
Retention time prediction and MRM validation reinforce the biomarker identification of LC-MS based phospholipidomics†
Jiangang
Zhang
 ,
Yu
Zhou
,
Juan
Lei
,
Xudong
Liu
,
Nan
Zhang
,
Lei
Wu
and
Yongsheng
Li
*
,
Yu
Zhou
,
Juan
Lei
,
Xudong
Liu
,
Nan
Zhang
,
Lei
Wu
and
Yongsheng
Li
*
Department of Medical Oncology, Chongqing University Cancer Hospital, Chongqing 400030, China. E-mail: lys@cqu.edu.cn
First published on 30th November 2023
Abstract
Dysfunctional lipid metabolism plays a crucial role in the development and progression of various diseases. Accurate measurement of lipidomes can help uncover the complex interactions between genes, proteins, and lipids in health and diseases. The prediction of retention time (RT) has become increasingly important in both targeted and untargeted metabolomics. However, the potential impact of RT prediction on targeted LC-MS based lipidomics is still not fully understood. Herein, we propose a simplified workflow for predicting RT in phospholipidomics. Our approach involves utilizing the fatty acyl chain length or carbon–carbon double bond (DB) number in combination with multiple reaction monitoring (MRM) validation. We found that our model's predictive capacity for RT was comparable to that of a publicly accessible program (QSRR Automator). Additionally, MRM validation helped in further mitigating the interference in signal recognition. Using this developed workflow, we conducted phospholipidomics of sorafenib resistant hepatocellular carcinoma (HCC) cell lines, namely MHCC97H and Hep3B. Our findings revealed an abundance of monounsaturated fatty acyl (MUFA) or polyunsaturated fatty acyl (PUFA) phospholipids in these cell lines after developing drug resistance. In both cell lines, a total of 29 lipids were found to be co-upregulated and 5 lipids were co-downregulated. Further validation was conducted on seven of the upregulated lipids using an independent dataset, which demonstrates the potential for translation of the established workflow or the lipid biomarkers.
1 Introduction
Perturbations in lipid metabolism are associated with various diseases, including cardiovascular disease, type 2 diabetes mellitus (T2DM), and cancer.1,2 The aggravation of lipids and their interaction with genes and proteins play a crucial role in the progression of these diseases.3,4 Over the past three decades, significant advancements in high throughput sequencing technology have facilitated the establishment of a well-defined regulatory network of genes and proteins.5,6 This has accelerated the translation of research findings from laboratory experiments to clinical applications, such as the early identification of susceptibility genes in diseases.7,8 In recent years, spatiotemporal multi-omics technology has expanded our understanding of the interactions between genes, proteins, and metabolites at the single-cell level.9,10 Additionally, lipids, which make up the lipidome in biological samples,11 serve as substrates for post-translational modification of proteins, contribute to the structure of cell membranes, and play a role in energy metabolism.12,13 The intricate interplay between lipids and signaling pathways highlights their potential as biomarkers for screening and development of novel therapeutics.Lipidomics is a powerful tool used for profiling lipidomes in a biological system. It can be divided into two categories: untargeted and targeted lipidomics.11,14 Untargeted lipidomics is utilized for the analysis of the comprehensive lipid profile, benefiting from its high resolution (HR) and wide ion coverage facilitated by mass analyzers such as time of flight (TOF) or Orbitrap. On the other hand, targeted lipidomics is suitable for determining a predefined list of targeted lipids using quadrupole (Q)-TOF and quadrupole ion trap (Q-TRAP) with high sensitivity, as indicated by relatively low limits of quantitation (LOQ) and detection (LOD).15 In both cases, liquid chromatography coupled with mass spectrometry (LC-MS) has become the most commonly used detection platform due to its high separation capacity and versatility in chromatography.16,17 However, interference peaks often occur when signals overlap in untargeted approaches or when one lipid generates a peak in another lipid's multiple reaction monitoring (MRM) transition within a small retention time (RT) range in targeted methods. This can result in inaccurate lipid annotation or quantification.18 While this issue can be partially addressed by using lipid library matching with an exact mass weight or tandem mass spectrometry (MS/MS) obtained mostly from untargeted methods, such as METLIN Gen2,19 MassBank database,20 the LIPID MAPS Web tools,21 LipidBlast,22etc., researchers still need to carefully consider the advantages and disadvantages of these methodologies before proceeding with their acquisition.
In addition to mass information, large-scale lipid identification requires supplementary information. RT from chromatography, included in lipid libraries, is crucial in supporting both untargeted and targeted strategies.23–25 The prediction of RT for lipids has gained popularity in untargeted lipidomics. Xu and Kohlbacher et al.23 introduced machine learning-based approaches to improve the assignment of lipid structures and automate annotation. Snel et al.26 developed an equivalent carbon number (ECN) model for predicting the RT of lipids with the same headgroup in reversed-phase separation. Other software packages such as LiPydomics,27 PredRet,24 quantitative structure-(chromatographic) retention relationship (QSRR) Automator,28 DynaStI,29etc., have also been developed for similar RT prediction purposes. Among these, QSRR models are the most commonly used theory for RT prediction.25,30 Artificial intelligence algorithms are used to enhance data integration and processing in iterative computing and data calibration. However, not all laboratories have a bioinformatics specialist proficient in applying these models and codes, leading to frequent occurrences of bugs and errors when data formats or default settings are inconsistent. Therefore, a modified workflow is often required to address such situations.
The interference of lipid signals may become more prominent and compromise the efficiency of detecting lipids in targeted lipidomics when a large number of MRM transitions are added to the methods, as suggested by previous research on targeted metabolomics.18 The use of the aforementioned model for RT prediction may help reduce false positive signal responses and improve lipid annotation to some extent. In our previous study, we utilized an equivalent carbon number and retention time prediction model to discover unusual odd chain fatty acyl lipids in targeted LC-MS lipidomics.31 However, such practices are still lacking in targeted lipidomics and may enhance the accuracy of lipid quantification, especially when researchers have limited diversity in mass spectrometry equipment. This prompts us to further investigate this field and explore the lipid phenotype in various disease contexts.
To gain further insight into the accurate measurement and analysis of lipids using targeted LC-MS lipidomics, here we developed a predictive model based on RT, validated it using MRM validation patterns, as well as used this model to investigate the changes in phospholipid dynamics. Furthermore, we verified these specific lipids using an independent clinical lipidomics dataset,32 to suggest the potential of these lipids as biomarkers for predicting the sensitivity of HCC treatment to sorafenib.
2 Methods
2.1 Chemicals and reagents
Acetonitrile (ACN, Cat#CAEQ-4-003306-4000), dichloroform (DCM, Cat#CAEQ-4-012002-4000), isopropanol (IPA, Cat#CAEQ-4-013493-1000) and methanol (MeOH, Cat#CAEQ-4-003302-4000) were purchased from Shanghai ANPEL Experimental Technology Co., Ltd (https://www.labsci.com.cn/). All solvents were HPLC grade. A Millipore Milli-Q purification system (Bedford, MA, UK) was used to produce deionized water and ammonium acetate (Cat#338818) was brought from Sigma-Aldrich (Taufkirchen, Germany). Sorafenib was obtained from Shanghai Beyotime Biotechnology Co., Ltd (Cat#SC0236, https://www.beyotime.com/index.htm). The authentic standards of PA(16:0/18:1) (Cat#840857C), PA(18:0/18:1) (Cat#840861P) and UltimateSPLASH™ ONE (Cat#330820L) were provided by Avanti Polar Lipids Inc. (AL, USA). In particular, UltimateSPLASH™ ONE for lipidomic analysis is a commercialized mixture of deuterium labeled internal standards (IS) containing 40 deuterium labeled phospholipids in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloromethane:methanol solution (1.2 mL per vial). The deuterated standards were provided at defined concentrations and were used directly (phospholipids are listed in Table S1†). The certificate of analysis for UltimateSPLASH™ ONE is available at https://avantilipids.com/.
:1 dichloromethane:methanol solution (1.2 mL per vial). The deuterated standards were provided at defined concentrations and were used directly (phospholipids are listed in Table S1†). The certificate of analysis for UltimateSPLASH™ ONE is available at https://avantilipids.com/.
