
A line-broadening free real-time 31P pure shift NMR method for phosphometabolomic analysis†
Karl Kristjan
Kaup
ab,
Lauri
Toom
 b,
Laura
Truu
a,
Sten
Miller
a,
Marju
Puurand
a,
Kersti
Tepp
a,
Tuuli
Käämbre
a and
Indrek
Reile
*a
b,
Laura
Truu
a,
Sten
Miller
a,
Marju
Puurand
a,
Kersti
Tepp
a,
Tuuli
Käämbre
a and
Indrek
Reile
*a
aNational Institute of Chemical Physics and Biophysics, Akadeemia tee 23, Tallinn 12618, Estonia. E-mail: indrek.reile@kbfi.ee
bInstitute of Chemistry, University of Tartu, Ravila 14A, 50411 Tartu, Estonia
First published on 20th August 2021
Abstract
Phosphometabolomics by 31P NMR can be challenging, since overlapping multiplets of homonuclear coupled phosphorus nuclei complicate spectral analysis. Pure shift NMR allows to simplify such spectra by collapsing multiplets into singlets, but most pure shift methods require substantially elongated measurement times or cause disturbing spectral line broadening. Herein, we combine established pure shift NMR and artefact suppression techniques to record 31P pure shift NMR spectra without penalties in measurement time or line width. Examples are demonstrated in resolution of a mixture of nucleotide triphosphates and a biological sample of 18O labelled ATP isotopomers.
NMR spectroscopy has established itself as a powerful and versatile tool in biochemical and biomedical research, giving access to easily interpretable and quantifiable information. It holds particular relevance for bioenergetics and phosphometabolomics, where 31P NMR is used to visualise the chemical energy carrying phosphometabolites that are involved in cellular energy production, transport and utilization.1
31P NMR is used to detect high-energy nucleotides, i.e. ATP (adenosine triphosphate), whose concentrations help to evaluate the energetic status and health of muscle tissue.2,3 The evaluation of nucleotides is also believed to have an impact in cancer research in the near future.4 Application of NMR in phosphometabolomics is, however, often held back by the general limitations of NMR spectroscopy: its sensitivity and resolution. Although 31P is one of the more sensitive MR active nuclei, measuring of dilute metabolites is time consuming and often takes multiple hours,3 limiting throughput.
Spectral resolution is a common limitation in 1D 1H NMR, but is usually less of an issue for other nuclei, including 31P NMR, where signals are dispersed across >400 ppm chemical shift range. In phosphometabolite analysis, however, ATP 31P signals are further split into multiplets by approximately 20 Hz 2J-couplings between adjacent phosphate residues. While this is not an issue for measuring pure ATP, it becomes a challenge when different nucleotide triphosphates are involved: their 31P chemical shift differences are of similar magnitude to their mutual J-coupling, giving rise to partially overlapping multiplets (Fig. 1b & c). Consequently, mixtures of different phosphates have also been studied by MS, which resolves them somewhat more easily, but requires more elaborate sample preparation and quantification routines.2
