
Structure, stability, reactivity and bonding in noble gas compounds
Ranita
Pal
a and
Pratim Kumar
Chattaraj
 *b
*b
aAdvanced Technology Development Centre, Indian Institute of Technology Kharagpur, Kharagpur 721302, India
bDepartment of Chemistry, Birla Institute of Technology Mesra, Ranchi, Jharkhand 835215, India. E-mail: pkc@chem.iitkgp.ac.in
First published on 7th March 2024
Abstract
Noble gases (Ngs) are recognized as the least reactive elements due to their fully filled valence electronic configuration. Their reluctance to engage in chemical bond formation necessitates extreme conditions such as low temperatures, high pressures, and reagents with high reactivity. In this Perspective, we discuss our endeavours in the theoretical prediction of viable Ng complexes, emphasizing the pursuit of synthesizing them under nearly ambient conditions. Our research encompasses various bonding categories of Ng complexes and our primary aim is to comprehend the bonding mechanisms within these complexes, utilizing state-of-the-art theoretical tools such as natural bond orbital, energy decomposition, and electron density analyses. These complex types manifest distinct bonding scenarios. In the non-insertion type, the donor–acceptor interaction strength hinges on the polarizing ability of the binding atom, drawing the electron density of the Ng towards itself. In certain instances, especially with heavier Ng elements, this interaction reaches a magnitude where it can be considered a covalent bond. Conversely, in most insertion cases, the Ng prefers to share electrons to form a covalent bond on one side while interacting electrostatically on the other side. In rare cases, both bonds may be portrayed as electron-shared covalent bonds. Furthermore, a host cage serves as an excellent platform to explore the limits of achieving Ng–Ng bonds (even for helium), under high pressure.
 Ranita Pal | Ranita Pal has recently obtained her PhD from the Indian Institute of Technology Kharagpur. She worked under the supervision of Professor Pratim Kumar Chattaraj, and has co-authored 25 journal articles and five book chapters. Her research activities include electronic structure theory, hydrogen storage, chemical reactivity, binding of small molecules and isomerization processes, studies on Adaptive Natural Density Partitioning method (AdNDP, investigation of nc-2e bond), machine learning and QSAR/QSPR/QSTR analyses in the domain of theoretical and computational chemistry. |
 Pratim Kumar Chattaraj | Pratim Kumar Chattaraj is a Distinguished Visiting Professor at BIT Mesra. He was an Institute Chair Professor at the IIT Kharagpur and a Distinguished Visiting Professor of IIT Bombay. His research interests include density functional theory, nonlinear dynamics, aromaticity in metal clusters, hydrogen storage, noble gas compounds, machine learning, chemical reactivity and quantum trajectories. He is a Fellow of The World Academy of Sciences and all three Indian Science Academics; West Bengal Academy of Science and Technology; and FWO, Belgium. He is a Sir J.C. Bose National Fellow. Several of his papers have become Editors’ choice/hot/most cited/most accessed/cover articles. |
Introduction
In the grand tapestry of chemical elements, noble gases (Ngs) have long occupied a unique and seemingly immutable position. Their full valence electron shells, a testament to the octet rule, were once thought to render them chemically inert and isolated from interatomic bonding. This perception, preserved in textbooks and reinforced by decades of seemingly inert behaviour, stood as a cornerstone of chemical understanding. However, high-pressure synthesis techniques and sophisticated theoretical calculations have cracked this facade. In terms of electronic configurations, they are characterized by fully occupied valence orbitals, ns2np6 for all except helium, which has 1s2. These electronic configurations offer exceptional stability and impede their proclivity to readily engage in chemical bonding with other elements, thereby designating them as “inert gases.” The pronounced elevation of their first ionization potentials (IPs), showcasing a gradual decrease from helium to radon,1 further reinforces their reputation for chemical inertness. Nonetheless, this decrease in IP signifies that the outermost electrons of the heavier noble gases, notably xenon, become progressively more vulnerable to external influences, paving the way for the plausible formation of noble gas compounds.Before the successful synthesis of noble gas compounds, several prominent chemists theorized the possibility of their formation based on atomic properties and periodic trends. In 1916, Kossel predicted the existence of xenon and krypton fluorides based on their loosely bound outermost electrons and fluorine's high electronegativity.2 Leveraging ionic radii data, Pauling predicted the potential formation of xenon fluorides (XeF6 or XeF8) and xenic acid (H4XeO6).3 In 1962, Bartlett's intuition regarding the resemblance between the first IPs of O2 and Xe yielded fruitful results. While undertaking a reaction between dioxygen and PtF6 to form O2+PtF6−,4 he made the said realization and immediately aimed to form an Xe+PtF6− complex by reacting Xe and PtF6.5 This reaction resulted in the precipitation of an orange-yellow solid. This landmark discovery shattered the longstanding notion of noble gas inertness, marking the inception of the “Noble Gas Chemistry” era. Initially presumed to be Xe+PtF6−, the formula of this first Ng compound underwent revision upon X-ray powder diffraction (XRPD) analysis. The actual product unveiled itself as XeF+Pt2F11−, a more intricate species than Bartlett's envisioned simple adduct.6 The reaction mechanism, as delineated by Christe's quantum-chemical calculations, implicated F− ion catalysis, yielding a mixture comprising XeF+PtF6−, PtF5, and the ultimate product, XeF+Pt2F11−.7,8
In the subsequent years, a surge of investigative endeavours resulted in the synthesis and characterization of numerous xenon compounds, encompassing XeF2, XeF4, XeF6, XeOF4, and XeO3. Krypton, too, participated in this chemical exploration with the introduction of KrF2.9,10 Xenon, distinguished by its loosely bound outer electrons, emerged as the focal point of scrutiny. A multitude of scholarly publications detailed the existence of species such as XeF2−6, XeOF4, XeO3, and others.11–16 Even the radioactive radon found recognition, forming compounds like RnF2 and [RnF][Sb2F11].17 The beginning of the 21st century marked another watershed moment: the isolation of the first argon compound, HArF, accomplished by Räsänen's research group within a low-temperature matrix.18,19 Subsequent to this achievement, the identification of various weak neon complexes, including NeAuF and (NeAr)Be2O2, expanded the horizons of Ng reactivity.20–23 In the year 1925, the HeH+ cation was observed by Hogness and Lunn.24 Later, Dong et al. demonstrated that, under high pressures, even helium can engage in chemical bonding, resulting in the formation of the Na2He compound.25
Presently, it is established that all Ng elements exhibit chemical reactivity, albeit contingent on specific conditions. The trajectory from inertness to reactivity persists, with ongoing research continuously expanding the frontiers of noble gas chemistry and revealing its diverse applications in domains such as anaesthesia, nuclear waste storage, and materials science.
Bonding pattern
The bonding in compounds involving Ng elements can be categorized into three primary types, namely, (a) non-insertion NgAB, (b) insertion ANgB, and (c) Ngn@cage complexes.Ng adducts, where the binding of the Ng atom occurs at an exposed end of a molecule (say, AB), constitute the non-insertion type of compounds. The interaction between Ng and A stems from the ability of the A atom to induce polarization in the electron cloud of the outermost orbital of the Ng, leading to the formation of an attractive donor–acceptor bond. This polarization is contingent upon the polarizing power of A, influenced by its size and charge. Smaller, highly charged A atoms result in more pronounced polarization and, consequently, stronger Ng–A interactions. Additionally, the difference in electronegativity between A and the counterion, B, plays a pivotal role. A substantial difference induces a dipole in the AB molecule, intensifying the polarization of the Ng atom. It is to be noted that there is a heightened effectiveness in inducing polarization by ionic counterions (B−) compared to that by the neutral ones. Finally, in neutral AB molecules with minimal electronegativity differences, dispersion forces contribute to bonding, albeit to a lesser degree. This intricate interplay of factors underscores the multifaceted nature of Ng–A interactions, rendering these adducts a compelling focal point in contemporary chemistry research.
For the insertion complex, the Ng atom inserts itself within the A–B bond to form an ANgB complex. The formation of this complex inherently disrupts the pre-existing A–B bond, presenting a thermodynamic challenge. While the collective interactions of A–Ng and Ng–B do contribute, they often fall short of fully compensating for the lost A–B stability. In contrast, the stability of NgAB primarily hinges on the thermodynamics of the Ng–A bond. Vigorous donor–acceptor interactions between the Ng atom and strongly polarizing centers in A contribute to their heightened stability. The insertion complex, ANgB species, tends to be metastable, and its existence is frequently contingent on kinetic factors. In-depth thermochemical analyses illuminate this kinetic reliance. The consideration of extensive dissociation pathways is imperative, with two predominant routes prevailing (others are mostly endergonic), ANgB → Ng + AB (highly exergonic) and ANgB → A + Ng + B (occasionally weakly exergonic). Evaluating the stability of ANgB entails assessing the activation energy barriers of these dissociation pathways.
