef CAS PubMed.
- H. Kawasaki, Y. Takeda and R. Arakawa, Anal. Chem., 2007, 79, 4182–4187 CrossRef CAS PubMed.
- H. Wang, X. Deng, H. Wu, A. Pi, J. Li and F. Huang, Def. Technol., 2019, 15, 875–884 CrossRef.
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- N. H. Anderson, J. M. Boncella and A. M. Tondreau, Organometallics, 2018, 37, 4675–4684 CrossRef CAS.
- A. Kumar, P. Daw, N. A. Espinosa-Jalapa, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Dalton Trans., 2019, 48, 14580–14584 RSC.
- S. Li, T. Rajeshkumar, J. Liu, L. Maron and X. Zhou, Org. Lett., 2023, 25, 163–168 CrossRef CAS PubMed.
- A. A. Kassie, I. Y. Castro De la Torre, M. S. Remy, S. Mukhopadhyay, J. Kampf, F. Qu and M. S. Sanford, Organometallics DOI:10.1021/acs.organomet.3c00175.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00284e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |