
Saline dry fixation for improved cell composition analysis using Raman spectroscopy†
Shreyas
Rangan
 ab,
Riley
Wong
a,
H. Georg
Schulze
c,
Martha Z.
Vardaki
d,
Michael W.
Blades
e,
Robin F. B.
Turner
*aef and
James M.
Piret
*abg
ab,
Riley
Wong
a,
H. Georg
Schulze
c,
Martha Z.
Vardaki
d,
Michael W.
Blades
e,
Robin F. B.
Turner
*aef and
James M.
Piret
*abg
aMichael Smith Laboratories, The University of British Columbia, 2185 East Mall, Vancouver, BC V6T 1Z4, Canada. E-mail: turner@msl.ubc.ca; james.piret@ubc.ca
bSchool of Biomedical Engineering, The University of British Columbia, 2222 Health Sciences Mall, Vancouver, BC V6T 1Z3, Canada
cMonte do Tojal, Caixa Postal 128, Hortinhas, Terena 7250-069, Portugal
dInstitute of Chemical Biology, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens, 11635, Greece
eDepartment of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada
fDepartment of Electrical and Computer Engineering, The University of British Columbia, 2332 Main Mall, Vancouver, BC V6T 1Z4, Canada
gDepartment of Chemical and Biological Engineering, The University of British Columbia, 2360 East Mall, Vancouver, BC V6T 1Z3, Canada
First published on 14th May 2023
Abstract
Raman spectroscopy enables the label-free assessment of cellular composition. While live cell analysis is the most accurate approach for cellular Raman spectroscopy, the analysis of fixed cells has proved to be very useful, particularly in collaborative projects where samples need to be serially examined by different laboratories or stored and reanalyzed at a later date. However, many chemicals that are widely used for cell fixation directly affect cellular biomolecules, yielding Raman spectra with missing or altered information. In this article, we compared the suitability of dry-fixation with saline versus chemical fixatives. We compared the Raman spectroscopy of saline dry-fixed cells with the more commonly used formaldehyde and methanol fixation and found that dry-fixed cell spectra preserved more cellular information than either chemical fixative. We also assessed the stability of dry-fixed cells over time and found that they were stable for at least 5 months. Finally, a comparison of dry-fixed and live cell spectra revealed effects due to the hydration state of the cells since they were recovered upon rehydrating dry-fixed samples. Thus, for fixed cell Raman spectroscopy, we recommend dry-fixation with unbuffered saline as a superior method to formaldehyde or methanol fixation.
1. Introduction
Raman spectroscopy is an information-rich, label-free analytical method that stimulated a wide range of research into applications for the analysis of biological systems.1,2 It has been shown to distinguish cancer cells and tissues,3,4 assess the differentiation status of stem cells,5,6 identify immune cell types and activation states,7 and distinguish modes of cell death.8,9 For cellular therapy manufacturing,6 we are working towards developing Raman spectroscopy as a non-destructive on-line live-cell analysis method. However, for many research and other applications there remains a need for the preservation of samples for off-line analysis.Chemical cell fixation is often used by biological researchers, both to preserve samples and prepare them for further analyses, particularly in studies making use of microscopic imaging techniques. While live cell imaging is possible, it is often only practical in situations where the timescale of imaging is short and minimal sample preparation is required, such as cell-line-based studies where a protein of interest has a fluorescent tag. When extensive sample preparation is required, live cell imaging becomes impractical and fixation is necessary. Common fixatives include aldehydes such as formaldehyde and glutaraldehyde, or solvents including methanol, ethanol, and acetone. Formaldehyde fixation (also known as paraformaldehyde or formalin fixation) is widely used, due to its ease of application and compatibility with fluorescence microscopy,10 while glutaraldehyde fixation is commonly used for scanning electron microscopy.11 Alcohol fixatives, such as methanol, are known to precipitate proteins and can be useful for quenching innate fluorescence and for preserving cytoskeletal structure, albeit at the cost of losing small molecules that are not precipitated.10 Physical cell fixation methods, such as desiccation and vitrification, have seen limited use in biological applications, although vitrification is sometimes used as an alternative to chemical fixation for electron microscopy.12
A summary of the most common fixatives evaluated in the context of cellular Raman spectroscopy is provided in Fig. 1, along with their adverse effects on one or more cellular components. This shows how, for Raman spectroscopy, the method of choice should depend on the information desired. For example, alcohol-based fixatives should be avoided when information about lipids is required, as alcohols solubilize cellular lipids and thereby can attenuate lipid-associated Raman peaks.13,14 There have also been reports that the impact of a particular fixative on Raman spectra depends on the cell type analyzed.14,15 These effects make Raman spectra from chemically fixed cells complex to interpret, as fixation sample changes should be accounted for in the data analysis. Also, sample preparation protocols need to be followed precisely to minimize sample variability. At worst, it may necessitate the time-consuming development of tailor-made fixation protocols for specific applications, such as could be done by formaldehyde fixation followed by OsO4 critical point drying to preserve the lipid droplet distribution in adipocytes.16 Ideally, a fixative should minimize induced variations in band intensities, have minimal or no Raman signal of its own, not induce autofluorescence, and preserve samples close to their biological state to maximize diagnostic potential.15
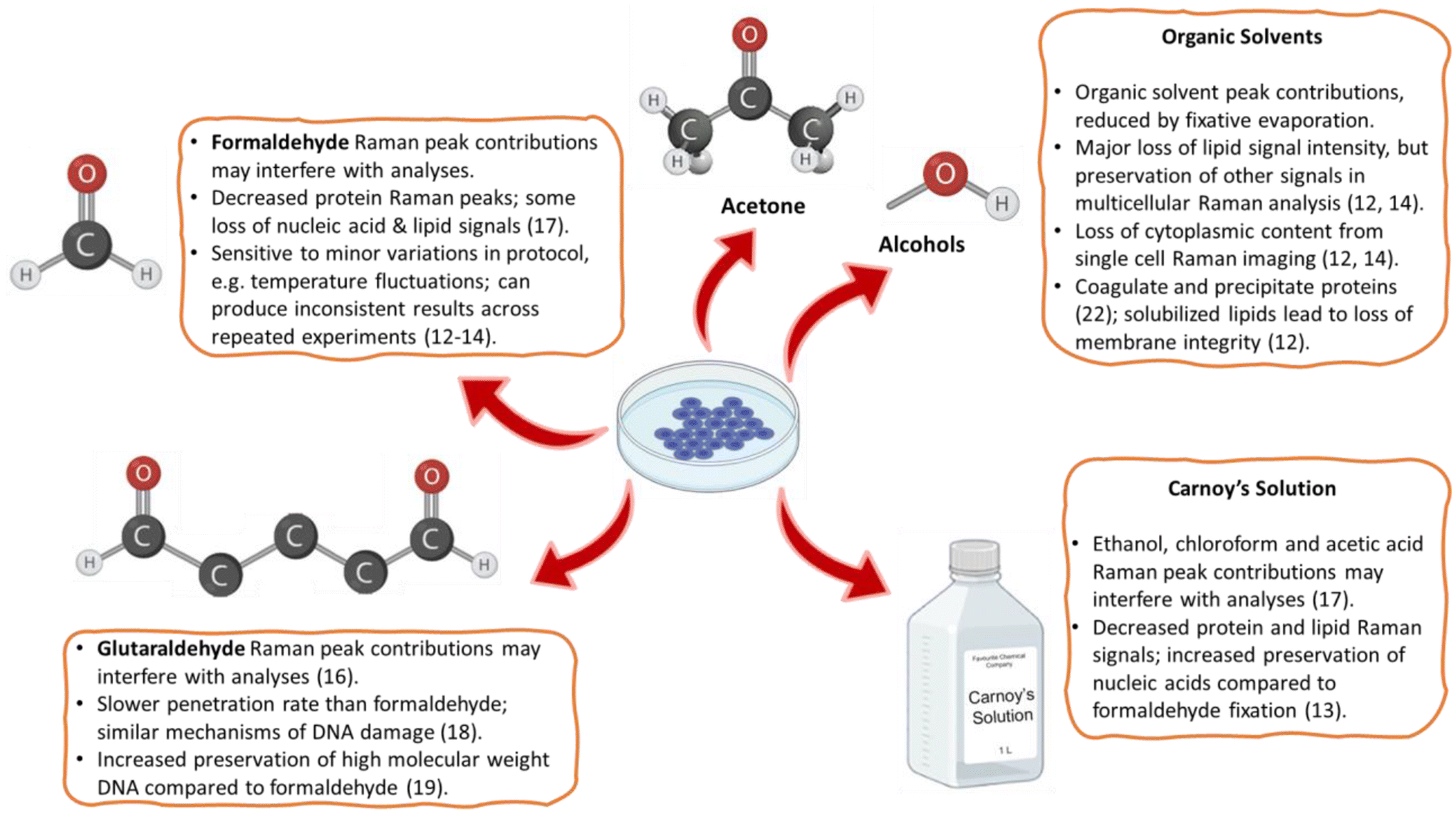 | ||
| Fig. 1 A summary of commonly used cell fixation methods for Raman spectroscopy, and their associated pitfalls.13–15,17–23 | ||
Most previously published literature examining cell fixation for Raman spectroscopy focuses on analyzing the effects of various chemical fixatives. Air-drying and desiccation of cells cultured on quartz substrates and washed with phosphate-buffered saline (PBS) are two physical cell fixation methods that have been tested with Raman spectroscopy. Previously reported results suggested that desiccation is preferable to air-drying as a fixation method for Raman spectroscopy, as it generated more consistent and higher quality spectra, with a greater effect observed in adherent cells compared to suspension cells. The contribution of residual phosphate crystals to these results remains unclear, with increased signals at 1002 cm−1 (phenylalanine) and the appearance of several peaks in the 1200–1500 cm−1 region observed in both air-dried and desiccated samples. These methods were compared to formaldehyde fixation, and it was found that desiccation yielded the most consistent results out of the three methods.15 Saline dry-fixation has the potential to both minimize alterations and interfering Raman signals generated by chemical fixatives and, since it does not include the phosphate buffer in PBS, any spectral distortions arising from the presence of phosphate crystals.