2.2 Sorafenib resistant HCC cell lines
MHCC97H and Hep3B cell lines were exposed to sorafenib for a duration of at least 12 months. This exposure was divided into an inducing phase and a steady phase, each lasting 6 months. During the inducing phase, MHCC97H cells were treated with a gradient concentration of 5, 7.5, and 10 μM of sorafenib, while Hep3B cells were treated with slightly lower concentrations of 2.5, 5, and 7.5 μM. Each gradient treatment lasted for two months. In the steady phase, MHCC97H and Hep3B cells were stimulated with 7.5 and 6 μM sorafenib, respectively, for the remaining 6 months of the experiment. As a result of this 12-month drug exposure, stable and acquired sorafenib resistant cell lines were obtained. The following drug resistant (DR) cell lines were referred to as MHCC97H-DR and Hep3B-DR, and the corresponding wildtype control (C) of MHCC97H-C and Hep3B-C respectively.2.3 Cell culture and collection
MHCC97H (Cat#SCSP-5092) and Hep3B (Cat#SCSP-5045) were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China), and both cell lines were authenticated and tested for mycoplasma. The wildtype and sorafenib resistant cells of MHCC97H and Hep3B were cultured in DMEM (4.5 mg mL−1D-glucose) from Gibco (Cat#C11995500BT, Invitrogen, Life Technologies). The culture medium was supplemented with 10% fetal bovine serum (FBS) (Cat#10091148, Gibco), 100 IU mL−1 penicillin, and 100 mg mL−1 streptomycin. Sorafenib (Cat#C0222, Beyotime) was added as described in section 2.2. Prior to collection, cells were counted using the Countstar Altair (Alit Biotech (Shanghai) Co., Ltd) and several aliquots of cells were seeded in culture dishes. After incubation in a 37 °C5% CO2 incubator, the culture flask was observed under an inverted microscope to confirm that the cells were ready to be collected for further experiments (80% confluence). Upon collection, the cells were washed with pre-warmed PBS at 37 °C, and then centrifuged to discard the supernatant. The cells were then snap-frozen in liquid nitrogen and stored at −80 °C until analysis. Additionally, two more wells of cells were scraped and extracted for BCA protein quantification using a BCA assay kit (Cat# P0009, Beyotime) and normalization.2.4 Lipid extraction
The lipid extraction process follows a modified Bligh and Dyer procedure33 as described in a previous study.31 Briefly, prior to opening, the ampule of the internal standard mixture was sonicated in a warm bath for approximately five minutes and repeated inversion or vortex mixing was performed to completely solubilize and mix the vial contents as per the manufacturer's protocols. Cell pellets were then warmed to ambient temperature and 0.9 mL of H2O was added into the samples. Subsequently, 5 μL of internal standards, 2 mL of MeOH, and 0.9 mL of DCM were successively added to the sample mixture. The extracts were gently vortexed for 5 seconds and left on the benchtop at room temperature for 30 minutes. An additional 1 mL of H2O and 0.9 mL of DCM were added to the extracts, followed by another gentle vortexing for 5 seconds. The extracts were then centrifuged for 10 minutes to separate them into a bilayer, and the bottom organic phase was transferred to a new glass tube for each extract. The original extracts underwent another isolation step by adding 1.8 mL of DCM, vortexing for 5 seconds, and centrifuging as mentioned earlier. The resulting extracts were combined with the previous aliquots. Finally, the sample extracts were concentrated under stable nitrogen flow and reconstituted in 100 μL of the running solution (1:1 DCM:MeOH containing 10 mM ammonium acetate).
2.5 Reverse phase LC-MS/MS-based lipidomics
LC-MS/MS analysis was performed on an Exion UHPLC-AB/SCIEX QTRAP 6500 Plus (Applied Biosystems, Foster City, CA, USA) platform. Data acquisition was performed using Analyst 1.6.3 software (AB SCIEX). LC separation was carried out using an Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm, Waters Corporation) and gradient elution was employed. Mobile phase A consisted of a solvent mixture of MeOH:ACN:H2O (1:1:1), while mobile phase B contained IPA, both with 5 mM ammonium acetate. The elution gradient of a total of 17 minutes was applied as follows: 0–1 min: 20% B, 1–2.5 min: 40% B, 2.5–4 min: 60% B, 4–14 min: 90% B, 14–15 min: 90% B, 15–15.1 min: 20% B, and 15.1–17 min: 20% B. The temperature and flow rate of the chromatography column were set at 40 °C and 0.3 mL min−1, respectively. An injection volume of 5 μL was used, and the negative ESI mode was employed throughout the analysis. Lipid quantitation was performed using scheduled MRM, and the parameters for ion spray voltage (ISV, −4500 V), curtain gas (CUR, 30 psi), nebulizer gas (GS1, 55 psi), turbo-gas (GS2, 55 psi), turbo ion spray source temperature (350 °C), and collision gas (CAD, medium) were optimized.
Additional parameters of the mass spectrometer, such as de-clustering potential (DP), entrance potential (EP), collision exit potential (CE), and collision exit potential (CXP), were optimized for each subclass of lipid species. Nitrogen was used as the collision gas with an auto-generator. Peak assignment and integration were performed using SCEX OS. The signals were automatically recognized and integrated based on the RT predictive value. Manual inspection was also conducted to ensure accurate peak integration by the software. All lipids were then normalized to the peak area of the internal standard within the same subclass (Table S4†).
2.6 Method validation and QC
The method validation was conducted following the procedures outlined in our previous study.31 Each batch consisted of six replicate samples for a group (n = 6), two method blanks, and a pooled sample (referred to as QC). Initially, three replicates of the QC were analyzed to condition the system, followed by the randomized analysis of the samples. The QC was injected every 6 samples throughout the batch sequence to serve as a reproducible and stable monitor of the system. The lipid signal responses were plotted against the measured samples, allowing for the identification of outliers due to sample preparation or instrumental failures. Additionally, principal component analysis (PCA) was employed to provide an overview of the outliers in all samples.2.7 Statistical analysis
Data normalization was performed using MetaboAnalyst (https://www.metaboanalyst.ca/). The concentrations of lipids were uploaded and filtered based on the interquartile range (IQR). Subsequently, the data were normalized through log transformation and Pareto scaling. The normalized data were then used for further analysis. Principal component analysis (PCA) and orthogonal projections to latent structures discriminant analysis (OPLS-DA) were performed using SIMCA version 14.1 (Umetrics, Umeå, Sweden). R2 was utilized to quantify the variation of lipid species, while Q2 was employed to evaluate the predictive capability of the model in SIMCA software. Statistical significance was determined at a P-value <0.05, and all P-values were tested for multiple corrections using the Bonferroni approach.Additional variable influence of projection (VIP) was calculated for each individual OPLS-DA model. Only lipids with P-values <0.05, VIP values >1, and fold changes ≥20% for relative abundance were considered statistically important. Data processing was also performed using MetaboAnalyst for cross-validation and reproducibility verification.
Other statistical analyses were conducted using the two-tailed, unpaired Student's t-test or one-way ANOVA with the Dunnett multiple comparison test, as specified in GraphPad Prism 9.0.0. P values (***P < 0.001, **P < 0.01, and *P < 0.05) were automatically labeled in the figures. The graphical abstract was created using BioRender (https://www.biorender.com/). Graphs were plotted using either OriginPro 2021 (64-bit) 9.8.0.200 or GraphPad Prism 9.0.0, and final figures were prepared using Adobe Illustrator CS4.
3 Results
3.1 RT predictive modeling of phospholipids in reverse phase liquid chromatography
In targeted LC-MS lipidomics, LC plays a central role due to its high separation power and versatility. The lipids first flow through the chromatographic column with elution solvents, and different polar fractions are successively brought to the MS apparatus. In soft ionization sources such as ESI or APCI, ionized lipids are carried into a quadrupole mass spectrometer for further scan and fragmentation. The MRM detective mode utilizes quadrupoles to select a precursor ion (Q1) of the lipid, which is usually found in a pseudo-molecular ion or adduct ion. It breaks the precursor ion into smaller ions (Q2) and then sequentially selects a lipid's characteristic product ion (Q3) before sending the ions to the detector. Due to the superior sensitivity and selectivity of MS, even compounds present at trace concentrations can be measured. The LC-MS technique separates different lipids based on their chromatographic RTs and the mass-to-charge ratio (m/z) of the precursor and product ions. Therefore, this workflow combines RT, Q1 m/z, and Q3 m/z for lipid-specific measurements.Using a limited sum of deuterated standards from commercially available products (Table S1†), referred to as the training lipid set, we initially generated the RT list of 40 phospholipids (Table S1†). The mean RTs of five replicates of LC-MS runs were calculated. The standard error of the RT was within an acceptable range and could be utilized for further computations. For both the internal standards and the phospholipid library (including PC, PE, phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidic acid (PA), Table S1†), we calculated the total number of carbon atoms and double bonds. The highest number of carbon atoms obtained was 43, which was consistent across the six subclasses of lipids. This value was then used to determine the relative carbon atom number using the equation y = C/Cmax, where C represents the number of carbon atoms of a specific lipid and Cmax is kept as 43. By defining Cmax as a constant, we simplified the comparison of interclass RTs of lipids and ensured the rational distribution of RTs among the six subclasses of phospholipids. Consequently, the relative carbon atom numbers of the lipids were naturally deduced. Similarly, the relative RT was calculated using the equation y = T/Tmax, where T represents the retention time of a specified lipid and Tmax was the elution time of the established chromatographic system (17 minutes in this case). This calculation allowed for the easy preparation of RT ratios for deuterated standards.