Beyond classical NgAB and ANgB complexes, a captivating domain unfolds where noble gas chemistry relies on the influences generated by the restricted environment facilitated by specific cages. Instances include Ng2 dimers enclosed within various hollow cages, such as fullerene, borospherene, C20H20, B12N12, B16N16, octa acids, cucurbit[n]uril, clathrate hydrates, and carbon nanotubes doped with BN. Our research group has extensively investigated these systems over the years. In this context, the confinement itself narrates the structure, bonding, and reactivity of the entrapped Ng2 guests. We systematically analyze them along with their confined movements within the cages, comparing them to their unconfined counterparts.
Computational details
Designing Ng compounds requires careful computational optimization. Selecting the right method is crucial, aiming for minimal errors in predicting geometry, electronic properties, and energy. For highly accurate calculations, especially on small systems, ab initio methods like CCSD(T) with advanced basis sets are ideal. However, for larger systems, computationally expensive CCSD(T) becomes impractical, necessitating DFT-based methods. Benchmark studies suggest specific DFT levels like MPW1B95/6-311+G(2df,2pd), BMK/aug-cc-pVTZ, DSD-BLYP/aug-cc-pVTZ, and B2GP-PLYP/aug-cc-pVTZ levels, which offer excellent accuracy in calculating Ng bond energies. Notably, MP2 with aug-cc-pVDZ shows less accuracy. Relativistic effects of heavier elements like Xe and Rn are often handled with effective core potentials or ZORA approximations. Interestingly, Ng insertion compounds are particularly sensitive to the chosen level of theory. Structures stable at DFT or MP2 levels can sometimes dissociate at the higher-level CCSD(T), making these high-level calculations essential for predicting truly stable Ng-inserted molecules. Conversely, non-insertion type systems are less sensitive, with only bond dissociation energies changing slightly across different levels of theory. It is highly unlikely for a stable species at one level to become unstable at another in these cases.The natural population analysis (NPA)26 scheme is used in NBO27 to determine atomic charges (q), followed by the computation of Wiberg bond indices (WBI)28 to assess the formation of potential bonds between atoms. Utilizing second-order perturbation allows for a qualitative understanding of interactions between filled Lewis-type NBOs (i, acting as donors) and empty non-Lewis NBOs (j, acting as acceptors). The stabilization energy of delocalization from i to j is given by eqn (1).
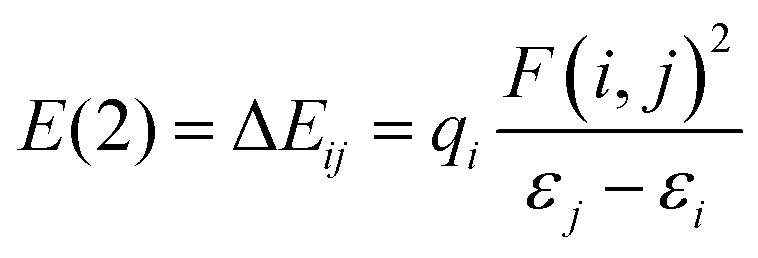 | (1) |
The bonding nature is further elucidated through electron density (ED, ρ(r)) topological analysis using Bader's quantum theory of atoms-in-molecules (QTAIM)32 method in the Multiwfn software.33 This analysis involves utilizing ∇ρ(r) and ∇2ρ(r) to delineate atomic regions and interatomic bond paths. Specifically, the bond path is identified as the one with the maximum gradient of the ED starting and ending in nuclei passing through the bond critical point (BCP). Covalent bonding is typically indicated by high ρ(rc) and negative ∇2ρ(rc) values, and non-covalent bonding by low values of ρ(rc) coupled with positive ∇2ρ(rc). In the case of heavy atoms, a negative total electron energy density (H(rc)) alongside G(rc)/ρ(rc) approximately equal to 1 suggests partial covalency (G(rc) denotes the local kinetic energy density).
Energy decomposition analysis (EDA)34 is conducted in conjunction with the natural orbitals for the chemical valence (NOCV)35 method, employing the ADF 2013.01 program36 package, to provide further insights into the bonding scenario. The interaction between the prepared fragments is quantified as the intrinsic interaction energy (ΔEint), consisting of four distinct energy components: electrostatic energy (ΔEelstat), orbital interaction energy (ΔEorb), dispersion interaction energy (ΔEdisp), and Pauli's repulsion energy (ΔEPauli). While the first three terms contribute to the total attractive interaction energy, the fourth one represents a repulsive force.
| ΔEint = ΔEelstat + ΔEorb + ΔEdisp + ΔEPauli | (2) |
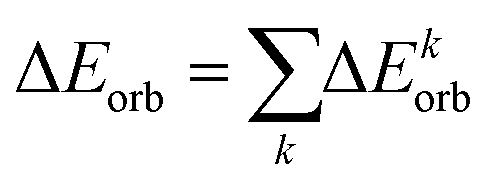 | (3) |
Discussion
Non-insertion complexes
Inspired by Pauzat et al.'s findings41–43 on triangular H3+ clusters interacting with noble gases, our research team has computationally demonstrated that H3+ can form remarkably strong bonds with a maximum of three Ng atoms.44 Notably, the first bond between H and Ng in H3(Ng)+ displayed significantly higher strength compared to larger clusters. This stability was linked to delocalization of the cationic charge, as revealed by conceptual DFT-based descriptors. Interestingly, Li3+ offered a contrasting picture. While each Li center can bind with an Ng atom forming Li3Ng3+, the bonds of Ng with Li are notably weaker than that with H atoms observed in the H3+ species. Comparing it to previously reported NgLiH and NgLiF, the Li–Ng bond strength turns out to be similar.45–49 Motivated by the intriguing interaction of Ng in such species and Li's potential for hydrogen storage, a comprehensive study explored Ng interactions with star-shaped C5Li7+50 and O2Li5+51 clusters to check how far Ng atoms can replace H2 molecules.52,53 The former can bind up to seven helium atoms, with other Ng atoms (Ne–Xe) reaching a maximum of 12. Similarly, O2Li5+ hosted up to seven Ng atoms (He–Xe). Further comparative analyses unveiled that, in contrast to helium and neon, which exhibit slightly weaker interactions with C5Li7+ and O2Li5+ clusters compared to hydrogen molecules, heavier noble gases display higher bond strength.47,48 This intriguing phenomenon extends beyond specific Li-decorated clusters. Our examination of various Li-decorated and super-alkali clusters consistently reaffirmed that clusters with highly positively charged Li centers tend to exhibit an inclination for bonding with noble gases.52,54 Applying an external electric field further strengthened the Li–Ng bonds.The higher ionic potential of Be compared to Li affords it a notably stronger affinity for these elusive Ng elements. As early as 1988, Frenking et al. theoretically predicted the potential formation of NgBeO complexes,55 which materialized into experimental reality in 1994 when Andrews et al. employed the pulsed-laser matrix isolation technique to detect these Ar, Kr and Xe analogues.56 Motivated by these revelations, we have explored some additional viable Ng–Be complex structures, viz., CN3Be3+, featuring two distinct types of beryllium centers. While each ring-connected beryllium can accommodate a single Ng atom, the lone beryllium outside the ring can interact with two, resulting in a total of four Ng partners. Our computational analyses suggest that the NgCN3Be3+ cluster may feasibly exist at room temperature for lighter noble gases (He–Ar), while colder temperatures may be required for the heavier ones (Kr–Rn). Significantly, the formation of a nearly half-bond between Be and heavier noble gases (Kr–Rn) is indicated by high Wiberg Bond Index (WBI) values. Apart from BeO, the entire class of NgBeX compounds, where X = O, S, Se, Te, and their positively charged counterparts are explored, where the latter forms exhibited a heightened affinity for Ng atoms compared to their neutral counterparts when considering a specific X atom. Moreover, within a given noble gas, the bond strength with Be diminished as we progressed from O to Te. Apart from these, the global minimum (GM) energy structures of Be2N2, Be3N2, and BeSiN2 species were explored.57 Markedly positive Be centers in these structures serve as attractive binding partners for the Ng atoms. Be2N2 accommodates two Ng atoms, Be3N2 hosts three, and BeSiN2 binds one, all through their Be centers. Notably, the presence of Ng can alter the energy rankings of these isomers. For instance, the linear (GM) structure of BeSiN2 has Be in the center, compared to the next higher energy isomer with Be situated terminally (Fig. 1a). This energy positioning changes in the presence of heavier Ngs (Ar–Rn), as the second isomer optimally positions the Be atom terminally (compared to the central position of Be in the former) for a more robust interaction with the Ng atoms.