In this article, we report on the feasibility of dry-fixation with saline for cellular Raman spectroscopy of Jurkat and primary T-cells. Primary T-cells are of interest due to the recent surge in their use for cell therapy, and the potential offered by Raman spectroscopy to serve as a process analytical technology during their manufacturing.6 We additionally tested H1 human embryonic stem cells, a stem cell line that has been used in guided differentiation studies towards pancreatic β-cells for the treatment of diabetes.24 We compared saline dry-fixation with two well-established chemical fixatives, formaldehyde, and methanol, to evaluate the consistency of the methods and the impact of different concentrations of these fixatives. We chose methanol and formaldehyde fixation as the comparators because they have been evaluated against several other fixation methods and have different deleterious effects on cell spectra, thus highlighting the need for improved methods.13–15 We also assessed how stable saline dry-fixed samples are over time. Finally, we compared saline dry-fixed and live cells to investigate the extent to which results from dry-fixed samples can be compared to live-cell measurements.
2. Materials and methods
2.1. Cells and cell culture
Buffy coats isolated from human blood were obtained from the Canadian Blood Services Blood4Research facility in Vancouver, Canada.25 Primary CD3+ T-cells were isolated using the RosetteSep Human T-Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada). Isolated T-cells were cryopreserved in Immunocult-XF medium (STEMCELL Technologies) supplemented with 10% DMSO.Cryopreserved primary T-cells or Jurkat cells were thawed in a 37 °C water bath, and ∼2 × 106 cells were seeded in T-75 flasks containing 15 mL Immunocult-XF medium supplemented with 1× antibiotic-antimycotic cocktail (GIBCO, Grand Island, NY). Primary cell culture medium was additionally supplemented with Immunocult Human CD3/CD28/CD2 T-Cell Activator (STEMCELL Technologies). The flasks were placed in a humidified incubator at 37 °C and 5% CO2 for 3 days (Jurkat cells) or 5 days (primary T-cells). Then, the exponentially growing cells were harvested and fixed for Raman spectroscopy.
H1 human embryonic stem cells were obtained from WiCell Research Institute, Inc. (Madison, WI) and maintained in complete mTeSR 1 medium (STEMCELL Technologies) using T-75 flasks coated with 0.27 mg mL−1 growth factor reduced Matrigel (Corning, Corning, NY). At ∼80% confluence, cells were treated with 1× TrypLE Express Enzyme (Life Technologies, Carlsbad, CA) at 37 °C for 5 minutes. Released single cells were washed with saline and fixed for Raman spectroscopy.
All cell handling was done in a sterile manner prior to measurement, including cell culture and cell fixation. We do not anticipate any possibility of contamination once the samples are dried.
2.2. Cell fixation
For each fixation method, approximately 2 × 106 cells were first collected and centrifuged at 300g for 5 minutes. The supernatant was removed, the cells were washed once with saline, then the washed cell pellets were treated as follows.For comparison with live cells, saline dry-fixed cells were rehydrated by gently aspirating 50 μL of distilled water, followed by incubation at 37 °C for 30 minutes.
Each dried spot represented one “sample”. Up to two samples were dried on one gold mirror while ensuring there was no contact between them. 3–5 biological replicate samples were tested for each experiment described; the exact number is indicated in the corresponding figure.
2.3. Raman spectroscopy
All fixed samples were stored at 4 °C until Raman spectra were collected. Raman spectroscopy was performed using an inVia™ Raman microscope (Renishaw, Gloucestershire, UK) equipped with a 785 nm diode laser that provided ∼100 mW power at the sample.26 The Raman microscope was calibrated using a silicon wafer standard at the start of each day of measurement. Raman spectra from the fixed-cell samples were acquired in map acquisition mode, a built-in acquisition mode in WiRE 4.4 (Renishaw) that enables collection of spectra along the points of a uniformly spaced grid on the sample surface. Spectra were collected using a 50× objective lens (Leica Microsystems, Wetzlar, Germany) with 10 s acquisition time per spectrum; 50–70 spectra were collected per sample and subsequently averaged. Each spectrum was estimated to contain information from 10–15 cells.For live cell spectroscopy, ∼20 × 106 Jurkat cells suspended in 50 μL of saline were pipetted onto a gold mirror. The cell suspension was covered with a CaF2 cover slip and Raman spectra were acquired as described previously but with a 40 s acquisition time per spectrum to improve the signal-to-noise ratio. While some cell movement was observed, the high concentration of cells yielded high-quality spectra.
2.4. Data analysis
Spectra were preprocessed using a suite of automated procedures, including baseline correction, smoothing, and cosmic ray spike removal, implemented in MATLAB (MathWorks, Natick, MA).27 Depending on the type of analysis performed, different normalization protocols were used due to the varied effects of the fixatives on cellular components. We normalized spectra to the nucleic acid peak at 782 cm−1 for univariate analyses to control for potential differences in sample thickness and resulting spot-to-spot variability in cell material as well as laser power fluctuation and enable comparisons between different samples. Raman spectra were constant-sum normalized for principal component analysis (PCA) as this often improves scores-based clustering. The PCA and paired t-tests for statistical analysis were performed using MATLAB.Standard deviations were calculated using MATLAB. Upper and lower standard deviations were calculated separately, to more accurately reflect their respective deviations from the mean spectra, using the following formulae –
N = number of points for a given wavenumber.
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) = the mean of these N points.
= the mean of these N points.
n H = number of points greater or equal to the mean.
n
L = number of points less than the mean.