The second degree polynomial regression (y = B0 + B1 × x + B2 × x2) was then used to fit the curve between fatty acyl chain length or DB number and RT. For example, in the case of PC, the compounds 17:0-14:1 PC-d5 (PC(31:1)-d5), 17:0-16:1 PC-d5 (PC(33:1)-d5), and 17:0-18:1 PC-d5 (PC(35:1)-d5), which had double bonds at carbon atom numbers 0.72, 0.77, and 0.81 respectively, exhibited relative RT values of 0.32, 0.34, and 0.37, respectively. Therefore, the regressive curve of PC(X:1) (X refers to the number of carbon atoms) could be fitted as y = 0.3047 − 0.4733x + 0.6801x2. Due to the limited number of standards available for model training, a simple rule regarding the variation of the RT values could be derived. This rule suggested a nearly constant RT ratio between PC(33:1)-d5/PC(31:1)-d5 (approximately 1.077151335) and PC(35:1)-d5/PC(33:1)-d5 (approximately 1.083677686), which was referred to as the carbon chain length constant (k). Based on this constant, the RT values of PC(37:1) and PC(39:1) were estimated to be 0.40 and 0.43, respectively.
The following experimental RT values confirmed our initial hypothesis. The fluctuation of RT is influenced by both the length of the carbon chain and the degree of unsaturation. In general, when a lipid has more double bonds or fewer carbon atoms, RT tends to decrease, while it tends to increase when there are fewer double bonds or more carbon atoms.34,35 We then investigated the relationship between lipids with the same chain length but varying double bonds, specifically 17:0-20:3 PC-d5 (PC(37:3)-d5), 17:0-22:4 PC-d5 (PC(39:4)-d5), and PC(37:1), PC(39:1). We found that the square root of the RT ratio between PC(37:3)-d5 and PC(37:1) was 0.941656387 (referred to as λ, the double bond constant). Surprisingly, the cube root of RTPC(39:4)-d5/RTPC(39:1) was 0.943296587, matching the previously obtained square root value. Based on the RT of PC(X:1) and these established results, we predicted the RT of PC(37:Y) (where Y refers to the number of double bonds, Y = 0, 1, 2, 3, 4, etc.) and PC(39:Y) in accordance with PC(X:1) (where X = 31, 33, 35, 37, 39, etc.). Subsequently, an array of relative RTs of PC were extrapolated and fitted with regressive curves simultaneously, as shown in Fig. 1A and Table S2.† Similarly, the RTs and fit curves of PE (Fig. 1B), PG (Fig. 1C), PI (Fig. 1D), and PS (Fig. 1E) were processed proficiently (Table S2†). However, due to a lack of deuterated PA standards in the kit, authentic standards of PA(16:0_18:1) and PA(18:0_18:1) were alternatively employed to estimate the k value of PA. Consistently, RT at 5.18 and 5.57 min yielded a k value of 1.075289575, illustrating the comparable value obtained from the other subclasses of phospholipids. Directly, the λ value from PC was used for the RT estimation of PA (Fig. 1F).
 | ||
| Fig. 1 Fatty acyl chain length or DB number-based RT predictive modeling of phospholipids. The predictive regression curve of PC (A), PE (B), PG (C), PI (D), PS (E) and PA (F) in the negative ESI mode. The X represents the total carbon chain length, x represents the relative fatty acyl chain length, and y represents the relative RT time. | ||
In addition, lysophospholipids, derived from the aforementioned lipid classes after hydrolysis to remove an acyl group, exhibited different properties compared to their original phospholipids. Therefore, the RT prediction of LPC (Fig. S1A†), LPE (Fig. S1B†), LPG (Fig. S1C†), LPI (Fig. S1D†), and LPS (Fig. S1E†) was also inferred using their respective deuterated standards containing 15:0, 17:0, and 19:0 fatty acyl moieties (Table S3†). In order to estimate the RT, the λ constant from the precursor lipids was approximately used. As a result, a library of different subclasses of phospholipids with predictive RTs was successfully constructed (Tables S4 and 5†).
3.2 Model evaluation and external validation
Experimental verification was conducted using RT matching and scheduled MRM to confirm the RT prediction. The phospholipids’ signal response was observed through the total ion chromatograph (TIC) using an optimal elution gradient (Fig. 2A). The difference between the actual reference RT and the predicted RT was less than 0.5 min (Fig. 2B), and there was a close linear relationship between the two factors (Fig. 2C). Additionally, there was a positive correlation between the RT and the mass range of lipids (Fig. 2D), and interclass RT comparison could be performed when X shared the same carbon chain length. For example, PE(X:1) had the longest RT, slightly longer than PC(X:1). The RTs of PG(X:1) and PI(X:1) were identical and significantly less than the RT value of PS(X:1). On the other hand, PA(X:1) exhibited the shortest retention in chromatography due to its relatively strong polarity (Fig. 1 and 2D). These findings are consistent with our previous research.31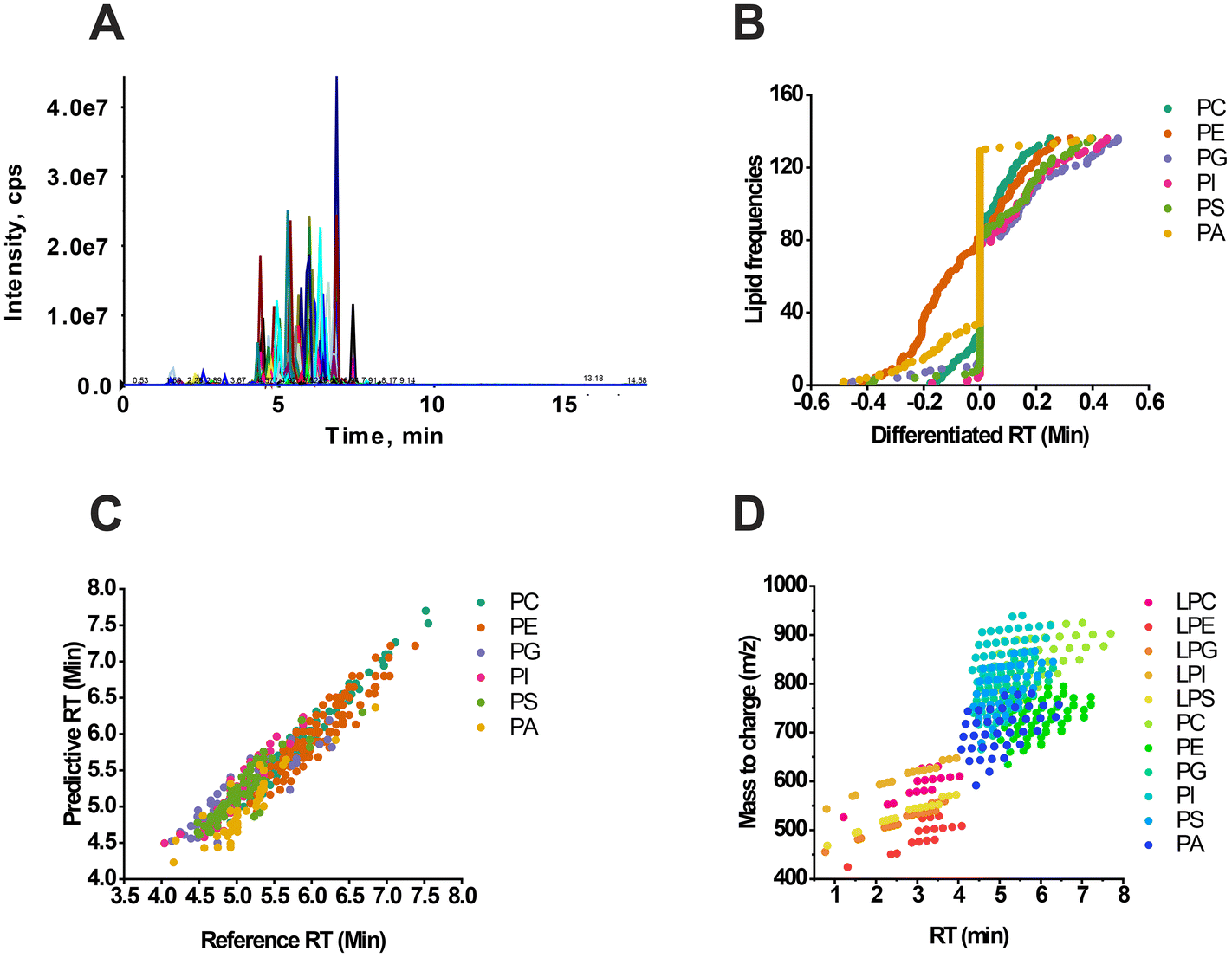 | ||
| Fig. 2 The method evaluation of RT predictive modeling. Total ion chromatography of phospholipids in the negative ESI mode (A), where x represents the RT (min) and y represents the intensity (cps). Differentiated RT between the predictive and real detective value (B), where x represents the RT difference value (min) and y represents the accumulated frequency of lipids. The regressive relationship between reference RT and predictive RT (C), where x represents the reference RT value (min) and y represents the predictive value (min). The distribution of both the mass range and RT from different subclasses of phospholipids (D). x represents the RT value (min), and y represents m/z. | ||
Recent studies have utilized machine learning techniques to predict RT. However, applying a published machine learning model to diverse research contexts and lab facilities may present challenges. Here we attempted to predict RT using various codes and packages, including QSRR Automator,28 a software package designed to automate RT prediction models. In this process, lipid IDs were first converted to the simplified molecular input line entry system (SMILE) format using MetaboAnalyst (https://www.metaboanalyst.ca/MetaboAnalyst/upload/ConvertView.xhtml). Subsequently, the RT values were added to the templates and the data were inputted, resulting in the smooth generation of predictive results for PC (Fig. S2A†) and PE (Fig. S2B†). The final models for PC and PE achieved R2 scores of 0.99 and 0.98, respectively. The mean absolute errors for PC and PE were 0.003 and 0.054, respectively. These results demonstrate the consistency of RT prediction between the QSRR Automator and our developed model.