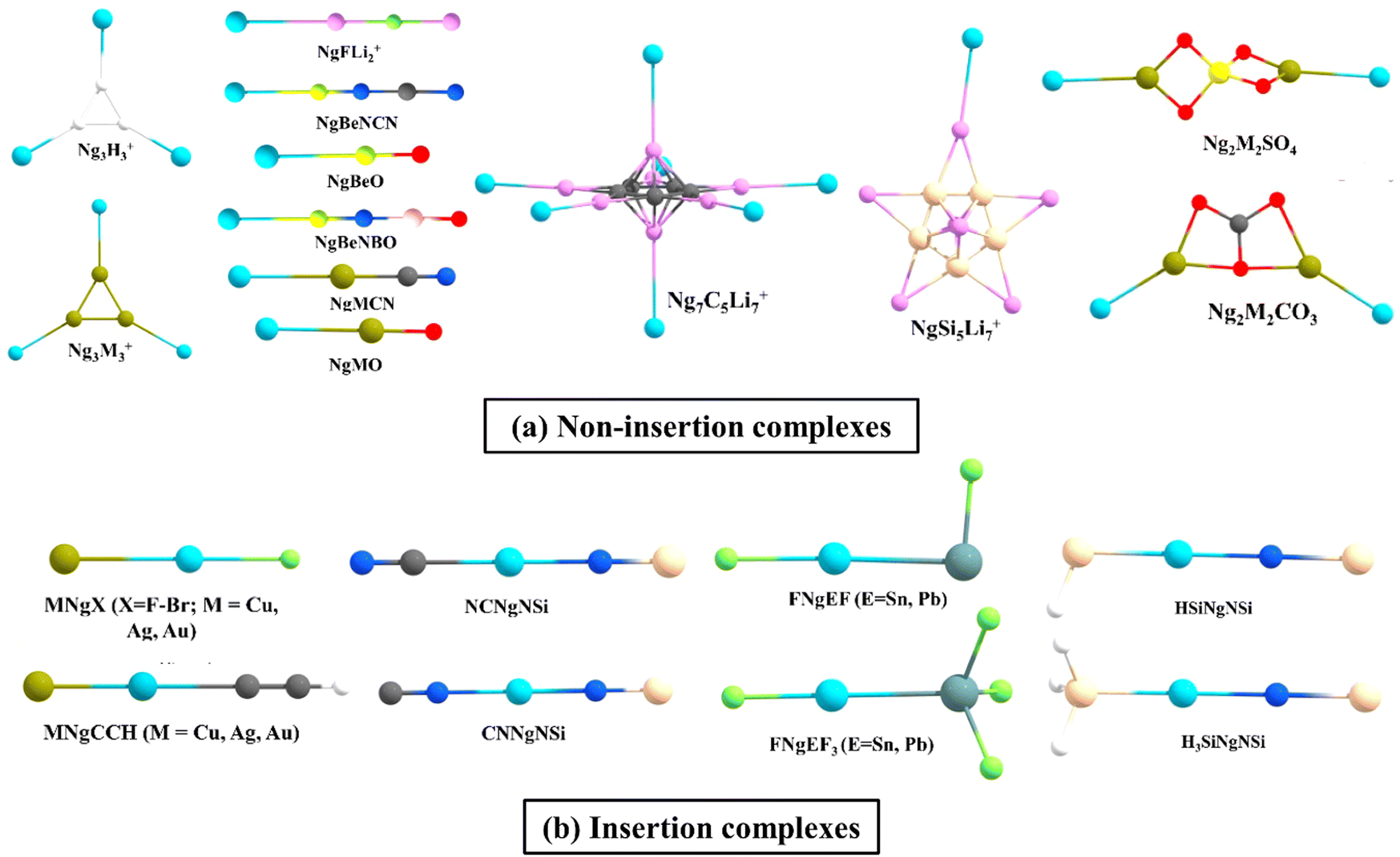 | ||
| Fig. 1 A few examples of the (a) non-insertion NgAB and (b) insertion ANgB type complexes [reproduced from ref. 88 with permission from MDPI, © 2019]. | ||
Attaching a Lewis acid, like BH3, to the oxygen center of BeO, as proposed by Grandinetti et al. enhances the interaction with Ng atoms. Similarly, substituting the H atom in BeNH with various groups, as observed by the same authors, proved effective in augmenting its Ng binding capabilities.58 Nevertheless, a trade-off was evident, as these modified systems did not represent the most stable configurations. Identifying two Be-containing molecules, BeNCN and BeNBO, proved pivotal as they achieved both exceptional stability (as both are GM structures) and robust Ng binding (highest and second-highest among reported neutral Be systems). These molecules, formed by replacing the H atom in BeNH with CN and BO, respectively, strategically position the Be atom terminally for optimal interaction with the Ng partners.59 In the pursuit of ideal Be-based salts for strong Ng–Be bonds, our investigation delved into the Ng-binding capacity of various BeX systems, where X = CO3, SO4, CrO4, and HPO4.60,61 A comparative analysis unveiled that NgBeSO4 forms the most potent bonds with Ngs in contrast to NgBeCO3 and NgBeO (with the exception of He–Be).
The interactions between Ngs and 1-tris(pyrazolyl)-borate salts of Be and Mg are also studied.62 Be demonstrates superior affinity for Ngs compared to Mg because of the former's smaller size and higher charge density, enabling a stronger polarizing force on the Ng electron cloud. Moving beyond the Be–Mg duo, we explored half-sandwich complexes where Ngs (He–Rn) reside atop metal cations (M = Be–Ba) with a cyclopentadienyl ring (Cp).63 These NgMCp+ complexes exhibited a hierarchy of bond strengths where, for a specific Ng, the interaction with Be was the most robust, followed by Mg, Ca, Sr, and Ba. Interestingly, for a fixed metal, the bond strength increased with the Ng atomic mass, from He to Rn. To understand the underlying mechanism of this interaction, we analyzed deformation densities related to the orbital contributors. The primary interaction was characterized by Ng pσ orbitals donating σ electron density to the BeCp+ complex, followed by the π-donation from Ng pπ orbitals to BeCp+.
We expanded our scope beyond beryllium and magnesium to explore the binding abilities of Ngs with group 14 elements as well. The EX3+ system, where E = elements of group 14 and X = H, F, Cl, Br, exhibited substantial yet comparatively lower attraction towards noble gases than boron in B3+.64,65 Notably, EH3+, for a given noble gas, showed the order C > Si > Ge in terms of binding strength. The substitution of a hydrogen atom in EH3+ with X (F, Cl, Br) introduced a complex interplay of two opposing factors: the inductive (–I) effect of X enhanced the Lewis acidity of E, while the π-backdonation from X to E had a negative effect on its ability to bind Ngs. Notably, while SiF3+ and GeF3+ exhibited higher affinity for Ngs compared to their EH3+ counterparts; due to the highly efficient F → C π-backdonation in CF3+, its Ng binding ability significantly diminished compared to CH3+. Further findings revealed that EH3+ (E = Si, Ge) and EF3+ (E = Si, Ge, Sn, Pb) can effectively bind two Ng atoms simultaneously, and their strong Lewis acidity extends to the Lewis base CO.66
Among the transition metal (TM) compounds, particularly superhalogens MF3 (M = Ru–Ag and Os–Au in the second and third row TM series, respectively), RuF3 and AuF3, exhibited the highest and the second highest affinity for binding with Xe, respectively.67 Subsequent studies revealed that the coinage metals (Cu, Ag, and Au) demonstrated very high Ng-binding abilities, generally surpassing those of other TMs. The σ-aromatic coinage metal clusters M3+ and their M3Ng3+68 complexes exhibited a bond dissociation energy range of 2.2–19.0 kcal mol−1 for the Ng–M bonds, following the order, Au > Cu > Ag. Unlike the orbital-dominated bonding observed in the main group elements, these Ng–M bonds derived nearly equal strength from both coulombic and covalent contributions. The crucial orbital factor stabilizing the Ng–M bonds originated from electron donation from a filled pNg to the LUMOM3+. The strong interactions between noble gases and coinage metals (also known as noble metals) uncovered within compounds like NgMNO3, NgCu(NO3)2, NgMSO4, Ng2M2SO4, NgCuCO3, Ng2M2CO3, NgMCN, NgMO, and [Ng–M(bipy)]+ complexes69–74 revealed a partial covalent interaction in the Ng–M bonds. For [Ng–M(bipy)]+ complexes, finding a suitable counterion like [SbF6]− that stabilizes the complex while maintaining the strength of the Ng–M bond becomes crucial for successful synthesis.