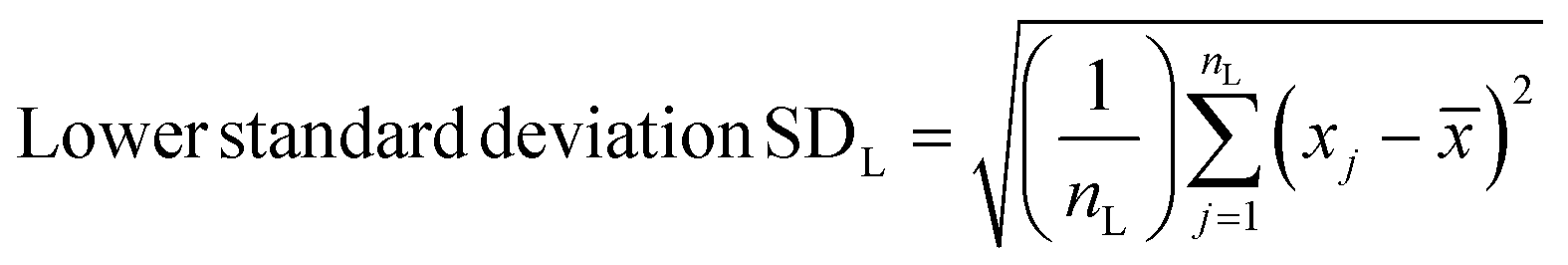
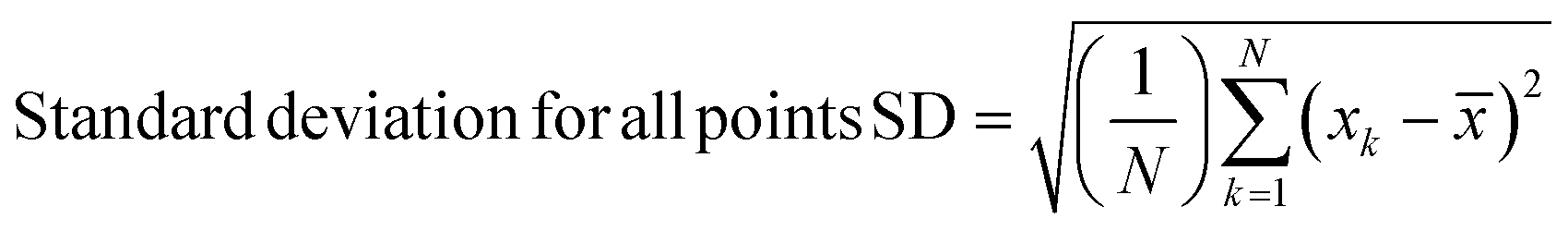
3. Results
3.1. Comparative evaluation of saline-dry fixation
We compared Raman spectra from saline dry-fixed, methanol-fixed, or formaldehyde-fixed primary T-cells (Fig. 2A and C), Jurkat cells (Fig. 2B and D), and H1 human embryonic stem cells (Fig. S1A and B†). Methanol-fixed cells of all cell types showed the greatest differences compared to the other two methods, most notably a distinct reduction or total loss of lipid-associated peaks, including 717 cm−1, 1065 cm−1, 1095 cm−1, 1255 cm−1, 1300–1305 cm−1 and 1450 cm−1. This loss of lipid signals also influenced peak shapes as shown by how a reduced 717 cm−1 phospholipid signal in methanol-fixed cells is likely responsible for the disappearance of the shoulder in the inset peaks, and the displacement of the peak maximum by ∼2 cm−1 (Fig. 2A and B).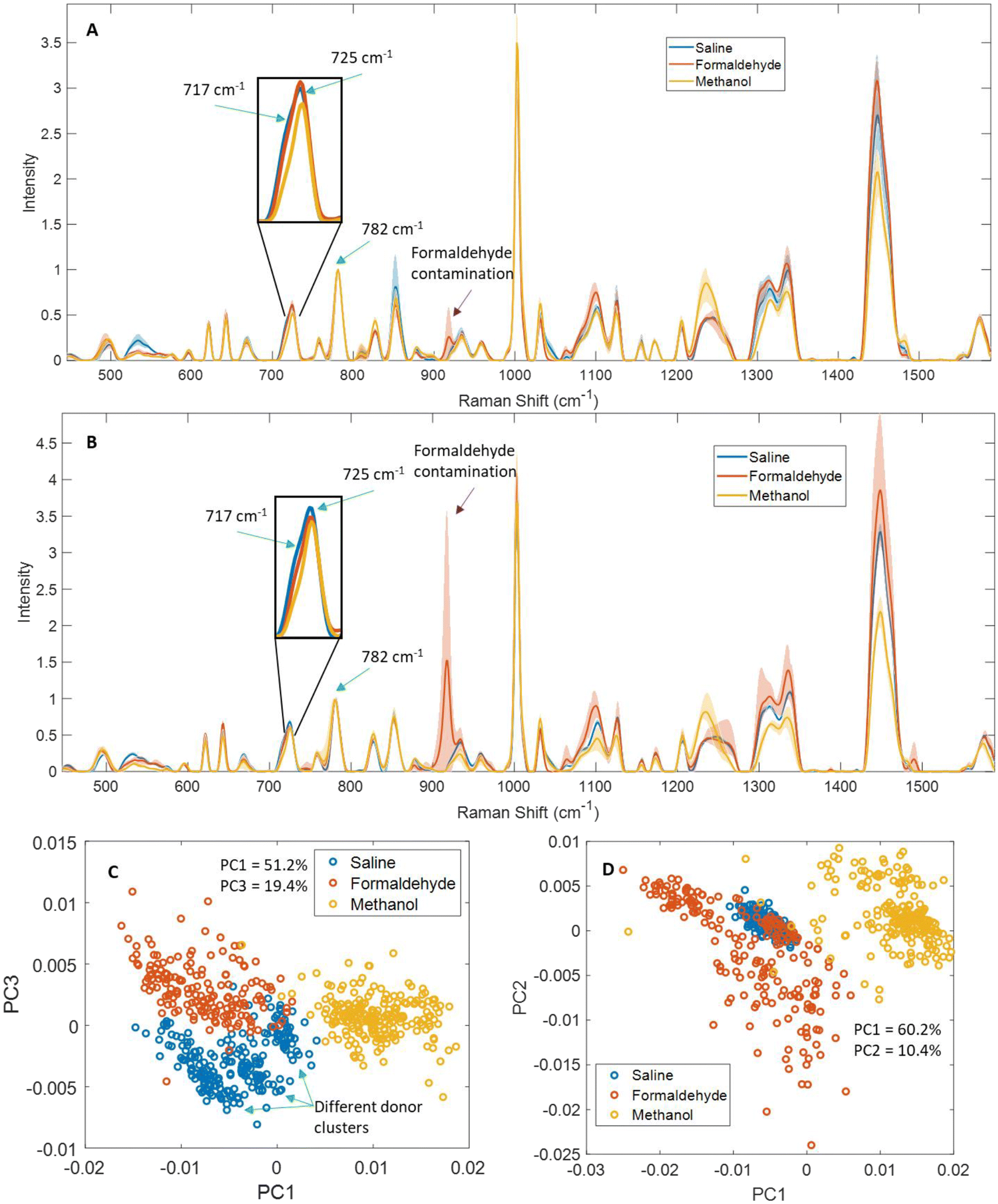 | ||
| Fig. 2 Normalized mean spectra from saline dry-fixed, methanol-fixed, and formaldehyde-fixed cells. (A) Mean spectra from primary human T-cells cells (n = 4 biological replicates) and (B) mean spectra from Jurkat cells (n = 4 biological replicates) showed expected differences between the fixation methods. Shaded regions indicate upper and lower standard deviations of biological replicates, calculated separately. PCA of (C) primary T-cell spectra and (D) Jurkat cell spectra showed clear separation between the different fixatives, with indications of donor-to-donor variability preserved in saline dry-fixed primary cell spectra. | ||
Methanol-fixed cells generated more intense protein signals, which is an expected outcome of protein precipitation. The formaldehyde-fixed cell spectra had an overall decrease in intensity, particularly in nucleic acid- and protein-associated peaks, as can be seen in unnormalized mean spectra (Fig. S2†). This was not clear in the (Fig. 2A and B) spectra since there were normalized to the 782 cm−1 nucleic acid peak.
Additional analysis of formaldehyde-fixed spectra proved troublesome due to the appearance of many formaldehyde peaks (918 cm−1, 1100 cm−1, 1335 cm−1, 1388 cm−1 and 1490 cm−1), which resulted in greater spectral inhomogeneity. We evaluated a spectrum of formaldehyde dissolved in saline and dried and observed the aforementioned peaks (Fig. S15†). We expect this inhomogeneity was amplified by the residual formaldehyde distributing unevenly in the samples during drying. Nevertheless, peaks not directly altered by the presence of formaldehyde also showed greater variability in formaldehyde-fixed samples, including 1002, 1030, 1300–1305, and 1450 cm−1. In contrast, saline dry-fixed cells showed greater spectral consistency across all replicates, while preserving most lipid-, protein-, and nucleic acid-associated peaks (Fig. 2A and B, S1A†). These observations were confirmed by PCA (Fig. 2C and D, S1B†), where methanol-fixed spectra showed clear separation along the first principal component, formaldehyde-fixed spectra showed the greatest variability across replicates in both principal components, and saline dry-fixed spectra overall showed greater consistency than the other two methods. Notably, saline dry-fixed spectra also preserve evidence of primary T-cell donor-to-donor variability in PCA (Fig. 2C and S13†), where the three indicated clusters formed correspond to spectra from different donor samples (scores from two donors overlap at the lowest PC1 values).
Lastly, we also compared air-drying in a biosafety cabinet to desiccation for saline dry-fixation and observed no significant differences between the methods (Fig. S11†).