3.3 MRM validation and signal interference diagnosis
According to the fragmentation rules, phospholipids exhibit two distinct patterns during collision-induced dissociation, except for lipids with the same sn-1 and sn-2 fatty acyl residues (Fig. S3†). Fragment ions resulting from this fragmentation process can be detected when the collision energy reaches a certain threshold. However, understanding the fragmentation rules alone is not sufficient. In negative ESI mode, MS/MS-based identification may be affected by various factors such as DP, CE, fatty acyl chain length, and unsaturation. To further evaluate, deuterated standards were created using two MRM transitions. The extracted ion chromatogram of both MRM channels showed a strong signal response under optimized ESI detection conditions with DP, EP, CE, and CXP set at 80, 10, 50, and 15 eV, respectively. This suggests that both MRMs are accessible for validation in our lipidomics instrumental setups. Subsequently, a library of 526 phospholipids with 1052 MRM transitions was generated, and de-replication was performed using the AB SCIEX Analyst 1.6.4 software prompts. Ultimately, a total of 1037 MRMs were listed for further assignment (Tables S4 and 5†).LC-MS analysis was conducted using sample extracts to identify peaks, as shown in Fig. 3. The peaks for PC(18:0_18:3), PE(16:0_20:1), PG(18:0_18:1), PI(18:0_20:5), PS(16:0_18:1), and PA(18:0_18:2) exhibited significant signal interference, complicating the lipid annotation process. However, by comparing peaks and considering predictive RT, it was possible to accurately identify the correct lipids even when one channel displayed a single peak and the other was affected by unknown chemicals.
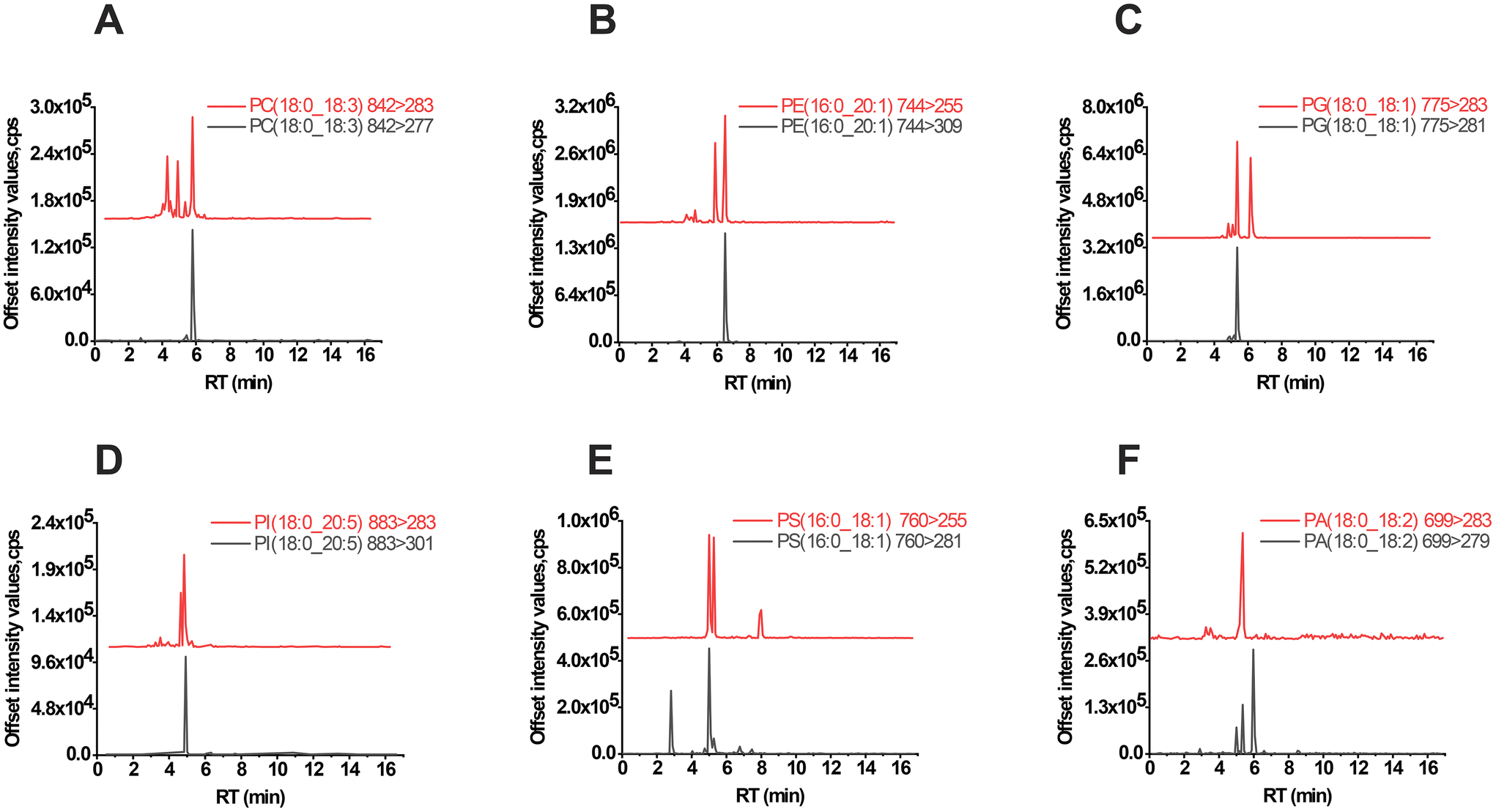 | ||
| Fig. 3 MRM validation for phospholipids. TIC of the representative subclass of phospholipids such as PC(18:0_18:3) (A), PE(16:0_20:1) (B), PG(18:0_18:1) (C), PI(18:0_20:5) (D), PS(16:0_18:1) (E) and PA(18:0_18:2) (F). x represents the RT value (min), and y represents the offset intensity values (cps). | ||
Moreover, there are two situations that can occur when annotating lipids with equivalent carbon atoms and degree of unsaturation. For instance, PC(38:5) can be split into PC(18:0_20:5) and PC(18:2_20:3) with retention times (RT) of 5.7 min and 5.4 min, respectively (Fig. S4A†). In this scenario, the two lipids can be automatically separated at baseline. However, in another case, lipids with the same molecular formula may not be resolved in chromatography, resulting in different fatty acyl moieties sharing nearly identical RTs. Examples of such lipids were PC(18:1_18:3), PC(16:0_20:4), and PC(18:2_18:2) of PC(36:4) (Fig. S4B†). Unfortunately, this technical limitation cannot be easily overcome during targeted lipidomics.
A recent study utilized the combination of hydrophilic interaction liquid chromatography (HILIC) and trapped ion mobility spectrometry (TIMS) to address the challenge of determining the double bond locations in phospholipids and sn-positions in phosphatidylcholine. This innovative approach offers a solution for differentiating lipid isomers within the phospholipidome.36 Despite the presence of remaining challenges facing the field, the validation of MRMs has proven to be an effective tool for lipid assignment. This was evident in the identification of various lipids, such as PC(16:0_18:1), PE(16:0_18:1), PG(16:0_18:1), PI(18:0_18:1), PS(18:0_18:1), and PA(16:0_18:1) (Fig. S5†), through robust identification in both MRM channels.