Insertion complexes
Continuing our investigation into noble gas interactions, we explore compounds, inspired by the works of Merino et al.,75,76 which provided insights into HNgY molecules (Y = CCH, CN, NC, F, Cl, Br, I and Ng = Xe, Rn), containing electron-sharing H–Ng bonds as well as ionic Ng–Y interactions. These systems can be conceptualized as Ng+ interacting with [H⋯Y]−, forming a polar covalent bond. Despite their metastable nature, they exhibit significant kinetic barriers against dissociation, preventing immediate breakdown into Ng and the parent molecule.We subsequently examined compounds like H3SiNgNSi and HSiNgNSi (Ng = Xe, Rn).77 In both the cases, the free energy change associated with dissociation pathways leading to free Ng and the parent compound is negative, implying thermodynamic stability. While the 2B path is exergonic, the 3B dissociation channel appears endergonic for the Rn analogues, and the Xe variant exhibits a marginally exergonic process, although calculations at lower temperatures suggest endergonicity. Negative ∇2ρ(rc) and H(rc) values, coupled with a high ELF at the Si–Ng bond critical point, reveal its covalent nature. In contrast, the Ng–N bond displays ionic characteristics. (H3SiNg)+(NSi)− and (HSiNg)+(NSi)− bonding representations turn out to be the most accurate. Energy decomposition analysis (EDA)78,79 further corroborates these findings, with ΔEorb as the dominant contributor to the Si–Ng bond and ΔEelstat playing a significant role in the Ng–N interaction. Computed activation energy barriers suggest that H3SiNgNSi possesses sufficient kinetic stability for detection at 250–300 K, whereas HSiNgNSi requires a relatively cold environment (150–200 K).
Expanding our exploration of these unique Ng compounds, we reported the first set featuring E–Ng covalent bonds (E = Sn, Pb and Ng = Kr, Xe, Rn) in FNgEF3 and FNgEF compounds.80 These systems also exhibit metastability, with only the Ng release path being highly exergonic with substantial energy barriers ensuring their protection. Natural bond orbital (NBO) analysis,81H(rc) values,82 and EDA calculations collectively indicate a covalent character in the Ng–E bonds and ionic in the Ng–F bonds. The significant role of superhalogens in stabilizing Ng insertion compounds was demonstrated in HNgY (Y = BO2, BF4) molecules by their enhanced stability compared to the halogen analogues.83 Building on this insight, we explored the efficacy of BeF3, a potent superhalogen, to stabilize HNgBeF3 (Ng = Ar–Rn) complexes.84 Interestingly, the 2B dissociation path (HNgBeF3 → Ng + HBeF3) is exergonic for all Ng variants, indicating thermodynamic stability. In contrast, the 3B path (HNgBeF3 → H + Ng + BeF3) and an additional identified route (HNgBeF3 → Ng + HF + BeF2) are endergonic, with the latter revealing a two-step process upon further scrutiny. Notably, Xe and Rn analogues might exhibit half-lives of up to 102 seconds at 100 K, further supporting their viability. Thorough bonding analysis reveals the covalent nature of H–Ng bonds and the ionic character of Ng–F interactions, presenting the most accurate representation of the molecule as (HNg)+(BeF3)−.
For a unique class of compounds, NCNgNSi (Ng = Kr–Rn), representing the first observed instance of the C–Ng–N unit with covalent bonds on either side of Ng, a deviation from the typical XNgY insertion molecules represented as X+(NgY)− was explored.85 The bonds of Ng with both C and N in NCNgNSi exhibit electron-sharing characteristics. All dissociation channels, except for NCNgNSi → Ng + CNSiN, are endergonic for Ng = Xe and Rn. Remarkably, the 3B dissociation of NCKrNSi has low exergonicity at room temperature, but lower temperatures will render it endergonic again. A gradual increase in free energy barriers of the 2B dissociation pathway from Kr to Rn (25.2 to 39.3 kcal mol−1) suggests that NCNgNSi (Ng = Xe, Rn) systems are viable candidates for detection under ambient conditions, while the Kr analogue might require colder temperatures. Covalent C–Ng and Ng–N bonds are confirmed by substantial WBI (>0.5), H(rc) < 0, and EDA, providing compelling evidence for this bonding picture. Additional analyses using AdNDP reinforce this description by identifying a delocalized 3c–2e π-bonding associated with the C–Ng–N unit and a completely delocalized 5c–2e π-bond. These findings solidify the unique bonding characteristics of this fascinating molecule. Intriguingly, Ng dissociation from NCNgNSi leads to CNSiN (the higher energy isomer), instead of the more stable NCNSi. However, the presence of Ngs significantly lowers the barrier for this transformation, facilitating the detection of the less stable isomer and emphasizing the role of Ngs in manipulating the energy landscape of these systems. More recently, another Ng insertion complex with the C–Ng–N motif, HCCNgNSi (Ng = Kr, Xe, Rn), has been studied.86 This Ng insertion within the parent compound HCCNSi compound helped realize the other isomer, HCCSiN, which was experimentally undetected. The most probable explanation behind this is that the Ng insertion within the C–N bond provided enough space between the HCC and NSi for the latter to rotate to SiN and form the HCCNgSiN complex, which dissociates to form Ng and HCCSiN. A DFT-based investigation was also performed on the stability and bonding of the novel complex, XNgOPO(OH)2 (X = F, Cl, Br; Ng = Kr, Xe, Rn).87
Confined complexes
The intriguing domain of noble gas chemistry takes a compelling turn when confinement factors into the equation. Fullerenes, particularly C60, stand out as versatile tools to investigate the impact of confinement on these traditionally inert elements. The encapsulation of two noble gas atoms within the C60 cage (Ng2@C60) provides an exceptional platform for examining bond formation within spatial constraints. Krapp and Frenking's ground-breaking discovery89 revealed that confinement within C60 substantially reduces the bond distance in Xe2 compared to the free molecule, surpassing even that of free Xe22+. Bonding analysis attributes this phenomenon to the potent steric pressure applied by the cage, bringing the Ng atoms into closer proximity, which encourages orbital overlap, leading to negative H(rc) values at the BCP for Ar, Kr, and Xe. The increasingly negative values with heavier elements suggest a more pronounced covalent character in the bond. A noteworthy conclusion emerged by further comparing encapsulated Ng2 reactivity with their free counterparts: confinement induces a true chemical bond between Ar–Xe, while for smaller He and Ne, the large C60 cavity allows for distant positions, minimizing Pauli repulsion and favoring weaker van der Waals interactions. To assess the kinetic stability of Ng2@C60, we have employed ab initio molecular dynamics, unveiling a remarkable precessional movement of the encapsulated pair, behaving as a unified entity.90 This synchronized movement provides additional support for the formation of a bond under confinement. Interestingly, the extent of precession reduces with increasing Ng atom size.In another interesting observation, encapsulating the Xe2 dimer necessitated a significant change in the structure of the host cage, C60. To accommodate the larger guest, the cage deviated from the isolated pentagonal rule (IPR),91 adopting a geometry with two adjacent pentalene units.92 Driven by the pursuit of He2 bonding, our investigation ventured into even smaller cages than C60.93 By trapping two helium atoms within the C20H20 cage, we achieved a He–He internuclear separation of 1.265 Å, which is less than half of the distance observed in the free He2 dimer. Despite this remarkable proximity, detailed analysis revealed very low charge transfer, a practically zero WBI, and molecular orbital plots indicative of a closed-shell interaction. This study underscores that a short internuclear distance alone does not guarantee a chemical bond; compelling evidence of significant electron sharing and orbital hybridization is necessary.94 Undeterred, our pursuit of a chemically bound He2 unit continued within B12N12 and B16N16 cages.95 Although these heteroatomic cages exhibited slightly improved charge transfer from He to the cage compared to C20H20, they remained thermochemically unstable. Nonetheless, ab initio simulations suggested their kinetic stability. More excitingly, He2@B12N12 exhibited negative H(rc) values at the He–He BCP, hinting at some degree of orbital involvement. Furthermore, EDA calculations revealed that 40.9% of the total attraction between the He atoms constitutes the ΔEorb term, supporting the observed orbital contribution.