3.2. Effects of fixative concentration
To further clarify the effects of these fixatives on specific peaks in Raman spectra, we tested four concentrations for each fixative. Deionized, distilled water was used to prepare diluted solutions of each fixative at concentrations of 100%, 75%, 50%, and 25% relative to the stock concentrations. For example, “100%” refers to the typically used concentration of the fixative −154 mM NaCl for saline, 5% w/v paraformaldehyde solution, or undiluted methanol. Mean spectra from saline dry-fixed samples showed relatively minor differences across the range of wavenumbers measured. A decrease was observed in some protein- and lipid-associated peaks at 25% and 50% saline – notably, 1002 cm−1, 1126 cm−1, 1155 cm−1, the amide III region from 1220–1260 cm−1, 1310 cm−1, 1337 cm−1, and 1448 cm−1, however the decrease is <10% of the maximum measured intensity in each peak (Fig. 3A). The inset in Fig. 3A shows excellent consistency across concentrations for lipid and adenine signals. This suggests that dry-fixation with saline does not systematically alter the Raman activity of these cellular biomolecules. The observed differences can likely be attributed to some lysis when cells were exposed to low osmotic pressures in samples fixed with diluted saline. PCA confirmed the minimal impact of these differences, with data from all concentrations overlapping and no evidence of separate clusters at the different saline concentrations (Fig. 3D).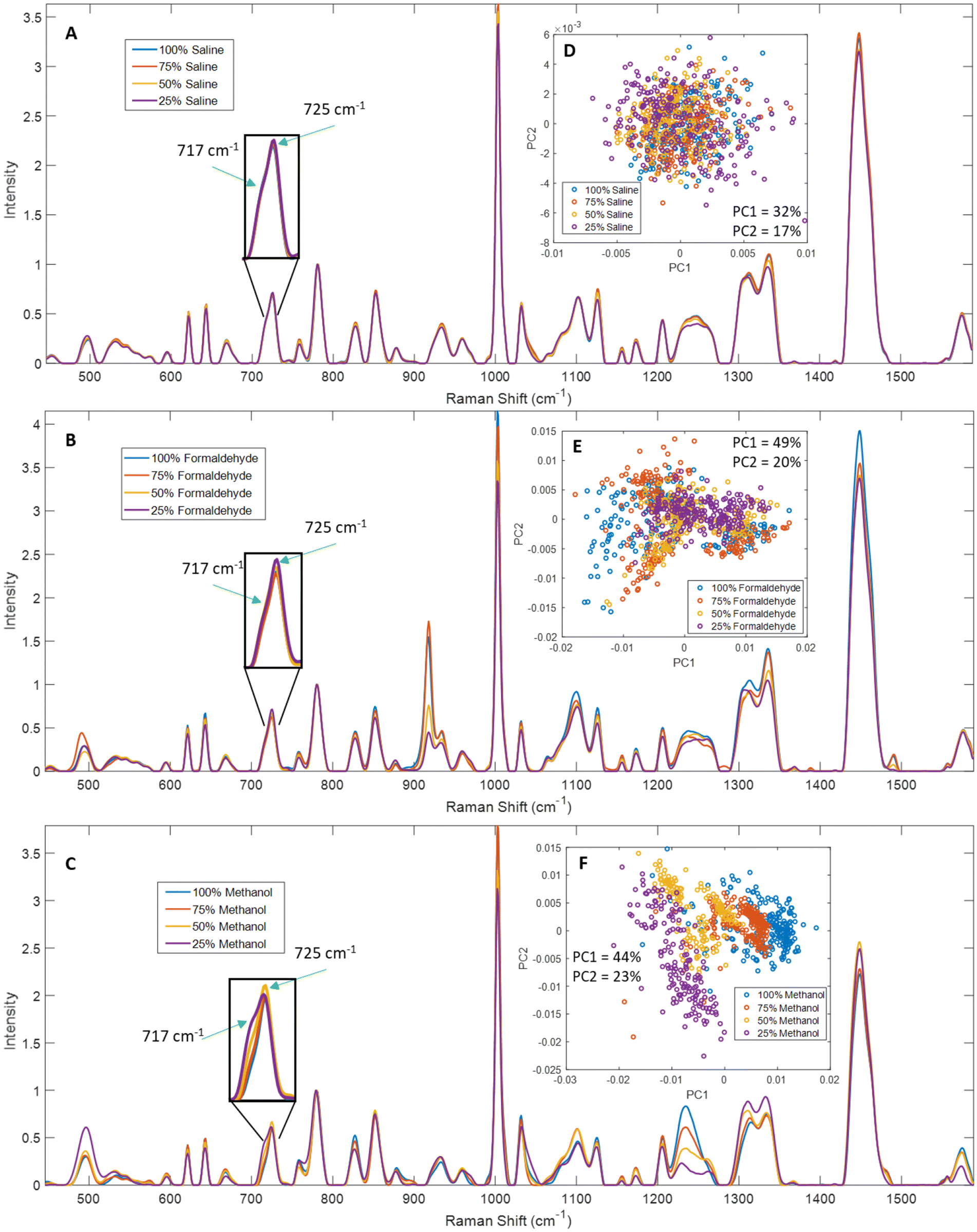 | ||
| Fig. 3 Normalized mean spectra from Jurkat cells fixed with different concentrations of saline, formaldehyde, and methanol. (A) and (D) Mean spectra and PCA from saline dry-fixed cells (n = 5 biological replicates) showed relatively minor differences across the spectral range, and all concentrations clustered together in PCA. (B) and (E) Mean spectra and PCA from formaldehyde-fixed cells (n = 4 biological replicates) showed differences in formaldehyde concentrations as well as protein-associated peaks, with unclear clustering in PCA that was influenced by formaldehyde signals despite removing the 895–935 cm−1 region before the PCA analysis. (C) and (F) Mean spectra and PCA from methanol-fixed cells (n = 4 biological replicates) showed a loss of spectral quality and better retention of lipid signals at the lower concentrations, with PCA showing separate clustering for each concentration. | ||
Spectra from formaldehyde-fixed cells showed numerous effects dependent on fixative concentration. Many formaldehyde peaks were observed to arise (918 cm−1, 1100 cm−1, 1335 cm−1, 1388 cm−1 and 1490 cm−1) and their relative intensities were directly correlated with formaldehyde concentration in 782 cm−1 normalized mean spectra (Fig. 3B), except for 75% formaldehyde which had slightly higher signals than 100% formaldehyde. Adding multiple wash steps may help reduce or eliminate the signal contributions of residual formaldehyde to a certain extent,28 however the effects of formaldehyde fixation on other peaks listed below are expected to persist. We tested this further by washing formaldehyde fixed cells up to three times; this experiment was performed by two different users in parallel (Fig. S16†). While additional wash steps reduced the signal contribution from formaldehyde in most cases, user #1 observed the highest average formaldehyde signal after two washes, indicating stochasticity of residual formaldehyde distribution on the sample surface. Additional wash steps also showed decreasing intensity of lipid signals (including 717, 1255, 1303 and 1450 cm−1), as well as loss of intensity in the 515–540 cm−1 region. Data from both users showed that three washes were insufficient to completely remove residual formaldehyde signals. The inset in Fig. 3B shows good consistency of the 717 cm−1 lipid signal across concentrations, but a slight decrease of the 725 cm−1 adenine signal at higher formaldehyde concentrations. Most protein-associated peaks (notably 621 cm−1, 643 cm−1, 827 cm−1, 851 cm−1, 1002 cm−1, 1125 cm−1 and 1220–1260 cm−1) also increased with increasing formaldehyde concentration, however comparing trends in peak intensity with 782 cm−1 normalized data is expected to be inaccurate as formaldehyde likely influences nucleic acid signals. We further analyzed these spectra using PCA, after removing the 895–935 cm−1 data containing the largest formaldehyde peak, to minimize the impact of formaldehyde signal (Fig. 3E). The data from all concentrations showed some overlap, but with varied clustering at the different formaldehyde concentrations. While the 25% formaldehyde formed a single cluster, the data from 100%, 75% and 50% formaldehyde each formed two or more clusters in the PCA. The first principal component showed strong contributions from 1440 cm−1, 1330 cm−1, 1300 cm−1 and 1260 cm−1, while the second principal component showed strong contributions from 780 cm−1, 1002 cm−1, 1093 cm−1, 1337 cm−1 and 1450 cm−1 (Fig. S3†), indicating that both peaks generated by formaldehyde as well as peaks impacted by formaldehyde fixation contribute to the observed clustering. These results suggest that formaldehyde concentrations typically used for cell fixation introduce unpredictable variability in cell Raman spectra through distortion of cell macromolecular peak intensities as well as potential formaldehyde signal contributions.
Methanol-fixed cell spectra showed relatively predictable changes in comparison to formaldehyde. The intensity of lipid-associated signals (717 cm−1, 1065–1080 cm−1, 1265–1270 cm−1, 1300 cm−1 and 1448 cm−1) was inversely correlated with methanol concentration in 782 cm−1 normalized spectra (Fig. 3C). The inset in Fig. 3C shows increasing lipid signals at 717 cm−1 with decreasing methanol concentration. Unexpectedly, decreased protein signals (621 cm−1, 643 cm−1, 1002 cm−1, 1031 cm−1) were observed in 25% and 50% methanol spectra, along with substantial distortion of the amide III region (1220–1260 cm−1). This may be caused by a loss of membrane integrity, perhaps due to the cells experiencing osmotic stress when exposed to methanol diluted with water, or due to partial freezing of cell suspensions in diluted methanol during incubation at −20 °C. PCA showed relatively clear separation of the data from the different concentrations (Fig. 3F). The two lower methanol concentrations showed greater variability, which is an expected outcome of the lower signal-to-noise ratio observed in spectra from those samples.
