
A universal electrochemical biosensor based on CRISPR/Cas12a and a DNA tetrahedron for ultrasensitive nucleic acid detection†
Jiangbo
Dong
a,
Xinyao
Li
a,
Wenxi
Hu
a,
Meilin
Liu
a,
Changjun
Hou
 ac,
Jingzhou
Hou
*ab,
Mei
Yang
*a and
Danqun
Huo
*ac
ac,
Jingzhou
Hou
*ab,
Mei
Yang
*a and
Danqun
Huo
*ac
aKey Laboratory for Biorheological Science and Technology (Chongqing University), Ministry of Education, College of Bioengineering, Chongqing University, Chongqing, 400044, P. R. China
bChongqing Engineering and Technology Research Center of Intelligent Rehabilitation and Eldercare, Chongqing City Management College, Chongqing, 401331, P. R. China
cChongqing Key Laboratory of Bio-perception & Intelligent Information Processing, School of Microelectronics and Communication Engineering, Chongqing University, Chongqing, 400044, P. R. China
First published on 4th June 2024
Abstract
Herein, a universal nucleic acid analysis platform was constructed for sensitive and accurate detection of miRNA-155 and ctDNA using isothermal amplification-assisted CRISPR/Cas12a and a tetrahedral DNA nanostructure (TDN) supported sensing interface. Under the optimal experimental conditions, the prepared sensor achieved specific detection of miRNA-155 and ctDNA at as low as aM levels in 2.6 h. Furthermore, the platform was also successfully applied to human serum sample recovery experiments and cancer cell lysates, demonstrating outstanding reliability and accuracy. We firmly believe that this work provides a universal, sensitive, and practical tool for early clinical diagnosis.
Nucleic acid analysis is of great significance for disease prevention, early screening, and prognosis monitoring. Commonly used techniques include northern blotting, next-generation sequencing, and the gold standard quantitative polymerase chain reaction (qPCR).1,2 Undeniably, these techniques have helped the diagnosis and treatment of patients to some extent, but often miss the optimal treatment window, resulting in high medical costs and significant physical and psychological harm to patients, especially cancer patients. The main reasons are that traditional nucleic acid analysis techniques have some limitations, such as limited sensitivity,3 complex and time-consuming operations,4 and expensive equipment,5 making it difficult to widely promote them in point-of-care (POC) molecular diagnostics applications, especially in resource-limited areas. Therefore, developing an efficient, sensitive, and accurate nucleic acid analysis method for detecting low-abundance biomarkers (such as disease-related DNA, miRNA) will break the limitations of traditional techniques, and promote the development of modern medicine.
Considering the extremely low early concentrations of disease-related biomarkers, it is necessary to introduce biological signal amplification techniques to enhance the sensitivity of detection. The discovery and application of the CRISPR-associated nuclease have rapidly become research hotspots in the arenas of gene editing and molecular diagnostics,6 leading to the development of various biosensors based on Cas9, Cas12a, and Cas13a for in vitro diagnostics. Among them, Cas12a, because of its simple crRNA design and unique DNase activity, exhibits unparalleled trans-cleavage ability once activated, thus amplifying the signal of target detection. However, in practical applications, due to suboptimal sensitivity and limited detection types (targeting only dsDNA or ssDNA),4,7 it is essential to introduce nucleic acid amplification strategies before the CRISPR/Cas12a system to enhance the sensitivity of the target and biological conversion. Additionally, a superior signal output platform can also greatly improve the sensitivity of sensor detection.
Electrochemical biosensors, known for their high sensitivity, rapid signal response, simplicity of operation, and ease of integration, have attracted considerable attention.8 Recently, a universal electrochemical biosensor based on CRISPR/Cas, called E-CRISPR, has been widely established for nucleic acid detection. For instance, Zhang's team designed an electrochemical strategy mediated by CRISPR/Cas12a for interface cleavage of hairpin DNA reporter genes, achieving detection sensitivity as low as 30 pM of HPV-16.9 Han and colleagues utilized activated CRISPR/Cas12a to cleave the linear polyA-MB signal probe at the electrode interface, enabling the development of a highly sensitive electrochemical sensor for detecting COVID-19 Np.10 However, in these methods, the reporter probes used on the electrode are based on one-dimensional linear chains11 or two-dimensional hairpin structures.12 Notably, these reporter probes often exhibit intertwining and collapsing phenomena,5 resulting in local crowding of probes on the electrode surface, exacerbating the effect of steric hindrance,13 and reducing the cleavage efficiency of CRISPR/Cas on the probes. As a result, the sensitivity of the sensor is decreased.