The adaptability of Ng interactions extends beyond confined fullerenes, encompassing a diverse array of host molecules. Two systems, namely, clathrate hydrates and cucurbit[n]uril (CB[n]) frameworks, both traditional and HF-doped, demonstrate their potential as hosts for Ng atoms.96–99 A comprehensive study involving 512, HF512 (doped), 51268, and HF51268 cages as hosts and He, Ne, and Ar as guests unveiled the stabilizing effect of HF doping. While 512 and HF512 cages can accommodate one Ng atom each, the larger HF51268 can house up to 10 He, 6 Ne, or 6 Ar atoms. Crucially, all interactions within these complexes manifest as purely non-covalent. The CB[n] macrocycle, notably, CB[6] accommodates up to three Ne atoms, while only two Ar or Kr atoms fit within its confines. This varying capacity arises from the larger sizes of Ar and Kr, causing distortion of the cage walls due to repulsive forces. Interestingly, the distances between the encapsulated Ng atoms fall below the sum of their van der Waals radii, indicating close proximity. However, electron density analysis and EDA calculations unveil a closed-shell, dispersion-dominated interaction between the Ng atoms within the CB[6] cage, revealing the non-covalent nature of the association. MD simulations further affirm the stability of these confined species at 77 K. Similar investigations have been conducted with octa acids,100 highlighting the diversity of potential host molecules.
The domain of Ng interactions transcends mere confinement, with the electronic charge distribution of the host emerging as a pivotal factor in stabilizing these enigmatic elements. Our scrutiny of BN-doped carbon nanotubes (BNCNTs) serves as an exemplification of this phenomenon.101 Helium dimers confined within BNCNTs exhibit an unexpectedly compact He–He bond distance of 1.824 Å, notably smaller than that observed in pristine CNTs (2.596 Å). This substantial disparity underscores the heightened polarization of He atoms within the BNCNT cavity compared to their behavior in CNTs. Analogous trends manifest in other Ng–Ng distances within Ngn@BNCNT systems (Ng = He, Ar, Kr; n = 3 for He, and 2 for Ar and Kr), with AIM analysis providing additional clarity on the nature of these interactions. Helium pairs display closed-shell interactions, while Ar–Ar and Kr–Kr interactions within the BNCNT cavity assume a partial covalent character.
Borospherene (B40) emerges as another interesting host for encapsulating Ng atoms. Utilizing DFT methods, we explored systems like Ngn@B40 (n = 1 and 2 for He–Kr, and only 1 for Xe and Rn).102,103 While lacking thermodynamic stability concerning dissociation, these complexes exhibit remarkable kinetic stability due to elevated free energy barriers ranging from 84.7 to 206.3 kcal mol−1 for the Ng@B40 complexes. The increasing size of the Ng atom induces greater distortion in the encapsulated B40 system, optimizing Ng2 dimer formation up to the Kr analogue within the B40 cavity. Notably, the Ng–Ng bonds in Ar2@B40 and Kr2@B40 acquire partial covalent character inside the cage, emphasizing the subtle influence of the host. The inherently fluxional nature of the B40 cage, involving continuous interconversion between the hexagonal and heptagonal boron rings, has some effect on the Ng encapsulation.104–109 The corresponding free energy barrier increases upon encapsulation, delineating the altered kinetic landscape. Moreover, the presence of Xe within the B40 cage augments its complexation ability with [Fe(η5-C5Me5)]+ compared to the free cage. This insightful observation underscores the potential for leveraging encapsulated Ng atoms to tailor the reactivity and functional properties of host molecules (Fig. 2).
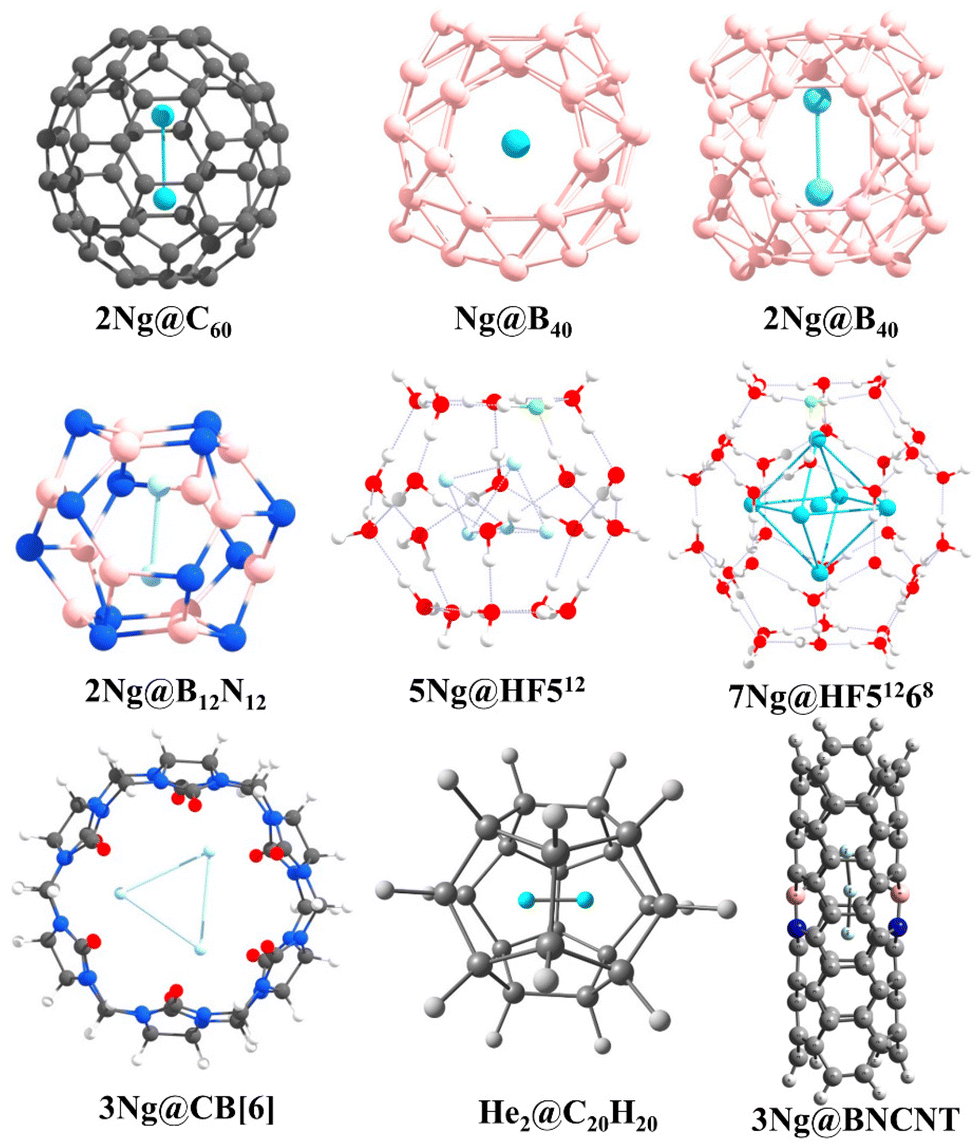 | ||
| Fig. 2 A few examples of the Ng-encapsulated systems [reproduced from ref. 88 with permission from MDPI, © 2019]. | ||
Summary
This Perspective discusses our research contributions concerning the prediction of novel noble gas (Ng) complexes and the associated bonding phenomena. Our investigations have spanned mainly three distinct categories of Ng compounds, namely, the non-insertion (NgAB), insertion (ANgB), and the cage complexes encapsulating Ng. Here, A and B can denote a single atom or a group of atoms. The thermochemical stability of NgAB hinges on the strength of the interaction, allowing stability at specific temperatures. Conversely, ANgB compounds have an exergonic dissociation channel, ANgB → Ng + AB, but are kinetically stable. ANgB molecules exhibit sensitivity to the level of theory employed. Instances have arisen where a system considered energetically stable at the DFT level may undergo dissociation while optimization at the coupled cluster [CCSD(T)] level. Consequently, we strongly recommend the latter for studying ANgB molecules. While bond dissociation energy may vary with changes in the level of theory for the other types of Ng compounds, stability remains relatively consistent. Analyzing the bonding scenario entails utilizing a comprehensive theoretical toolkit, which includes NBO, AIM, EDA, and AdNDP analyses. In NgAB systems, the extent of interaction between the donor and the acceptor is contingent on the polarizing ability of the A center, potentially leading to covalent bonds, especially for heavier Ng elements. Within ANgB complexes, Ng often shares electrons to form covalent bonds with A while engaging in electrostatic interactions with B, portraying itself as [ANg]+B−. Uncommon instances, exemplified by NCNgNSi, exhibit bonds on either side of Ng as electron-shared covalent bonds. Cage hosts serve as excellent entities to explore the limits of achieving Ng–Ng bonding, even for helium, under the condition of high pressure. The constraining influence exerted by the small B12N12 cage can induce some degree of covalency between two He atoms in He2@B12N12. Our investigations into BN-doped nanotubes and fluxional borospherene contribute compelling chapters to the ongoing narrative of noble gas interactions. These studies not only unveil the nuanced interplay between host electronics and Ng atom stabilization, but also illuminate how confinement induces intriguing alterations in behaviour and reactivity. As we delve deeper into this captivating realm, exciting possibilities for manipulating and harnessing the unique properties of noble gas systems beckon on the horizon.Conflicts of interest
The authors declare that they have no conflict of interest regarding the publication of this article, financial, and/or otherwise.Acknowledgements
P. K. C. thanks Professor Michael Rowan for kindly inviting him to contribute an article to the PCCP 25th Anniversary Issue. He also thanks DST, New Delhi for the J. C. Bose National Fellowship, grant number SR/S2/JCB-09/2009. RP thanks CSIR for her fellowship.References
- L. R. Arny, The Search for Data in the Physical and Chemical Sciences, Special Libraries Association, Alexandria, VA, USA, 1984 Search PubMed.