3.3. Temporal stability of saline dry-fixed cells
One of the major advantages of chemical fixatives is their ability to preserve samples for long periods of time, up to several years in some cases. We wanted to establish how long saline dry-fixed samples could be stored without substantial loss of Raman spectral quality. To assess this, we dry-fixed Jurkat cells with saline and measured them over time, with multiple measurements over the first week, followed by weekly measurements for the first ∼1.5 months, and bi-weekly measurements up to 7 months (n = 3 biological replicates). Samples were stored in a 4 °C refrigerator between measurements. No specific light parameters were considered for this study. Mean spectra from each replicate showed good consistency, with standard deviations <10% of the mean across the measurement range (Fig. 4A). We further examined this data by comparing standard normal variate (SNV) profiles of unnormalized representative peaks for proteins (1002 cm−1 phenylalanine), total nucleic acids (782 cm−1), lipids (1303 cm−1 CH3/CH2 twisting and bending modes), and carbohydrates (937 cm−1) (Fig. 4B, S4A and S5A†). SNV profiles showed variations over time that correlated well with unnormalized peak intensities and did not show a consistent decline over time for peaks from any macromolecule (Fig. 4B and C, Fig. S4A, B and S5A, B†). Normalized intensities for the same peaks (with 725 cm−1 replacing 782 cm−1) showed excellent consistency over the range of measurement, with <10% differences over the time-course and no monotonic decline observed in any of the peaks tested (Fig. 4D). We visualized the time-course data using surface plots of 782 cm−1 normalized mean spectra across the range of measurement days, that showed few changes across the spectral range (Fig. S6†). While fluctuations are observed in many peaks throughout the measurement range, these differences were not monotonic and can likely be attributed to cell–cell variations in the specific regions of the sample that were measured. Paired t-tests were performed on selected normalized peaks between days 0–49 and 50–220 of the time course of measurement and showed that the change in intensity was small but statistically significant for the normalized peaks tested (725 cm−1, 937 cm−1, 1002 cm−1 and 1303 cm−1) across three replicates, suggesting that some sample degradation cannot be ruled out over 7 months of storage (Fig. S12†). However, the same peaks did not show a statistically significant difference between days 0–28 and 29–90 of measurement in any of the replicates tested. PCA showed some overlap for data from days 0–60 and 61–150, with greater separation observable in PC2 from days 151–210 (Fig. 4E). An examination of principal component loadings revealed derivative-like features in three sharp protein-associated peaks (621 cm−1, 643 cm−1 and 1002 cm−1) in PC2, suggesting that the observed separation arises from slight wavenumber drifts over time combined with the large number of sampling days, and not from systematic macromolecular changes (Fig. 4F).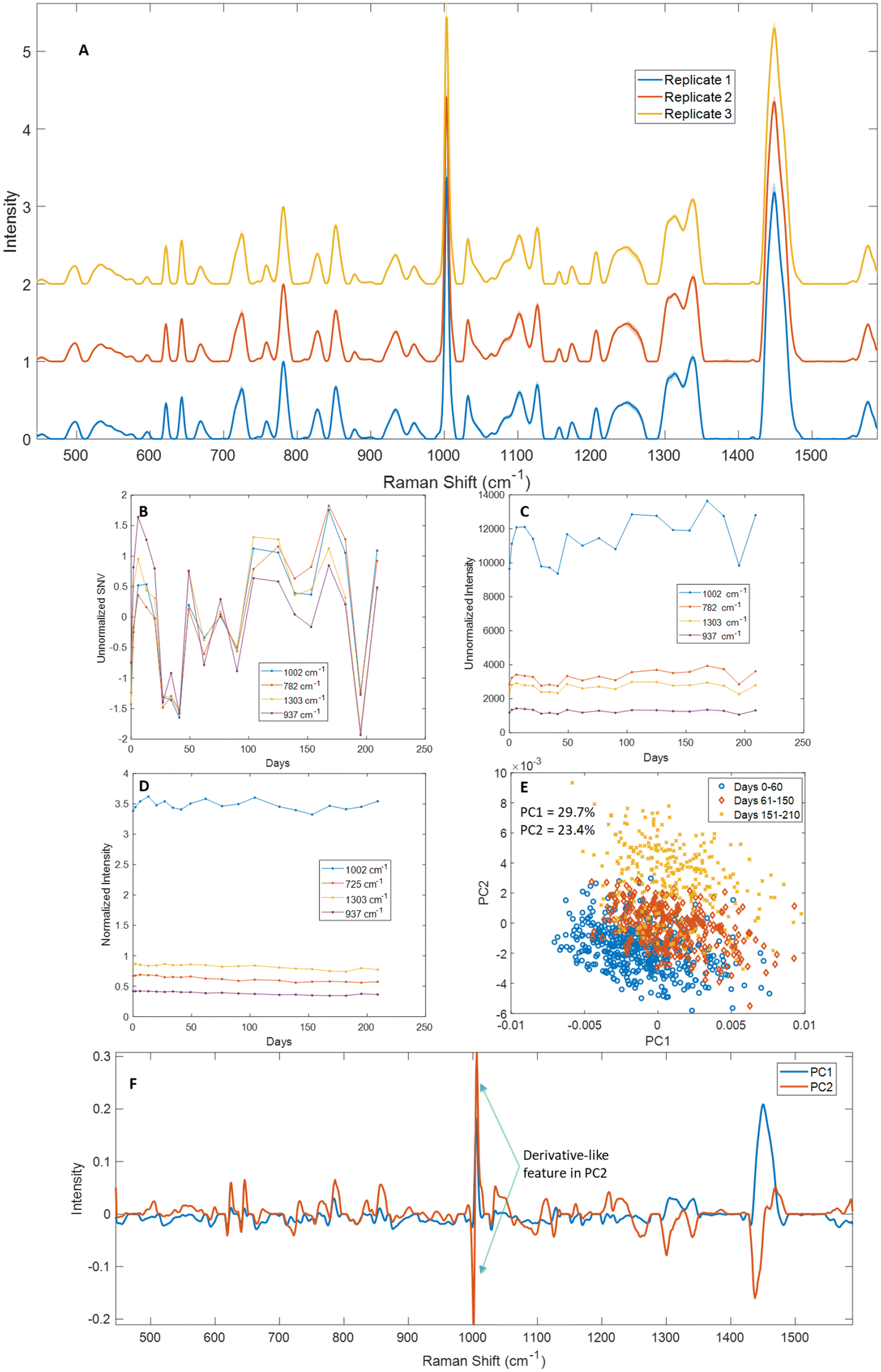 | ||
| Fig. 4 Temporal stability of saline dry-fixed Jurkat cells. (A) Mean spectra from 3 biological replicates of Jurkat cells measured over ∼220 days; shaded regions indicate standard deviations across the time-course of measurement. For replicate 1: (B) SNV profiles of peaks representative of different macromolecules over the measurement range and (C) unnormalized mean intensity profiles of the same peaks. (D) Normalized mean intensity profiles of peaks corresponding to different macromolecules showed <5% variation across the measurement range. (E) PCA showed tight clustering for the first ∼150 days, with some separation observed from days 151–210. (F) PC loadings showed derivative features in PC2, suggesting that the observed variability arose from instrument drift over time. | ||
A similar examination was performed for the other two replicates (Fig. S4–S6†). While replicate 2 (Fig. S4 and S6B†) showed excellent agreement with Fig. 4, the data from replicate 3 (Fig. S5 and S6C†) showed a greater decline in intensity for some macromolecules. A 10–20% decrease was observed in many protein-associated peaks in the latter half of the time course of measurement (621 cm−1, 643 cm−1, 757 cm−1, 827 cm−1, 834 cm−1, 934 cm−1, 1002 cm−1, 1100 cm−1, 1220–1260 cm−1 and 1337 cm−1). A decrease of 15–20% was observed in lipid-associated peaks at similar timescales to protein signals (596 cm−1, 717 cm−1, 1082 cm−1, 1101 cm−1, 1265 cm−1, 1303 cm−1, 1315 cm−1 and 1448 cm−1). Lastly, a decrease was also observed in nucleic acid-associated peaks – adenine (725 cm−1), RNA (811 cm−1) and thymine and guanine (667 cm−1). However, since these differences over time were largely not monotonic and were not observed in either of the other two replicates, we expect it is a result of uneven sampling of Raman spectra from the sample over the time course, and not a systematic change in the macromolecular content of the cell samples.