Tetrahedral DNA nanostructures (TDNs) are a rigid three-dimensional scaffold formed by self-assembly of folded complementary double-stranded DNA, which can provide a feasible solution for space control based on interface detection. Compared with the probes with linear-stranded or hairpin structure, TDNs not only enable precise control of surface density at the sensing interface, mitigating surface crowding effects,5 but also increase the accessibility of the target,14 thus improving the sensitivity of the sensor. Based on their superior performance, probes based on TDNs have found extensive application in the detection of nucleic acid and non-nucleic acid targets together with electrochemical methods.15,16
Herein, we constructed an ultra-sensitive universal nucleic acid analysis platform based on isothermal amplification-assisted CRISPR/Cas12a combined with a DNA tetrahedron framework. As a proof of concept, miRNA-155 and ctDNA KRAS were used as detection objects to assess the sensor's performance (Scheme 1). In Scheme 1A, the presence of the target can trigger endonuclease-mediated rolling circle amplification (E-RCA) reaction, generating a large number of single-stranded DNA activators after several rounds of amplification, providing an ample supply of raw materials for the subsequent activation of the CRISPR/Cas12a system. Once activated, the CRISPR/Cas12a system cleaved the single-stranded DNA signal probes fixed on the sensing electrode interface at the TDN vertices, causing the detachment of MB-labeled electroactive signal molecules from the electrode surface, thereby inducing signal attenuation (Scheme 1B).
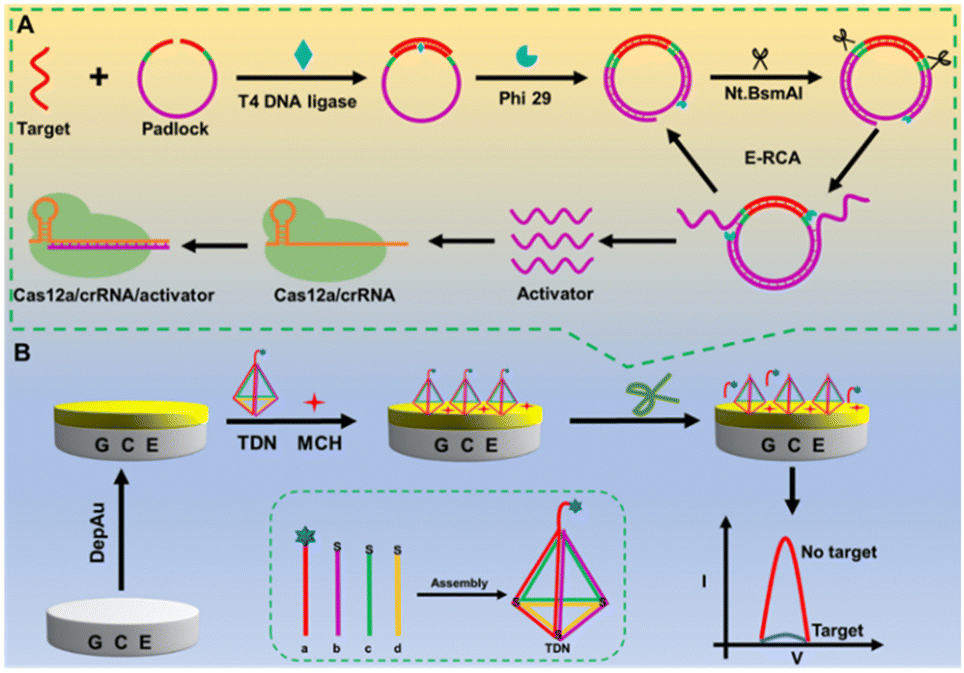 | ||
| Scheme 1 Schematic illustration of the target-triggered E-RCA strategy (A), and construction of the CRISPR/Cas12a-driven TDNA framework-based electrochemical biosensor (B). | ||
Four carefully designed single-stranded DNAs (a, b, c and d) can be self-assembled to form TDNs by annealing in a thermal cycler (Fig. 1A). Due to the excellent controllability and high precision orientation of TDNs, it not only avoided entanglement between probes and local aggregation of self-assembled monolayers, thus resisting nonspecific adsorption, but also accelerated electron transfer, thereby improving detection efficiency. Atomic force microscopy (AFM) was used to characterize the formation of TDNs. In Fig. 1B and C, the prepared TDN was uniformly distributed without aggregation, providing strong evidence for the visual confirmation of TDN formation. Subsequently, we further characterized the self-assembly process of the TDN using 10% polyacrylamide gel electrophoresis (PAGE). As illustrated in Fig. 1D, lanes 1–4 corresponded to the single strands a, b, c, and d that formed TDN, respectively. With the addition of strands in lanes 5 to 7, the migration speed of the DNA bands gradually slowed down, indicating the formation of a larger molecular weight and more intricate spatial configuration TDN.