- W. Kossel, Über Molekülbildung als Frage des Atombaus, Ann. Phys., 1916, 354, 229–362 CrossRef.
- L. Pauling, The formulas of antimonic acid and the antimonates, J. Am. Chem. Soc., 1933, 55, 1895–1900 CrossRef CAS.
- N. Bartlett and D. Lohmann, 1005. Fluorides of the noble metals. Part II. Dioxygenyl hexafluoroplatinate(V), O2+[PtF6]-, J. Am. Chem. Soc., 1962, 5253–5261 RSC.
- N. Bartlett, Xenon hexafluoroplatinate(V) Xe+[PtF6]-, Proc. Chem. Soc. Lond., 1962, 218 CAS.
- L. Graham, O. Graudejus, N. K. Jha and N. Bartlett, Concerning the nature of XePtF6, Coord. Chem. Rev., 2000, 197, 321–334 CrossRef CAS.
- R. Craciun, D. Picone, R. T. Long, S. Li, D. A. Dixon, K. A. Peterson and K. O. Christe, Third row transition metal hexafluorides, extraordinary oxidizers, and Lewis acids: Electron affnities, fluoride affnities, and heats of formation of WF6, ReF6, OsF6, IrF6, PtF6, and AuF6, Inorg. Chem., 2010, 49, 1056–1070 CrossRef CAS PubMed.
- K. O. Christe, Bartlett's discovery of noble gas fluorides, a milestone in chemical history, Chem. Commun., 2013, 49, 4588–4590 RSC.
- A. Streng, A. Kirshenbaum, L. Streng and A. Grosse, Preparation of Rare-Gas Fluorides and Oxyfluorides by the Electric Discharge Method and their Properties, in Noble Gas Compounds, ed. H. H. Hyman, The University of Chicago Press, Chicago, IL, USA, 1963, pp. 73–80 Search PubMed.
- J. F. Lehmann, H. P. Mercier and G. J. Schrobilgen, The chemistry of krypton, Coord. Chem. Rev., 2002, 233, 1–39 CrossRef.
- H. H. Claassen, H. Selig and J. G. Malm, Xenon tetrafluoride, J. Am. Chem. Soc., 1962, 84, 3593 CrossRef CAS.
- J. Slivnik, B. Brcic, B. Volavsek, J. Marsel, V. Vrscaj, A. Smalc, B. Frlec and Z. Zemljic, Über die Synthese von XeF6, Croat. Chem. Acta, 1962, 34, 253 CAS.
- J. Turner and G. C. Pimentel, Krypton fluoride: Preparation by the matrix isolation technique, Science, 1963, 140, 974–975 CrossRef CAS PubMed.
- L. Y. Nelson and G. C. Pimentel, Infrared detection of xenon dichloride, Inorg. Chem., 1967, 6, 1758–1759 CrossRef CAS.
- N. Bartlett and M. Wechsberg, The Xenon Difluoride Complexes XeF2·XeOF4; XeF2·XeF6·AsF5 and XeF2·2 XeF6·2AsF5 and Their Relevance to Bond Polarity and Fluoride Ion Donor Ability of XeF2 and XeF6, Z. Anorg. Allg. Chem., 1951, 455, 5–17 Search PubMed.
- J. H. Holloway and E. G. Hope, Recent advances in noble-gas chemistry, Adv. Inorg. Chem., 1998, 46, 51–100 CrossRef CAS.
- L. Stein, Ionic radon solutions, Science, 1970, 168, 362–364 CrossRef CAS PubMed.
- L. Khriachtchev, M. Pettersson, N. Runeberg, J. Lundell and M. Räsänen, A stable argon compound, Nature, 2000, 406, 874 CrossRef CAS PubMed.
- G. Frenking, Another noble gas conquered, Nature, 2000, 406, 836 CrossRef CAS PubMed.
- Q. Wang and X. Wang, Infrared Spectra of NgBeS (Ng = Ne, Ar, Kr, Xe) and BeS2 in Noble-Gas Matrices, J. Phys. Chem. A, 2013, 117, 1508–1513 CrossRef CAS PubMed.
- Q. Zhang, M. Chen, M. Zhou, D. M. Andrada and G. Frenking, Experimental and Theoretical Studies of the Infrared Spectra and Bonding Properties of NgBeCO3 and a Comparison with NgBeO (Ng = He, Ne, Ar, Kr, Xe), J. Phys. Chem. A, 2014, 119, 2543–2552 CrossRef PubMed.
- W. Yu, X. Liu, B. Xu, X. Xing and X. Wang, Infrared Spectra of Novel NgBeSO2 Complexes (Ng = Ne, Ar, Kr, Xe) in Low Temperature Matrixes, J. Phys. Chem. A, 2016, 120, 8590–8598 CrossRef CAS PubMed.
- Q. Zhang, W. L. Li, L. Zhao, M. Chen, M. Zhou, J. Li and G. Frenking, A Very Short Be-Be Distance but No Bond: Synthesis and Bonding Analysis of Ng-Be2O2-Ng′ (Ng, Ng′ = Ne, Ar, Kr, Xe), Chem. – Eur. J., 2017, 23, 2035–2039 CrossRef CAS PubMed.
- T. R. Hogness and E. G. Lunn, The ionization of hydrogen by electron impact as interpreted by positive ray analysis, Phys. Rev., 1925, 26, 44–55 CrossRef CAS.
- X. Dong, A. R. Oganov, A. F. Goncharov, E. Stavrou, S. Lobanov, G. Saleh, G.-R. Qian, Q. Zhu, C. Gatti and V. L. Deringer, et al., A stable compound of helium and sodium at high pressure, Nat. Chem., 2017, 9, 440–445 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- B. Yin, G. Wang, N. Sa and Y. Huang, Bonding analysis and stability on alternant B16N16 cage and its dimers, J. Mol. Model., 2008, 14, 789–795 CrossRef CAS PubMed.
- B. Yin, Y. Huang, G. Wang and Y. Wang, Combined DFT and NBO study on the electronic basis of Si··· N-β-donor bond, J. Mol. Model., 2010, 16, 437–446 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. F. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- K. Morokuma, Molecular Orbital Studies of Hydrogen Bonds. III. C = O···H–O Hydrogen Bond in H2CO···H2O and H2CO···2H2O, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, A combined charge and energy decomposition scheme for bond analysis, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- E. Baerends, T. Ziegler, J. Autschbach, D. Bashford, A. Berces, F. Bickelhaupt, C. Bo, P. Boerrigter, L. Cavallo and D. Chong, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2013 Search PubMed.
- T. Thonhauser, V. R. Cooper, S. Li, A. Puzder, P. Hyldgaard and D. C. Langreth, van der Waals density functional: Self-consistent potential and the nature of the van der Waals bond, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 125112 CrossRef.
- T. Clark, Halogen bonds and σ-holes, Faraday Discuss., 2017, 203, 9–27 RSC.
- T. Clark, J. S. Murray and P. Politzer, A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions, Phys. Chem. Chem. Phys., 2018, 20, 30076–30082 RSC.
- P. Politzer and J. S. Murray, The neglected nuclei, Molecules, 2021, 26, 2982 CrossRef CAS PubMed.
- F. Pauzat and Y. Ellinger, H3+ as a trap for noble gases: 1—The case of Argon, Planet. Space Sci., 2005, 53(13), 1389–1399 CrossRef CAS.
- F. Pauzat and Y. Ellinger, H3+ as a trap for noble gases-2: structure and energetics of XH3+ complexes from X = neon to xenon, J. Chem. Phys., 2007, 127, 014308 CrossRef CAS PubMed.
- F. Pauzat, Y. Ellinger, J. Pilmé and O. Mousis, H3+ as a trap for noble gases-3: Multiple trapping of neon, argon, and krypton in XnH3+ (n = 1–3), J. Chem. Phys., 2009, 130, 174313 CrossRef CAS PubMed.