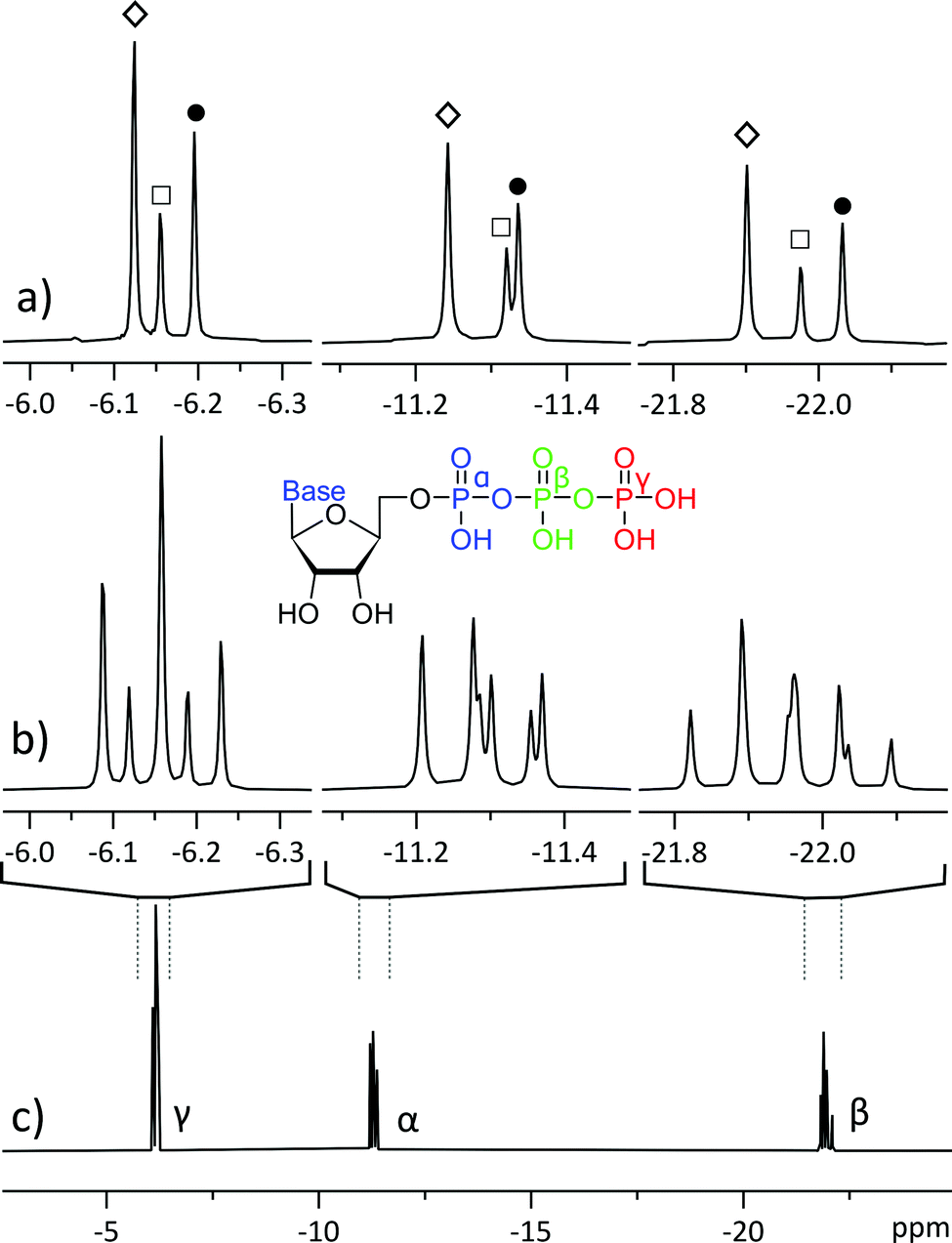 | ||
| Fig. 1 31P NMR spectra of a mixture of 20 mM ATP (adenosine triphosphate, ◊), 10 mM CTP (cytidine triphosphate, ●) and 5 mM UTP (uridine triphosphate, □): (a) 31P pure shift spectra, acquired with the method reported in this study; (b) magnification of the three phosphate sites from trace c; (c) normal 31P 1D NMR spectrum. All spectra were acquired using 256 scans, with trace (a) acquired in four 64-scan experiments of different SAPPHIRE orders and the FIDs were added after LP processing. | ||
We encountered a similar problem in mapping the concentrations and turnover rates (fluxes) of different ATP synthesis and utilization sites.1,3 These experiments rely on the small (approx. 0.025 ppm) chemical shift difference between ATP isotopomers: dynamic 18O labelling introduces isotopomer signals into the 31P spectra (Fig. 2b & c), forming a complex pattern of overlapping multiplets that can be difficult to interpret.5 The biochemical meaning of all isotopomers is known6 and the relative intensities of 16O/18O related 31P signals at any ATP phosphate (α, β, γ) can be related to activities of different ATP processing pathways.5 However, isotope shifts are of similar magnitude to J-coupling and can overlap with right-hand multiplet components, complicating analysis.6 Furthermore, signal intensity is dispersed among numerous isotopomers, increasing necessary measurement time.
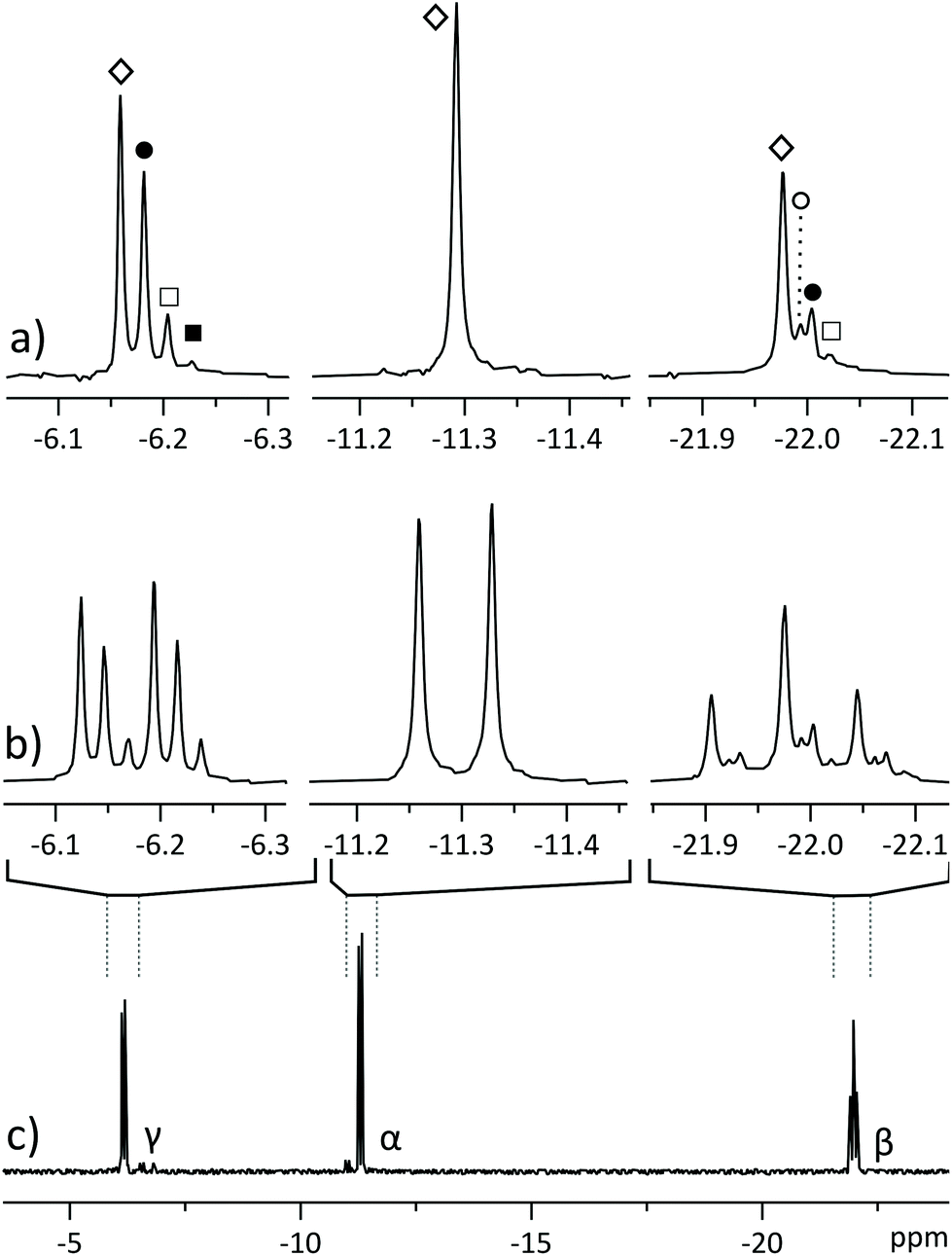 | ||
| Fig. 2 31P NMR spectra of partially 18O-labelled ATP, isolated from a perfused rat heart. (a) Pure shift spectrum displays a singlet for each isotopomer: ◊ for all 16O isotopomer, ● for phosphate with one oxygen 18O-labelled, □ for two labelled oxygens and ■ for three labelled oxygens. β-Phosphate also yields a signal (○) corresponding to a 18O label in the bridging position between adjacent phosphates;6 (b) segments from regular 31P spectrum displaying overlapping multiplets; (c) regular 31P spectrum of the sample. All spectra were acquired in 2048 scans, with spectrum (a) acquired in four 512-scan experiments of different SAPPHIRE orders and the FIDs were added after LP extrapolation. | ||
Such spectra could be resolved by 2D 31P spectroscopy, but this experiment would also be undesirably long for dilute biological samples.5–7 Herein we demonstrate a different approach, where resolution is achieved by pure shift NMR methodology, yielding better resolved and easier to interpret spectra.
Experimental