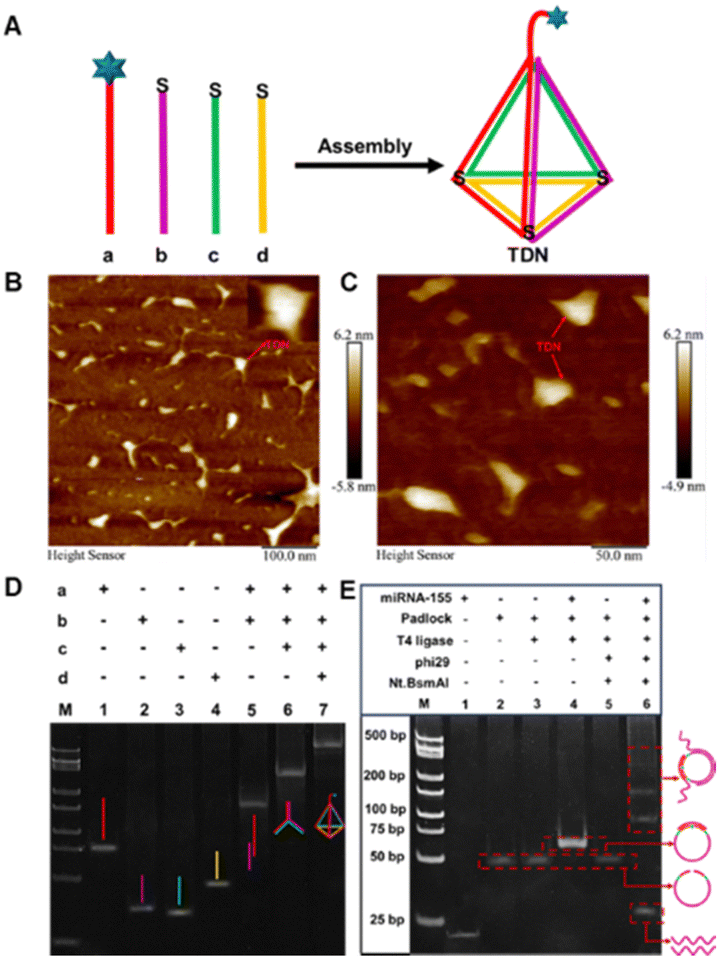 | ||
| Fig. 1 (A) Self-assembly of TDN. (B) and (C) AFM images of TDN. 10% PAGE characterization of the formation of TDN (D), and the E-RCA strategy (E). | ||
Next, we used 10% PAGE to characterize the target-triggered E-RCA amplification products. As shown in Fig. 1E, lanes 1 and 2 corresponded to miRNA-155 and the padlock probe, respectively. Assisted by T4 DNA ligase, new bands appeared in lane 4, which was caused by the formation of a new structure after the padlock probe was cyclized. Lanes 5 and 6 represent the control and experimental groups of E-RCA amplification with and without the target, respectively. In lane 6, a series of bands with different electrophoretic mobilities were observed, attributed to intermediate products of varying molecular weights generated during the E-RCA amplification, which were absent in lane 5. As anticipated, the fastest migrating DNA band in lane 6 had a molecular weight of approximately 26 bp, consistent with the designed E-RCA target product for activating the CRISPR/Cas12a system.