- A. Chakraborty, S. Giri and P. K. Chattaraj, Trapping of noble gases (He–Kr) by the aromatic H3+ and Li3+ species: a conceptual DFT approach, New J. Chem., 2010, 34, 1936–1945 RSC.
- G. Jana, S. Pan, P. L. Rodríguez-Kessler, G. Merino and P. K. Chattaraj, Adsorption of Molecular Hydrogen on Lithium-Phosphorus Double-Helices, J. Phys. Chem. C, 2018, 122, 27941–27946 CrossRef CAS.
- S. Pan, S. Giri and P. K. Chattaraj, A Computational study on the hydrogen adsorption capacity of various lithium-doped boron hydrides, J. Comput. Chem, 2012, 33, 425–434 CrossRef CAS PubMed.
- S. Pan, S. Banerjee and P. K. Chattaraj, Role of Lithium Decoration on Hydrogen Storage, J. Mex. Chem. Soc., 2012, 56, 229–240 CAS.
- S. Pan, G. Merino and P. K. Chattaraj, Hydrogen Trapping Potential of Some Li-doped Star-like Clusters and Super-alkali Systems, Phys. Chem. Chem. Phys., 2012, 14, 10345–10350 RSC.
- R. Saha, S. Pan and P. K. Chattaraj, Hydrogen storage in all-metal and non-metal aromatic clusters, in Emerging Materials for Energy Conversion and Storage, ed. K. Y. Cheong, G. Impellizzeri and M. A. Fraga, Elsevier: Amsterdam, The Netherlands, 2018, pp. 329–362 Search PubMed.
- N. Perez-Peralta, M. Contreras, W. Tiznado, J. Stewart, K. J. Donald and G. Merino, Stabilizing carbon-lithium stars, Phys. Chem. Chem. Phys., 2011, 13, 12975–12980 RSC.
- J. Tong, Y. Li, D. Wu, Z.-R. Li and X.-R. Huang, Ab Initio Investigation on a New Class of Binuclear Superalkali Cations M2Li2k+1+ (F2Li3+, O2Li5+, N2Li7+, and C2Li9+), J. Phys. Chem. A, 2011, 115, 2041–2046 CrossRef CAS PubMed.
- S. Pan, M. Contreras, J. Romero, A. Reyes, P. K. Chattaraj and G. Merino, C5Li7+ and O2Li5+ as Noble-Gas-Trapping Agents, Chem. – Eur. J., 2013, 19, 2322–2329 CrossRef CAS PubMed.
- S. Pan, S. Jalife, J. Romero, A. Reyes, G. Merino and P. K. Chattaraj, Attractive Xe-Li Interaction in Li-Decorated Clusters, Comput. Theor. Chem., 2013, 1021, 62–69 CrossRef CAS.
- S. Pan, R. Saha, A. Gupta and P. K. Chattaraj, Modeling of 1-D Nanowires and analyzing their Hydrogen and Noble Gas Binding Ability, J. Chem. Sci., 2017, 129, 849–858 CrossRef CAS.
- G. Frenking, W. Koch, J. Gauss and D. Cremer, Stabilities and nature of the attractive interactions in HeBeO, NeBeO, and ArBeO and a comparison with analogs NgLiF, NgBN, and NgLiH (Ng = He, Ar). A theoretical investigation, J. Am. Chem. Soc., 1988, 110, 8007–8016 CrossRef CAS.
- C. A. Thompson and L. Andrews, Noble gas complexes with BeO: infrared spectra of Ng-BeO (Ng = Ar, Kr, Xe), J. Am. Chem. Soc., 1994, 116, 423–424 CrossRef CAS.
- S. Pan, D. Moreno, J. L. Cabellos, G. Merino and P. K. Chattaraj, Ab initio study on the stability of NgnBe2N2, NgnBe3N2 and NgBeSiN2 Clusters, Chem. Phys. Chem., 2014, 15, 2618–2625 CrossRef CAS PubMed.
- S. Borocci, N. Bronzolino and F. Grandinetti, From OBeHe to H3BOBeHe: Enhancing the stability of a neutral helium compound, Chem. Phys. Lett., 2005, 406, 179–183 CrossRef CAS.
- S. Pan, D. Moreno, J. L. Cabellos, J. Romero, A. Reyes, G. Merino and P. K. Chattaraj, In quest of strong Be–Ng bonds among the neutral Ng–Be complexes, J. Phys. Chem. A, 2013, 118, 487–494 CrossRef PubMed.
- R. Saha, S. Pan, G. Merino and P. K. Chattaraj, Comparative Study on the Noble-Gas Binding Ability of BeX Clusters (X = SO4, CO3, O), J. Phys. Chem. A, 2015, 119, 6746–6752 CrossRef CAS PubMed.
- S. Pan, M. Ghara, S. Ghosh and P. K. Chattaraj, Noble gas bound beryllium chromate and beryllium hydrogen phosphate: a comparison with noble gas bound beryllium oxide, RSC Adv., 2016, 6, 92786–92794 RSC.
- S. Pan, R. Saha and P. K. Chattaraj, On the stability of noble gas bound 1-tris (pyrazolyl) borate beryllium and magnesium complexes, New J. Chem., 2015, 39, 6778–6786 RSC.
- R. Saha, S. Pan and P. K. Chattaraj, NgMCp+: Noble Gas Bound Half-Sandwich Complexes (Ng= He–Rn, M= Be–Ba, and Cp = η5-C5H5), J. Phys. Chem. A, 2017, 121, 3526–3539 CrossRef CAS PubMed.
- S. Pan, D. Moreno, G. Merino and P. K. Chattaraj, Stability of Noble-Gas-Bound SiH3+ Clusters, Chem. Phys. Chem., 2014, 15, 3554–3564 CrossRef CAS PubMed.
- S. Pan, D. Moreno, S. Ghosh and P. K. Chattaraj, G. Merino. Structure and stability of noble gas bound compounds (E = C, Ge, Sn, Pb; X= H, F, Cl, Br), J. Comput. Chem., 2016, 37, 226–236 CrossRef CAS PubMed.
- M. Ghara, S. Pan, A. Kumar, G. Merino and P. K. Chattaraj, Structure, Stability, and Nature of Bonding in Carbon Monoxide bound EX3+ Complexes (E = Group 14 element; X = H, F, Cl, Br, I), J. Comput. Chem., 2016, 37, 2202–2211 CrossRef CAS PubMed.
- D. Chakraborty and P. K. Chattaraj, In quest of a superhalogen supported covalent bond involving a noble gas atom, J. Phys. Chem. A, 2015, 119, 3064–3074 CrossRef CAS PubMed.
- S. Pan, R. Saha, S. Mandal and P. K. Chattaraj, π-Aromatic cyclic M3+ (M = Cu, Ag, Au) clusters and their complexation with dimethyl imidazol-2-ylidene, pyridine, isoxazole, furan, noble gases and carbon monoxide, Phys. Chem. Chem. Phys., 2016, 18, 11661–11676 RSC.
- M. Ghara, S. Pan, J. Deb, A. Kumar, U. Sarkar and P. K. Chattaraj, A computational study on structure, stability and bonding in Noble Gas bound metal Nitrates, Sulfates and Carbonates (Metal = Cu, Ag, Au), J. Chem. Sci., 2016, 128, 1537–1548 CrossRef CAS.
- S. Pan, R. Saha, A. Gupta, G. Merino and P. K. Chattaraj, A coupled-cluster study on the noble gas binding ability of metal cyanides versus metal halides (metal = Cu, Ag, Au), J. Comput. Chem., 2015, 36, 2168–2176 CrossRef CAS PubMed.
- S. Pan, R. Saha, A. Kumar, A. Gupta, G. Merino and P. K. Chattaraj, A noble interaction: An assessment of noble gas binding ability of metal oxides (metal = Cu, Ag, Au), Int. J. Quantum Chem., 2016, 116, 1016–1024 CrossRef CAS.
- G. Jana, S. Pan and P. K. Chattaraj, Binding of Small Gas Molecules by Metal–Bipyridyl Monocationic Complexes (Metal = Cu, Ag, Au) and Possible Bond Activations Therein, J. Phys. Chem. A, 2017, 121, 3803–3817 CrossRef CAS PubMed.
- G. Jana, R. Saha, S. Pan, A. Kumar, G. Merino and P. K. Chattaraj, Noble Gas Binding Ability of Metal-Bipyridine Monocationic Complexes (Metal = Cu, Ag, Au): A Computational Study, ChemistrySelect, 2016, 1, 5842–5849 CrossRef CAS.
- R. Pal and P. K. Chattaraj, On the Nature of the Partial Covalent Bond between Noble Gas Elements and Noble Metal Atoms, Molecules, 2023, 28, 3253 CrossRef CAS PubMed.