Spectra were acquired with a 5 mm BBO (broad band observe) probe on a 700 MHz Bruker Avance III spectrometer, corresponding to 283 MHz 31P frequency. All experiments were recorded in 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz bandwidth with 1H decoupling. 16k complex points were recorded in a 0.82 s acquisition time, with a relaxation delay of 2 s. Sample temperature was regulated at 6 °C (2 °C above the freezing point of D2O) and RF pulses were calibrated for each sample (typically around 14.7 μs hard 90° pulse). Samples were measured either in 5 mm tubes or in Shigemi tubes (18O labelled samples). Samples were prepared by dissolving solid reagents or lyophilized biological material in D2O and adjusting the pH to 9.5 with NaOH solution. 18O labelled sample was prepared by perfusion of rat heart,8 followed by preparative LC separation of ATP (details in ESI†). Animal experiments were approved by the Estonian National Board of Animal Experiments in accordance with the European Community Directive (86/609/EEC). LP extrapolation of data chunks was performed in Bruker Topspin and final spectral processing was performed in Topspin or MestreNova software packages. Pulse sequence code, processing macro and further details are available in ESI.†
000 Hz bandwidth with 1H decoupling. 16k complex points were recorded in a 0.82 s acquisition time, with a relaxation delay of 2 s. Sample temperature was regulated at 6 °C (2 °C above the freezing point of D2O) and RF pulses were calibrated for each sample (typically around 14.7 μs hard 90° pulse). Samples were measured either in 5 mm tubes or in Shigemi tubes (18O labelled samples). Samples were prepared by dissolving solid reagents or lyophilized biological material in D2O and adjusting the pH to 9.5 with NaOH solution. 18O labelled sample was prepared by perfusion of rat heart,8 followed by preparative LC separation of ATP (details in ESI†). Animal experiments were approved by the Estonian National Board of Animal Experiments in accordance with the European Community Directive (86/609/EEC). LP extrapolation of data chunks was performed in Bruker Topspin and final spectral processing was performed in Topspin or MestreNova software packages. Pulse sequence code, processing macro and further details are available in ESI.†
Pure shift NMR
Pure shift,9,10 or homonuclear decoupling, techniques have been extensively developed11–13 and are included in the modern NMR toolbox. They gain in resolution by sacrificing information embedded in multiplet patterns, collapsing them into singlets. This can also yield added sensitivity, as the intensities of multiplet components add. Methodology has been published for simplification of 1D spectra of small molecules,9 diastereomers14 or peptides15 and for added resolution in 2D NMR.16–18 While the majority of pure shift methods focus on 1H NMR and there are a few examples available for heteronuclei,19 phosphometabolomics by 31P NMR presents an opportunity to expand the application scope of pure shift.The first10 and best quality20,21 pure shift techniques rely on interferogram (or pseudo-2D)9 acquisition where the homonuclear decoupled FID is constructed from “chunks” from tens of individual 1D measurements. This generally yields good quality spectra but is not applicable to