Initially, we analyzed and evaluated whether the target-triggered E-RCA product could activate the CRISPR/Cas12a system. To visually observe the activity of Cas12a ssDNase, the fluorescent reporter probe (FQ-reporter) was employed as its cleavage substrate. As shown in Fig. 2A, we tested the CRISPR/Cas12a system under different conditions. The results indicated a significant fluorescence change only when the E-RCA product, Cas12a, and crRNA were present simultaneously (curve a), suggesting the effective cleavage of the FQ-reporter. The above-mentioned fluorescence signal response with strong contrast showed that the target-triggered E-RCA product could bind with the Cas12a–crRNA dimer to create a ternary complex with ssDNase activity. Although we demonstrated the indiscriminate trans-cleavage capability of activated CRISPR/Cas12a toward ssDNA, whether it has the same effect on the pendant probe on the TDN was further investigated. The analysis results from 10% PAGE provided us with the answer (Fig. S1, ESI†). After incubating the TDN and E-RCA products with the Cas12a system, the DNA bands in lane 3 exhibited a faster migration compared to those in lane 2, robustly demonstrating that the activated CRISPR/Cas12a could still cleave the pendant probe on the TDN.
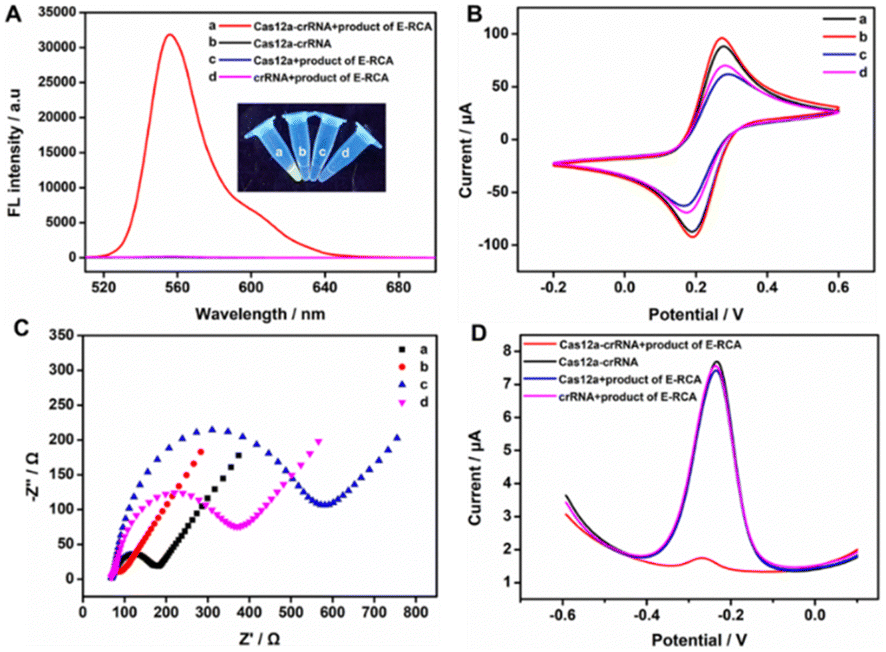 | ||
| Fig. 2 (A) Fluorescence spectra of the FQ-reporter before and after Cas12a-mediated cleavage. (B) CV and EIS characterization of the modification process of the sensor: (a) bare GCE, (b) AuNPs/GCE, (c) TDN/AuNPs/GCE, and (d) MCH/TDN/AuNPs/GCE in 0.1 M KCl containing 5 mM [Fe(CN)6]3−/4−. (D) DPV responses of MB before and after Cas12a-mediated interfacial cleavage of the TDN. | ||
Before the trans-cleavage of the pendant probe on the TDN mediated by CRISPR/Cas12a on the sensing electrode surface, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were employed to analyze the electrode modification process. As shown in Fig. 2B and C, after the electrodeposition of AuNPs, the AuNPs/GCE exhibited a larger CV peak current and smaller electron transfer resistance (curve b), which was attributed to the enhanced conductivity of AuNPs accelerating the electron transfer rates. Upon sequential modification of the TDN (curve c) and MCH (curve d) onto the electrode surface, the CV peak current progressively decreased, while the electron transfer resistance gradually increased. This phenomenon was attributed to the negatively charged phosphate backbone of TDN and the compact MCH layer hindering the diffusion of [Fe(CN)6]3−/4− to the sensing surface. The above changes of CV and EIS (curves a–c) gradually characterized the successful modification of the electrode. Furthermore, DPV testing was conducted to further verify the viability of this strategy. As demonstrated in Fig. 2D, when the sensor was exposed to E-RCA products without Cas12a or crRNA, a high oxidation peak current of MB was observed at −0.25 V. When E-RCA products and the Cas12a–crRNA complex were present, the peak current of MB fell significantly, offering additional confirmation of the effective cleavage of the pendant probe on TDN and the subsequent release of the MB signal molecule on the electrode surface. This phenomenon was also reflected in the CRISPR/Cas12a-mediated fluorescence spectra and DPV responses with or without the presence of the target miRNA-155 (Fig. S2, ESI†).