3.4. Comparison of live and saline dry-fixed cells
Live cell spectroscopy is the gold standard for cellular Raman spectroscopy, and ideally would enable the sample-free analysis of cells cultured for therapy.6 We compared live and saline dry-fixed Jurkat cell Raman spectroscopy to characterize the impacts of dry-fixation and thereby assess how effectively fixed-cell sample analysis can predict live cell results. Saline dry-fixed cells were also rehydrated with deionized water after spectral acquisition, to help clarify the hydration effects on the spectra.Mean spectra from live and saline dry-fixed samples were normalized to the total nucleic acid peak at 782 cm−1 in each spectrum (Fig. 5A). Several differences were observed across the spectral range tested. Most protein- and lipid-associated peaks showed greater intensity in dry-fixed cells compared to live cells, including 515–540 cm−1, 621 cm−1, 643 cm−1, 828 cm−1, 853 cm−1, 879 cm−1, 1002 cm−1, 1031 cm−1, 1155 cm−1, 1173 cm−1, 1205 cm−1, 1220–1260 cm−1, 1337 cm−1 and 1448 cm−1 (proteins) and 596 cm−1, 717 cm−1, 1101 cm−1, 1265 cm−1, 1303 cm−1, 1315 cm−1 and 1448 cm−1 (lipids). Some nucleic acid-associated peaks showed greater intensity in saline dry-fixed samples (667 cm−1, 725 cm−1 and 1577 cm−1), while others showed greater intensity in live cells, including the RNA peak at 811 cm−1, and the low intensity peaks at 1373 cm−1, 1421 cm−1 and 1513 cm−1.
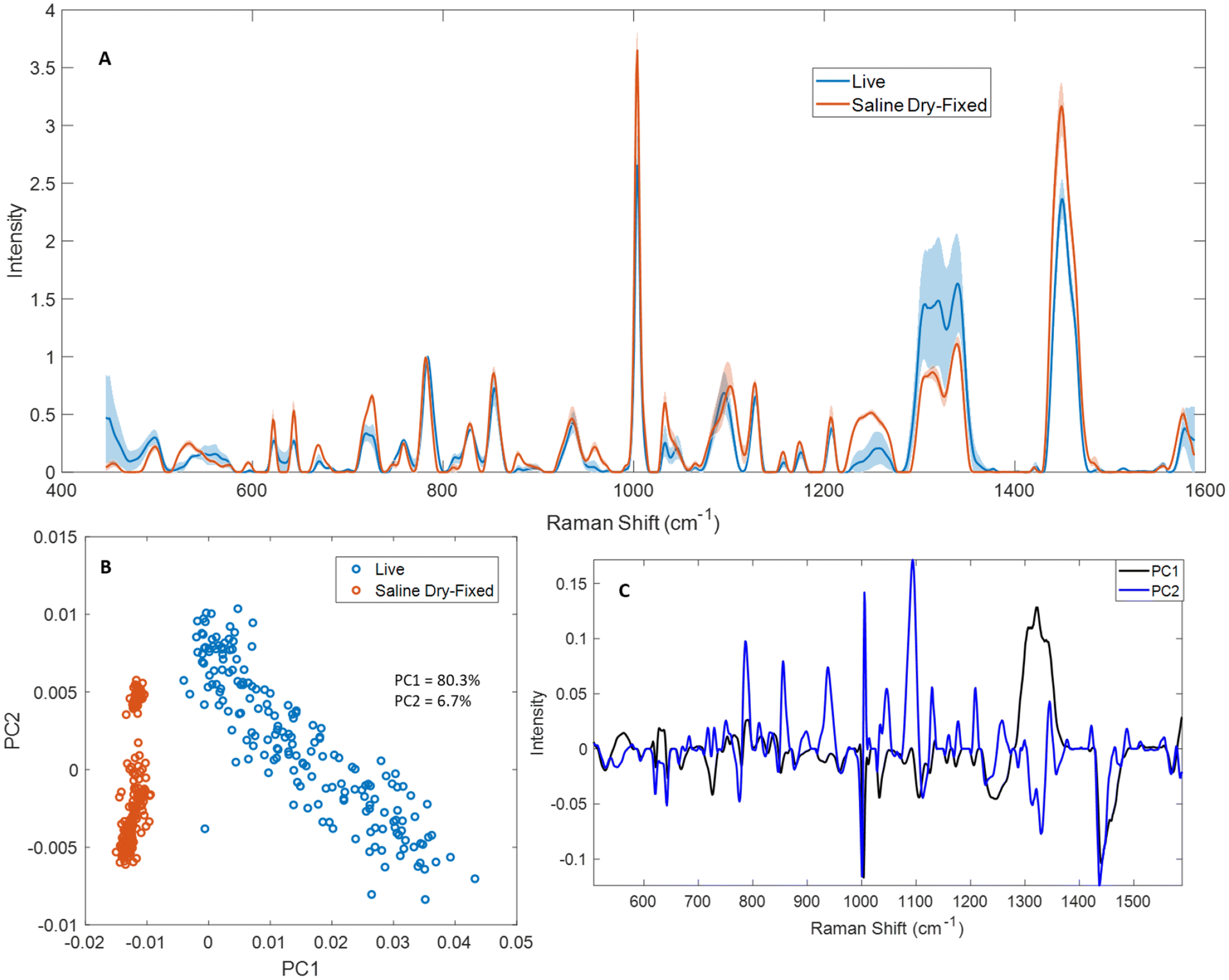 | ||
| Fig. 5 Normalized mean spectra from live and saline dry-fixed Jurkat cells. (A) Mean spectra (n = 4 biological replicates) showed many different features in protein- and lipid-associated peaks between live and saline dry-fixed cells, both in peak shape and intensity. The shaded regions indicate upper and lower standard deviations of biological replicates, calculated separately. (B) The samples clustered separately in PCA, with saline dry-fixed samples showing less variability than live cells. (C) PC loadings show the basis of separation, with differences in the 1300–1340 cm−1 region dominating the separation of live cells along PC1, and differences in nucleic acids and 1100 cm−1 dominating the separation along PC2. | ||
Saline dry-fixed samples were subsequently rehydrated, to understand which spectral changes occurred because of the hydration state of the sample (Fig. S14†). Rehydrated samples generally showed lower intensity in many protein- and lipid-associated peaks, suggesting sample changes during the fixation–rehydration process. Substantial peak shape changes were observed in saline dry-fixed cells compared to live cells at 530 cm−1, 725 cm−1, 749 cm−1, 1045 cm−1, 1080–1102 cm−1, 1220–1260 cm−1, 1304 cm−1, 1320 cm−1 and 1490 cm−1. These shape changes were recovered upon rehydrating saline dry-fixed samples at all peaks listed above except 530 cm−1 and 1318 cm−1, suggesting that the changes in the shapes of other peaks is a result of the hydration state of the sample (Fig. S14A†). A peak shift was observed in dry-fixed samples that produced a peak with maximum intensity at 782 cm−1, a total nucleic acid peak, while hydrated samples produced a maximum intensity at 784 cm−1. No such difference was observed in other sharp peaks including 621 cm−1, 643 cm−1 and 1002 cm−1.
Live and saline dry-fixed samples clustered separately in PCA (Fig. 5B) along the first principal component; this is an expected result based on the large variability observed in the mean spectra across multiple peaks. Adding rehydrated samples to this analysis produced a similar result, with live and dry-fixed rehydrated samples clustering separately along PC2 (Fig. S14B†). Saline dry-fixed samples showed the least variability in PCA and formed a tight cluster, while the rehydrated samples showed more variability and live cells showed the greatest variability along PC1. An examination of PC loadings revealed that dried and hydrated samples separate primarily based on the 1300–1340 cm−1 region, along with strong contributions from 1002 cm−1 and 1448 cm−1 (Fig. 5C and S14C†). It is expected that much of the variability observed in live samples along PC1 is also based on differences in 1300–1340 cm−1, as an examination of raw and preprocessed unnormalized spectra indicated substantial variability in that spectral region (Fig. S7†). The dry-fixed, rehydrated spectra were distinguished along PC2 based on the amplified signal at 1100 cm−1, along with strong contributions from 1002 cm−1 and 782 cm−1, suggesting possible reorganization or degradation of the rehydrated nucleic acid and protein cellular components.