phosphometabolomics, where acquiring a single 1D spectrum takes hours and recording tens of spectra would make the experiment prohibitively long. The alternative is real-time (RT) pure shift, where a single FID is recorded,12 but acquisition is periodically interrupted to refocus the J-evolution and chemical shifts by RF pulses.22 While the measurement time penalty of interferogram pure shift is overcome, undesired spectral line broadening occurs. This is caused by relaxation between recorded “data chunks” as the time during RF pulses is omitted from the acquired signal, causing shortening of the FID.22
The best current solution is called semi-RT pure shift,23 which involves conducting two 1D measurements and combining the results into one continuous FID. Consequently, it introduces a two-fold increase in measurement time, a potential issue in dilute samples where even a single 1D measurement requires thousands of scans to be acquired. Here, we build on the principles of semi-RT, but instead of two experiments, we record only one discontinuous FID and fill in the gaps by linear prediction (LP) from adjacent data chunks to obtain a 31P pure shift spectrum with natural linewidth (Fig. 1 & 2a).
Pulse sequence and data processing
The pulse sequence (Fig. 3) is based on the 1st row of the band selective semi-RT experiment.23 Spins are divided into two subgroups: active spins, which are the spins whose signal we want to measure, and passive spins, which cause the J-evolution of active spins. A series of pulsed field gradient spin echoes select the active spins with a band-selective soft pulse and set the starting point of J-evolution in the FID. Chemical shift is allowed to evolve unperturbed throughout acquisition, while the receiver is blanked periodically to invert passive spins with band-selective soft pulses to refocus the J-evolution in the mid-point of the following data chunk. Note that when measuring the β-ATP signal, two band-selective pulses are applied at the same time at passive α and γ phosphate frequencies. Data points are acquired during the FID gaps, but since the receiver is blanked, they do not carry meaningful information. This process is repeated multiple times during acquisition, resulting in a chunked FID (Fig. 4a).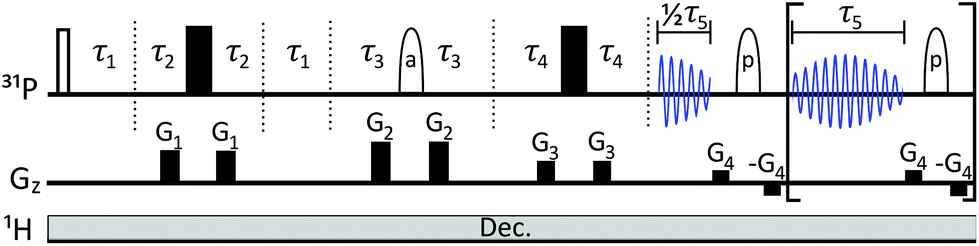 | ||
| Fig. 3 The pulse sequence is effectively a combination of SAPPHIRE20 and semi-RT23 sequences, with one additional delay τ2. Gz describes gradient pulses. Filled rectangles stand for 180° hard pulses, unfilled rectangle for 90° hard pulse. Ellipses denote 180° RSnob soft pulses for (a) active spins (that will be detected) and (p) for passive spins. Hence, prior knowledge of the spectral position of passive and active spins is required. Following the semi-RT example,23 Tycko-7/MLEV-4 supercycle is used for passive spin RSnob pulses. Explanations for delays τ1–τ5 and further details are given in ESI.† | ||
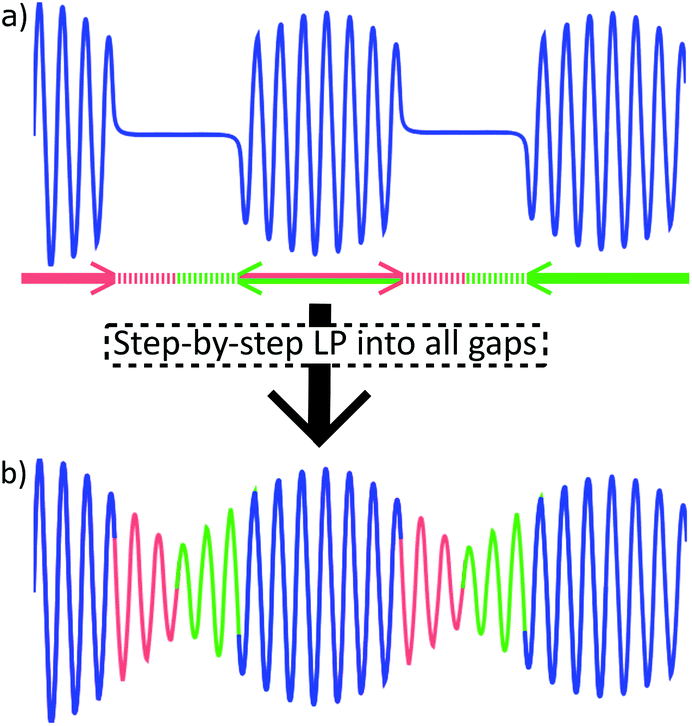 | ||