In the optimal experimental conditions (Fig. S3, ESI†), we evaluated the performance of the constructed sensor for miRNA-155 detection. As shown in Fig. 3A, the response of DPV gradually declined with the increasing miRNA-155 concentration. This decline was attributed to the higher miRNA-155 concentration, leading to the production of more E-RCA products. Consequently, more Cas12a was activated, resulting in a greater number of signal probes being cleaved on the electrode. In Fig. 3B, the change value (ΔI) of the DPV signal had a strong linear correlation with the logarithm (IgC) of the target concentration within the range of 10 fM to 1 nM. This relationship was described by the equation: ΔI = −0.991![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IgCmiRNA-155/pM-2.841 (R2 = 0.995), with a detection limit (LOD) of 0.5 fM. Compared to other strategies for miRNA-155 detection (Table S2, ESI†), our biosensor showed superior sensitivity and a broader linear range.
IgCmiRNA-155/pM-2.841 (R2 = 0.995), with a detection limit (LOD) of 0.5 fM. Compared to other strategies for miRNA-155 detection (Table S2, ESI†), our biosensor showed superior sensitivity and a broader linear range.
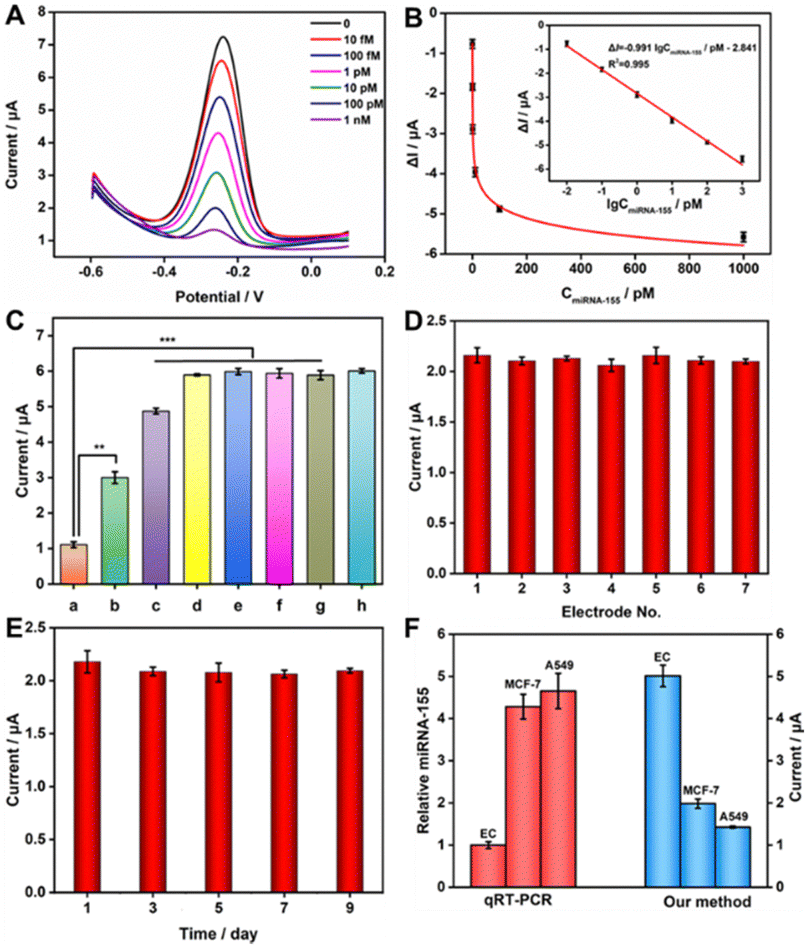 | ||
| Fig. 3 (A) DPV responses of the biosensor across varying miRNA-155 concentrations. (B) The calibration curve for the ΔI versus the logarithm of miRNA-155. (C) Specificity investigation of the biosensor (a → h, miRNA-155, M1, M3, Let-7a, miRNA-21, miRNA-141, random RNA, and blank). (D) The reproducibility and (E) stability of the biosensor in the presence of 10 pM target miRNA-155. (F) Confirmation of miRNA155 expression in different cells using qRT-PCR and our strategy. | ||
The specificity of the sensing platform was evaluated using 100 pM of different kinds of interfering RNAs, including Let-7a, miRNA-21, miRNA-141, random RNA, as well as single-base mismatch (M1) and three-base mismatch (M3). As shown in Fig. 3C, compared to the DPV responses generated by control group interfering RNAs (histogram b → g), the experimental group containing the same concentration of target miRNA-155 exhibited a lower DPV signal (histogram a), and the difference was statistically significant, suggesting that the prepared biosensor had outstanding specificity in detecting miRNA-155, even when a single-base mismatch was present. Subsequently, the reproducibility and stability of the prepared biosensor were investigated. With 10 pM target miRNA-155 present, the biosensor's reproducibility was assessed through seven independent measurements. As depicted in Fig. 3D, the relative standard deviation (RSD) obtained from the seven electrodes was 2.74%, signifying good manufacturing reproducibility for the sensor. In addition, the DPV response of the prepared sensor was measured every other day, and then stored at 4 °C. On the ninth day, the DPV signal intensity measured still kept 96% of the initial signal intensity (Fig. 3E), robustly demonstrating the satisfactory stability of the prepared sensing platform.