- N. Perez-Peralta, R. Juarez, E. Cerpa, F. M. Bickelhaupt and G. Merino, Bonding of Xenon Hydrides, J. Phys. Chem. A, 2009, 113, 9700–9706 CrossRef CAS PubMed.
- R. Juarez, C. Zavala-Oseguera, J. O. C. Jimenez-Halla, F. M. Bickelhaupt and G. Merino, Radon hydrides: structure and bonding, Phys. Chem. Chem. Phys., 2011, 13, 2222–2227 RSC.
- S. Pan, R. Saha and P. Chattaraj, Exploring the nature of silicon-noble gas bonds in H3SiNgNSi and HSiNgNSi compounds (Ng = Xe, Rn), Int. J. Mol. Sci., 2015, 16, 6402–6418 CrossRef CAS PubMed.
- A. Michalak, M. Mitoraj and T. Ziegler, Bond orbitals from chemical valence theory, J. Phys. Chem. A, 2008, 112, 1933–1939 CrossRef CAS PubMed.
- M. P. Mitoraj, A. Michalak and T. Ziegler, A combined charge and energy decomposition scheme for bond analysis, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- S. Pan, A. Gupta, S. Mandal, D. Moreno, G. Merino and P. K. Chattaraj, Metastable behavior of noble gas inserted tin and lead fluorides, Phys. Chem. Chem. Phys., 2015, 17, 972–982 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO6.0: Natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- R. F. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- D. Samanta, Prediction of superhalogen-stabilized noble gas compounds, J. Phys. Chem. Lett., 2014, 5, 3151–3156 CrossRef CAS PubMed.
- R. Saha, B. Mandal and P. K. Chattaraj, HNgBeF3 (Ng= Ar-Rn): Superhalogen-supported noble gas insertion compounds, Int. J. Quantum Chem., 2018, 118, e25499 CrossRef.
- S. Pan, G. Jana, E. Ravell, X. Zarate, E. Osorio, G. Merino and P. K. Chattaraj, Stable NCNgNSi (Ng = Kr, Xe, Rn) Compounds with Covalently Bound C-Ng-N Unit: Possible Isomerization of NCNSi through the Release of the Noble Gas Atom, Chem. – Eur. J., 2018, 24, 2879–2887 CrossRef CAS PubMed.
- G. Jana, R. Pal and P. K. Chattaraj, XNgNSi (X= HCC, F; Ng= Kr, Xe, Rn): A new class of metastable insertion compounds containing Ng–C/F and Ng–N bonds and possible isomerization therein, J. Phys. Chem. A, 2021, 125, 10514–10523 CrossRef CAS PubMed.
- R. Pal, G. Jana and P. K. Chattaraj, Structure and stability of a new set of noble gas insertion compounds, XNgOPO(OH)2 (X = F, Cl, Br; Ng = Kr, Xe, Rn): an in silico investigation, Theor. Chem. Acc., 2023, 142, 34 Search PubMed.
- R. Saha, G. Jana, S. Pan, G. Merino and P. K. Chattaraj, How Far Can One Push the Noble Gases Towards Bonding?: A Personal Account, Molecules, 2019, 24, 2933–2955 Search PubMed.
- A. Krapp and G. Frenking, Is this a chemical bond? A theoretical study of Ng2@C60 (Ng= He, Ne, Ar, Kr, Xe), Chem. – Eur. J., 2007, 13, 8256–8270 CrossRef CAS PubMed.
- M. Khatua, S. Pan and P. K. Chattaraj, Movement of Ng2 molecules confined in a C60 cage: an ab initio molecular dynamics study, Chem. Phys. Lett., 2014, 610, 351–356 CrossRef.
- H. W. Kroto, The Stability of the Fullerenes Cn, with n = 24, 28, 32, 25, 36, 50, 60 and 70, Nature, 1987, 329, 529–531 CrossRef CAS.
- S. Jalife, S. Mondal, J. L. Cabellos, S. Pan, M. A. Méndez-Rojas, I. Fernández, G. Frenking and G. Merino, Breaking the isolated pentagon rule by encapsulating Xe2 in C60: The guest defines the shape of the host, ChemistrySelect, 2016, 1, 2405–2408 CrossRef CAS.
- E. Cerpa, A. Krapp, R. Flores-Moreno, K. J. Donald and G. Merino, Influence of endohedral confinement on the electronic interaction between He atoms: A He2@C20H20 case study, Chem. – Eur. J., 2009, 15, 1985–1990 CrossRef CAS PubMed.
- E. Cerpa, A. Krapp, A. Vela and G. Merino, The implications of symmetry of the external potential on bond paths, Chem. – Eur. J., 2008, 14, 10232–10234 CrossRef CAS PubMed.
- M. Khatua, S. Pan and P. K. Chattaraj, Confinement induced binding of noble gas atoms, J. Chem. Phys., 2014, 140, 164306 CrossRef PubMed.
- S. Mondal and P. K. Chattaraj, Noble gas encapsulation: clathrate hydrates and their HF doped analogues, Phys. Chem. Chem. Phys., 2014, 16, 17943–17954 RSC.
- S. Pan, G. Jana, A. Gupta, G. Merino and P. K. Chattaraj, Endohedral Gas Adsorption by Cucurbit[7]uril: A Theoretical Study, Phys. Chem. Chem. Phys., 2017, 19, 24448–24452 RSC.
- S. Pan, R. Saha, S. Mandal, S. Mondal, A. Gupta, H. M. Fernández, G. Merino and P. K. Chattaraj, Selectivity in Gas Adsorption by Molecular Cucurbit[6]uril, J. Phys. Chem. C, 2016, 120, 13911–13921 CrossRef CAS.
- S. Pan, S. Mondal and P. K. Chattaraj, Cucurbiturils as Promising Hydrogen Storage Materials: A Case Study of Cucurbit[7]uril, New J. Chem., 2013, 37, 2492–2499 RSC.
- D. Chakraborty, S. Pan and P. K. Chattaraj, Encapsulation of small gas molecules and rare gas atoms inside the octa acid cavitand, Theor. Chem. Acc., 2016, 135, 119 Search PubMed.
- D. Chakraborty and P. K. Chattaraj, Confinement induced binding in noble gas atoms within a BN-doped carbon nanotube, Chem. Phys. Lett., 2015, 621, 29–34 CrossRef CAS.
- S. Pan, M. Ghara, S. Kar, X. Zarate, G. Merino and P. K. Chattaraj, Noble gas encapsulated B40 cage, Phys. Chem. Chem. Phys., 2018, 20, 1953–1963 RSC.
- R. Pal and P. K. Chattaraj, Can the Fluxionality in Borospherene Influence the Confinement-Induced Bonding between Two Noble Gas Atoms?, Molecules, 2022, 27, 8683 CrossRef CAS PubMed.
- Z. Hua-Jin, G. Jin-Chang, F. Lin-Yan, W. Ying-Jin, S. Jalife, A. Vásquez-Espinal, J. L. Cabellos, S. Pan and G. Merino, Triple Coaxial-layered versus helical Be6B11− cluster: dual structural fluxionality and multifold aromaticity, Angew. Chem., Int. Ed., 2017, 129, 10308–10311 CrossRef.
- R. Saha, S. Kar, S. Pan, G. Martínez-Guajardo, G. Merino and P. K. Chattaraj, A spinning umbrella: carbon monoxide and dinitrogen bound MB12− clusters (M = Co, Rh, Ir), J. Phys. Chem. A, 2017, 121, 2971–2979 CrossRef CAS PubMed.
- S. Jalife, L. Liu, S. Pan, J. L. Cabellos, E. Osorio, C. Lu, T. Heine, K. J. Donald and G. Merino, Dynamical behavior of boron clusters, Nanoscale, 2016, 8, 17639–17644 RSC.
- L. Liu, D. Moreno, E. Osorio, A. C. Castro, S. Pan, P. K. Chattaraj, T. Heine and G. Merino, Structure and bonding of IrB12-: Converting a rigid boron B12 platelet to a Wankel motor, RSC Adv., 2016, 6, 27177–27182 RSC.
- D. Moreno, S. Pan, G. Martínez-Guajardo, L. Lei-Zeonjuk, R. Islas, E. Osorio, P. K. Chattaraj, T. Heine and G. Merino, B182-: A quasi-planar bowl member of the Wankel motor family, Chem. Commun., 2014, 50, 8140–8143 RSC.
- G. Martinez-Guajardo, J. L. Cabellos, A. Diaz-Celaya, S. Pan, R. Islas, P. K. Chattaraj, T. Heine and G. Merino, Dynamical behavior of borospherene: A nanobubble, Sci. Rep., 2015, 5, 11287 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2024 |