| Fig. 4 Linear prediction to fill the FID gaps in trace (a): red segments are predicted forwards, based on the full length of the preceding data chunk (blue). Green segments are predicted backwards. Resulting continuous FID (b) can be processed as usual. In present work, data chunk and gap durations were 20 ms and 5.6 ms, respectively. | ||
Gaps in the discontinuous FID are filled computationally (Fig. 4b). A similar problem has been solved previously in EXACT16,24 experiments by algorithmic reconstruction of the missing data points, requiring specialized software not always available at NMR laboratories. Instead, we extrapolate the information from within chunks into FID gaps by using standard linear prediction (LP) methodology that is commonly available in the software toolbox of modern spectrometers.
Every chunk is used to extrapolate half of the duration of the following and half of the preceding gap (with the exception of the 1st chunk that predicts only into the following gap). This way, J-coupling evolution is kept symmetrical about the centre of data chunks. It also relieves the conditions set on the relative lengths of chunks and gaps, as it allows to predict less points from the limited number of experimental points available. Repeating this procedure for all gaps produces a continuous FID that can be processed by usual means.
As is common in pure shift, the resulting FID will be periodically modulated by J-evolution that is centred around the midpoint of every data chunk (Fig. 4b).13,22 Fourier transform of such FID creates a spectrum with “chunking sideband” artefacts (Fig. 5b–e) positioned around the signals at distances inversely proportional to the sum of the data chunk and gap lengths (the periodically repeating unit in the FID). This issue is alleviated by the SAPPHIRE20 method that shifts the phase of the J-modulation across different scans (Fig. 5b–e; discussion in ESI†). Accordingly, the desired number of scans should be recorded in an even number of experiments where the artefacts are positioned in opposite phases. Adding the FIDs of such experiments will add the desired signals constructively, while “cancelling out” artefacts (Fig. 5a).
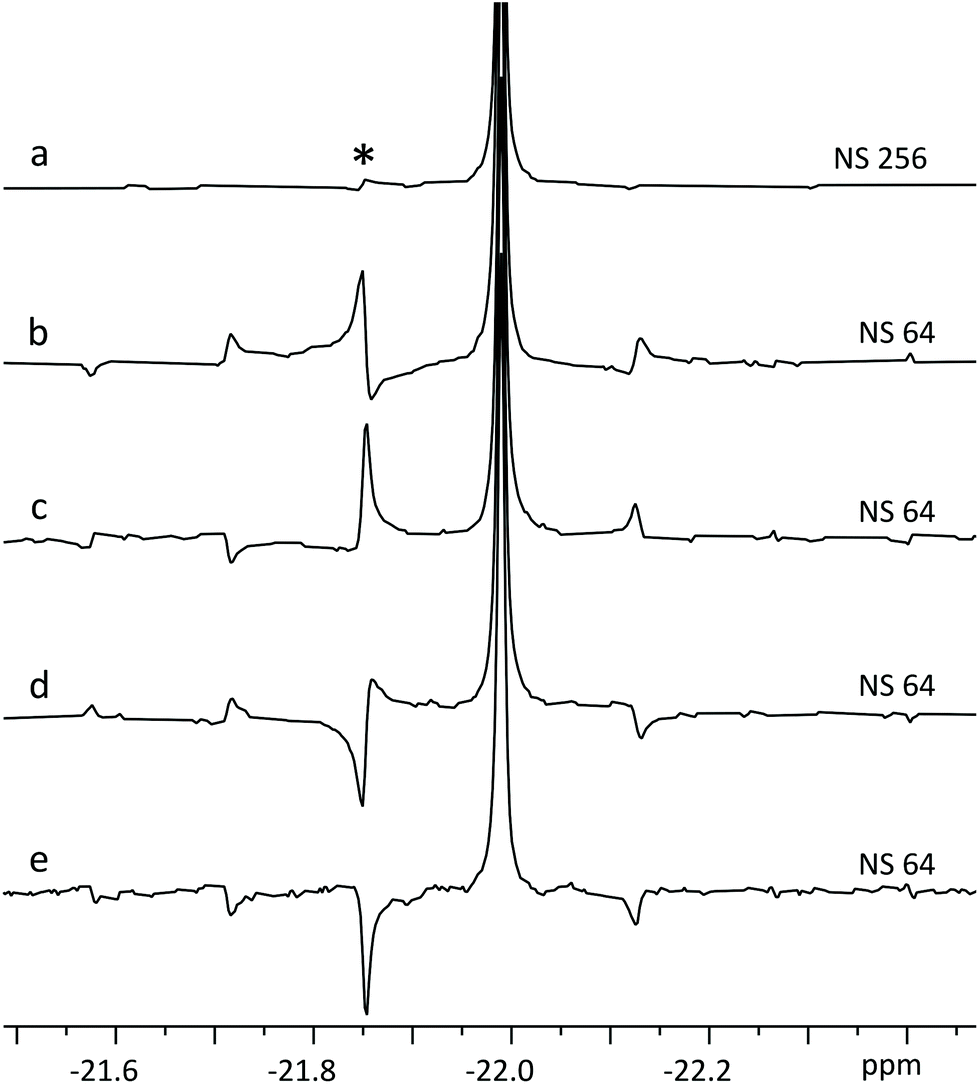 | ||
| Fig. 5 Fourier transform after LP of four SAPPHIRE experiments (b–e) on ATP β-phosphate (20 mM). Note how the artefacts are pairwise in opposite phases in traces b & d and c & e. Trace (a) displays FT of the sum of FID-s from b–e, where the chunking artefacts have been largely cancelled out,20 with the most prominent artefact (*) having <2% intensity of the main signal. | ||