To assess the potential of the prepared biosensor in practical applications, the standard addition recovery experiment was conducted. Various concentrations of target miRNA-155 (0.1 pM, 1 pM, 10 pM) were introduced into 20-fold diluted human serum samples, and the recovery rates were calculated based on the obtained DPV signal intensities. As shown in Table S4 (ESI†), the recovery rates ranged from 99.7% to 119.33%, with RSDs not exceeding 3.16%, indicating the potential practical value of the constructed sensing platform in clinical detection. Furthermore, we measured the expression levels of miRNA-155 extracted from different cell samples to assess the practicality of this strategy in complex biological matrices. Human normal endothelial cells (Ec) were used as the negative control group, while human lung cancer cells (A549) and breast cancer cells (MCF-7) served as positive controls. As observed in Fig. 3F, the current intensity from Ec cell samples was significantly higher compared to that from A549 and MCF-7 cell samples, indicating overexpression of miRNA-155 in these two cancer cells, consistent with the results of qRT-PCR detection. These results once again demonstrated that the prepared sensor exhibited high accuracy and excellent analytical performance in clinical applications.
Furthermore, to validate the universality of the proposed sensing platform, circulating tumor DNA (ctDNA), an effective non-invasive biomarker, was employed as the detection object. The results showed that the sensor also had good performance in ctDNA detection (Fig. S4, ESI†).
In summary, we successfully constructed a superior electrochemical biosensor for highly sensitive nucleic acid analysis. The introduction of E-RCA signal amplification converts trace targets into a large number of activators for CRISPR/Cas12a system activation, significantly enhancing the sensor's sensitivity. Moreover, the use of 3D TDN as a scaffold not only precisely controls the nanoscale spacing of the signal probes on the electrode surface, preventing mutual aggregation between probes, but also improves the efficiency of Cas12a reverse cleavage, further enhancing the sensor's sensitivity. Consequently, the developed electrochemical sensor can specifically detect miRNA-155 or ctDNA with a concentration as low as aM levels in the range of 10 fM to 1 nM. Importantly, the sensor demonstrates good versatility, easily extendable to the analysis and detection of other disease-related biomarkers by simply replacing the target-responsive padlock probe. In future work, we can use a more efficient amplification method to shorten the time and develop a miniaturized electrochemical detection device for field testing.