3.4 Phospholipidomics of sorafenib resistant HCC cell lines
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related death worldwide. The first line systemic and traditional therapies for HCC are sorafenib and lenvatinib. However, resistance to sorafenib poses a significant challenge in achieving HCC regression. Previous research has indicated that dysregulated lipid metabolism is involved in sorafenib resistance. However, the dynamics of phospholipids during acquired sorafenib resistance and the potential use of specialized lipids for predicting treatment sensitivity are still not well understood. To investigate these persistent issues, we conducted phospholipidomics using our established method in sorafenib resistant HCC cell lines, specifically MHCC97H and Hep3B. The overall workflow of building drug resistant cell lines and the simplified processes of lipidomics are illustrated in Fig. 4A. Initially, we performed principal component analysis (PCA) to analyze the data dispersion of each group based on the lipidomic profile (Fig. 4B and Table S6†). The results showed no outliers and negligible differences within groups. However, there was a clear difference in clustering between the control (C) group and the drug resistance (DR) group in both cell lines (Fig. 4B). The orthogonal partial least squares-discriminant analysis (OPLS-DA) also clustered group C and DR for either MHCC97H (Fig. 4C) or Hep3B (Fig. 4D). The volcano plot, which combines the fold change (FC) analysis and T-tests, allows for the intuitive screening of featured lipids based on their statistical significance. The data were then processed using volcano plot analysis, revealing a set of lipids such as PG(16:0_16:0), PG(16:0_16:1), PA(16:0_16:0), PA(16:0_16:1), PI(18:0_22:6), etc., which were found to be more abundant in the DR group of MHCC97H (Fig. 4E). Additionally, a majority of PC lipids like PC(18:0_20:4), PC(16:0_20:4), PC(18:0_20:5), and PC(18:2_20:5) showed considerable decay after sorafenib resistance (Fig. 4E). In contrast, the volcano plot from Hep3B showed a notable accumulation of PC(18:1_20:4), PC(18:1_22:4), PC(16:0_20:4), PE(18:1_20:4), PE(18:0_16:0), PE(18:2_22:5), etc., and a significant decrease in PA(16:0_14:0), PA(16:0_16:0), PA(16:0_18:1), PA(16:0_18:2), etc. during sorafenib resistance (Fig. 4F). | ||
| Fig. 4 Phospholipidomics of sorafenib resistant HCC cell lines. A simplified process of sorafenib resistant cell line construction and lipidomic analysis (A). PCA analysis of grouped samples (B) as indicated in the graph. OPLS-DA analysis of control and drug resistant MHCC97H (C) and Hep 3B cell lines (D). Volcano plot of control and drug resistant MHCC97H (E) and Hep3B cell lines (F), where blue stands for downregulated lipids, gray stands for non-significant lipids and red stands for upregulated lipids. | ||
The distribution of phospholipids was analyzed in cell extracts, and a total of 307 lipids were detected. Among these, PE was found to be the most abundant, with a total of 110 lipids, nearly twice the amount compared to PC which had a total of 59 lipids. The abundance of PG, PI, PS, and PA varied, with the number of lipids ranging from 23 to 45. When comparing the total phospholipid abundance between the control group (C) and the DR in the MHCC97H cell line, no significant difference was observed (Fig. 5B). However, a significant deficiency in phospholipid abundance was observed in the DR group of the Hep3B cell line. Specifically, when analyzing the individual subtypes of phospholipids, a decrease in PC and PI abundance and an increase in PG, PI, and PA abundance were observed in the DR group of the MHCC97H cell line (Fig. 5C). In contrast, there was no significant divergence in the abundance of these phospholipid subtypes in the Hep3B cell line (Fig. 5D). Additionally, the fatty acyl residues in the phospholipids were divided into saturated (SFA), monounsaturated (MUFA), and polyunsaturated (PUFA) moieties. The degree of unsaturation in the phospholipids of both DR groups of cell lines significantly increased, as exemplified by an increase in MUFA in the MHCC97H cell line (Fig. 5E) and an increase in PUFA in the Hep3B cell line (Fig. 5F).
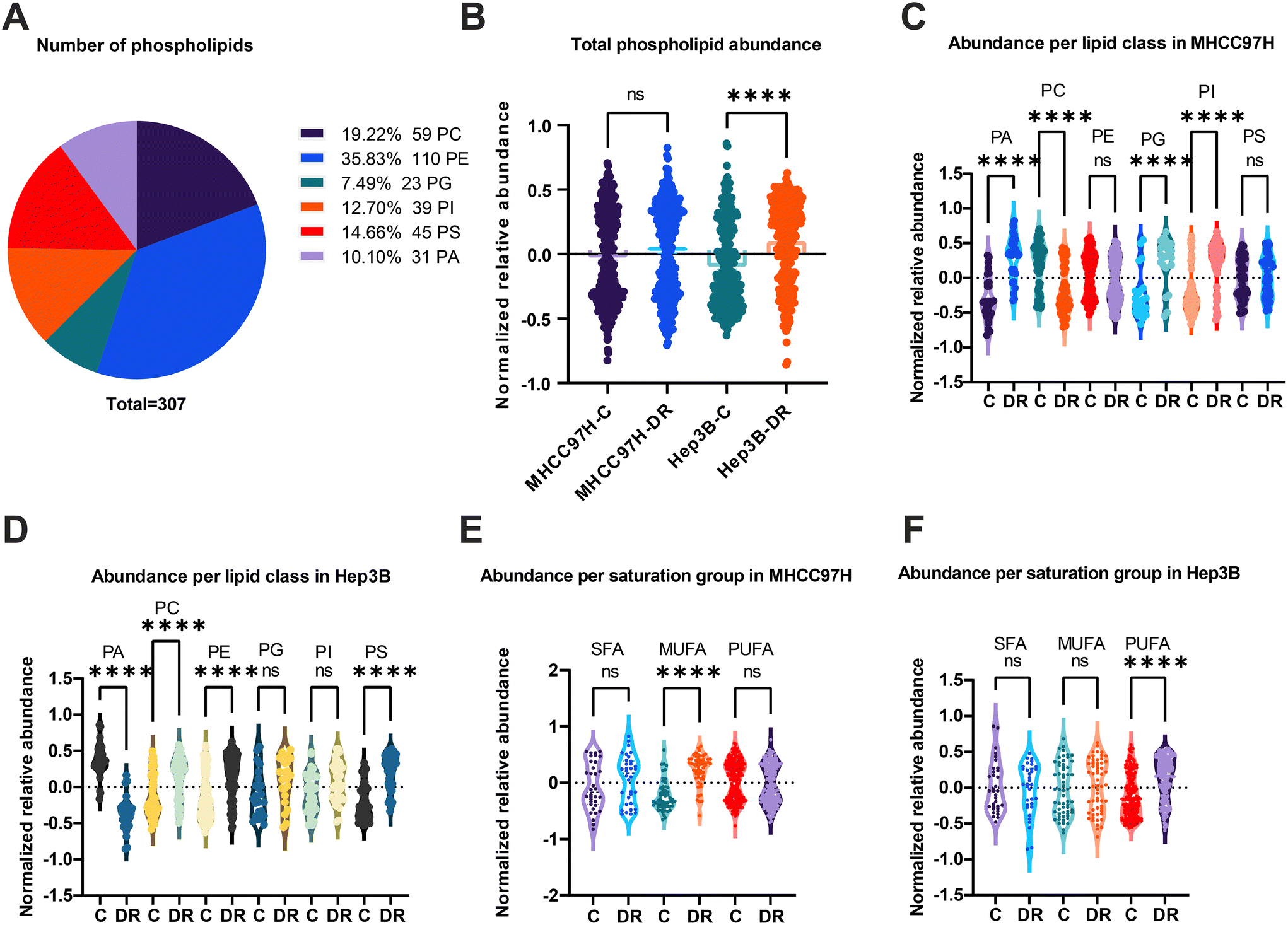 | ||
| Fig. 5 The dynamics of phospholipids after sorafenib resistance. Different subclass of lipids detected in the method and its fraction (A). The total phospholipid abundance from MHCC97H and Hep3B (B). The abundance of different subclasses of phospholipids in MHCC97H (C) and Hep3B (D). The distinct SFA, MUFA and PUFA containing phospholipids from MHCC97H (E) and Hep3B (F). | ||
To further filter the co-regulated lipids or biomarkers, OPLS-DA models were used to reanalyze the data from both cell lines. The VIP values greater than 1 were considered statistically relevant. This analysis identified 183 lipids with VIP > 1 in MHCC97H cells and 181 lipids with VIP > 1 in Hep3B cells. There were 109 lipids that were common to both cell lines (Fig. 6A and Table S7†). Among these 109 lipids, those with a correlation coefficient (p(cor)) greater than 0.6 or less than −0.5 were considered to have a positive or negative association with the model, respectively. As a result, 29 lipids (Fig. 6B and Table S8†) were identified as co-upregulated, and five lipids (Fig. 6C and Table S9†) were identified as co-downregulated in both MHCC97H and Hep3B cells.