Theoretically, it is possible to extend SAPPHIRE to an arbitrary order (measured in an infinite number of experimental pairs), cancelling out all sidebands and producing clean spectra. However, since the experiment herein requires LP from the 1st data chunk (half the duration of following chunks) into the 1st gap, it cannot be implemented beyond the second order – there would not be enough data points available for reliable LP. Even so, artefact intensity decreases rapidly and it is seldom necessary to use higher order canceling.20 In the experiment of Fig. 5a, SAPPHIRE reduces artefact intensity approx. thirtyfold, yielding a pure shift spectrum with virtually identical linewidth to the original spectra and increased sensitivity as multiplet components add constructively. The resulting experiment compares favourably to other pure shift approaches, especially when time efficiency is considered (Fig. S4 and Table S1†).
Phosphometabolomics analysis
Applying the method to a mixture of similar concentrations of nucleotide triphosphates, a realistic scenario in phosphometabolomics of biological material,2 allows to resolve signals of different nucleotides (Fig. 1), whose multiplets would otherwise overlap. The method gives excellent spectral quality, with line shape being indistinguishable from regular 1D NMR (Fig. 6). The experiment being band selective, two separate measurements must be conducted to cover all ATP phosphorus sites. This does not necessarily cost additional measurement time since decoupling provides a slight sensitivity increase, allowing to accumulate less scans to achieve similar SNR. Moreover, in the case of a mixture of Fig. 1, acquiring pure shift spectra of only one phosphate site suffices to characterize the ratio of all components.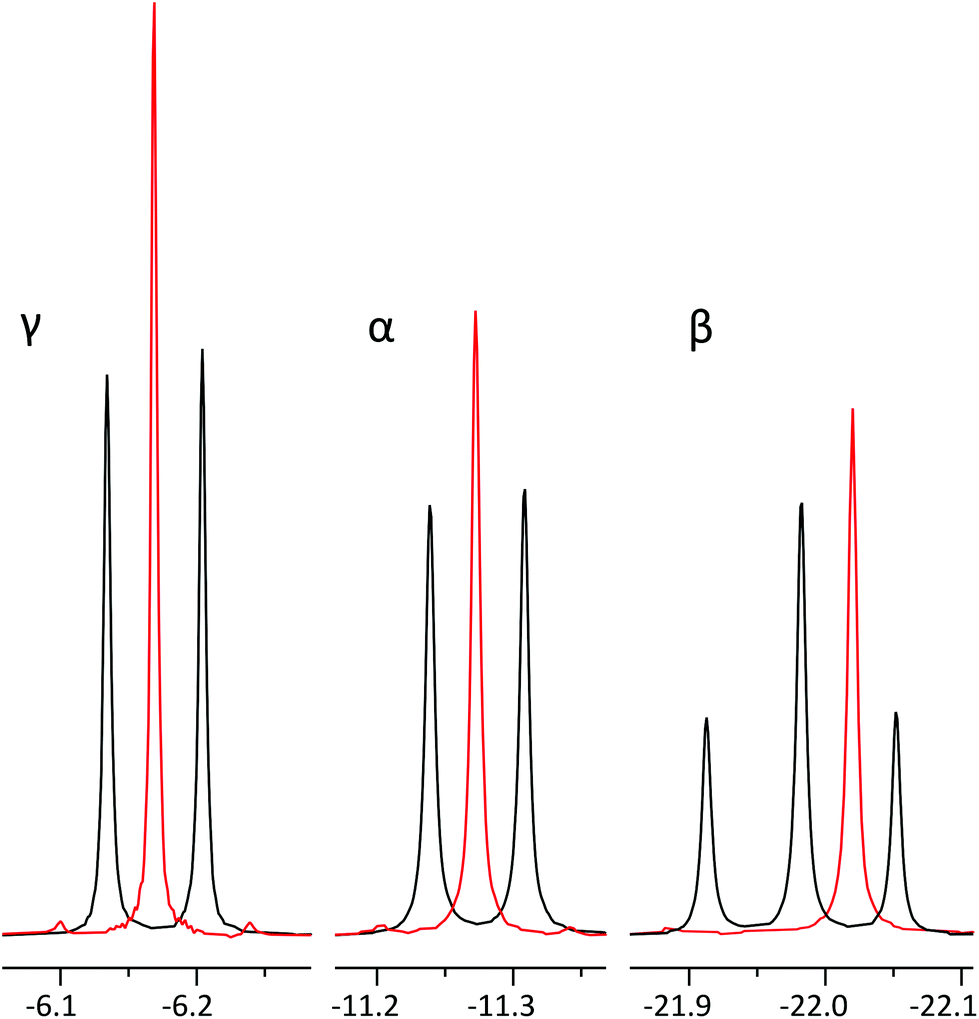 | ||
| Fig. 6 Comparison of regular 31P spectra of ATP α, β and γ phosphates (black) and their corresponding pure shift spectra (red) with identical line width. The β phosphate signal has been offset slightly to allow for better comparison of signals. In order to acquire all three positions, two separate experiments have to be recorded: one for α and γ phosphate and one for β phosphate. Signal intensities increase in pure shift spectra: 1,6-fold for γ, 1,4-fold for α and 1,2-fold for β. We suggest the differences are due to relaxation weighting during the pulse sequence. γ-Phosphate displays the largest artefact intensity of 1.5–2% of main signal. | ||
In parallel, we applied the methodology to analysis of a mixture of 16O/18O labelled ATP isotopomers that were extracted from a perfused rat heart (Fig. 2).1 Such experiments are usually sample limited and analysis has to be performed on dilute mixtures that are prepared from animal tissue and require thousands of scans to achieve sufficient SNR. Applying the method reported in this study reduces overlapping ATP isotopomer signals into singlets, making the spectra easier to interpret (Fig. 2a). One could argue that overlap is not an issue in this case and it is possible to simply count the rightmost signals of the multiplets. This might, however, cause misinterpretation, since left hand multiplet components of some isotopomers may overlap with those of the right multiplet components of other isotopomers. This issue would be corroborated if one moves to an even higher field spectrometer, where the frequency differences of isotopomers increase while coupling remains constant.