4. Discussion and conclusions
Compared to the rapid and intensive development of Raman spectroscopy data pre-processing and data analytics, relatively little analysis has focused on developing improved methods for cell sample processing. In this article, we examined the effects of unbuffered saline dry-fixation on the Raman spectra of cells and contrasted these effects with the commonly used chemical fixatives methanol and formaldehyde. While chemical fixatives have many applications in other biological research, in Raman spectroscopy applications their tendency to introduce or alter molecular vibrations impacts their interpretability, often producing spectra with diminished or confounded signals.Methanol fixation solubilizes lipids but precipitates proteins; precipitated proteins produce strong Raman bands with excellent signal-to-noise ratio, leading to very high overall spectral quality, albeit with drastically reduced lipid signals and rearranged lipid distribution across the sample. This observation was also consistent with the observation that methanol concentration was inversely correlated with lipid signal intensity. Indeed, methanol fixation may be a suitable method in situations where the required information does not include contributions from lipids, and intracellular resolution is not required as the distribution of biomolecules is altered by lipid solubilization and protein precipitation. However, methanol diluted with water results in spectra with unexpected peak changes and worse signal-to-noise ratio as evidenced by Fig. 2C, and as such it makes sense that diluted methanol is not used for Raman spectroscopy cell fixation.
Formaldehyde-fixed cell spectra proved to be the most troublesome to deal with out of the methods tested. We recommend avoiding formaldehyde fixation, when possible, as this method was the least reproducible across all our tests. Comparing raw and preprocessed spectra from a single biological sample for each fixative showed that cells fixed with formaldehyde produced the greatest variability within a sample (Fig. S8†). From the same figure, it is also evident that this variability arises from both greater sample inhomogeneity as well as differing strengths of formaldehyde signal, despite a saline washing step. Additional washing steps did generally reduce residual formaldehyde signals but did not eliminate them, and instead also led to diminished lipid peak intensities, suggesting that formaldehyde signals cannot be eliminated without loss of macromolecular signal intensity (Fig. S16†). Further, we observed differences between reported Raman signals from formaldehyde-fixed tissues and our formaldehyde-fixed cells. Typically reported formaldehyde signals from tissues include 907 cm−1, 1041 cm−1, 1254 cm−1, 1490 cm−1 and 1492 cm−1.28,29 In contrast, we observed strong formaldehyde contributions at 917 cm−1, 1094 cm−1, 1335 cm−1, 1387 cm−1 and 1490 cm−1 that were highlighted in a formaldehyde-fixed Jurkat cell sample that had been improperly washed (Fig. S9†). These differences may arise from inherent variability between cells and tissues, and from fixation protocol differences. For example, tissues are usually fixed for longer time periods (several hours) to enable formaldehyde to penetrate into the tissue, while we fixed cells using a 10-minute incubation in formaldehyde. Additionally, in addition to formaldehyde, commercially purchased formalin typically contains 10% methanol as an additive to facilitate storage and prevent degradation, which may further influence Raman spectra. We avoided this confounding factor by using freshly prepared formaldehyde from paraformaldehyde powder.
In aqueous solutions, formaldehyde is hydrated to form methylene glycol. Carbonyl formaldehyde (also called “dehydrated” formaldehyde) and methylene glycol exist in equilibrium in aqueous solution which favors methylene glycol, and more carbonyl formaldehyde is generated as the existing formaldehyde is removed from the solution by the fixation reaction. Both methylene glycol and carbonyl formaldehyde can form cross-links and fix cells and tissues. The most important parameter influencing fixation rate is reported to be the shift in equilibrium towards the formation of more carbonyl formaldehyde, which can be achieved by using higher temperatures or a slightly alkaline pH of 7–8.22 It has also been reported that Raman spectra of formaldehyde solutions primarily correspond to methylene glycol; the presence of carbonyl formaldehyde may thus also contribute to the observed differences between previous reports of formaldehyde fixation and the peaks observed in our dehydrated samples.22
We chose unbuffered saline as the cell suspension solution for physical fixation as phosphate crystals produced upon drying phosphate-buffered saline (PBS) generates substantial distortions in the biological fingerprint region of Raman shifts, notably at 1002 cm−1, multiple peaks from 1200–1340 cm−1, 1418 cm−1, 1430 cm−1 and 1454 cm−1 (Fig. S10†). Saline dry-fixation produced more consistent spectra than formaldehyde or methanol fixation over a similar scanned area in multiple samples, suggesting that the samples produced by dry-fixation were more homogeneous. This was further supported by saline dry-fixed samples having the lowest overall standard deviation from the mean and with the least spread in PCA across both Jurkat and primary T-cells. Notably, the evidence of donor-to-donor cell variability was preserved in the PCA of saline dry-fixed primary T-cells, whereas it was not for the formaldehyde and methanol fixation methods. Donor-to-donor variability is a feature of primary cells isolated from different donors.30,31 Since investigating this variability is of great interest, the preservation of these inherent differences is an important advantage of the saline method.
Dry-fixing with different dilutions of saline did not produce systematic alterations in spectra, while similar tests with methanol and formaldehyde did produce systematic alterations, further supporting the conclusion that saline dry-fixation has minimal direct impacts on samples. We examined saline concentrations as low as 10% and did not observe a large effect on collected spectra, and they remained comparable with spectra collected from 25% and 50% saline concentrations (data not shown). Though low salt concentrations would also be expected to produce osmotic stress, it is likely less severe than would occur in diluted methanol where no salt is present. We also tested saline dry-fixation for single cell spectroscopy (data not shown) and generated high-quality spectra from single cells; however, we would recommend using diluted saline (25%) as using the 100% saline concentration makes individual cells difficult to visualize due to the presence of salt crystals. Some cell shrinkage is observed during the drying process, which is an expected outcome of increasing salt concentration in the extracellular environment.
Previous reports have indicated that desiccation is preferred to air-drying for physical fixation.15 We compared our method of slow air-drying in a biosafety cabinet to drying in a desiccator as previously recommended and observed no differences, suggesting that either drying approach is acceptable for saline dry-fixation (Fig. S11†). However, loss of spectral quality was observed when we used a fast stream of air to rapidly dry a sample (data not shown).
It is important to know how long samples can be stored without substantial changes to their Raman spectra, including in cell therapy clinical trials where adverse outcomes may arise months after cells are administered. Measurements of stored cell samples are particularly useful for retrospective biomarker discovery, once therapeutic or other process outcomes are known. We assessed the stability of saline dry-fixed samples after up to ∼7 months of storage. Minor variability (<5%) was observed in sharp protein-associated peaks in the first several measurements, up to ∼90 days of storage. Subsequently, a 10–20% decrease was observed in protein- and lipid-associated peaks compared to fresh samples, as well as a larger decrease in RNA at 811 cm−1, with the greatest decline occurring during the last ∼40 days of storage. These observations suggest that the decline in signal intensity begins around two months of storage, but saline dry-fixed samples produce reasonable quality spectra till at least the 6-month mark. The small, systematic changes observed over the period of storage should not substantially interfere with analysis in a therapeutic cell manufacturing context. We expect that useful measurements of these samples can be made even beyond the recommended storage period, as long as temporal peak stability for the cell types in question are tested, and the user is aware of the slight losses in peak intensity over time.
Live cell spectroscopy is generally considered the gold standard for cellular Raman spectroscopy and it enables the potential of non-destructive, label-free analysis. This is particularly relevant in applications such as cell therapy manufacturing, where Raman spectroscopy implemented in a non-invasive manner may be one of the few methodologies that can provide near continuous information about the state of cells during the manufacturing process.6 In practice, however, Raman facilities generally do not have the equipment or expertise to handle complex cell culture protocols. In such cases, fixed cell analysis is an essential intermediate step to establish the suitability of Raman spectroscopy towards monitoring a cell culture process and its outcomes. To this end, we compared live and saline dry-fixed cells to establish what systematic changes occur in dried samples, and rehydrated the dry-fixed samples to determine which spectral features can be recovered by rehydration. While several changes were observed between dry-fixed and live samples, most peak shapes were recovered upon rehydration. There was a loss of intensity in some peaks of the rehydrated sample, suggesting sample changes during the fixation–rehydration process. Dry-fixed samples showed greater consistency than live cells in PCA, likely due to the live cell suspension movement, as opposed to dried samples which formed a thinner, static layer during the drying process. The recovery of some aspects of live cell spectra suggests that rehydration could be used to approximate live cell spectra as rehydration appears to provide an intermediate condition between saline dry-fixed and live cells. We expect this intermediate condition arises from a reorganization of cellular macromolecular content during the rehydration process, where hydrophilic nucleic acids and some proteins would readily dissolve in water, while hydrophobic lipids and proteins would remain insoluble, leading to differing concentrations of these macromolecules in the Raman sampling volume between live and rehydrated samples. Given these observations, we expect that univariate analyses can be translated from dry-fixed to live cell settings with relative consistency, as long as the user is aware of the systematic differences arising from hydration. Multivariate analyses may require greater effort for adaptation, as dry-fixed samples produced spectra with greater consistency than live samples (Fig. S7†).