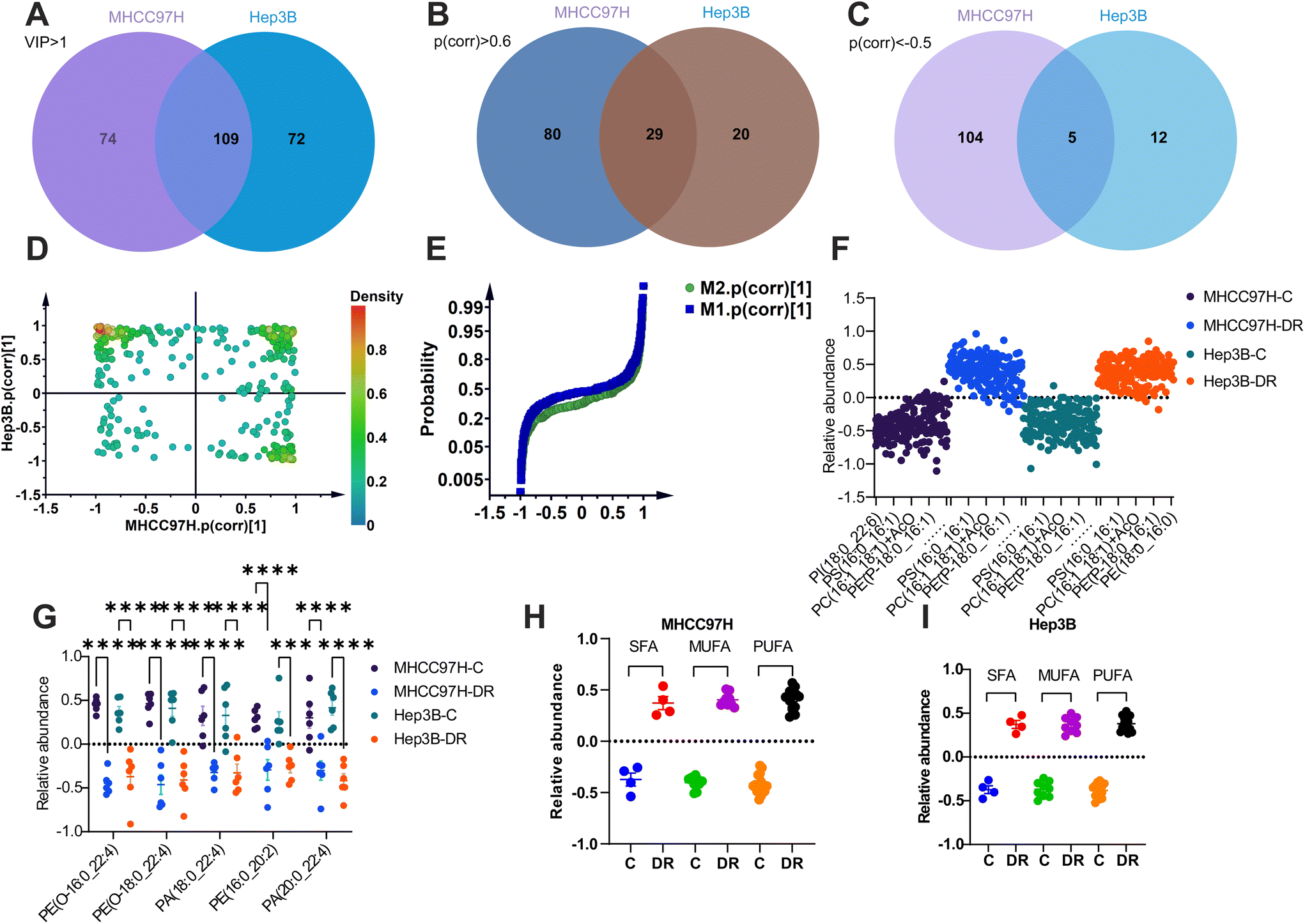 | ||
| Fig. 6 Co-regulated lipid screening. Phospholipids with VIP > 1 from OPLS-DA analysis of both cell lines and their overlapped lipids as shown in the Venn diagram (A). Phospholipids with p(corr) > 0.6 or <−0.5 from OPLS-DA analysis of MHCC97H (B) and Hep3B (C) and their overlapped lipids as shown in the Venn diagram. The SUS plot of OPLS-DA analysis from both cell lines (D). The probability curve of lipids linked with their p(corr) coefficient, M1 and M2 refers to OPLS-DA analysis of MHCC97H and Hep3B cells, respectively (E). The shared upregulated (F and Table S8†) and downregulated (G) phospholipids were sorted out from both cell lines after sorafenib resistance. The accumulated abundance of SFA, MUFA, and PUFA containing co-upregulated phospholipids from MHCC97H (H) and Hep3B cells (I). | ||
The shared and unique structures plot (SUS-plot) was used to correlate the two OPLS-DA models in order to identify appropriate lipids for discovering biomarkers from sorafenib resistant cells. The plot confirmed the previous findings, showing that 29 co-upregulated lipids were located in the first quadrant of the coordinate, while five co-downregulated lipids were located in the third quadrant (Fig. 6D). This suggests that these lipids share common characteristics, with either increased or decreased abundance. In contrast, the lipids scattered in the second and fourth quadrants exhibit unique fluctuations (Fig. 6D). The p(cor) plot of OPLS-DA analysis from both cell lines was used to confirm the co-regulated lipids at specific angles of the curve (Fig. 6E). The overlapping lipid set from enriched and decreased lipids in both sorafenib resistant cell lines was then plotted, representing specialized lipid biomarkers regardless of the cell line (Fig. 6F and G). Additionally, the fatty acyl moieties in the 29 lipids were re-analyzed, showing enhanced properties for either SFA, MUFA, or PUFA in the two sorafenib resistant HCC cell lines (Fig. 6H and I).
3.5 Clinical lipidomics validation
To validate our findings from in vitro experiments, we reviewed previous research and unexpectedly came across a study that analyzed the lipid profiles in plasma samples from 44 HCC patients before sorafenib therapy. The aim of this study was to understand the lipid profiles associated with the efficacy and safety of sorafenib treatment.32 The results of the study indicated that the levels of phosphatidylcholine, specifically PC(34:2), PC(34:3), PC(35:2), and PC(36:4), were significantly lower in the responder group.32 This finding prompted us to compare the 29 co-upregulated lipids from sorafenib resistant HCC cell lines with the published data. Interestingly, we found that seven lipids, including PC(32:1), PC(34:2), PC(36:2), PI(34:2), PI(36:2), PI(38:4), and PI(40:6), exhibited reduced abundance in drug responders (Fig. 7B), matching the lipids identified in the plasma samples (Fig. 7A). These results suggest that these intersected lipids could potentially serve as candidate biomarkers for predicting the sensitivity of sorafenib treatment.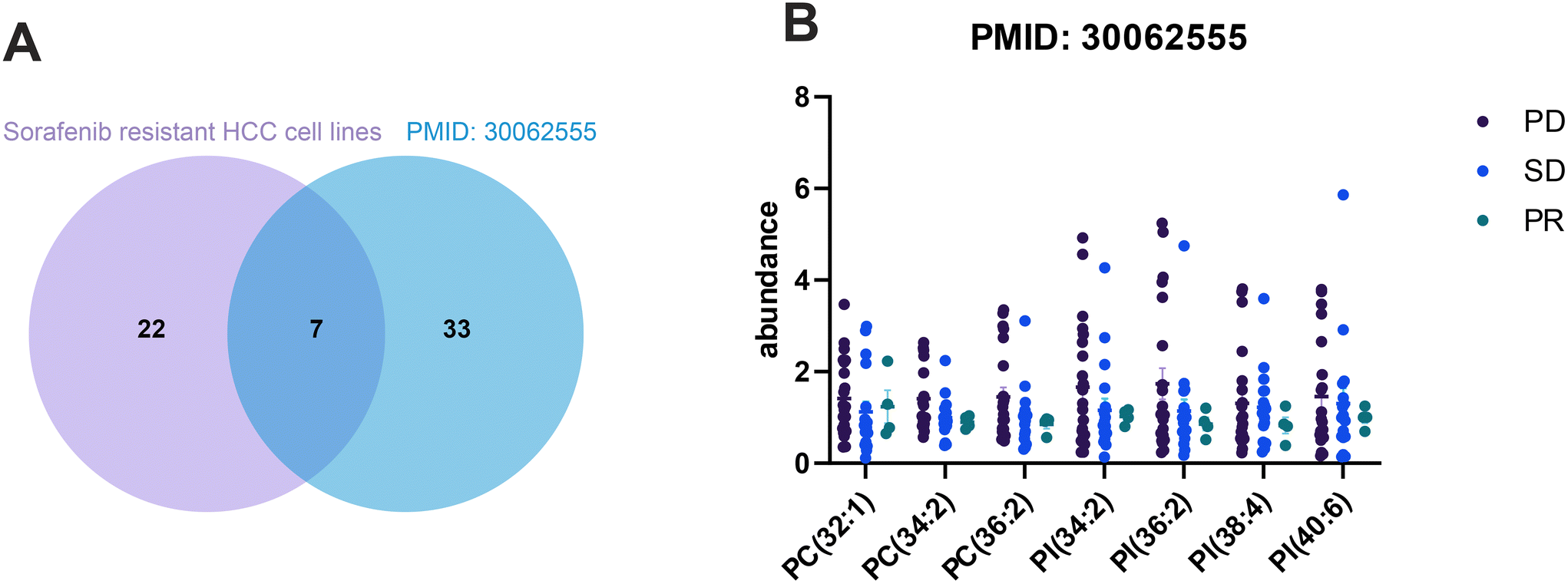 | ||
| Fig. 7 Lipid biomarker validation from an independent data set. The co-upregulated phospholipids from this study and a previous study with PMID:300062555 and intersected lipids are shown in the Venn diagram (A). The abundance of the co-existing lipids was re-analyzed in three different groups (B) as shown in the graph. PD: progressive disease. PR: partial response, and SD: stable disease. | ||
4 Discussion
In this study, we proposed a workflow that combines RT prediction with MRM validation for LC-MS based phospholipidomics. We used the fatty acyl chain length or DB number and the second degree polynomial regression to model the RT prediction of six subtypes of phospholipids and five subtypes of lysophospholipids. The relationship between RT and the carbon chain length or carbon–carbon double bond was represented by coefficients k and λ, respectively. These coefficients facilitated the construction of an RT fit curve with either varying carbon chain length or unsaturation. We evaluated our model by comparing the RT values obtained experimentally with the predicted values, as well as using the QSRR Automator, which demonstrated the excellent predictive capability of our model. To validate our model further, we performed sequential MRM validation that integrated the fragmentation rule of phospholipids and scheduled MRM. This validation involved two MRM transitions from different fatty acyl residues and diagnosed signal interference.To investigate the phospholipidomics of sorafenib resistant HCC cell lines, we conducted experiments on two cell lines, MHCC97H and Hep3B. The analysis revealed the accumulation of unsaturated lipids in both cell lines after acquiring resistance to sorafenib. Previous studies have shown that stearoyl-CoA desaturase-1 (SCD1) plays a crucial and rate-limiting role in the synthesis of MUFA. In line with this, we found that the expression of SCD1 was significantly upregulated in the sorafenib resistant PLC/PRF/5 HCC cell line and patient-derived tumor xenograft (PDTX) model.37 This suggests that SCD1 may also have a similar role in our sorafenib resistant cell lines, as we detected an increase in the abundance of MUFA or PUFA. Moreover, the inhibition of SCD1 was found to mitigate the sorafenib sensitivity in HCC,37 indicating a potential combinatorial strategy of targeting SCD1 along with sorafenib to combat HCC. Our in vitro experiments provided further support for these findings.