Analysing these isotopomer singlets gives a time-freeze overview of the phosphometabolomic state of the cells at the time of labelling, as has been described by Nemutlu et al.25 The slightly added sensitivity allows to spot the less abundant isotopomers that can go unnoticed in simple 1D NMR in equal number of scans (■, 18O3 in Fig. 2). In the case of β-ATP we observe very closely resonating signals from bridging and peripheral labelled isotopomers (○, 18O1 in Fig. 2), but still benefit from simplification of multiplet patterns along with an increase in sensitivity. While the sample of Fig. 2 was prepared by LC purification of a perfused rat heart extract, the method is also applicable directly to the extract, yielding simultaneous pure shift spectra of nearby ATP and ADP signals (Fig. S5†).
Conclusions
Combining the concepts of semi-RT-pure shift, SAPPHIRE and chunk extrapolation by LP allowed to demonstrate a heteronuclear application of pure shift NMR in phosphometabolomic analysis. Although spectral resolution is rarely as prominent an issue in 31P NMR as it is in 1H NMR, resolution of highly similar nucleotides and their isotopomers presents a strong case for developing methodology and expanding the application envelope of pure shift methodology for heteronuclei. We foresee using this and related methods in the metabolomic study of challenging diseases,26 including cancer,4 in the near future. We suggest the method reported in this study is also applicable to 1H NMR, especially when additional resolution is sought after for very dilute samples, where line broadening and sensitivity penalties have to be avoided. The developed method gives access to homonuclear decoupled spectra in real-time, with natural linewidth, and utilizes only software and functionalities that are commonly built into any modern NMR software package.Author contributions
K. K. K. developed pulse program and processing software, performed NMR measurements. L. Truu, M. P., K. T., S. M. and T. K. carried out biological sample preparation. The project was conceptualized and supervised by L. Toom, T. K. and I. R. The manuscript was written by K. K. K. and I. R. and edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the personal research grant PSG11, the mobility grant MOBTP51 and institutional funding scheme IUT23-1 of the Estonian Research Council, as well as from the Center of Excellence TK134 of the Archimedes Foundation. The authors are grateful to Cambridge Laboratories, Inc. for their support with H218O and D2O. NMR work was carried out using the instrumentation at the Estonian Center of Analytical Chemistry (http://www.akki.ee).Notes and references
- D. Pucar, P. P. Dzeja, P. Bast, N. Juranic, S. Macura and A. Terzic, J. Biol. Chem., 2001, 276, 44812–44819 CrossRef CAS PubMed.