In summary, we recommend physical dry-fixation with unbuffered saline as a superior alternative to chemical fixatives. While a more thorough examination of fixation effects would be valuable, the apparently minimal direct impact on biomolecules and consequently on Raman spectra enables easier interpretation of spectra and translation to live-cell settings. Consistent, high-quality spectra generated from dry-fixation enable further univariate and multivariate analyses to proceed with greater confidence and without difficult, often subjective corrections. The opportunity for increased collaborations between Raman spectroscopists and biologists should lead to more rapid advancements in the applications of Raman spectroscopy to biological research.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research for financial support. We also acknowledge the Canadian Foundation for Innovation and the British Columbia Knowledge Development Foundation for funding the purchase of instrumentation that was required for this work, including through the UBC Laboratory for Molecular Biophysics.References
- T. Huser and J. Chan, Raman spectroscopy for physiological investigations of tissues and cells, Adv. Drug Delivery Rev., 2015, 89, 57–70 CrossRef CAS PubMed.
- C. Krafft and J. Popp, The many facets of Raman spectroscopy for biomedical analysis, Anal. Bioanal. Chem., 2015, 407(3), 699–717 CrossRef CAS PubMed.
- G. W. Auner, S. K. Koya, C. Huang, B. Broadbent, M. Trexler and Z. Auner, et al., Applications of Raman spectroscopy in cancer diagnosis, Cancer Metastasis Rev., 2018, 37(4), 691–717 CrossRef CAS PubMed.
- M. Paraskevaidi, B. J. Matthew, B. J. Holly, B. J. Hugh, C. P. V. Thulya and C. Loren, et al., Clinical applications of infrared and Raman spectroscopy in the fields of cancer and infectious diseases, Appl. Spectrosc. Rev., 2021, 56(8–10), 804–868 CrossRef CAS.
- S. O. Konorov, H. G. Schulze, B. K. Gage, T. J. Kieffer, J. M. Piret and M. W. Blades, et al., Process Analytical Utility of Raman Microspectroscopy in the Directed Differentiation of Human Pancreatic Insulin-Positive Cells, Anal. Chem., 2015, 87(21), 10762–10769 CrossRef CAS PubMed.
- S. Rangan, H. G. Schulze, M. Z. Vardaki, M. W. Blades, J. M. Piret and R. F. B. Turner, Applications of Raman spectroscopy in the development of cell therapies: state of the art and future perspectives, Analyst, 2020, 145(6), 2070–2105 RSC.
- T. Ichimura, L.-D. Chiu, K. Fujita, H. Machiyama, T. Yamaguchi and T. M. Watanabe, et al., Non-label immune cell state prediction using Raman spectroscopy, Sci. Rep., 2016, 6(1), 37562 CrossRef CAS PubMed.
- E. Brauchle, S. Thude, S. Y. Brucker and K. Schenke-Layland, Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy, Sci. Rep., 2014, 4(1), 4698 CrossRef PubMed.
- S. Rangan, S. Kamal, S. O. Konorov, H. G. Schulze, M. W. Blades and R. F. B. Turner, et al., Types of cell death and apoptotic stages in Chinese Hamster Ovary cells distinguished by Raman spectroscopy, Biotechnol. Bioeng., 2018, 115(2), 401–412 CrossRef CAS PubMed.
- S. J. Rodig, Fixing Attached Cells for Staining, Cold Spring Harb. Protoc., 2020, 2020(8) DOI:10.1101/pdb.prot099689.
- S. Golinejad and M. H. Mirjalili, Fast and cost-effective preparation of plant cells for scanning electron microscopy (SEM) analysis, Anal. Biochem., 2020, 609, 113920 CrossRef CAS PubMed.
- J. R. Harris, Transmission electron microscopy in molecular structural biology: A historical survey, Arch. Biochem. Biophys., 2015, 581, 3–18 CrossRef CAS PubMed.
- A. J. Hobro and N. I. Smith, An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging, Vib. Spectrosc., 2017, 91, 31–45 CrossRef CAS.
- A. D. Meade, C. Clarke, F. Draux, G. D. Sockalingum, M. Manfait and F. M. Lyng, et al., Studies of chemical fixation effects in human cell lines using Raman microspectroscopy, Anal. Bioanal. Chem., 2010, 396(5), 1781–1791 CrossRef CAS PubMed.
- M. M. Mariani, P. Lampen, J. Popp, B. R. Wood and V. Deckert, Impact of fixation on in vitro cell culture lines monitored with Raman spectroscopy, Analyst, 2009, 134(6), 1154–1161 RSC.
- E. Gazi, P. Gardner, N. P. Lockyer, C. A. Hart, M. D. Brown and N. W. Clarke, Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy, J. Lipid Res., 2007, 48(8), 1846–1856 CrossRef CAS PubMed.
- E. Bik, A. Dorosz, L. Mateuszuk, M. Baranska and K. Majzner, Fixed versus live endothelial cells: The effect of glutaraldehyde fixation manifested by characteristic bands on the Raman spectra of cells, Spectrochim. Acta, Part A, 2020, 240, 118460 CrossRef CAS PubMed.
- F. S. R. Carvalho, M. M. L. Verde, K. F. Viana, T. M. M. Bezerra, S. G. D. C. Fonseca and K. M. A. Pereira, et al., Pharmacological Characterization and Raman Spectroscopy Evaluation of Oral and Maxillofacial Surgery-Related Carnoy'S Solution Modified by Different Viscosity Agents, Asian Pac. J. Cancer Prev., 2019, 20(11), 3335–3339 CrossRef CAS PubMed.
- M. P. Douglas and S. O. Rogers, DNA damage caused by common cytological fixatives, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1998, 401(1), 77–88 CrossRef CAS PubMed.
- S. M. Hykin, K. Bi and J. A. McGuire, Fixing Formalin: A Method to Recover Genomic-Scale DNA Sequence Data from Formalin-Fixed Museum Specimens Using High-Throughput Sequencing, PLoS One, 2015, 10(10), e0141579 CrossRef PubMed.
- M. Srinivasan, D. Sedmak and S. Jewell, Effect of Fixatives and Tissue Processing on the Content and Integrity of Nucleic Acids, Am. J. Pathol., 2002, 161(6), 1961–1971 CrossRef CAS PubMed.
- R. Thavarajah, V. K. Mudimbaimannar, J. Elizabeth, U. K. Rao and K. Ranganathan, Chemical and physical basics of routine formaldehyde fixation, J. Oral Maxillofac. Pathol., 2012, 16(3), 400–405 CrossRef PubMed.
- N. W. Troiano, W. A. Ciovacco and M. A. Kacena, The Effects of Fixation and Dehydration on the Histological Quality of Undecalcified Murine Bone Specimens Embedded in Methylmethacrylate, J. Histotechnol., 2009, 32(1), 27–31 CrossRef CAS PubMed.
- A. Rezania, J. E. Bruin, P. Arora, A. Rubin, I. Batushansky and A. Asadi, et al., Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells, Nat. Biotechnol., 2014, 32(11), 1121–1133 CrossRef CAS PubMed.
- https://www.blood.ca/en/research/products-and-services-researchers/products-research/donate-blood-research .
- H. G. Schulze, S. O. Konorov, N. J. Caron, J. M. Piret, M. W. Blades and R. F. B. Turner, Assessing Differentiation Status of Human Embryonic Stem Cells Noninvasively Using Raman Microspectroscopy, Anal. Chem., 2010, 82(12), 5020–5027 CrossRef CAS PubMed.
- H. G. Schulze, S. Rangan, J. M. Piret, M. W. Blades and R. F. B. Turner, Developing Fully Automated Quality Control Methods for Preprocessing Raman Spectra of Biomedical and Biological Samples, Appl. Spectrosc., 2018, 72(9), 1322–1340 CrossRef CAS PubMed.
- Z. Huang, A. McWilliams, S. Lam, J. English, D. I. McLean and H. Lui, et al., Effect of formalin fixation on the near-infrared Raman spectroscopy of normal and cancerous human bronchial tissues, Int. J. Oncol., 2003, 23(3), 649–655 Search PubMed.
- A. C. S. Talari, Z. Movasaghi, S. Rehman and I. U. Rehman, Raman Spectroscopy of Biological Tissues, Appl. Spectrosc. Rev., 2015, 50(1), 46–111 CrossRef CAS.
- R. E. Burnham, J. T. Zoine, J. Y. Story, S. N. Garimalla, G. Gibson and A. Rae, et al., Characterization of Donor Variability for γδ T Cell ex vivo Expansion and Development of an Allogeneic γδ T Cell Immunotherapy, Front. Med., 2020, 7, 588453 CrossRef PubMed.
- C. L. Daniel Gibson and A. Madrigal, Strategies for Dealing with Donor Variability, Cell Gene Ther. Insights, 2018, 4(9), 901–909 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01916g |
| This journal is © The Royal Society of Chemistry 2023 |