In our study, we discovered that 29 co-upregulated and 5 co-downregulated lipids were found in both models of the OPLS-DA or SUS plot. These lipids were evaluated from an external lipidomic dataset obtained from the plasma of sorafenib pretreatment HCC patients,32 with the aim of predicting sorafenib sensitivity. Out of these, seven lipids were found to have reduced abundance in drug responders, which aligns with our observation that these lipids were present in higher concentrations in sorafenib resistant cells. This finding supports the potential use of these lipids as biomarkers for predicting sorafenib sensitivity. However, the underlying mechanisms behind the increased abundance of these lipids and how they can be manipulated to enhance the killing effect of sorafenib are still largely unknown. Our results provide valuable insights into the lipid phenotype and may contribute to developing strategies to overcome sorafenib resistance in HCC.
However, our proposed RT prediction modeling may also have some drawbacks, such as overfitting and limited coverage of metabolites, like metabolomics. Regarding overfitting, we quantified the difference in RT between in silico and experimental parameters, and found that the largest gap in RT was less than 0.5 min. This value was considered acceptable, especially when MRM validation was taken into account. To improve the accuracy of RT prediction, we also explored the use of iterative data to train the model, since the availability of deuterated or authentic standards was limited. However, our efforts were hindered by the inconsistent criteria used by different labs. Additionally, the inclusion of a comprehensive library of metabolites and corresponding standards would undoubtedly enhance the training and prediction capabilities of RT. Furthermore, the use of machine learning algorithms, based on theoretical calculations using chemical structure formulas like SMILE, has significantly accelerated the fidelity of RT prediction and the discovery of novel compounds. Our model simplifies the predictive process and demonstrates good performance in RT prediction, making it a viable strategy for customized phospholipidomics.
The understanding of lipid fragmentation rules is still incomplete, and the identification of lipids using MRM is complicated by the interference of multiple signals at different sample contexts in lipidomics. This interference affects lipid assignment in various ways. In the case of phospholipids, there are two fatty acyl residues located either at the sn-1 or sn-2 position of the glycerol backbone, resulting in the need for two MRM transitions for target identification. This leads to the generation of nearly a thousand ion pairs in one setting, which increases the cycle time, decreases data acquisition, and results in poorer peak shapes. To address this issue, scheduled MRMs were employed instead of using a constant dwell time. By incorporating one additional MRM transition in LC-MS analysis, the peak annotation was corrected, and the confidence in lipid identification was improved. It is important to note that the multiplex information of lipids, which depends on their own fragmentation pattern, cannot be easily transferred to other classes of lipids.
Overall, this study demonstrates the application of the fatty acyl chain length or DB number as a model for developing a lipidomics library. Using MRM validation in conjunction with this workflow, it significantly strengthened confidence in targeted lipidomics and provided comprehensive coverage of the phospholipidome. Furthermore, the study generated RTs for 526 lipids by selecting a limited number of deuterated or authentic standards. The determination of RTs greatly accelerated scheduled MRM for high throughput analysis, with a difference of less than 0.5 minutes between measured and predicted RT values. As a result, the utility of this workflow was demonstrated in sorafenib resistant HCC cell lines, we found an increase in MUFA or PUFA in sorafenib-resistant cells, with seven co-upregulated lipids being identified and validated using an independent data set. This workflow will be potentially applied for RT prediction and lipid biomarker identification in diseases.
Data availability
All data supporting this study are included in the manuscript and ESI.†Author contributions
Jiangang Zhang and Yongsheng Li: conceptualization, methodology, and software. Jiangang Zhang: data curation and writing– original draft preparation. Yu Zhou, Juan Lei, Xudong Liu, Nan Zhang, and Lei Wu: visualization and investigation. Yongsheng Li: supervision. Xudong Liu, Nan Zhang, and Lei Wu: software and validation. Yongsheng Li: writing, reviewing and editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.Acknowledgements
This work was supported by the Major International (Regional) Joint Research Program of the National Natural Science Foundation of China (no. 81920108027 to Yongsheng Li), the Chongqing Outstanding Youth Science Foundation (no. cstc2020jcyj-jqX0030 to Yongsheng Li), the Funding for University Innovation Research Group of Chongqing (to Yongsheng Li), the Science and Technology Research Program of Chongqing Municipal Education Commission (no. KJQN202300112 to Jiangang Zhang) and the Chongqing Medicinal Biotech Association (no. cmba2022kyym-zkxmQ0007 to Jiangang Zhang).References
- L. Chen, X. W. Chen, X. Huang, B. L. Song, Y. Wang and Y. Wang, Regulation of glucose and lipid metabolism in health and disease, Sci. China: Life Sci., 2019, 62, 1420–1458 CrossRef.
- H. Yoon, J. L. Shaw, M. C. Haigis and A. Greka, Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity, Mol. Cell, 2021, 81, 3708–3730 CrossRef CAS.
- T. Peng, X. Yuan and H. C. Hang, Turning the spotlight on protein-lipid interactions in cells, Curr. Opin. Chem. Biol., 2014, 21, 144–153 CrossRef CAS.
- J. P. Pégorier, Regulation of gene expression by fatty acids, Curr. Opin. Clin. Nutr. Metab. Care, 1998, 1, 329–334 CrossRef.