This work was supported by the Chongqing Natural Science Foundation (CSTB2023NSCQ-BHX00789), Open Research Fund of State Key Laboratory of Digital Medical Engineering (2023-K08), Sichuan Science and Technology Program (2022YFSY0013), Fundamental Research Funds for the Central Universities (2022CDJYGRH-013), Chongqing Graduate Tutor Team Construction Project, Analytical and Testing Center of Chongqing University for (AFM) and sharing fund of Chongqing University's large equipment.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Q. Wang, T. Zhou, D. Xue, H. Yang, Z. Sui, X. Yuan and J. Xu, Chem. Commun., 2024, 60, 2910–2913 RSC.
- J. Liu, J. Shi, Q. Feng, W. Fan and C. Liu, Chem. Commun., 2023, 59, 11851–11854 RSC.
- Y. Xue, H. Xie, Y. Wang, S. Feng, J. Sun, J. Huang and X. Yang, Biosens. Bioelectron., 2022, 218, 114762 CrossRef CAS PubMed.
- B. Shen, L. Li, C. Liu, X. Li, X. Li, X. Cheng, H. Wu, T. Yang, W. Cheng and S. Ding, Anal. Chem., 2023, 95, 4486–4495 CrossRef CAS PubMed.
- Y. Xu, C. Wang, G. Liu, X. Zhao, Q. Qian, S. Li and X. Mi, Biosens. Bioelectron., 2022, 217, 114671 CrossRef CAS PubMed.
- X. Chen, X. Liu, Y. Yu, H. Wang, C. Li, I. Vallée, M. Liu, L. Zhao and X. Bai, Sens. Actuators, B, 2023, 395, 134493 CrossRef CAS.
- W. Feng, A. M. Newbigging, J. Tao, Y. Cao, H. Peng, C. Le, J. Wu, B. Pang, J. Li, D. L. Tyrrell, H. Zhang and X. C. Le, Chem. Sci., 2021, 12, 4683–4698 RSC.
- C. He, J. Zhao, Y. Long, H. Yang, J. Dong, H. Liu, Z. Hu, M. Yang, D. Huo and C. Hou, Chem. Commun., 2023, 59, 350–353 RSC.
- D. Zhang, Y. Yan, H. Que, T. Yang, X. Cheng, S. Ding, X. Zhang and W. Cheng, ACS Sens., 2020, 5, 557–562 CrossRef CAS PubMed.
- C. Han, W. Li, Q. Li, W. Xing, H. Luo, H. Ji, X. Fang, Z. Luo and L. Zhang, Biosens. Bioelectron., 2022, 200, 113922 CrossRef CAS PubMed.
- W. Heo, K. Lee, S. Park, K.-A. Hyun and H.-I. Jung, Biosens. Bioelectron., 2022, 201, 113960 CrossRef CAS PubMed.
- H. Yu, Q. Pu, Z. Weng, X. Zhou, J. Li, Y. Yang, W. Luo, Y. Guo, H. Chen, D. Wang and G. Xie, Biosens. Bioelectron., 2021, 194, 113625 CrossRef CAS PubMed.
- Y. Wen, H. Pei, Y. Wan, Y. Su, Q. Huang, S. Song and C. Fan, Anal. Chem., 2011, 83, 7418–7423 CrossRef CAS PubMed.
- J. Guan, K. He and S. Gunasekaran, Talanta, 2022, 243, 123292 CrossRef CAS PubMed.
- Q. Zhang, H. Zheng, X. Zhang, C. Du and J. Chen, Sens. Actuators, B, 2023, 396, 134639 CrossRef CAS.
- T. Yao, J. Chen, L. Kong, Y. Liu, R. Yuan and Y. Chai, Anal. Chem., 2023, 95, 13211–13219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01646g |
| This journal is © The Royal Society of Chemistry 2024 |