- E. Nemutlu, S. Zhang, A. Gupta, N. O. Juranic, S. I. Macura, A. Terzic, A. Jahangir and P. Dzeja, Physiol. Genomics, 2012, 44, 386–402 CrossRef CAS PubMed.
- D. Pucar, P. P. Dzeja, P. Bast, R. J. Gumina, C. Drahl, L. Lim, N. Juranic, S. Macura and A. Terzic, Mol. Cell. Biochem., 2004, 256, 281–289 CrossRef PubMed.
- N. N. Pavlova and C. B. Thompson, Cell Metab., 2016, 23, 27–47 CrossRef CAS PubMed.
- E. Nemutlu, N. Juranic, S. Zhang, L. E. Ward, T. Dutta, K. S. Nair, A. Terzic, S. Macura and P. P. Dzeja, Anal. Bioanal. Chem., 2012, 403, 697–706 CrossRef CAS PubMed.
- N. Juranić, E. Nemutlu, S. Zhang, P. Dzeja, A. Terzic and S. MacUra, J. Biomol. NMR, 2011, 50, 237–245 CrossRef PubMed.
- A. Majumdar, Y. Sun, M. Shah and C. L. Freel Meyers, J. Org. Chem., 2010, 75, 3214–3223 CrossRef CAS PubMed.
- D. Pucar, E. Janssen, P. P. Dzeja, N. Juranic, S. Macura, B. Wieringa and A. Terzic, J. Biol. Chem., 2000, 275, 41424–41429 CrossRef CAS PubMed.
- J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901–3903 CrossRef CAS PubMed.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS.
- K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1–20 CrossRef CAS PubMed.
- L. Castañar, Magn. Reson. Chem., 2017, 55, 47–53 CrossRef PubMed.
- R. W. Adams, in eMagRes, John Wiley & Sons, Ltd, Chichester, UK, 2014, vol. 3, pp. 295–310 Search PubMed.
- R. W. Adams, L. Byrne, P. Király, M. Foroozandeh, L. Paudel, M. Nilsson, J. Clayden and G. A. Morris, Chem. Commun., 2014, 50, 2512–2514 RSC.
- J. Ying, J. Roche and A. Bax, J. Magn. Reson., 2014, 241, 97–102 CrossRef CAS PubMed.
- I. E. Ndukwe, A. Shchukina, V. Zorin, C. Cobas, K. Kazimierczuk and C. P. Butts, ChemPhysChem, 2017, 18, 2081–2087 CrossRef CAS PubMed.
- M. Foroozandeh, P. Giraudeau and D. Jeannerat, Magn. Reson. Chem., 2013, 51, 808–814 CrossRef CAS PubMed.
- C. Mauve, S. Khlifi, F. Gilard, G. Mouille and J. Farjon, Chem. Commun., 2016, 52, 6142–6145 RSC.
- A. B. Jones, G. C. Lloyd-Jones and D. Uhrín, Anal. Chem., 2017, 89, 10013–10021 CrossRef CAS PubMed.
- P. Moutzouri, Y. Chen, M. Foroozandeh, P. Kiraly, A. R. Phillips, S. R. Coombes, M. Nilsson and G. A. Morris, Chem. Commun., 2017, 53, 10188–10191 RSC.
- M. Foroozandeh, R. W. Adams, N. J. Meharry, D. Jeannerat, M. Nilsson and G. a. Morris, Angew. Chem., Int. Ed., 2014, 53, 6990–6992 CrossRef CAS PubMed.
- L. Castañar and T. Parella, Magn. Reson. Chem., 2015, 53, 399–426 CrossRef PubMed.
- P. Kiraly, M. Nilsson and G. A. Morris, J. Magn. Reson., 2018, 293, 19–27 CrossRef CAS.
- I. E. Ndukwe, A. Shchukina, K. Kazimierczuk, C. Cobas and C. P. Butts, ChemPhysChem, 2016, 17, 2799–2803 CrossRef CAS PubMed.
- E. Nemutlu, S. Zhang, N. O. Juranic, A. Terzic, S. Macura and P. Dzeja, Croat. Med. J., 2012, 53, 529–534 CrossRef CAS.
- M. A. Siddiqui, S. Pandey, A. Azim, N. Sinha and M. H. Siddiqui, Biophys. Chem., 2020, 267, 106462 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an01198g |
| This journal is © The Royal Society of Chemistry 2021 |