- M. Wilhelm, J. Schlegl, H. Hahne, A. M. Gholami, M. Lieberenz, M. M. Savitski, E. Ziegler, L. Butzmann, S. Gessulat, H. Marx, T. Mathieson, S. Lemeer, K. Schnatbaum, U. Reimer, H. Wenschuh, M. Mollenhauer, J. Slotta-Huspenina, J. H. Boese, M. Bantscheff, A. Gerstmair, F. Faerber and B. Kuster, Mass-spectrometry-based draft of the human proteome, Nature, 2014, 509, 582–587 CrossRef CAS.
- R. M. Sherman and S. L. Salzberg, Pan-genomics in the human genome era, Nat. Rev. Genet., 2020, 21, 243–254 CrossRef CAS.
- Y. Xie, G. Li, M. Chen, X. Guo, L. Tang, X. Luo, S. Wang, W. Yi, L. Dai and J. Wang, Mutation screening of 10 cancer susceptibility genes in unselected breast cancer patients, Clin. Genet., 2018, 93, 41–51 CrossRef CAS PubMed.
- J. Weber, C. J. Braun, D. Saur and R. Rad, In vivo functional screening for systems-level integrative cancer genomics, Nat. Rev. Cancer, 2020, 20, 573–593 CrossRef CAS PubMed.
- Y. Wu, Y. Cheng, X. Wang, J. Fan and Q. Gao, Spatial omics: Navigating to the golden era of cancer research, Clin. Transl. Med., 2022, 12, e696 CrossRef.
- M. Babu and M. Snyder, Multi-omics profiling for health, Mol. Cell. Proteomics, 2023, 22, 100561 CrossRef CAS.
- X. Han and R. W. Gross, The foundations and development of lipidomics, J. Lipid Res., 2022, 63, 100164 CrossRef CAS.
- G. van Meer, Cellular lipidomics, EMBO J., 2005, 24, 3159–3165 CrossRef CAS.
- J. Finkelstein, M. T. Heemels, S. Shadan and U. Weiss, Lipids in health and disease, Nature, 2014, 510, 47 CrossRef CAS PubMed.
- X. Han, Lipidomics for studying metabolism, Nat. Rev. Endocrinol., 2016, 12, 668–679 CrossRef.
- T. Cajka and O. Fiehn, Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics, Anal. Chem., 2016, 88, 524–545 CrossRef.
- D. J. Stephenson, L. A. Hoeferlin and C. E. Chalfant, Lipidomics in translational research and the clinical significance of lipid-based biomarkers, J. Lab. Clin. Med., 2017, 189, 13–29 Search PubMed.
- H. C. Köfeler, R. Ahrends, E. S. Baker, K. Ekroos, X. Han, N. Hoffmann, M. Holčapek, M. R. Wenk and G. Liebisch, Recommendations for good practice in MS-based lipidomics, J. Lipid Res., 2021, 62, 100138 CrossRef.
- Z. Jia, Q. Qiu, R. He, T. Zhou and L. Chen, Identification of metabolite interference is necessary for accurate LC-MS targeted metabolomics analysis, Anal. Chem., 2023, 95, 7985–7992 CrossRef PubMed.
- C. Guijas, J. R. Montenegro-Burke, X. Domingo-Almenara, A. Palermo, B. Warth, G. Hermann, G. Koellensperger, T. Huan, W. Uritboonthai, A. E. Aisporna, D. W. Wolan, M. E. Spilker, H. P. Benton and G. Siuzdak, METLIN: a technology platform for identifying knowns and unknowns, Anal. Chem., 2018, 90, 3156–3164 CrossRef CAS PubMed.
- H. Horai, M. Arita, S. Kanaya, Y. Nihei, T. Ikeda, K. Suwa, Y. Ojima, K. Tanaka, S. Tanaka, K. Aoshima, Y. Oda, Y. Kakazu, M. Kusano, T. Tohge, F. Matsuda, Y. Sawada, M. Y. Hirai, H. Nakanishi, K. Ikeda, N. Akimoto, T. Maoka, H. Takahashi, T. Ara, N. Sakurai, H. Suzuki, D. Shibata, S. Neumann, T. Iida, K. Tanaka, K. Funatsu, F. Matsuura, T. Soga, R. Taguchi, K. Saito and T. Nishioka, MassBank: a public repository for sharing mass spectral data for life sciences, J. Mass Spectrom., 2010, 45, 703–714 CrossRef CAS.
- M. Sud, E. Fahy, D. Cotter, E. A. Dennis and S. Subramaniam, LIPID MAPS-nature lipidomics gateway: an online resource for students and educators interested in lipids, J. Chem. Educ., 2012, 89, 291–292 CrossRef CAS PubMed.
- T. Kind, Y. Okazaki, K. Saito and O. Fiehn, LipidBlast templates as flexible tools for creating new in-silico tandem mass spectral libraries, Anal. Chem., 2014, 86, 11024–11027 CrossRef CAS.
- F. Aicheler, J. Li, M. Hoene, R. Lehmann, G. Xu and O. Kohlbacher, Retention time prediction improves identification in nontargeted lipidomics approaches, Anal. Chem., 2015, 87, 7698–7704 CrossRef CAS PubMed.
- J. Stanstrup, S. Neumann and U. Vrhovšek, PredRet: prediction of retention time by direct mapping between multiple chromatographic systems, Anal. Chem., 2015, 87, 9421–9428 CrossRef CAS.
- R. Put and Y. Vander Heyden, Review on modelling aspects in reversed-phase liquid chromatographic quantitative structure-retention relationships, Anal. Chim. Acta, 2007, 602, 164–172 CrossRef CAS PubMed.
- J. B. White, P. J. Trim, T. Salagaras, A. Long, P. J. Psaltis, J. W. Verjans and M. F. Snel, Equivalent carbon number and interclass retention time conversion enhance lipid identification in untargeted clinical lipidomics, Anal. Chem., 2022, 94, 3476–3484 CrossRef CAS PubMed.
- D. H. Ross, J. H. Cho, R. Zhang, K. M. Hines and L. Xu, LiPydomics: a python package for comprehensive prediction of lipid collision cross sections and retention times and analysis of ion mobility-mass spectrometry-based lipidomics data, Anal. Chem., 2020, 92, 14967–14975 CrossRef CAS PubMed.
- B. C. Naylor, J. L. Catrow, J. A. Maschek and J. E. Cox, QSRR automator: a tool for automating retention time prediction in lipidomics and metabolomics, Metabolites, 2020, 10(6), 237 CrossRef CAS.
- S. Codesido, G. M. Randazzo, F. Lehmann, V. González-Ruiz, A. García, I. Xenarios, R. Liechti, A. Bridge, J. Boccard and S. Rudaz, DynaStI: a dynamic retention time database for steroidomics, Metabolites, 2019, 9(5), 85 CrossRef CAS.
- K. Héberger, Quantitative structure-(chromatographic) retention relationships, J. Chromatogr. A, 2007, 1158, 273–305 CrossRef.
- J. Zhang, S. Yang, J. Wang, Y. Xu, H. Zhao, J. Lei, Y. Zhou, Y. Chen, L. Wu and Y. Li, Equivalent carbon number-based targeted odd-chain fatty acyl lipidomics reveals triacylglycerol profiling in clinical colon cancer, J. Lipid Res., 2023, 64, 100393 CrossRef CAS.
- K. Saito, M. Ikeda, Y. Kojima, H. Hosoi, Y. Saito and S. Kondo, Lipid profiling of pre-treatment plasma reveals biomarker candidates associated with response rates and hand-foot skin reactions in sorafenib-treated patients, Cancer Chemother. Pharmacol., 2018, 82, 677–684 CrossRef CAS PubMed.
- E. G. Bligh and W. J. Dyer, A rapid method of total lipid extraction and purification, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS.
- Y. Nakagawa and L. A. Horrocks, Separation of alkenylacyl, alkylacyl, and diacyl analogues and their molecular species by high performance liquid chromatography, J. Lipid Res., 1983, 24, 1268–1275 CrossRef CAS.
- M. Ovčačíková, M. Lísa, E. Cífková and M. Holčapek, Retention behavior of lipids in reversed-phase ultrahigh-performance liquid chromatography-electrospray ionization mass spectrometry, J. Chromatogr. A, 2016, 1450, 76–85 CrossRef.
- T. Xia, F. Zhou, D. Zhang, X. Jin, H. Shi, H. Yin, Y. Gong and Y. Xia, Deep-profiling of phospholipidome via rapid orthogonal separations and isomer-resolved mass spectrometry, Nat. Commun., 2023, 14, 4263 CrossRef CAS.
- M. K. F. Ma, E. Y. T. Lau, D. H. W. Leung, J. Lo, N. P. Y. Ho, L. K. W. Cheng, S. Ma, C. H. Lin, J. A. Copland, J. Ding, R. C. L. Lo, I. O. L. Ng and T. K. W. Lee, Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation, J. Hepatol., 2017, 67, 979–990 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01735d |
| This journal is © The Royal Society of Chemistry 2024 |